
1
Budowa polimerów
POLITECHNIKA ŁÓDZKA
POLITECHNIKA ŁÓDZKA
WYDZIAŁ INŻYNIERII PROCESOWEJ I OCHRONY ŚRODOWISKA
WYDZIAŁ INŻYNIERII PROCESOWEJ I OCHRONY ŚRODOWISKA
Prof. dr hab. Maria Mucha
POLIMERY
3
2
Pojęcia podstawowe
Polimer – związek chemiczny składający się z powtarzających się jednostek,
różniący się własnościami od odpowiadających mu monomerów.
Monomer – związek chemiczny zdolny do polimeryzacji.
Oligomer – 1000<M<10000.
Klasyfikacja polimerów
Polimery naturalne (kauczuk, celuloza, kwasy nukleinowe, proteiny –
kolagen, kazeina),
Polimery syntetyczne,
Homołańcuchowe – łańcuch główny zbudowany wyłącznie z atomów
węgla: PE, PP, PCW, poliakrylonitryl, itd.
Heterołańcuchowe – heteroatomy w łańcuchu głównym (energia
wiązania w łańcuchu gł. jest wyższa niż u polimerów
homołańcuchowych) – duża trwałość chemiczna i termiczna i
mechaniczna, poliestry, poliwęglany, poliamidy, polisiloksany, żywice,
Polimery liniowe i rozgałęzione,
Polimery usieciowane.
Kopolimery (przemienny, blokowy, statystyczny).

3
Skład i budowa cząsteczek polimerowych
1. Budowa Meru
a. Skład elementarny
b. Rodzaje wiązań między atomami
c. Rodzaj i długość podstawników
d. Obecność pierścieni w łańcuchu lub w grupach bocznych
2. Odległość i kąty walencyjne, efektywne promienie atomów,
(dyfrakcja: X, elektronów, neutronów).
3. Siły wewnątrzcząsteczkowe wiążące atomy są kowalentnymi
wiązaniami
chemicznymi o energii dysocjacji 50 – 140 kcal/mol.
4. Rodzaj i charakter grup końcowych
5. Charakterystyka rozgałęzień
6. Masa cząsteczkowa (pojęcie średniej masy cząsteczkowej) i
polimolekularność
(stopień polidyspersji Mw/Mn).
7. Oddziaływania międzycząsteczkowe
- siły wiążące makrocząsteczki w agregaty, kryształy

4
I. Siły van der Waalsa – powstają w wyniku:
- orientacji trwałych dipoli (zależna od temp.)
- efektu indukcyjnego – polaryzacja elektryczna (mniej zależny od temp.)
- efektu dyspersyjnego (zaburzenia gęstości ładunku elektronowego w cząsteczce
chwilowy dipol).
II. Oddziaływania donorowo – akceptorowe (charge transfer)
- tworzenie się kompleksów donorowo – akceptorowych.
III. Oddziaływania rezonansowe (fotochemia)
- przejście energii ze stanu wzbudzonego jednej cząsteczki do drugiej w
stanie podstawowym.
IV. Wiązania wodorowe
A – H(+) …B(-) atom z wolną parą elektronową (atom silnie
elektroujemny: tlen, azot)
H…..B – wiązanie elektrostatyczne:
- są to wiązania słabe;
- powodują wzrost uporządkowania;
- np. alkohol poliwinylowy, celuloza.
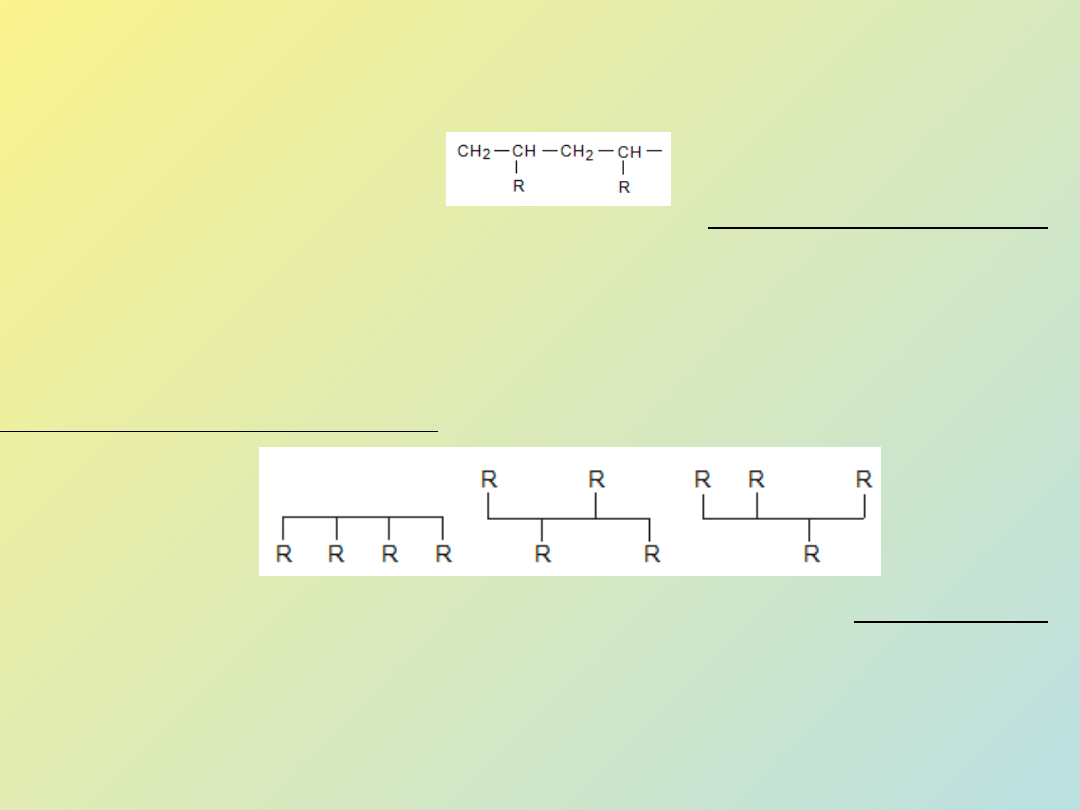
5
9. Ukształtowanie łańcucha głównego
a) struktura I-rzędowa jest związana z podstawową budową łańcucha
- następstwo merów, np. głowa do ogona i td.
Konfiguracja łańcucha
Przestrzenne ułożenie atomów i grup atomów w makrocząsteczce
wynikające z długości wiązań i kątów walencyjnych, np. konfiguracja
podstawników wokół asymetrycznego atomu węgla prowadzi do
taktyczności polimeru (polimery stereo regularne – polimeryzacja stereo
specyficzna).
Struktury taktyczne polimeru:
izotaktyczna, syndiotaktyczna, ataktyczna
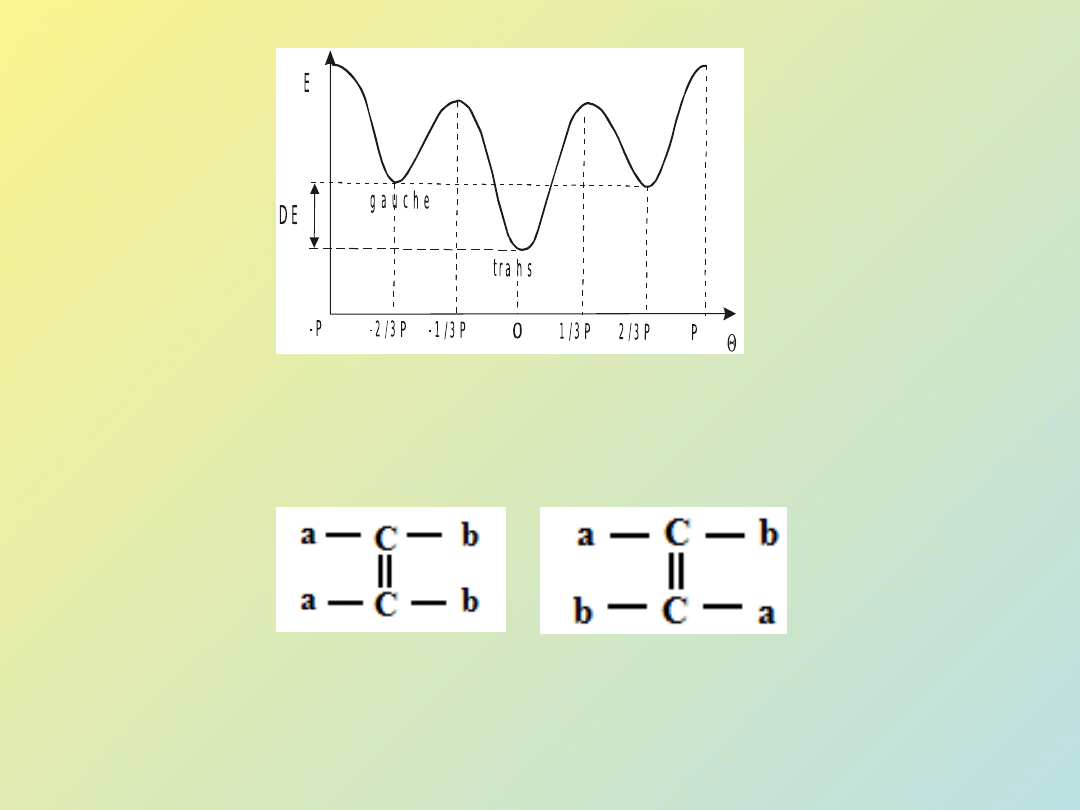
Konformacja
Jest to jedno z nieidentycznych położeń w przestrzeni atomów danej
cząsteczki tworzące się
podczas rotacji wokół wiązania pojedynczego. Pełna rotacja jest ograniczona
występowaniem bariery energetycznej swobodnej rotacji. Uprzywilejowane
położenia atomów spełniające warunek minimum energii potencjalnej
prowadzą do stabilnych izomerów.
6
W przypadku parafin i PE uprzywilejowana jest konformacja trans
W przypadku polimerów o asymetrycznej budowie CH2 – CHX preferowaną konformacją
jest trans – gauche.
Konformacja cis-trans powstaje w wyniku istnienia wiązania podwójnego
cis
trans
Izomery cis i trans różnią się:
Temp. topnienia, rozpuszczalnością, reaktywnością, widmem IR, st . krystaliczności
.
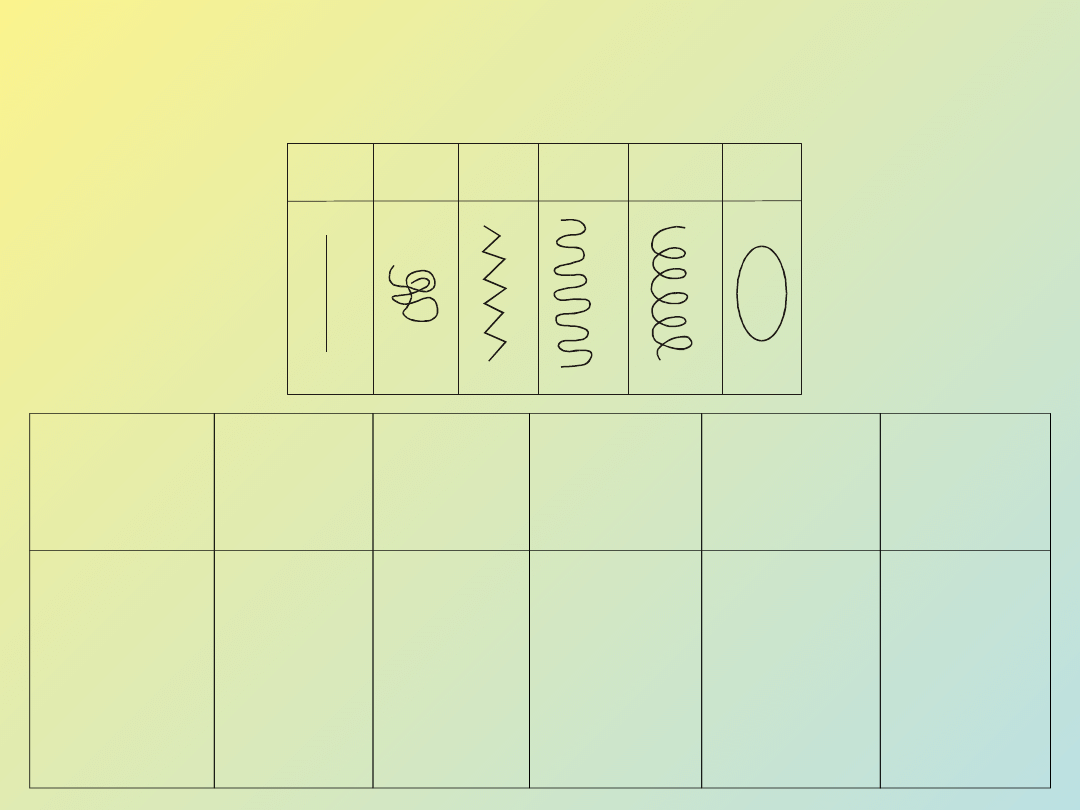
7
Struktury II rzędowe - są związane z ukształtowaniem łańcucha głównego polimeru:
Makrocząsteczki mogą występować w następujących postaciach konformacyjnych:
Łańcuch
całkowicie
wyprostowany
A
Globularna
(kłębek)
B
Rektalna
(linia
zygzakowat
a)
C
Lamelarna
(sfałdowana)
D
Helikoidalna
(spiralna)
E
Cykliczna
F
rzadko
występuje;
- polimery
włóknotwórcze
naturalne;
-
krystalizowane
pod wysokim
ciśnieniem;
w obszarach
amorficznyc
h,
rozcieńczon
ych
roztworach
w
polimerach
taktycznych;
W polimerach
krystalicznyc
h
- spirale
prawo lub
lewo skrętne,
polimery
naturalne,
izotaktyczne
(rzadka);
Silikony;
pierścienie
peptydowe;
A
B
C
D
E
F
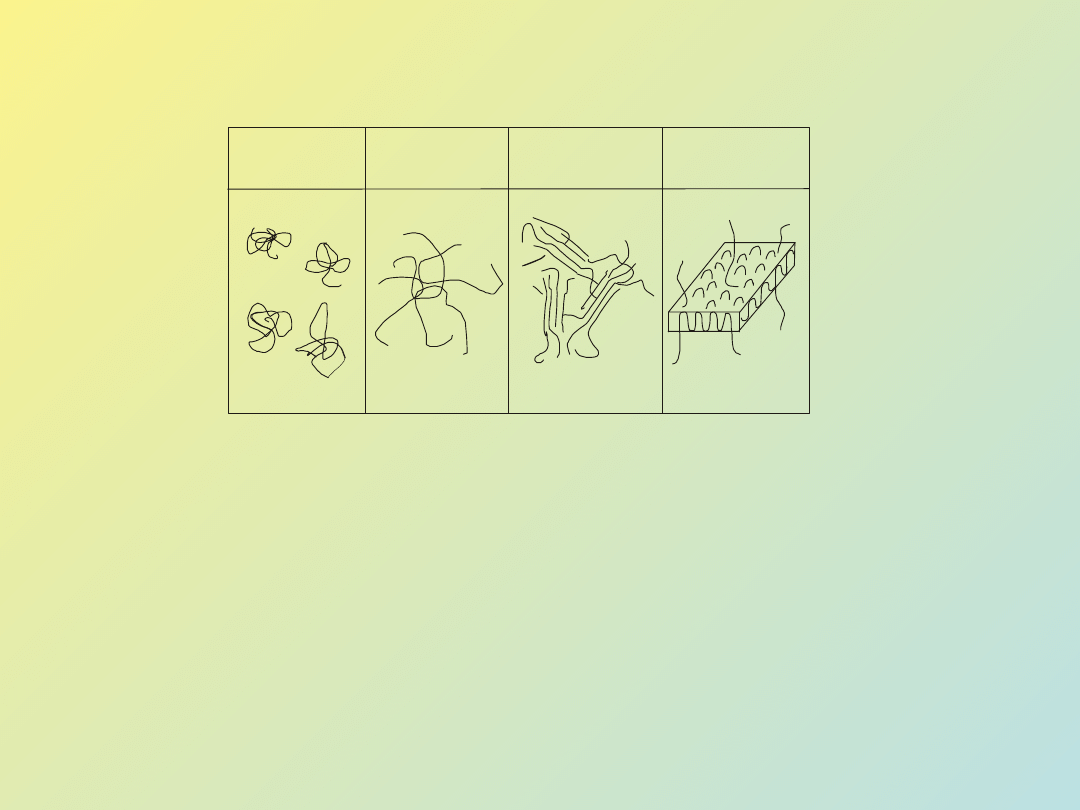
8
Struktury III rzędowe - są związane z tworzeniem się agregatów
A
B
C
D
A. Postać globularna (hemoglobina)
B. Struktura spaghetti
C. Micele frędzlowate
D. Płytkowe struktury lamelarne
E. Struktura superhelikoidalna

9
Makrocząsteczki w roztworach rozcieńczonych
1. Rola rozpuszczalnika w procesie rozpuszczania polega na
przezwyciężaniu sił van der Waalsa. Informacje o oddziaływaniach
międzycząsteczkowych uzyskuje się zatem z
badań rozpuszczalności.
2. Roztwór rozcieńczony < 1% (nie występują zderzenia
makrocząsteczek).
3. Termodynamiczny warunek rozpuszczalności to ujemna wartość
swobodnej
energii rozcieńczenia:
ΔF = ΔH – TΔS < 0
Gdzie:
ΔH – zmiana entalpii
ΔS – zmiana entropii > 0 i małe ΔS = k
lnW
ΔH powinno być małe
2
1
2
2
/
1
2
2
2
/
1
1
1
v
v
V
E
V
E
H
Gdzie:
E
1
, E
2
– energia parowania (kohezji) rozpuszczalnika i polimeru
V
1
, V
2
– objętości molowe rozpuszczalnika i polimeru
v
1
, v
2
– ułamki objętościowe rozpuszczalnika i polimeru
E/V – gęstość energii kohezji (CED)
(E/V)
1/2
=
–
parametr rozpuszczalności

10
H = (
1
-
2
)
1/2
v
1
v
2
H
min
gdy
1
2
[(kcal/cm
2
)
1/2
]
Rozpuszczalnik
1
Polimer
2
Heksan 7,3
PE 7,9
Ksylen 8,8
PP 8,1
Benzen 9,2
PS 9,1
Aceton 10,0
PCW 9,7
Etanol 12,7
PW 9,8
Woda 23,4
Poliamid 13,6
Najbardziej prawdopodobnym kształtem pojedynczej makrocząsteczki w roztworze są
statystyczne zbiory kłębków polimerowych.
Określa się ich:
a. średnie wymiary (h)
b. kształt
c. stopień skłębienia
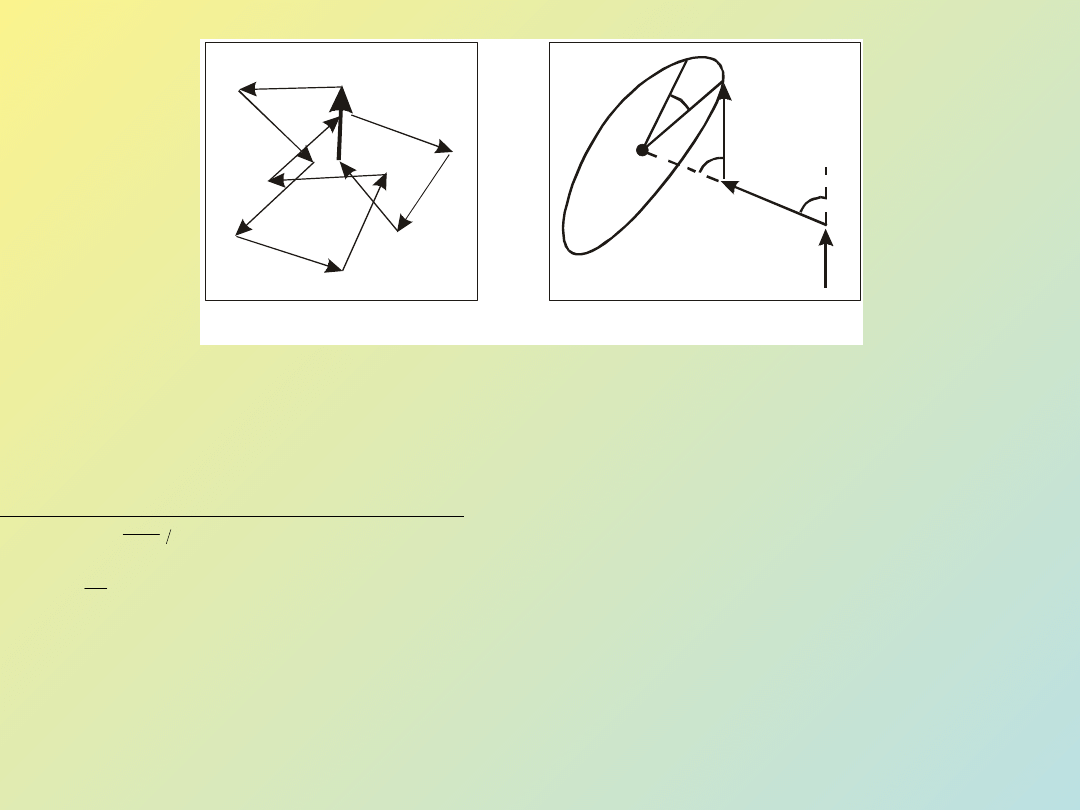
11
l
i + 2
l
i+ 1
l
i
l
i
h
Q
a )
b )
Q
Rys. 1 Struktura kłębka w roztworze: a) łańcucha swobodnie związanego, b)
łańcucha o ograniczonej giętkości l
i
– średnia długość wiązania, h – średnia
wielkość kłębka, odległość między końcami łańcucha,
, – kąty: walencyjne,
rotacji
1. Łańcuch swobodnie związany
2
1
2
h
h
2
nl
h
gdzie:
l – średnia długość wiązania,
n – liczba wiązań.
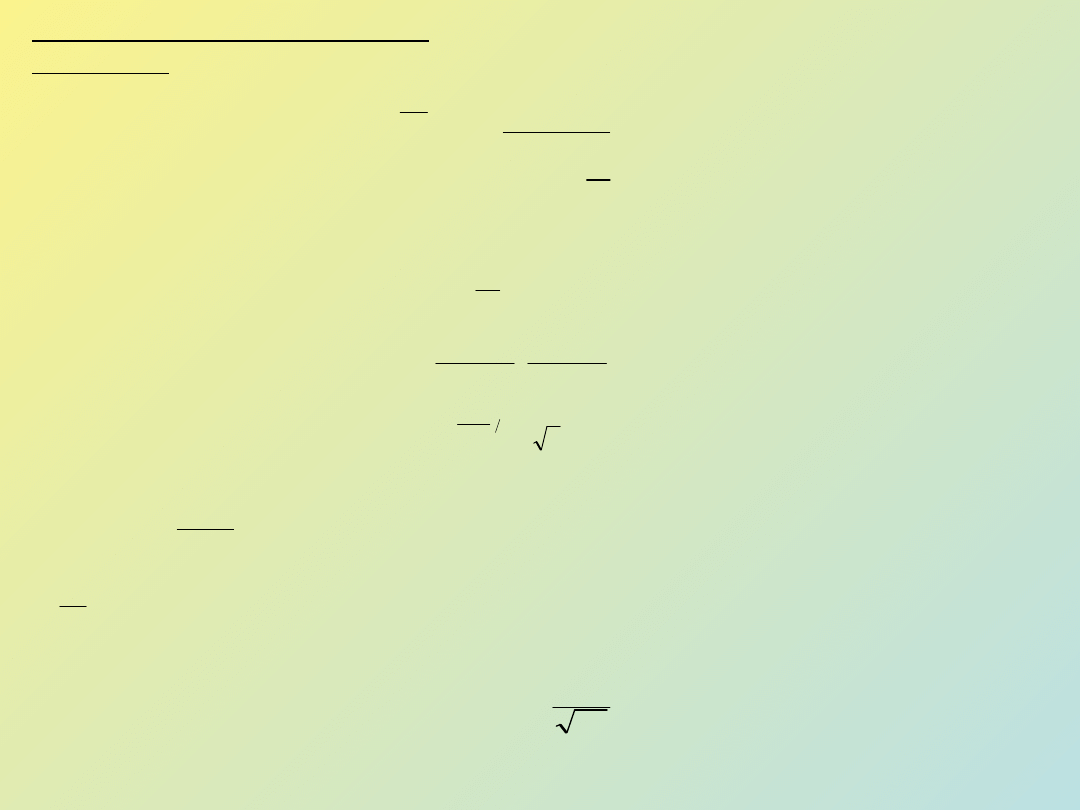
12
2. Łańcuch o ograniczonej
giętkości
gdy stałe, dowolne, wtedy:
cos
1
cos
1
2
2
nl
h
np. dla PE = 109
o
28’
2
2
2nl
h
gdy stałe, stałe lub przyjmuje określone wartości
2
2
2
n
h
cos
1
cos
1
cos
1
cos
1
2
2
l
G
R
h
6
2
1
2
gdzie:
2
/
1
2
i
i
i
G
m
r
m
R
średni promień bezwładności, który określa się metodą rozpraszania światła.
2
h
= const M
1/2
(dla rozpuszczalnika teta),
średnia gęstość kłębka
M
1
const

13
Dobry rozpuszczalnik:
1. Oddziaływania polimer – rozpuszczalnik są większe niż polimer
– polimer
2. Skłębiona cząsteczka ulega rozprostowaniu
3. Temperatura Q, w której oba oddziaływania są równe
4. - współczynnik oddziaływania polimer – rozpuszczalnik (mały)
Układy skondensowane polimerów
(Żele, stopy,
stanstały)
a )
b )
c )
Rys. 2 Schemat procesu powstawania żelu
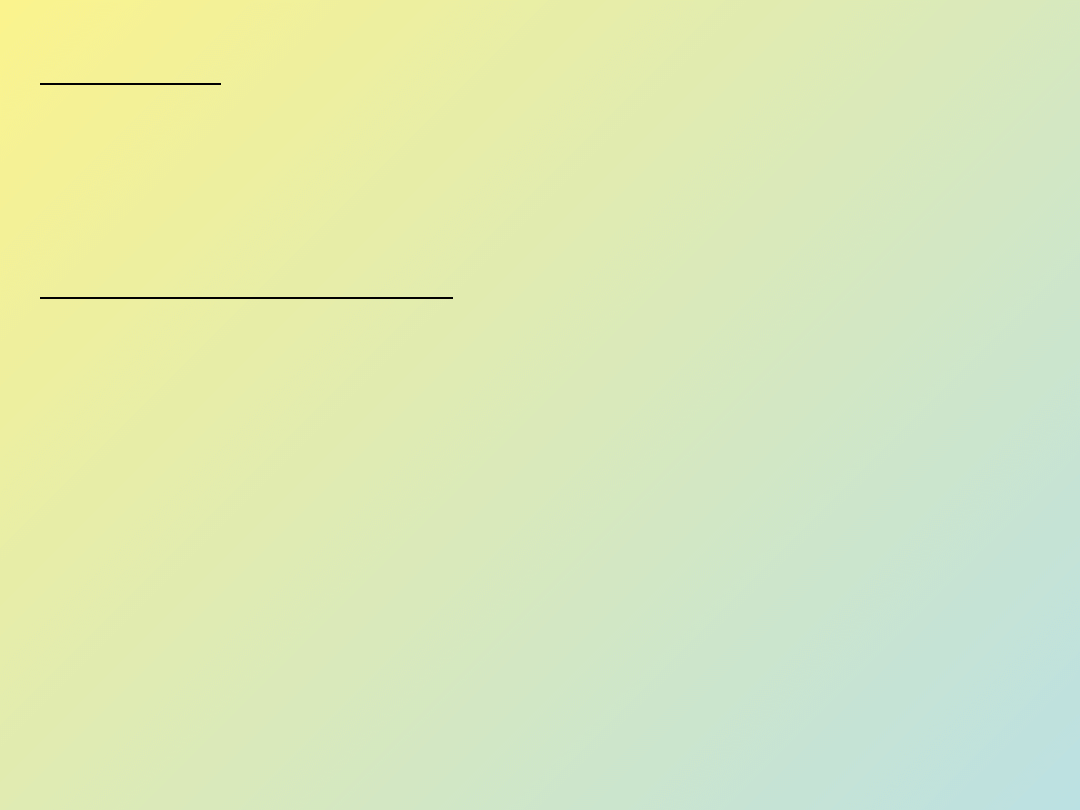
14
Ad. 1 - Żele
Jest to stan, w którym stężenie polimeru w roztworze jest tak duże, że
makrocząstaczki nie mogą poruszać się swobodnie. Odbywają się ruchy
segmentalne w przestrzeniach wypełnionych rozpuszczalnikiem.
Sposoby otrzymywania żelu
• wzajemne przenikanie i splątanie kłębków (PE)
• kontrakcja kłębków – struktury komórkowe (PS)
Usieciowanie żelu – żel makroskopowy
siły van der Waalsa – żele nietrwałe
wiązania chemiczne (kowalentne) – żele trwałe
(np. PS usieciowany dwu-winylobenzenem).
15
próbki
masa
czona
nierozpusz
masa
próbki
j
wysuszone
objętość
próbki
j
spęcznione
objętość
Charakterystyka żelu – pęcznienie żelu
a. Frakcja żelu =
b. Współczynnik pęcznienia =
c. Gęstość sieciowania wyraża liczbę moli segmentów łańcucha między dwoma sieciującymi
wiązaniami na 1 cm
3
.
d. Współczynnik sieciowania, wskaźnik sieciowania - liczba jednakowo usieciowanych na
wagowo i liczbowo
średnią cząsteczkę polimeru.
Pęcznienie usieciowanego polimeru jest związane z liczbowo średnią masą cząsteczkową
segmentu polimeru zawartego między dwoma sieciującymi wiązaniami.
Własności mechaniczne żeli uzależnione są od budowy chemicznej cząsteczek
Ad. 2 - Stopy
a. liczne zetknięcia i splątania - sieci nieskończone
b. węzły sieci
• energetyczne – trwałe wiązania (zlokalizowane)
• typu splątanego (nie zlokalizowane)
• kontaktowe
16
Ad. 3 - Stan stały
1. Polimery amorficzne (bezpostaciowe) – stan nieuporządkowany
2. Polimery krystaliczne – istnienie obszarów dużego uporządkowania
– krystality, monokryształy (b. rzadko)
Ad.1. Polimery bezpostaciowe występują w trzech stanach:
• szklistym (zamrożenie ruchów segmentalnych) > 10
11
• wysokoelastycznym (kauczukopodobnym) = 10
7
• lepkopłynnym (ciekłym) η<10
7
T
g
– temperatura przejścia z a do b (T
g
jest właściwością fazy amorficznej).
T
m
– temperatura płynięcia (lub topnienia) z b do c (T
m
jest właściwością fazy krystalicznej)
Temperatury T
g
i T
m
zależą od:
• giętkości łańcucha (skład i budowa łańcucha) – oddziaływania wewnątrzcząsteczkowe.
• oddziaływania międzycząsteczkowe (energia kohezji).
Segment jest elementem łańcucha o pewnej liczbie merów zdolnym do wykonywania
niezależnych ruchów kinetycznych powyżej T
g
.

17
W T
g
nie jest osiągany stan równowagi termodynamicznej.
Tg przejście termodynamiczne II rzędu
Entalpia swobodna G
1
= G
2
S
1
= S
2
V
1
= V
2
T
C
T
S
T
G
p
p
p
2
2
V
p
V
p
G
T
T
2
2
V
T
V
T
p
G
p
2
*
T
m
to przejście termodynamiczne I rzędu G
1
= G
2
S
T
G
p
2
1
S
S
V
p
G
T
2
1
V
V
S oraz V – pierwsze pochodne G zmniejszają się skokowo.
Parametry, takie jak C
p
(ciepło właściwe), β
(ściśliwość), α (współczynnik
rozszerzalności cieplnej)
– drugie pochodne G zmieniają się skokowo.
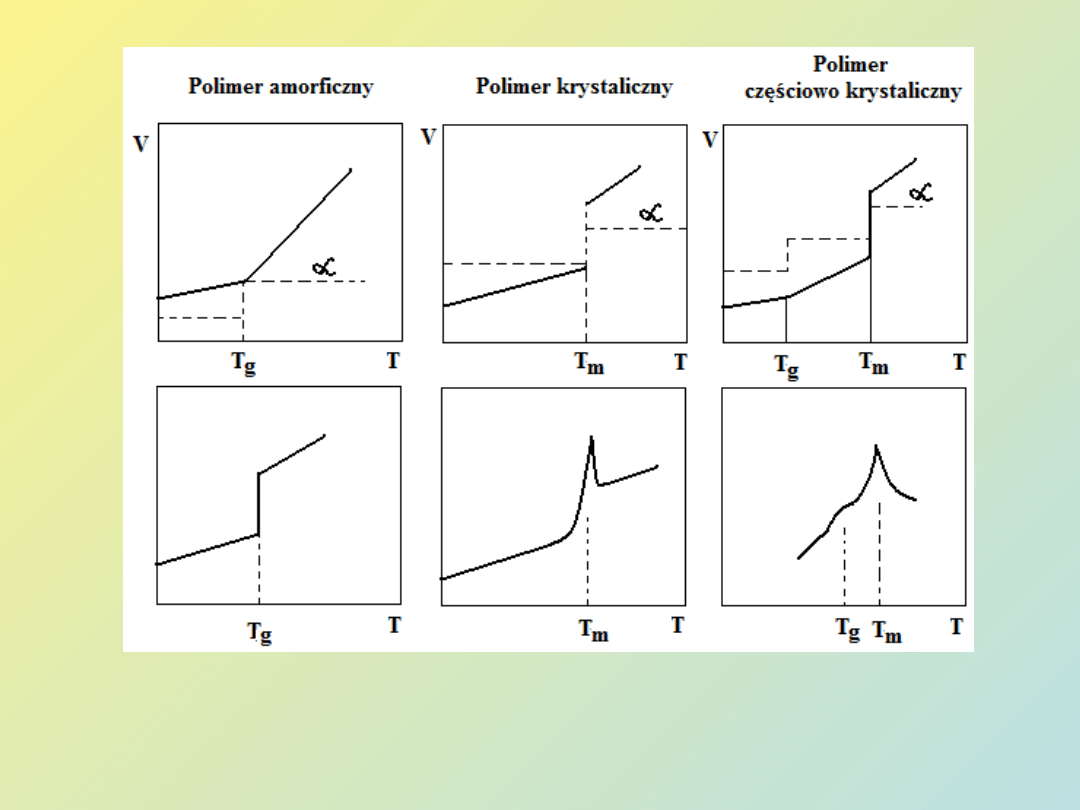
18
Rys. 3 Schematy opisujące zależność objętości V oraz ciepła właściwego C
p
od temperatury
T pokazujące oznaczenie temperatur przeisc fazowych T
g
i T
m
.
C
p
C
p
C
p
19
Poniżej T
g
polimery są twarde i kruche (tylko drgania rotacyjne i
oscylacyjne poszczególnych grup). Powyżej T
g
polimery są miękkie i
giętkie.
Temperatury T
g
i T
m
polimeru zależą od:
• Usieciowania polimeru liniowego (wzrost T
g
i T
m
)
• Kopolimeryzacji
• Zastosowania plastyfikatorów i antyplastyfikatorów
• Od masy cząsteczkowej M.
• Usztywnienia łańcucha (np. grupy fenylowe).
• Występowania grup polarnych i dużych podstawników.
Polimery krystaliczne
Charakterystyka struktury krystalicznej
1. budowa sieci przestrzennej krystalitów tj. rodzaj i wielkość komórki elementarnej
2. wymiary krystalitów i innych form morfologicznych (sferolitów, fibryl).
3. orientacja przestrzenna krystalitów
4. stopień krystaliczności polimeru (ułamek wagowy lub objętościowy fazy krystalicznej) – X
20
Zdolność polimerów do krystalizacji
1. Regularność budowy łańcucha (łańcuchy stereoregularne),
symetryczność podstawników, polarność.
2. Obecność rozgałęzień obniża zdolność do krystalizacji, np. PE
wysokociśnieniowy
– rozgałęziony ma niższe X niż PE niskociśnieniowy, liniowy.
3. Brak usieciowania
4. Wystąpienie sił zewnętrznych – orientujących
5. Skłonność do tworzenia wiązań wodorowych
Krystalizacja z roztworu lub ze stopu
Krystalizacja pierwotna i wtórna
Krystality – kryształy molekularne
Występują: siły van der Waalsa
Wiązania wodorowe
Siły jonowe
Kinetyka krystalizacji obejmuje dwa procesy
• nukleację (powstawanie zarodków krystalicznych)
• wzrost kryształu (równanie Avramiego)

21
Zależność własności polimeru od struktury i morfologii.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
Wyszukiwarka
Podobne podstrony:
2008 pojecia podstawoweid 26547 ppt
1b Pojęcia podstawoweid 18683 ppt
1a Pojęcia podstawoweid 18648 ppt
1 podstawowe pojecia i definicjeid 9573 ppt
01 Podstawowe pojęcia patofizjologiiid 2628 ppt
03 Wykład - Statystyka WZ, PODSTAWOWE POJĘCIA STATYSTYKI
01 E CELE PODSTAWYid 3061 ppt
Metodyka Obiektowa pojęcia podstawowe
Pojęcia podstawowe, excel
Pojęcia podstawowe, Studia, Prawo, Prawo Kanoniczne
1 pojecia podstawoweid 8796
Podstawy turystyki 10.03.12, II semestr, Podstawy turystyki
Podstawy turystyki 10.03.12, II semestr, Podstawy turystyki
Szablon 03, WAT, SEMESTR VI, podstawy zabezpieczeń sieci, lab
1 Kancelaria współczesna pojęcia podstawowe
Pojęcie, Podstawy ubezpieczeń, Podstawy ubezpieczeń
Pojęcia podstawowe w układach trójfazowych, POLITECHNIKA LUBELSKA w LUBLINIE_
Z chaosu, Ergonomia-pojecia podstawowe, 1-1
więcej podobnych podstron