
FENYLOKETONURIA
FENYLOKETONURIA
należy do monogenowych wrodzonych błędów
należy do monogenowych wrodzonych błędów
metabolizmu dziedziczących się w sposób AR
metabolizmu dziedziczących się w sposób AR
częstość występowania w Polsce – 1:7000-8000 urodzeń
częstość występowania w Polsce – 1:7000-8000 urodzeń
(co 46 osoba jest nosicielem zmutowanego genu PAH)
(co 46 osoba jest nosicielem zmutowanego genu PAH)
przyczyną – brak lub częściowy deficyt hydroksylazy
przyczyną – brak lub częściowy deficyt hydroksylazy
fenyloalaniny
fenyloalaniny
gen hydroksylazy fenyloalaniny – 12q22-24.1
gen hydroksylazy fenyloalaniny – 12q22-24.1
zidentyfikowano ponad 400 mutacji genu PAH
zidentyfikowano ponad 400 mutacji genu PAH
główny szlak przemiany fenyloalaniny – hydroksylacja
główny szlak przemiany fenyloalaniny – hydroksylacja
do tyrozyny w obecności hydroksylazy fenyloalaniny,
do tyrozyny w obecności hydroksylazy fenyloalaniny,
molekularnego tlenu i tetrahydrobiopteryny (BH4)
molekularnego tlenu i tetrahydrobiopteryny (BH4)
duże stężenie fenyloalaniny jest przyczyną uszkodzenia
duże stężenie fenyloalaniny jest przyczyną uszkodzenia
oun
oun
wczesne i systematyczne leczenie dietą
wczesne i systematyczne leczenie dietą
niskofenyloalaninową zapewnia prawidłowy rozwój
niskofenyloalaninową zapewnia prawidłowy rozwój
fizyczny i umysłowy pacjentów
fizyczny i umysłowy pacjentów

KLASYCZNA POSTAĆ
KLASYCZNA POSTAĆ
FENYLOKETONURII:
FENYLOKETONURII:
dziecko rodzi się pozornie zdrowe, w badaniu
dziecko rodzi się pozornie zdrowe, w badaniu
klinicznym nie stwierdza się nieprawidłowości
klinicznym nie stwierdza się nieprawidłowości
bezpośrednio po urodzeniu stężenie fenyloalaniny
bezpośrednio po urodzeniu stężenie fenyloalaniny
we krwi jest prawidłowe wyrównane w okresie
we krwi jest prawidłowe wyrównane w okresie
płodowym przez enzym matki)
płodowym przez enzym matki)
Objawy wczesne
Objawy wczesne
– niecharakterystyczne:
– niecharakterystyczne:
•
nawracające uporczywe wymioty
nawracające uporczywe wymioty
•
zmiany skórne – od tendencji do suchości i
zmiany skórne – od tendencji do suchości i
nadwrażliwości skóry do rozległych zmian
nadwrażliwości skóry do rozległych zmian
przypominających wyprysk alergiczny lub
przypominających wyprysk alergiczny lub
łojotokowy
łojotokowy
•
„
„
rozcieńczenie” barwnika – jasna karnacja,
rozcieńczenie” barwnika – jasna karnacja,
niebieskie tęczówki, jasne włosy
niebieskie tęczówki, jasne włosy
•
ok. 2 m-ca życia – „mysi zapach” – spowodowany
ok. 2 m-ca życia – „mysi zapach” – spowodowany
wydzielaniem kwasu ortohydroksyfenylooctowego
wydzielaniem kwasu ortohydroksyfenylooctowego

KLASYCZNA POSTAĆ
KLASYCZNA POSTAĆ
FENYLOKETONURII:
FENYLOKETONURII:
Opóźnienie rozwoju psychoruchowego
Opóźnienie rozwoju psychoruchowego
:
:
•
narasta stopniowo i w różnym tempie u
narasta stopniowo i w różnym tempie u
rożnych chorych
rożnych chorych
•
wczesny objaw – opóźnienie rozwoju
wczesny objaw – opóźnienie rozwoju
ruchowego
ruchowego
•
u nieleczonych dzieci starszych dominuje
u nieleczonych dzieci starszych dominuje
upośledzenie umysłowe w stopniu znacznym
upośledzenie umysłowe w stopniu znacznym
•
IQ 20-40
IQ 20-40
•
nie występują uszkodzenia innych narządówi
nie występują uszkodzenia innych narządówi
układów poza oun
układów poza oun
•
okres przeżycia jest takiego samego rzędu co
okres przeżycia jest takiego samego rzędu co
u osób zdrowych
u osób zdrowych

KLASYCZNA POSTAĆ
KLASYCZNA POSTAĆ
FENYLOKETONURII:
FENYLOKETONURII:
Objawy neurologiczne
Objawy neurologiczne
– niecharakterystyczne:
– niecharakterystyczne:
•
małogłowie – stała cecha (68-94%)
małogłowie – stała cecha (68-94%)
•
hipotonia mięśniowa
hipotonia mięśniowa
•
wygórowanie odruchów głębokich
wygórowanie odruchów głębokich
•
u starszych nieleczonych dzieci – stereotypie
u starszych nieleczonych dzieci – stereotypie
ruchowe, zespoły spastyczne, chód
ruchowe, zespoły spastyczne, chód
atetotyczny
atetotyczny
•
drgawki – u 25% przed ukończeniem 1 rż
drgawki – u 25% przed ukończeniem 1 rż
•
nieprawidłowy zapis EEG
nieprawidłowy zapis EEG
•
zmiany charakterologiczne
zmiany charakterologiczne
•
zaburzenia emocjonalne – nadpobudliwość,
zaburzenia emocjonalne – nadpobudliwość,
skłonność do agresji, stany lękowe, psychozy
skłonność do agresji, stany lękowe, psychozy

BADANIA PRZESIEWOWE
BADANIA PRZESIEWOWE
NOWORODKÓW:
NOWORODKÓW:
od 1964 – badania przesiewowe w IMiD
od 1964 – badania przesiewowe w IMiD
od 1994 wszystkie noworodki w Polsce objęte są
od 1994 wszystkie noworodki w Polsce objęte są
obligatoryjnie testami przesiewowymi w
obligatoryjnie testami przesiewowymi w
kierunku fenyloketonurii i hipotyreozy
kierunku fenyloketonurii i hipotyreozy
test Guthriego lub test kalorymetryczny–
test Guthriego lub test kalorymetryczny–
oznaczenie stężenia fenyloalaniny we krwi
oznaczenie stężenia fenyloalaniny we krwi
włościczkowej pobranej w 4-7 dobie życia z
włościczkowej pobranej w 4-7 dobie życia z
nakłucia pięty
nakłucia pięty
noworodki z dodatnim testem przesiewowym
noworodki z dodatnim testem przesiewowym
wzywane są do specjalistycznych klinik, gdzie
wzywane są do specjalistycznych klinik, gdzie
wykonuje się oznaczenie fenyloalaniny i tyrozyny
wykonuje się oznaczenie fenyloalaniny i tyrozyny
duże stężenie fenyloalaniny – diagnostyka
duże stężenie fenyloalaniny – diagnostyka
różnicowa hiperfenyloalaninemii i badanie
różnicowa hiperfenyloalaninemii i badanie
genetyczne
genetyczne

CHOROBA HUNTINGTONA
CHOROBA HUNTINGTONA
postępująca choroba
postępująca choroba
neurodegeneracyjna uwarunkowana
neurodegeneracyjna uwarunkowana
genetycznie
genetycznie
dziedziczenie AD
dziedziczenie AD
częstość występowania w krajach
częstość występowania w krajach
europejskich 4-8/100 000
europejskich 4-8/100 000
pierwsze objawy – 4 dekada życia
pierwsze objawy – 4 dekada życia
typowy obraz kliniczny – ruchy
typowy obraz kliniczny – ruchy
mimowolne o charakterze pląsawiczym i
mimowolne o charakterze pląsawiczym i
postępujący zespół psychoorganiczny
postępujący zespół psychoorganiczny

GENETYKA:
GENETYKA:
gen na chromosomie 4
gen na chromosomie 4
(4p16.3)
(4p16.3)
charakteryzuje się zwielokrotnieniem replikacji tripletu
charakteryzuje się zwielokrotnieniem replikacji tripletu
CAG
CAG
w warunach prawidłowych – 11-34 powtórzeń
w warunach prawidłowych – 11-34 powtórzeń
choroba Huntingtona > 37 powtórzeń
choroba Huntingtona > 37 powtórzeń
grupa pośrednia – 35-37 powtórzeń – duże ryzyko
grupa pośrednia – 35-37 powtórzeń – duże ryzyko
wystąpienia choroby u potomstwa
wystąpienia choroby u potomstwa
zdarzają się mutacje
zdarzają się mutacje
de novo
de novo
zjawisko antycypacji – w kolejnych pokoleniach objawy
zjawisko antycypacji – w kolejnych pokoleniach objawy
występują wcześniej i są siniej wyrażone
występują wcześniej i są siniej wyrażone
jeżeli mutacja jest przekazywana przez matkę - liczba
jeżeli mutacja jest przekazywana przez matkę - liczba
powtórzeń CAG nie ulega większym zmianom
powtórzeń CAG nie ulega większym zmianom
jeżeli mutacja jest przekazywana przez ojca - dochodzi
jeżeli mutacja jest przekazywana przez ojca - dochodzi
do zwiększenia liczby powtórzeń tej sekwencji
do zwiększenia liczby powtórzeń tej sekwencji

GENETYKA:
GENETYKA:
prawidłowy gen koduje huntingtynę
prawidłowy gen koduje huntingtynę
w wyniku błędu genetycznego dochodzi do
w wyniku błędu genetycznego dochodzi do
zwiększenia ilości poliglutaminy, co
zwiększenia ilości poliglutaminy, co
prowadzi do agregacji huntingtyny w
prowadzi do agregacji huntingtyny w
cytoplazmie i jądrze komórkowym;
cytoplazmie i jądrze komórkowym;
następstwem – zanik komórek nerwowych
następstwem – zanik komórek nerwowych
proces chorobowy dotyczy głownie jądra
proces chorobowy dotyczy głownie jądra
ogoniastego, skorupy i kory mózgowej
ogoniastego, skorupy i kory mózgowej
zanik GABA-ergicznych wypustek,
zanik GABA-ergicznych wypustek,
działających hamująco na gałkę bladą jest
działających hamująco na gałkę bladą jest
odpowiedzialny za pląsawicze ruchy
odpowiedzialny za pląsawicze ruchy
mimowolne
mimowolne

OBJAWY KLINICZNE:
OBJAWY KLINICZNE:
Zaburzenia ruchowe
Zaburzenia ruchowe
szybkie, nieskoordynowane ruchy, które nakładają
szybkie, nieskoordynowane ruchy, które nakładają
się na ruchy dowolne lub występują w spoczynku
się na ruchy dowolne lub występują w spoczynku
emocje i ruchy czynne – nasilają ruchy pląsawicze
emocje i ruchy czynne – nasilają ruchy pląsawicze
zanikają we śnie
zanikają we śnie
dotyczą mięśni twarzy, języka, kończyn i tułowia
dotyczą mięśni twarzy, języka, kończyn i tułowia
ruchy pląsawicze nie zawsze są symetryczne
ruchy pląsawicze nie zawsze są symetryczne
dysfagia, dyzartria
dysfagia, dyzartria
zaburzenia gałkoruchowe
zaburzenia gałkoruchowe
zmniejszenie napięcia mięśniowego
zmniejszenie napięcia mięśniowego
niemożność utrzymania wyciągniętego języka na
niemożność utrzymania wyciągniętego języka na
brodzie – bardzo czuły test
brodzie – bardzo czuły test
zaburzenia chodu – chód taneczny
zaburzenia chodu – chód taneczny
w końcowym stadium choroby – sztywność
w końcowym stadium choroby – sztywność
pozapiramidowa
pozapiramidowa

OBJAWY KLINICZNE:
OBJAWY KLINICZNE:
Zaburzenia funkcji poznawczych:
Zaburzenia funkcji poznawczych:
zespół otępienny o cechach otępienia
zespół otępienny o cechach otępienia
podkorowego ze spowolnieniem
podkorowego ze spowolnieniem
myślowym przy braku korowych
myślowym przy braku korowych
objawów otępiennych
objawów otępiennych
ma charakter postępujący
ma charakter postępujący
w miarę progresji choroby – zaburzenia
w miarę progresji choroby – zaburzenia
pamięci, upośledzenie funkcji
pamięci, upośledzenie funkcji
intelektualnych i pogłębiająca się apatia
intelektualnych i pogłębiająca się apatia

OBJAWY KLINICZNE:
OBJAWY KLINICZNE:
Zaburzenia emocjonalne:
Zaburzenia emocjonalne:
drażliwość, wybuchowość i
drażliwość, wybuchowość i
nietrzymanie afektu
nietrzymanie afektu
objawy psychotyczne (niestałe)
objawy psychotyczne (niestałe)
depresja, skłonności samobójcze
depresja, skłonności samobójcze
zaburzenia cyklu snu i czuwania z
zaburzenia cyklu snu i czuwania z
nadmierną sennością w ciągu dnia i
nadmierną sennością w ciągu dnia i
nocną bezsennością
nocną bezsennością

PORADNITWO
PORADNITWO
GENETYCZNE:
GENETYCZNE:
1/3 mutacji de novo
1/3 mutacji de novo
późny początek
późny początek
informacje o chorobie
informacje o chorobie
badania molekularne
badania molekularne
badanie osoby chorej (identyfikacja mutacji)
badanie osoby chorej (identyfikacja mutacji)
badanie osoby podejrzanej – stwierdzenie tej samej
badanie osoby podejrzanej – stwierdzenie tej samej
mutacji
mutacji
diagnostyka prenatalna
diagnostyka prenatalna
– jedynym ośrodkiem
– jedynym ośrodkiem
– Instytut Psychiatrii i Neurologii w Warszawie
– Instytut Psychiatrii i Neurologii w Warszawie
przeprowadza się tylko wtedy, gdy jedno z rodziców
przeprowadza się tylko wtedy, gdy jedno z rodziców
choruje
choruje

LECZENIE – objawowe:
LECZENIE – objawowe:
neuroleptyki – haloperidol, pimozyd
neuroleptyki – haloperidol, pimozyd
sztywność pozapiramidowa –
sztywność pozapiramidowa –
lewodopa, agoniści receptora
lewodopa, agoniści receptora
dopaminergicznego
dopaminergicznego
próby przeszczepu tkanki płodowej
próby przeszczepu tkanki płodowej
pochodzącej z prążkowia chorym z
pochodzącej z prążkowia chorym z
pląsawicą Huntingtona
pląsawicą Huntingtona

ZESPÓŁ RETTA
ZESPÓŁ RETTA
- jest postępującym schorzeniem
- jest postępującym schorzeniem
neurologicznym, przebiegającym z
neurologicznym, przebiegającym z
ataksją, zachowaniami
ataksją, zachowaniami
autystycznymi, stereotypiami rąk,
autystycznymi, stereotypiami rąk,
utratą mowy, napadami
utratą mowy, napadami
drgawkowymi i deceleracją wzrostu
drgawkowymi i deceleracją wzrostu
obwodu głowy
obwodu głowy

ZESPÓŁ RETTA
ZESPÓŁ RETTA
I opis – Andreas Rett – 1966
I opis – Andreas Rett – 1966
1983 – Hagberg i wsp. – opisali 35
1983 – Hagberg i wsp. – opisali 35
dziewczynek z regresem w rozwoju
dziewczynek z regresem w rozwoju
psychomotorycznym, z
psychomotorycznym, z
charakterystycznymi stereotypiami,
charakterystycznymi stereotypiami,
utratą zdolności celowych ruchów
utratą zdolności celowych ruchów
rąk, ataksją i wtórnym małogłowiem
rąk, ataksją i wtórnym małogłowiem

ZESPÓŁ RETTA
ZESPÓŁ RETTA
dziedziczenie w sposób dominujący
dziedziczenie w sposób dominujący
sprzężony z chromosomem X
sprzężony z chromosomem X
częstość występowania 1:10 000-1:15 000
częstość występowania 1:10 000-1:15 000
stanowi jedną z najważniejszych po
stanowi jedną z najważniejszych po
zespole Downa przyczyn znacznego
zespole Downa przyczyn znacznego
upośledzenia umysłowego u płci żeńskiej
upośledzenia umysłowego u płci żeńskiej
odmienny tor rozwoju dziecka – po
odmienny tor rozwoju dziecka – po
okresie prawidłowego rozwoju do wieku
okresie prawidłowego rozwoju do wieku
7-18 m-cy występują pierwsze oznaki jego
7-18 m-cy występują pierwsze oznaki jego
zahamowania oraz zaburzenia ze strony
zahamowania oraz zaburzenia ze strony
układu nerwowego
układu nerwowego

Gen MECP2
Gen MECP2
zlokalizowany na Xq
zlokalizowany na Xq
gen regulatorowy – aktywuje lub hamuje
gen regulatorowy – aktywuje lub hamuje
czynność kolejnych genów
czynność kolejnych genów
produktem – białko MeCP2 – pomaga utrzymać
produktem – białko MeCP2 – pomaga utrzymać
ekspresję genów tkankowo swoistych, jego
ekspresję genów tkankowo swoistych, jego
ulega inaktywacji wraz z danym chromosomem
ulega inaktywacji wraz z danym chromosomem
rodzicielskim
rodzicielskim
99,5% mutacje de novo
99,5% mutacje de novo
nosicielstwo związane jest z całkowitą
nosicielstwo związane jest z całkowitą
inaktywacją chromosomu z mutacją MECP2 lub
inaktywacją chromosomu z mutacją MECP2 lub
mozaiką germinalną (podejrzewamy, gdy w
mozaiką germinalną (podejrzewamy, gdy w
rodzinie jest co najmniej 2 dzieci z tym samym
rodzinie jest co najmniej 2 dzieci z tym samym
schorzeniem, a wynik badania mutacji
schorzeniem, a wynik badania mutacji
rozpatrywanego genu pozostaje u rodziców
rozpatrywanego genu pozostaje u rodziców
negatywny)
negatywny)
mutacje punktowe
mutacje punktowe

CZTEROETAPOWY MODEL
CZTEROETAPOWY MODEL
ROZWOJU ZESPOŁU RETTA:
ROZWOJU ZESPOŁU RETTA:
Stadium I – 6-18m-ąc życia:
Stadium I – 6-18m-ąc życia:
wczesne zahamowanie rozwoju
wczesne zahamowanie rozwoju
obraz kliniczny – niespecyficzny
obraz kliniczny – niespecyficzny
możliwe jest nabywanie dodatkowych
możliwe jest nabywanie dodatkowych
umiejętności przez dziecko, jednak zawsze z
umiejętności przez dziecko, jednak zawsze z
pewnym opóźnieniem
pewnym opóźnieniem
dziewczynki rodzą się najczęściej z ciąży
dziewczynki rodzą się najczęściej z ciąży
niepowikłanej, rozwiązanej o czasie prawidłowym
niepowikłanej, rozwiązanej o czasie prawidłowym
porodem
porodem
nie zauważa się żadnych zaburzeń, choć rodzice
nie zauważa się żadnych zaburzeń, choć rodzice
retrospektywnie określają chore dziewczynki jako
retrospektywnie określają chore dziewczynki jako
spokojniejsze, niezbyt aktywne i nieco bardziej
spokojniejsze, niezbyt aktywne i nieco bardziej
wiotkie
wiotkie

CZTEROETAPOWY MODEL
CZTEROETAPOWY MODEL
ROZWOJU ZESPOŁU RETTA:
ROZWOJU ZESPOŁU RETTA:
Stadium II – 1-2 rok życia:
Stadium II – 1-2 rok życia:
charakteryzuje się nagłym, specyficznym regresem nabytych
charakteryzuje się nagłym, specyficznym regresem nabytych
zdolności, występującym jednak ze zmiennością osobniczą
zdolności, występującym jednak ze zmiennością osobniczą
u wielu okres ten jest wyraźnie odgraniczony gwałtownym
u wielu okres ten jest wyraźnie odgraniczony gwałtownym
wystąpieniem zaburzeń kontaktu emocjonalnego,
wystąpieniem zaburzeń kontaktu emocjonalnego,
komunikacji oraz utratą umiejętności celowego
komunikacji oraz utratą umiejętności celowego
wykorzystania rąk i regresem w zakresie mowy
wykorzystania rąk i regresem w zakresie mowy
Jednym z najwcześniej występujących zaburzeń jest utrata
Jednym z najwcześniej występujących zaburzeń jest utrata
zdolności zachowania równowagi i postawy
zdolności zachowania równowagi i postawy
antygrawitacyjnej
antygrawitacyjnej
utrata zainteresowania otoczeniem i kontaktu z rodzicami
utrata zainteresowania otoczeniem i kontaktu z rodzicami
szczególny regres dotyczy zręczności manipulacyjnej palców,
szczególny regres dotyczy zręczności manipulacyjnej palców,
zanika zdolność celowego używania rąk
zanika zdolność celowego używania rąk
stereotypowe ruchy rąk – początkowo wkładanie rąk do buzi,
stereotypowe ruchy rąk – początkowo wkładanie rąk do buzi,
ssanie palców, pociąganie języka, potem – typowe ruchy
ssanie palców, pociąganie języka, potem – typowe ruchy
„myjące”, zaciskanie, klaskanie, uderzanie ręką o rękę
„myjące”, zaciskanie, klaskanie, uderzanie ręką o rękę

CZTEROETAPOWY MODEL
CZTEROETAPOWY MODEL
ROZWOJU ZESPOŁU RETTA:
ROZWOJU ZESPOŁU RETTA:
Stadium III – 3-4 rok życia:
Stadium III – 3-4 rok życia:
rozpoczyna się, gdy dzieci, choć już z niewątpliwym
rozpoczyna się, gdy dzieci, choć już z niewątpliwym
uu odzyskują indywidualne umiejętności
uu odzyskują indywidualne umiejętności
poprawia się kontakt z dzieckiem, ze zmniejszeniem
poprawia się kontakt z dzieckiem, ze zmniejszeniem
cech autystycznych, aż do ich ustąpienia włącznie
cech autystycznych, aż do ich ustąpienia włącznie
etap ten określa się jako rzekomostacjonarny –
etap ten określa się jako rzekomostacjonarny –
rodzice i opiekunowie zaprzeczają dalszemu
rodzice i opiekunowie zaprzeczają dalszemu
regresowi
regresowi
opóźnienie rozwoju w tym czasie ocenia się już
opóźnienie rozwoju w tym czasie ocenia się już
jednak jako znaczne
jednak jako znaczne
często dołączają się napady padaczkowe
często dołączają się napady padaczkowe
ok. 15-20% osób pozostaje w tym stadium aż do
ok. 15-20% osób pozostaje w tym stadium aż do
wieku średniego
wieku średniego

CZTEROETAPOWY MODEL
CZTEROETAPOWY MODEL
ROZWOJU ZESPOŁU RETTA:
ROZWOJU ZESPOŁU RETTA:
Stadium IV:
Stadium IV:
okres, w którym osłabienie funkcji
okres, w którym osłabienie funkcji
nerwowo-mięśniowych i wyniszczenie
nerwowo-mięśniowych i wyniszczenie
zmuszają zdolną wcześniej do
zmuszają zdolną wcześniej do
samodzielnego poruszania się pacjentkę
samodzielnego poruszania się pacjentkę
do pełnego uzależnienia od wózka
do pełnego uzależnienia od wózka
inwalidzkiego
inwalidzkiego
skolioza
skolioza
zaniki mięśniowe w obrębie kończyn,
zaniki mięśniowe w obrębie kończyn,
zwłaszcza podudzi i stóp (zaburzenia
zwłaszcza podudzi i stóp (zaburzenia
troficzne, małe i zimne stopy)
troficzne, małe i zimne stopy)

OBJAWY W ZESPOLE
OBJAWY W ZESPOLE
RETTA:
RETTA:
STEREOTYPIE – prawie ciągle występujące
STEREOTYPIE – prawie ciągle występujące
automatyzmy podczas czuwania, pojawiające
automatyzmy podczas czuwania, pojawiające
się w okresie regresu (stadium II)
się w okresie regresu (stadium II)
specyficzne dla każdej pacjentki
specyficzne dla każdej pacjentki
przypominają wykręcanie, ściskanie,
przypominają wykręcanie, ściskanie,
wyżynanie, klaskanie, poklepywanie,
wyżynanie, klaskanie, poklepywanie,
zaciskanie, ugniatanie lub przyjmują bardziej
zaciskanie, ugniatanie lub przyjmują bardziej
złożone formy
złożone formy
w młodszym wieku są intensywniejsze i
w młodszym wieku są intensywniejsze i
bardziej złożone, z czasem stają się wolniejsze,
bardziej złożone, z czasem stają się wolniejsze,
bardziej „usztywnione” i mniej skomplikowane
bardziej „usztywnione” i mniej skomplikowane

OBJAWY W ZESPOLE
OBJAWY W ZESPOLE
RETTA:
RETTA:
ZAHAMOWANIE PRZYROSTU OBWODU GŁOWY
ZAHAMOWANIE PRZYROSTU OBWODU GŁOWY
prowadzi do małogłowia
prowadzi do małogłowia
wprost proporcjonalna zależność między
wprost proporcjonalna zależność między
obwodem głowy, a stopniem ciężkości
obwodem głowy, a stopniem ciężkości
przebiegu choroby
przebiegu choroby
u dziewczynek z odmianą rettopodobną można
u dziewczynek z odmianą rettopodobną można
zaobserwować tylko niewielkiego stopnia
zaobserwować tylko niewielkiego stopnia
zmniejszenie obwodu głowy
zmniejszenie obwodu głowy
ZAHAMOWANIE WZROSTU – początek we
ZAHAMOWANIE WZROSTU – początek we
wczesnym niemowlęctwie
wczesnym niemowlęctwie
nieproporcjonalne znacznego stopnia wczesne
nieproporcjonalne znacznego stopnia wczesne
zatrzymanie wzrastania stóp (są małe, zimne, o
zatrzymanie wzrastania stóp (są małe, zimne, o
sinawym zabarwieniu skóry ze skłonnością do
sinawym zabarwieniu skóry ze skłonnością do
zmian troficznych i zniekształceń paznokci)
zmian troficznych i zniekształceń paznokci)

OBJAWY W ZESPOLE
OBJAWY W ZESPOLE
RETTA:
RETTA:
ZABURZENIA ODDYCHANIA
ZABURZENIA ODDYCHANIA
objawiają się hiperwentylacją i okresowym bezdechem
objawiają się hiperwentylacją i okresowym bezdechem
bezdechy występują u 2/3 chorych, także niezależnie
bezdechy występują u 2/3 chorych, także niezależnie
od epizodów hiperwentylacji
od epizodów hiperwentylacji
podczas snu oddychanie jest zwykle prawidłowe
podczas snu oddychanie jest zwykle prawidłowe
ATAKSJA z ruchami „zrywającymi” tułowia
ATAKSJA z ruchami „zrywającymi” tułowia
są to szczególne ruchy drżenia tułowia będące
są to szczególne ruchy drżenia tułowia będące
przejawem ogólnej apraksji
przejawem ogólnej apraksji
jest to zjawisko przejściowe, widoczne w okresie, w
jest to zjawisko przejściowe, widoczne w okresie, w
którym dziewczynki osiągają zdolność samodzielnego
którym dziewczynki osiągają zdolność samodzielnego
siadania
siadania
w kolejnych stadiach objaw ten jest zwykle
w kolejnych stadiach objaw ten jest zwykle
przesłaniany bardziej widocznymi zaburzeniami
przesłaniany bardziej widocznymi zaburzeniami
neuronu ruchowego i zwojów podstawnych
neuronu ruchowego i zwojów podstawnych

OBJAWY W ZESPOLE
OBJAWY W ZESPOLE
RETTA:
RETTA:
ZABURZENIA W OBRĘBIE OBWODOWEGO
ZABURZENIA W OBRĘBIE OBWODOWEGO
NEURONU RUCHOWEGO
NEURONU RUCHOWEGO
manifestują się najczęściej atrofią mięśnia
manifestują się najczęściej atrofią mięśnia
strzałkowego, co powoduje obraz stopy
strzałkowego, co powoduje obraz stopy
„opadającej” z cechami dystonii
„opadającej” z cechami dystonii
SKOLIOZA o etiologii neurogennej
SKOLIOZA o etiologii neurogennej
powoduje liczne problemy i prowadzi do
powoduje liczne problemy i prowadzi do
inwalidztwa
inwalidztwa
jest przyczyną głębokich deformacji –
jest przyczyną głębokich deformacji –
wydłużona, podwójna krzywizna piersiowo-
wydłużona, podwójna krzywizna piersiowo-
lędźwiowa
lędźwiowa
OSŁABENIE CZUCIA BÓLU – u 80%
OSŁABENIE CZUCIA BÓLU – u 80%

OBJAWY W ZESPOLE
OBJAWY W ZESPOLE
RETTA:
RETTA:
ZGRZYTANIE ZĘBAMI – u 90%
ZGRZYTANIE ZĘBAMI – u 90%
WZDĘCIA – 5-10%
WZDĘCIA – 5-10%
NOCNE NAPADY ŚMIECHU – nie są
NOCNE NAPADY ŚMIECHU – nie są
objawem stałym, mają przebieg
objawem stałym, mają przebieg
okresowy, występują u 75%
okresowy, występują u 75%
GWAŁTOWNE NAPADY KRZYKU
GWAŁTOWNE NAPADY KRZYKU

KRYTERIA DIAGNOSTYCZNE DLA
KRYTERIA DIAGNOSTYCZNE DLA
POSTACI KLASYCZNEJ ZESPOŁU
POSTACI KLASYCZNEJ ZESPOŁU
RETTA:
RETTA:
KRYTERIA KONIECZNE:
KRYTERIA KONIECZNE:
nieobciążony wywiad okołoporodowy
nieobciążony wywiad okołoporodowy
prawidłowy rozwój psychoruchowy w
prawidłowy rozwój psychoruchowy w
pierwszych 6 mż (może być zaburzony od
pierwszych 6 mż (może być zaburzony od
urodzenia)
urodzenia)
prawidłowy obwód głowy przy urodzeniu
prawidłowy obwód głowy przy urodzeniu
utrata zdolności zamierzonego posługiwania
utrata zdolności zamierzonego posługiwania
się rękami między 6-30 mż
się rękami między 6-30 mż
stereotypowe ruchy rąk po utracie
stereotypowe ruchy rąk po utracie
zamierzonego posługiwania się rękami
zamierzonego posługiwania się rękami
zaburzenia komunikacji, utrata zdolności
zaburzenia komunikacji, utrata zdolności
mówienia i rozumienia mowy
mówienia i rozumienia mowy
nieprawidłowy chód (ataksja/apraksja)
nieprawidłowy chód (ataksja/apraksja)

KRYTERIA DIAGNOSTYCZNE DLA
KRYTERIA DIAGNOSTYCZNE DLA
POSTACI KLASYCZNEJ ZESPOŁU
POSTACI KLASYCZNEJ ZESPOŁU
RETTA:
RETTA:
KRYTERIA DODATKOWE:
KRYTERIA DODATKOWE:
zaburzenia oddychania (okresowy bezdech
zaburzenia oddychania (okresowy bezdech
w czasie czuwania, hiperwentylacja)
w czasie czuwania, hiperwentylacja)
bruksizm
bruksizm
nocne napady śmiechu
nocne napady śmiechu
wzmożone napięcie mięśniowe, często z
wzmożone napięcie mięśniowe, często z
towarzyszącymi zanikami mięśniowymi i
towarzyszącymi zanikami mięśniowymi i
dystonią
dystonią
zaburzenia ukrwienia obwodowego
zaburzenia ukrwienia obwodowego
skolioza lub kifoza
skolioza lub kifoza
małe, hipotroficzne i zimne stopy i/lub ręce
małe, hipotroficzne i zimne stopy i/lub ręce

KRYTERIA DIAGNOSTYCZNE DLA
KRYTERIA DIAGNOSTYCZNE DLA
POSTACI KLASYCZNEJ ZESPOŁU
POSTACI KLASYCZNEJ ZESPOŁU
RETTA:
RETTA:
KRYTERIA WYKLUCZAJĄCE:
KRYTERIA WYKLUCZAJĄCE:
organomegalia
organomegalia
retinopatia, zaćma lub atrofia nerwów
retinopatia, zaćma lub atrofia nerwów
wzrokowych
wzrokowych
okołoporodowe uszkodzenia mózgu
okołoporodowe uszkodzenia mózgu
rozpoznana choroba metaboliczna lub
rozpoznana choroba metaboliczna lub
inne postępujące zaburzenia
inne postępujące zaburzenia
neurologiczne
neurologiczne
nabyte schorzenia neurologiczne po
nabyte schorzenia neurologiczne po
infekcjach lub urazach głowy
infekcjach lub urazach głowy
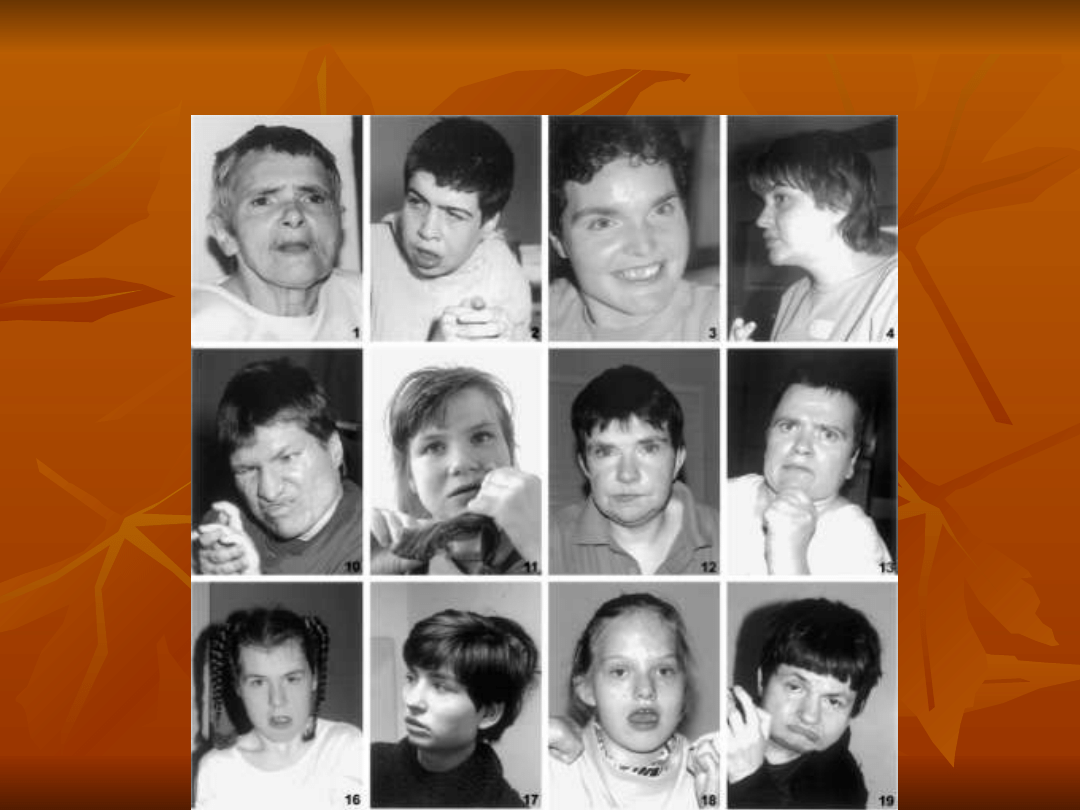
ZESPÓŁ RETTA
ZESPÓŁ RETTA

DYSTROFIA MIĘŚNIOWA
DYSTROFIA MIĘŚNIOWA
DUCHENNE’A
DUCHENNE’A
najczęstsza i najcięższa postać dystrofii
najczęstsza i najcięższa postać dystrofii
1:3000-3500 nowo narodzonych chłopców
1:3000-3500 nowo narodzonych chłopców
dziedziczenie recesywne sprzężone z chromosomem X
dziedziczenie recesywne sprzężone z chromosomem X
1/3 przypadków stanowią nowe mutacje
1/3 przypadków stanowią nowe mutacje
gen DMD - zidentyfikowany w 1987r
gen DMD - zidentyfikowany w 1987r
produktem genu jest dystrofina – białko błony
produktem genu jest dystrofina – białko błony
komórkowej
komórkowej
w DMD stwierdza się brak lub śladowej ilości
w DMD stwierdza się brak lub śladowej ilości
dystrofiny w mięśniach
dystrofiny w mięśniach
w dystrofii Beckera poziom dytsrofiny jest obniżony, w
w dystrofii Beckera poziom dytsrofiny jest obniżony, w
70-80% przypadków masa cząsteczkowa jest niższa od
70-80% przypadków masa cząsteczkowa jest niższa od
prawidłowej
prawidłowej
w mięśniach pozbawionych dystrofiny pojawia się
w mięśniach pozbawionych dystrofiny pojawia się
urofina
urofina
w pewnym stopniu istnieją zależności między ilością
w pewnym stopniu istnieją zależności między ilością
dystrofiny a ciężkością objawów
dystrofiny a ciężkością objawów
łagodny fenotyp kojarzy się z poziomem dystrofiny
łagodny fenotyp kojarzy się z poziomem dystrofiny
>20% normy
>20% normy

OBJAWY KLINICZNE:
OBJAWY KLINICZNE:
Pierwsze objawy zauważane przez rodziców:
Pierwsze objawy zauważane przez rodziców:
niezręczny, „kaczkowaty” chód
niezręczny, „kaczkowaty” chód
pierwsze kroki na palcach
pierwsze kroki na palcach
bóle nóg
bóle nóg
tendencja do padania
tendencja do padania
trudności we wstawaniu z pozycji leżącej
trudności we wstawaniu z pozycji leżącej
objaw Gowersa – tzw. wspinanie się po sobie
objaw Gowersa – tzw. wspinanie się po sobie
kłopoty we wchodzeniu na schody
kłopoty we wchodzeniu na schody
nadmierna lordoza lędźwiowa
nadmierna lordoza lędźwiowa
zanik mięśni – najpierw obręczy miedniczej
zanik mięśni – najpierw obręczy miedniczej
potem barkowej
potem barkowej

OBJAWY KLINICZNE:
OBJAWY KLINICZNE:
w okresie późniejszym zostają osłabione
w okresie późniejszym zostają osłabione
inne mięśnie, ale zawsze pozostaje
inne mięśnie, ale zawsze pozostaje
przewaga osłabienia zginaczy mięśni
przewaga osłabienia zginaczy mięśni
rzadkością jest zanik mięśni języka
rzadkością jest zanik mięśni języka
często zanik błony śluzowej żołądka
często zanik błony śluzowej żołądka
przerost prawdziwy lub rzekomy łydek
przerost prawdziwy lub rzekomy łydek
osłabienie a potem zanik odruchów
osłabienie a potem zanik odruchów
głębokich, zwłaszcza kolanowych (dłużej
głębokich, zwłaszcza kolanowych (dłużej
utrzymują się odruchy skokowe)
utrzymują się odruchy skokowe)
dzieci są albo bardzo szczupłe, albo otyłe
dzieci są albo bardzo szczupłe, albo otyłe
(otyłość może maskować zanik mięśni)
(otyłość może maskować zanik mięśni)

OBJAWY KLINICZNE:
OBJAWY KLINICZNE:
wady twarzowo-zgryzowe w postaci zgryzu
wady twarzowo-zgryzowe w postaci zgryzu
krzyżowego i całkowicie otwartego
krzyżowego i całkowicie otwartego
od 8-9 r. ż rozwijają się przykurcze, głównie ścięgna
od 8-9 r. ż rozwijają się przykurcze, głównie ścięgna
Achillesa
Achillesa
ok. 6 r.ż. może wystąpić krótkotrwała i dość pozorna
ok. 6 r.ż. może wystąpić krótkotrwała i dość pozorna
poprawa, wynikająca z przewagi tempa rozwoju
poprawa, wynikająca z przewagi tempa rozwoju
ruchowego nad tempem progresji procesu
ruchowego nad tempem progresji procesu
chorobowego(ang.honeymoon)
chorobowego(ang.honeymoon)
średni wiek unieruchomienia – 12-13 r.ż. – pojawia się
średni wiek unieruchomienia – 12-13 r.ż. – pojawia się
zniekształcenie klatki piersiowej i kręgosłupa, ciężka
zniekształcenie klatki piersiowej i kręgosłupa, ciężka
niewydolność oddechowa i zaburzenia krążenia
niewydolność oddechowa i zaburzenia krążenia
wielu autorów stwierdziło w DMD objawy opóźnienia
wielu autorów stwierdziło w DMD objawy opóźnienia
rozwoju umysłowego w 30-50%
rozwoju umysłowego w 30-50%

BADANIA DODATKOWE:
BADANIA DODATKOWE:
badanie dystrofiny mięśniowej
badanie dystrofiny mięśniowej
- w DMD
- w DMD
dystrofina występuje w śladowych ilościach lub
dystrofina występuje w śladowych ilościach lub
wcale
wcale
EMG
EMG
– można za jego pomocą zakwalifikować
– można za jego pomocą zakwalifikować
dany przypadek do chorób pierwotnie
dany przypadek do chorób pierwotnie
mięśniowych, ale nie można potwierdzić
mięśniowych, ale nie można potwierdzić
rozpoznania dystrofii (bogaty miogenny zapis
rozpoznania dystrofii (bogaty miogenny zapis
wysiłkowy wykazujący interferencję
wysiłkowy wykazujący interferencję
patologiczną, przewodzenie ruchowe i
patologiczną, przewodzenie ruchowe i
czuciowe jest prawidłowe)
czuciowe jest prawidłowe)
EKG
EKG
– u 50% - skrócenie odstępu PQ
– u 50% - skrócenie odstępu PQ
Badanie holterowskie
Badanie holterowskie
– wykazuje często
– wykazuje często
okresy tachykardii zatokowej
okresy tachykardii zatokowej
ECHO
ECHO
– cechy kardiomiopatii rozstrzeniowej
– cechy kardiomiopatii rozstrzeniowej
lub umiarkowanej kardiomiopatii przerostowej
lub umiarkowanej kardiomiopatii przerostowej

BADANIA BIOCHEMICZNE:
BADANIA BIOCHEMICZNE:
CPK
CPK
w DMD i DMB przekracza nawet 200x
w DMD i DMB przekracza nawet 200x
normę
normę
wysoka aktywność enzymu jest szczególnie
wysoka aktywność enzymu jest szczególnie
częsta u chorych <10r.ż
częsta u chorych <10r.ż
w miarę postępu choroby aktywność CPK
w miarę postępu choroby aktywność CPK
stopniowo obniża się
stopniowo obniża się
oznaczenie CPK pozwala także na wykrycie
oznaczenie CPK pozwala także na wykrycie
stanów przedklnicznych – aktywność często
stanów przedklnicznych – aktywność często
już po urodzeniu jest niezwykle wysoka
już po urodzeniu jest niezwykle wysoka
ALDOLAZA, AMINOTRANSFERAZY,
ALDOLAZA, AMINOTRANSFERAZY,
DEHYDROGENAZA MLECZANOWA
DEHYDROGENAZA MLECZANOWA
niewielki wzrost aktywności u chorych na
niewielki wzrost aktywności u chorych na
dystrofię Beckera
dystrofię Beckera

DIAGNOSTYKA
DIAGNOSTYKA
MOLEKULARNA:
MOLEKULARNA:
delecję wykrywa się w 65% przypadków DMD
delecję wykrywa się w 65% przypadków DMD
w 85% DMB
w 85% DMB
30% przypadków pozostaje
30% przypadków pozostaje
niezidentyfikowanych
niezidentyfikowanych
brak korelacji między wielkością delecji a
brak korelacji między wielkością delecji a
ciężkością fenotypu
ciężkością fenotypu
najczęstsze miejsca delecji- między eksonami
najczęstsze miejsca delecji- między eksonami
2-20 (30%) oraz 44-55 (70%)
2-20 (30%) oraz 44-55 (70%)
mutacja występuje częściej tam, gdzie intron
mutacja występuje częściej tam, gdzie intron
jest długi np.44-55
jest długi np.44-55

DYSTROFIA MIĘŚNOWA BECKERA
DYSTROFIA MIĘŚNOWA BECKERA
częstość występowania 3-6 przypadków na 100 000 urodzonych
częstość występowania 3-6 przypadków na 100 000 urodzonych
chłopców
chłopców
uważa się, że postać Beckera występuje znacznie rzadziej (prawie
uważa się, że postać Beckera występuje znacznie rzadziej (prawie
10-krotnie) niż postać Duchenne’a – ostatnie badania temu
10-krotnie) niż postać Duchenne’a – ostatnie badania temu
zaprzeczają
zaprzeczają
postać Beckera wg jednych autorów obejmuje zarówno postać
postać Beckera wg jednych autorów obejmuje zarówno postać
łagodniejszej dystrofii dosiebnej, jak też kardiomiopatii izolowanej
łagodniejszej dystrofii dosiebnej, jak też kardiomiopatii izolowanej
lub współistniejącej z uszkodzeniem mięśni szkieletowych,
lub współistniejącej z uszkodzeniem mięśni szkieletowych,
kurczów mięśniowych, mialgii, a także zwiększonej aktywności
kurczów mięśniowych, mialgii, a także zwiększonej aktywności
kinazy kreatynowej jako jedynego objawu
kinazy kreatynowej jako jedynego objawu
objawy chorobowe zaczynają się znacznie później niż w DMD, ok.
objawy chorobowe zaczynają się znacznie później niż w DMD, ok.
11r.ż, ale zdarza się, że zauważalny początek przypada na 30 r. z
11r.ż, ale zdarza się, że zauważalny początek przypada na 30 r. z
choroba przebiega łagodniej, chorzy długo są sprawni ruchowo,
choroba przebiega łagodniej, chorzy długo są sprawni ruchowo,
unieruchomienie przed 40r.ż. jest rzadkie, a okres przeżycia jest
unieruchomienie przed 40r.ż. jest rzadkie, a okres przeżycia jest
prawie taki sam jak w całej populacji
prawie taki sam jak w całej populacji
jednym z pierwszych objawów na długo poprzedzającym
jednym z pierwszych objawów na długo poprzedzającym
wystąpienie innych jest przerost łydek i kurcze mięśniowe
wystąpienie innych jest przerost łydek i kurcze mięśniowe
słabość i ew. zanik mięśni rozkłada się tak jak w postaci
słabość i ew. zanik mięśni rozkłada się tak jak w postaci
Duchene’a, ale nie dotyczy aż tylu mięśni i nie w takim stopniu
Duchene’a, ale nie dotyczy aż tylu mięśni i nie w takim stopniu
przykurcze i skrzywienie kręgosłupa są rzadkie
przykurcze i skrzywienie kręgosłupa są rzadkie

NOSICIELKI:
NOSICIELKI:
objawowe i bezobjawowe
objawowe i bezobjawowe
10-15% nosicielek wykazuje objawy ujawniające się przy
10-15% nosicielek wykazuje objawy ujawniające się przy
bardzo dokładnym badaniu
bardzo dokładnym badaniu
objawy w znacznie łagodniejszej formie
objawy w znacznie łagodniejszej formie
zmiany chorobowe przypominające dystrofię o różnym
zmiany chorobowe przypominające dystrofię o różnym
nasileniu
nasileniu
przerosłe łydki
przerosłe łydki
skrzywienie kręgosłupa
skrzywienie kręgosłupa
osłabienie mięśni dosiebnych
osłabienie mięśni dosiebnych
zmiany w mięśniu sercowym (kardiomiopatia)
zmiany w mięśniu sercowym (kardiomiopatia)
znacznie podwyższona aktywność kinazy kreatynowej
znacznie podwyższona aktywność kinazy kreatynowej
zmiany czynności bioelektrycznej mięśni szkieletowych
zmiany czynności bioelektrycznej mięśni szkieletowych
zmiany w biopsji mięśniowej, polegające na
zmiany w biopsji mięśniowej, polegające na
nierównomiernej średnicy włókien, centralnym położeniu
nierównomiernej średnicy włókien, centralnym położeniu
jąder i często rozszczepianiu włókien
jąder i często rozszczepianiu włókien
badanie nosicielstwa opiera się głownie na ilościowym i
badanie nosicielstwa opiera się głownie na ilościowym i
jakościowym badaniu dystrofiny mięśniowej oraz na
jakościowym badaniu dystrofiny mięśniowej oraz na
badaniu DNA
badaniu DNA

NOSICIELKI:
NOSICIELKI:
wzrost aktywności kinazy i zmiana ilości
wzrost aktywności kinazy i zmiana ilości
dystrofiny są szczególnie nasilone u bardzo
dystrofiny są szczególnie nasilone u bardzo
młodych nosicielek – często, żeby wyjaśnić
młodych nosicielek – często, żeby wyjaśnić
nosicielstwo matki, wykonuje się badania jej
nosicielstwo matki, wykonuje się badania jej
młodej córce
młodej córce
prawidłowe wartości CK przed 20Hbd należy
prawidłowe wartości CK przed 20Hbd należy
taktować ostrożnie z uwagi na obniżanie się w tym
taktować ostrożnie z uwagi na obniżanie się w tym
czasie stężeń tego enzymu
czasie stężeń tego enzymu
badania skirningowe aktywności CK u nowo
badania skirningowe aktywności CK u nowo
narodzonych chłopców mają służyć
narodzonych chłopców mają służyć
ukierunkowaniu ewentualnych badań nosicielstwa
ukierunkowaniu ewentualnych badań nosicielstwa
w rodzinie, są niejako formą diagnostyki
w rodzinie, są niejako formą diagnostyki
prenatalnej dla następnego dziecka płci męskiej,
prenatalnej dla następnego dziecka płci męskiej,
jeżeli urodzi się ono zanim u pierwszego dziecka
jeżeli urodzi się ono zanim u pierwszego dziecka
staną się zauważalne kliniczne objawy choroby
staną się zauważalne kliniczne objawy choroby

ROZPOZNANIE DMD i DMB:
ROZPOZNANIE DMD i DMB:
Opiera się na następujących
Opiera się na następujących
kryteriach:
kryteriach:
sposobie dziedziczenia
sposobie dziedziczenia
elektromiografii miogennej
elektromiografii miogennej
podwyższonej aktywności CK
podwyższonej aktywności CK
biopsji mięśniowej
biopsji mięśniowej
wykryciu delecji
wykryciu delecji
braku lub niedoborze dystrofiny
braku lub niedoborze dystrofiny

PROFILAKTYKA I TERAPIA DMD i
PROFILAKTYKA I TERAPIA DMD i
DMB
DMB
małe dawki prednizonu (0,3-0,75 mg/kg dziennie) po 5 r.ż.
małe dawki prednizonu (0,3-0,75 mg/kg dziennie) po 5 r.ż.
środki wzmacniających i obfitująca w witaminy dieta
środki wzmacniających i obfitująca w witaminy dieta
postępowanie rehabilitacyjne - przeciwdziałanie
postępowanie rehabilitacyjne - przeciwdziałanie
przykurczom i zniekształceniom kręgosłupa, utrzymanie
przykurczom i zniekształceniom kręgosłupa, utrzymanie
jak najdłużej zdolności chodzenia
jak najdłużej zdolności chodzenia
tendencje do powstawania stopy końskiej lub końsko-
tendencje do powstawania stopy końskiej lub końsko-
szpotawej ze skróceniem ścięgna Achillesa, rozwijają się
szpotawej ze skróceniem ścięgna Achillesa, rozwijają się
przykurcze zgięciowe uda i zgninaczy kolan – prewencją
przykurcze zgięciowe uda i zgninaczy kolan – prewencją
jest bierne rozciąganie, długie leżenie dziecka na podłodze,
jest bierne rozciąganie, długie leżenie dziecka na podłodze,
zakładanie na noc longet, które utrzymują staw skokowy w
zakładanie na noc longet, które utrzymują staw skokowy w
zgięciu 90º
zgięciu 90º
każda narkoza zwiększa możliwość powstania
każda narkoza zwiększa możliwość powstania
mioglobinurii i hipertermii złośliwej
mioglobinurii i hipertermii złośliwej
od 11r.ż - opieką pulmonologiczna (nocna, przerywana,
od 11r.ż - opieką pulmonologiczna (nocna, przerywana,
pozytywna wentylacja)
pozytywna wentylacja)

ZESPÓŁ ŁAMLIWEGO
ZESPÓŁ ŁAMLIWEGO
CHROMOSOMU X
CHROMOSOMU X
jest drugą co do częstości po zespole Downa
jest drugą co do częstości po zespole Downa
genetyczną postacią uu
genetyczną postacią uu
chorobowość jest oceniana: 0,2-0,6 na 1000
chorobowość jest oceniana: 0,2-0,6 na 1000
kobiet i 0,4-0,9 na 1000 mężczyzn
kobiet i 0,4-0,9 na 1000 mężczyzn
dziedziczenie i penetracja zespołu
dziedziczenie i penetracja zespołu
łamliwego chromosomu X różnią się od
łamliwego chromosomu X różnią się od
innych zaburzeń sprzężonych z
innych zaburzeń sprzężonych z
chromosomem X
chromosomem X
mężczyźni z tym genem mogą być klinicznie
mężczyźni z tym genem mogą być klinicznie
normalni, ale mogą przekazać zaburzenie
normalni, ale mogą przekazać zaburzenie
przez wszystkie swoje córki na wnuki
przez wszystkie swoje córki na wnuki
zdrowy mężczyzna – nosiciel łamliwego X
zdrowy mężczyzna – nosiciel łamliwego X
jest nazywany transmitting male-TM
jest nazywany transmitting male-TM

ZESPÓŁ ŁAMLIWEGO
ZESPÓŁ ŁAMLIWEGO
CHROMOSOMU X
CHROMOSOMU X
podstawą molekularną zespołu jest wydłużenie
podstawą molekularną zespołu jest wydłużenie
ciągów powtórzeń trinukleotydowych CGG genu
ciągów powtórzeń trinukleotydowych CGG genu
FMR1
FMR1
w rodzinach z łamliwym X region ulega wydłużeniu
w rodzinach z łamliwym X region ulega wydłużeniu
poprzez serię międzypokoleniowych zmian od
poprzez serię międzypokoleniowych zmian od
premutacji, które są nieme, do pełnych mutacji,
premutacji, które są nieme, do pełnych mutacji,
które wpływają na transkrypcję przyległego genu
które wpływają na transkrypcję przyległego genu
Mogą wystąpić 3 podstawowe stany wydłużenia:
Mogą wystąpić 3 podstawowe stany wydłużenia:
normalny (50 tripletów) – inteligencja jest normalna,
normalny (50 tripletów) – inteligencja jest normalna,
a długość taka sama w każdym pokoleniu
a długość taka sama w każdym pokoleniu
premutacje (50-200 tripletów) – inteligencja jest
premutacje (50-200 tripletów) – inteligencja jest
normalna, ale możliwe są różnice między
normalna, ale możliwe są różnice między
generacjami o zwiększającym się nasileniu
generacjami o zwiększającym się nasileniu
pełne mutacje (>200) – są masywnymi, somatycznie
pełne mutacje (>200) – są masywnymi, somatycznie
niestabilnymi zmianami u chorych osobników
niestabilnymi zmianami u chorych osobników

GEN FMR1:
GEN FMR1:
w zespole fraX nie dochodzi do
w zespole fraX nie dochodzi do
transkrypcji genu FMR1 z powodu
transkrypcji genu FMR1 z powodu
dużej ekspansji powtórzeń CGG w
dużej ekspansji powtórzeń CGG w
pobliskim regionie
pobliskim regionie

REGUŁY DZIEDZICZENIA FraX:
REGUŁY DZIEDZICZENIA FraX:
każde chore dziecko ma matkę nosicielkę
każde chore dziecko ma matkę nosicielkę
nowe mutacje nigdy nie powstają
nowe mutacje nigdy nie powstają
bezpośrednio od stanu normalnego do pełnej
bezpośrednio od stanu normalnego do pełnej
mutacji
mutacji
matki chorych dzieci zawsze posiadają
matki chorych dzieci zawsze posiadają
wydłużenia sekwencji
wydłużenia sekwencji
matki nosicielki mogą posiadać premutację
matki nosicielki mogą posiadać premutację
lub pełną mutację
lub pełną mutację
matki z uu związanym z łamliwym X mają
matki z uu związanym z łamliwym X mają
pełne mutacje
pełne mutacje
jeśli matka posiada pełną mutację, babka ze
jeśli matka posiada pełną mutację, babka ze
strony matki jest obligatoryjną nosicielką, ale
strony matki jest obligatoryjną nosicielką, ale
jeśli matka posiada premutację, źródłem może
jeśli matka posiada premutację, źródłem może
być którekolwiek z dziadków ze strony matki
być którekolwiek z dziadków ze strony matki

REGUŁY DZIEDZICZENIA
REGUŁY DZIEDZICZENIA
FraX:
FraX:
prawdopodobieństwo, że kobieta nosicielka będzie
prawdopodobieństwo, że kobieta nosicielka będzie
miała dziecko z pełną mutacją jest proporcjonalne
miała dziecko z pełną mutacją jest proporcjonalne
do wielkości wydłużenia, które posiada
do wielkości wydłużenia, które posiada
małe matczyne premutacje (mniej niż 90
małe matczyne premutacje (mniej niż 90
powtórzeń) nie zawsze stają się mutacjami pełnej
powtórzeń) nie zawsze stają się mutacjami pełnej
długości podczas przekazywania potomstwu
długości podczas przekazywania potomstwu
przekazanie premutacji większej niż 90 powtórzeń
przekazanie premutacji większej niż 90 powtórzeń
prawie zawsze powoduje pełną mutację
prawie zawsze powoduje pełną mutację
premutacje są dziedziczone bezobjawowo na
premutacje są dziedziczone bezobjawowo na
przestrzeni lat, nigdy nie odnotowano przejścia
przestrzeni lat, nigdy nie odnotowano przejścia
między pokoleniami od stałego allela przeciętnej
między pokoleniami od stałego allela przeciętnej
długości do premutacji –dlatego dalecy krewni
długości do premutacji –dlatego dalecy krewni
obarczeni są ryzykiem
obarczeni są ryzykiem

FENOTYP WYGLĄDU
FENOTYP WYGLĄDU
ZEWNĘTRZNEGO:
ZEWNĘTRZNEGO:
W pełni wyrażony fenotyp chorych mężczyzn
W pełni wyrażony fenotyp chorych mężczyzn
obejmuje:
obejmuje:
podłużna twarz z dużą żuchwą
podłużna twarz z dużą żuchwą
duże i/lub odstające uszy
duże i/lub odstające uszy
powiększone jądra
powiększone jądra
objawy te są często nieobecne lub nieznacznie
objawy te są często nieobecne lub nieznacznie
wyrażone przed pokwitaniem i nie zawsze są
wyrażone przed pokwitaniem i nie zawsze są
obecne u dojrzałych chorych mężczyzn
obecne u dojrzałych chorych mężczyzn
łagodnie wielkogłowie i wzrost powyżej średniej
łagodnie wielkogłowie i wzrost powyżej średniej
mogą zwracać uwagę w dzieciństwie, ale
mogą zwracać uwagę w dzieciństwie, ale
dorośli mężczyźni z łamliwym X wykazują
dorośli mężczyźni z łamliwym X wykazują
tendencję do wzrostu niższego od przeciętnego
tendencję do wzrostu niższego od przeciętnego

FENOTYP WYGLĄDU
FENOTYP WYGLĄDU
ZEWNĘTRZNEGO:
ZEWNĘTRZNEGO:
kobiety z pełną mutacją wykazują
kobiety z pełną mutacją wykazują
zwykle cechy fizyczne zespołu –
zwykle cechy fizyczne zespołu –
podłużna twarz, odstające uszy i
podłużna twarz, odstające uszy i
przeprost w stawach palców
przeprost w stawach palców
kobiety nosicielki premutacji mogą
kobiety nosicielki premutacji mogą
wejść w menopauzę we wcześniejszym
wejść w menopauzę we wcześniejszym
wieku niż normalnie i częściej rodzą
wieku niż normalnie i częściej rodzą
bliźnięta dizygotyczne (sugeruje to, że
bliźnięta dizygotyczne (sugeruje to, że
gen FMR1 ma pewien wpływ na funkcję
gen FMR1 ma pewien wpływ na funkcję
gonady żeńskiej) – nie jest to
gonady żeńskiej) – nie jest to
obserwowane u kobiet z pełnymi
obserwowane u kobiet z pełnymi
mutacjami
mutacjami

FENOTYP POZNAWCZY I
FENOTYP POZNAWCZY I
BEHAWIORALNY:
BEHAWIORALNY:
mężczyźni przed pokwitaniem mogą funkcjonować
mężczyźni przed pokwitaniem mogą funkcjonować
na poziomie lekkiego upośledzenia, chociaż
na poziomie lekkiego upośledzenia, chociaż
opóźnienie psychomotoryczne jest ewidentne od
opóźnienie psychomotoryczne jest ewidentne od
urodzenia
urodzenia
większość dorosłych jest umiarkowanie
większość dorosłych jest umiarkowanie
upośledzona umysłowo, a 30% znacznie lub głęboko
upośledzona umysłowo, a 30% znacznie lub głęboko
IQ obniża się z wiekiem
IQ obniża się z wiekiem
osłabienie zdolności arytmetycznych, pamięci
osłabienie zdolności arytmetycznych, pamięci
krótkoterminowej dla bodźców znaczących,
krótkoterminowej dla bodźców znaczących,
słownictwa, zdolności ekspresji werbalnej i
słownictwa, zdolności ekspresji werbalnej i
pojmowania
pojmowania
mają opóźniony rozwój mowy i języka i często
mają opóźniony rozwój mowy i języka i często
wykazują perseweracje, mowę bezładną,
wykazują perseweracje, mowę bezładną,
skandowaną i monotonną
skandowaną i monotonną
chłopcy mają tendencję do niskiego głosu z
chłopcy mają tendencję do niskiego głosu z
cechami ochrypłości lub chropowatości
cechami ochrypłości lub chropowatości

FENOTYP POZNAWCZY I
FENOTYP POZNAWCZY I
BEHAWIORALNY:
BEHAWIORALNY:
Typowy fenotyp zachowania
Typowy fenotyp zachowania
występujący od dzieciństwa obejmuje:
występujący od dzieciństwa obejmuje:
hiperaktywność
hiperaktywność
unikanie towarzystwa i lęk
unikanie towarzystwa i lęk
zachowania stereotypowe –
zachowania stereotypowe –
wymachiwanie rękami, gryzienie rąk,
wymachiwanie rękami, gryzienie rąk,
unikanie wzroku i obrona przed
unikanie wzroku i obrona przed
dotykiem
dotykiem

FENOTYP POZNAWCZY I
FENOTYP POZNAWCZY I
BEHAWIORALNY:
BEHAWIORALNY:
35% kobiet z cytogenetyczną ekspresją
35% kobiet z cytogenetyczną ekspresją
łamliwego chromosomu X jest upośledzona
łamliwego chromosomu X jest upośledzona
umysłowo, a przynajmniej 50% ma zaburzenia
umysłowo, a przynajmniej 50% ma zaburzenia
uczenia się
uczenia się
kobiety z pełną mutacją mają niższe IQ niż
kobiety z pełną mutacją mają niższe IQ niż
nosicielki premutacji
nosicielki premutacji
IQ młodych kobiet z łamliwym X mają tendencję
IQ młodych kobiet z łamliwym X mają tendencję
do obniżania się w czasie, chociaż mogą
do obniżania się w czasie, chociaż mogą
pozostawać na stałym poziomie u kobiet >18 rż
pozostawać na stałym poziomie u kobiet >18 rż
fenotyp behawioralny pełnej mutacji jest
fenotyp behawioralny pełnej mutacji jest
podobny u kobiet i mężczyzn
podobny u kobiet i mężczyzn
kobiety z premutacją wydają się nie różnić w
kobiety z premutacją wydają się nie różnić w
testach psychometrycznych od grupy kontrolnej
testach psychometrycznych od grupy kontrolnej

NIEPRAWIDŁOWOŚCI
NIEPRAWIDŁOWOŚCI
NEUROLOGICZNE:
NEUROLOGICZNE:
Zawsze obecne są:
Zawsze obecne są:
niezgrabny chód
niezgrabny chód
upośledzona koordynacja
upośledzona koordynacja
Częste są:
Częste są:
hipotonia
hipotonia
dysfunkcja okoruchowa (zez, opadanie powiek)
dysfunkcja okoruchowa (zez, opadanie powiek)
drgawki – u 25% - typowe są rzadko
drgawki – u 25% - typowe są rzadko
występujące i uogólnione drgawki, które po raz
występujące i uogólnione drgawki, które po raz
pierwszy pojawiają się w dzieciństwie, reagują
pierwszy pojawiają się w dzieciństwie, reagują
na leczenie p/drgawkowe i zanikają po okresie
na leczenie p/drgawkowe i zanikają po okresie
dojrzewania
dojrzewania
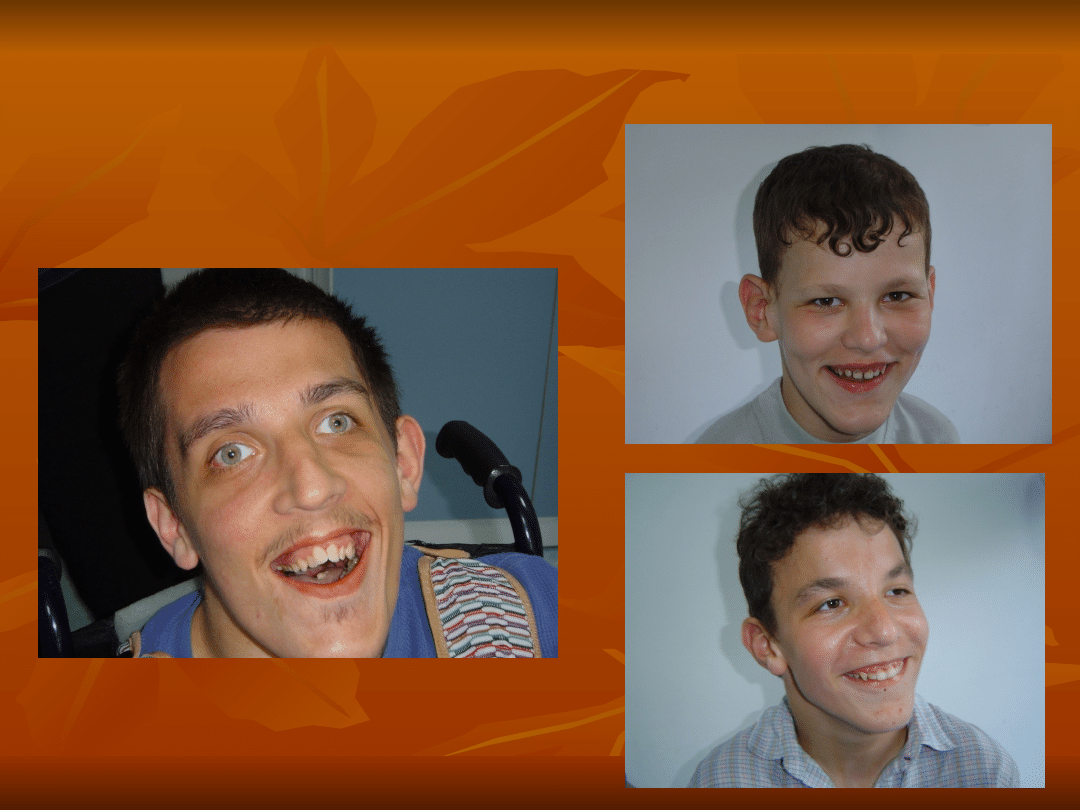
ZESPÓŁ ŁAMLIWEGO
ZESPÓŁ ŁAMLIWEGO
CHROMOSOMU X
CHROMOSOMU X

DIAGNOSTYKA MOLEKULARNA:
DIAGNOSTYKA MOLEKULARNA:
znalezienie pełnej mutacji jakiejkolwiek wielkości u
znalezienie pełnej mutacji jakiejkolwiek wielkości u
osobnika opóźnionego w rozwoju uważane jest za
osobnika opóźnionego w rozwoju uważane jest za
potwierdzenie fraX
potwierdzenie fraX
jeśli pełna mutacja jest stwierdzona u kobiety przed
jeśli pełna mutacja jest stwierdzona u kobiety przed
postawieniem diagnozy klinicznej – np. w analizie
postawieniem diagnozy klinicznej – np. w analizie
prenatalnej – nie ma pewności czy będzie ona chora, ani
prenatalnej – nie ma pewności czy będzie ona chora, ani
jak duże będzie nasilenie choroby
jak duże będzie nasilenie choroby
premutacja wykrywana u osobnika opóźnionego w rozwoju
premutacja wykrywana u osobnika opóźnionego w rozwoju
przy nieobecności mozaikowości dla pełnej mutacji nie
przy nieobecności mozaikowości dla pełnej mutacji nie
potwierdza diagnozy zespołu fra X, ale oznacza, że pacjent
potwierdza diagnozy zespołu fra X, ale oznacza, że pacjent
i jego krewni są nosicielami
i jego krewni są nosicielami
normalny wynik stwierdzony u osobnika z opóźnieniem
normalny wynik stwierdzony u osobnika z opóźnieniem
rozwoju przy nieobecności mozaikowości dla pełnej
rozwoju przy nieobecności mozaikowości dla pełnej
mutacji definitywnie wyklucza zespól łamliwego
mutacji definitywnie wyklucza zespól łamliwego
chromosomu X
chromosomu X
prenatalne badanie DNA jest wskazane głównie, jeśli nie
prenatalne badanie DNA jest wskazane głównie, jeśli nie
wyłącznie w rodzinach, o których wiadomo, że matka jest
wyłącznie w rodzinach, o których wiadomo, że matka jest
nosicielką udowodnionej ekspansji CGG istotnej wielkości
nosicielką udowodnionej ekspansji CGG istotnej wielkości
nie ma wskazań do badań płodów, których matki nie są
nie ma wskazań do badań płodów, których matki nie są
nosicielkami
nosicielkami
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
Wyszukiwarka
Podobne podstrony:
Choroby monogenowe, VI rok Lekarski CM UMK, Genetyka CMUMK 2015 VI rok, Genetyka, GENETYKA VI, !Mate
choroby naczyn i serca(1)
ŻYWIENIE A CHOROBY 4b
Choroby układu nerwowego ppt
Produkty przeciwwskazane w chorobach jelit II
Choroba niedokrwienna serca
CZLOWIEK I CHOROBA – PODSTAWOWE REAKCJE NA
Choroby nerwów czaszkowych
Choroby Cywilizacyjne 5
3 Seminarium Patofizjologia chorób rozrostowych
Replikacja DNA i choroby związane
Choroby ukadu pok
Żywienie a choroby
Krew i choroby układu krwionośnego
więcej podobnych podstron