
Choroby tkanki łącznej

Choroby układowe tkanki łącznej
Definicja:grupa luźno powiązanych ze sobą chorób wielonarządowych,
wtórnych do procesów autoimmunologicznych o nieustalonych
przyczynach, charakteryzujących się występowaniem zwyrodnienia
włóknikowatego w obrazie histopatologicznym
Podział:
Toczeń rumieniowaty układowy (Systemic lupus \
erythematodes - SLE)
Twardzina układowa (sclerodermia systemica – SSc)
Zapalenie wielomięśniowe dorosłych (Polymyositis – PM)
Zapalenie skórno-mięśniowe (Dermatomyosistis –DM)
Mieszana choroba tkanki łącznej (Mixed connective tissue
disease –MCTD)
Reumatoidalne zapalenie stawów (Rheumatoid arthritis –
RA)
Podgrupa seronegatywnych spondyloartropatii
Zespół Sjögrena

Toczeń rumieniowaty układowy (SLE)
– modelowa choroba autoimmunologiczna
Etiopatogeneza
Europa – zachorowalność roczna 8 przypadków na 100 000 mieszkańców
USA – 5.56 przypadków na 100 000 mieszkańców w latach 1980-1992
1.51 – 1950-1979
7.3 - 196 5-1995
90% kobiety między 25 a 40 rokiem życia ; K:M = 9:1
Czynniki wiodące do zachorowania
Hormonalny (estrogeny)
Genetyczny
Środowiskowy

Toczeń rumieniowaty układowy
Uwarunkowania genetyczne
Haplotypy:
A10/B8/DR2/C4A4/C4B2/BFS ( związane z C2 –niedoborem heterozygotą)
B8/DR3/DQw2/C4QO (początek po 35 rż, związane z anty-Ro i brakiem C4)
B7/Cw7/DR2/DQw1 (początek przed 25 rż, zajęcie nerek, ciązki przebieg
Pojedyńcze geny HLA
Homozygoty z niedoborem wczesnych frakcji dopełniacza
C2, C1q, C1r, C4, C1 Inh DR3 i DR2
Brak C4A
Dr2 z DQw1 lub DQB1*0502
TNF-alfa – allele kodujące niskie wydzielanie (DR2, DQw1)
Nie HLA geny predysponujące do SLE
Geny dla TCR (związane z anty-Ro)
Geny łańcuchów ciężkich (Gm) i łańcuchów lekkich (Km) Ig
Delecja genu Ig Vh Hkumhv 3005
3 – 4 geny predysponuja do zachorowania na SLE

SLE
Czynniki środowiskowe
Potwierdzone:
Promieniowanie ultrafioletowe
Prawdopodobne
Dieta: wysokokaloryczna, bogata w tłuszcze nasycone
Infekcje: retrowirusy, liposacharydy bakteryjne
Ekspozycja na leki:
Hydralazyna
Procainamid
Izoniazid
Hydantoina
Chlorpromazine
Methyldopa
d-penicillamina
Interferon - alfa

SLE
Promieniowanie ultrafioletowe B
Działanie UV na DNA zwiększa powstawanie dimerów tymidyny i stąd DNA
bardziej immunogenne
Przesunięcie antygenów Ro, La i U1RNP na powierzchnię keratynocytów,
gdzie mogą być związane przez uczulone limfocyty T lub przeciwciała
Uszkodzenie komórek przez UV powoduje uwalnianie białek szoku
termicznego, które biorą udział w aktywacji autoreaktywnych limfocytów
T.
Interakcja między czynnikami genetycznymi i środowiskowymi wywołuje
nieprawidłową odpowiedź immunologiczną obejmująca:
Aktywność nadreaktywnych limfocytów B i T
Niesprawność układów regulujących odpowiedź immunologiczna
Efekt: produkcja patogennych przeciwciał, kompleksów immunologicznych i
limfocytów T , uszkadzających tkanki i wywołujących objawy kliniczne SLE

Toczeń rumieniowaty układowy
Właściwości nadreaktywnych limfocytów B
Liczba limfocytów B produkujących immunoglobuliny znacznie
podwyższona (nawet do 50x)
Podwyższona całkowita liczba
Zaburzenia mogą wyprzedzać objawy SLE
Produkowane Ig reagują z licznymi własnymi antygenami
Odpowiedź na antygeny egzogenne może być obniżona
Limfocyty B łatwo ulegają poliklonalnej aktywacji, co może wyprzedzać
autoantygenową-swoistą aktywację
Niektóre limfocyty B są aktywowane przez swoiste autoantygeny
(produkcja autoprzeciwciał jest ograniczona uwarunkowaniami
genetycznymi)
Nadreaktywności limfocytów B sprzyja podwyższony poziom cytokin (IL-6,
IL-10)

SLE
Geny HLA a autoprzeciwciała
Anty –dsDNA
DR2, DR3
Anty-Ro, anty-La
DR2, DR3, DQw1/DQw2
Anty-U1-RNP
DR4
Anty-U1-70-kd
DR4, DR2
Anty-Sm
DR2
Antyfosfolipidowe
DR4, DR7

SLE
Autoprzeciwciała
Zasadniczy immunogen: nukleosom
Nukleosomy uwalniane do krążenia w trakcie apoptozy komórek stymulują
Th1 i limfocyty B, co w efekcie prowadzi do produkcji anty-DNA, wchodząc
w skład kompleksów immunologicznych zapoczatkowują zmiany w
nerkach przez wiązanie się z proteoglikanami błony podstawnej kłębuszka
oraz penetrację do wnętrza komórek
uszkodzenie i apoptoza komórek kłębuszka

SLE
Właściwości nadreaktywnych limfocytów T
Obniżona liczba całkowita limfocytów T
Obecność autoprzeciwciał wiążących się z limfocytami T
i upośledzającymi ich aktywność
Zaburzenie wczesnej aktywacji
Ekspresja markerów aktywacji (IL-2R, DR, DP) a też wyższy
poziom w surowicy IL-2, s-IL-2, IFN-gamma
Upośledzona odpowiedź proliferacyjna na mitogeny
Udział w ciągłej produkcji przeciwciał
Autoprzeciwciała po przetworzeniu na peptydy są
prezentowane na limfocytach B wraz z HLA i aktywują
własne limfocyty Th do dalszej produkcji przeciwciał

SLE
patogenne przeciwciała
Należą do klasy G (zwykle), wykazują dużą awidność do własnych
antygenów, ograniczoną swoistość
Wykazują zdolność bezpośredniego wiązania z tkanką docelową
(błony erytrocytów, płytki........)
Mają zdolność wiązania komplementu
Posiadają ładunek dodatni, ułatwiający przyleganie dp
polianionowych regionów błony
Przeciwciała naturalne:
IgM, słaba awidność do własnych antygenów, wielospecyficzne,
reagują krzyżowo z licznymi antygenami.

Kompleksy immunologiczne a SLE
Nadmiar KI (duża ilość przeciwciał lub antygenów)
Właściwa wielkość KI
duże – fagocytowane przez makrofagi
bardzo małe – wydzielane z moczem
średniej wielkości – najłatwiej odkładają się w tkankach
Tropizm tkanek do KI
Ładunek dodatni KI + ładunek ujemny tkanek
Reakcje przeciwciał in situ
Zmniejszony klirens i katabolizm KI

SLE
Kryteria klasyfikacyjne ACR zaktualizowane w 1997
roku
(Lupus 1999, 8, 8, 586-595)
1.
Rumień na twarzy
2.
Rumień krążkowy
3.
Nadwrażliwość na światło słoneczne
4.
Owrzodzenia w jamie ustnej
5.
Zapalenie stawów bez nadżerek
6.
Zapalenie opłucnej lub osierdzia
7.
Zmiany w nerkach (białkomocz >0.5g/d lub wałeczki w moczu)
8.
Zmiany w układzie nerwowym
9.
Zaburzenia hematologiczne (niedokrwistość hemolityczna z
retikulocytozą/leukopenia <4000/mm
3
/ limfopenia <1500/mm
3
/małopłytkowość <100 000/mm
3
10.
Zaburzenia immmunologiczne (Abs: nDNA, Sm, ACA, LAC,
VDRL)
11.
Obecność przeciwciał przeciwjądrowych

SLE
Podgrupy kliniczne
Podostry toczeń skóry (subacute cutaneus lupus erythematosus –
SCLE)
Objawy typowe:
wysypka grudkowa, łuszcząca się lub rumień krążkowy
Objawy rzadkie:
zajęcie narządów wewnętrznych
2/3 chorych – przeciwciała anty-SSA/Ro

Podgrupy kliniczne tocznia
Toczeń indukowany lekami
Obraz kliniczny: dominacja zmian skórnych, ból stawów,
zapalenie błon surowiczych
Rzadkie: zajęcie nerek i układu nerwowego
Anty-nDNA – (-), przeciw białkom histonowym – (+), C3, C4 –
norma
Toczeń noworodków
SSA/Ro – w klasie G Ig; przejściowe: zmiany skórne (wysypka /
rumień), zaburzenia hematologiczne, powiększenie wątroby,
śledziony
Blok przedsionkowo-komorowy różnego stopnia!

SLE
Nerki – 90% chorych
(klasyfikacja zmian zapalnych wg Światowej Organizacji Zdrowia)
Klasa I
- zmiany minimalne/norma
Klasa II
- zmiany ograniczone do mezangium
Klasa III
- ogniskowe, rozplemowe zapalenie
klębuszków nerkowych
Klasa IV
- rozlane, rozplemowe zapalenie kłębuszków
nerkowych
Klasa V
- błoniaste zapalenie kłębuszków nerkowych
Klasa VI
- postępujące zapalenie kłębuszków
nerkowych ze stwardnieniem
Typ I – III – ustępują po włączeniu sterydów

SLE
Zmiany w ośrodkowym układzie nerwowym (75% chorych)
Od 1999 roku 19 różnych zespołów zostało włączonych do postaci
neuropsychiatrycznej SLE. (Lancet 2001, 357, 1027)
Jawna psychoza 5-15%
Udar mózgu 10-35%
Neuropatie nerwów czaszkowych
Poprzeczne zapalenie rdzenia
Jałowe zapalenie opon mózgowo-rdzeniowych
Ból głowy – 30 – 50%

Toczeń z zespołem Sjögrena
Zespół Pierwotny
-
klasyfikacja z 2002 roku
1. Objawy oczne
codzienne uczucie suchości > 3 miesięcy
nawracające uczucie piasku
sztuczne łzy . 3 x dziennie
2. Objawy oralne
codzienne uczucie suchości > 3 miesięcy
stały/ okresowy obrzęk ślinianek
popijanie suchych kęsów
Twierdząca odpowiedź na minimum jedno pytanie z kryteriów 1 lub
2

Toczeń z zespołem Sjögrena
Zespół Pierwotny
-
klasyfikacja z 2002 roku
3. Obiektywne objawy oczne = dodatni wynik 1 z 2 prób
test Schirmera 1 (<5mm/5min)
test z różem bengalskim (>4 punktów)
4. Badanie histopatologiczne ślinianek
(> 1. ognisko nacieku kk. limfoidalnych/4mm
3
tkanki)

Toczeń z zespołem Sjögrena
Zespół Pierwotny
-
klasyfikacja z 2002 roku
5. Dowody zajęcia ślinianek = dodatni wynik 1 z 3 testów
niestymulowana sialometria (<1.5ml / 15min)
sialografia ślinianek przyusznych – rozstrzenia przewodów (punkcikowe,
jamiste lub destrukcja)
scyntygrafia ślinianek – opóźnienie wychwytu lub wydalania, zmniejszenie
stężenia znacznika
6. Autoprzeciwciała w surowicy –przeciw: SS-A/Ro, SSB/La

Toczeń z zespołem antyfosfolipidowym
Kryteria diagnostyczne zespołu antyfosfolipidowego (1998r.)
Kryteria kliniczne:
Duże: zakrzepica naczyń, straty ciąż
Małe: Livedo reticularis, zmiany na zastawkach serca, pląsawica,
małopłytkowość, krwotok do nadnerczy i inne
Kryteria laboratoryjne
Duże: aCL-IgG – wysoki poziom, beta2GPI, LAC
Małe: aCL-IgM, aCL-IgG – średni, niski poziom, VDRL (+)

Lupus antykoagulant (LA)
Toczniowy czynnik przeciwkrzepliwy
Charakterystyka
Przeciwciało w klasie G lub M immunoglobulin przeciw ujemnie
naładowanym fosfolipidom występującym w formie
heksagonalnej.
W pojęciu LA zawiera się bardzo heterogenna grupa przeciwciał
antyfosfolipidowych (APL), które cechuje to, że in vitro
przedłużają czasy krzepnięcia osocza zależne od fosfolipidów np.
czas koalinowo-kefalinowy (APTT)
Skład
Najczęściej: przeciwciała przeciw kardiolipinie
Rzadziej: przeciwciała przeciw: fosfatydyloserynie,
fosfatydylocholinie, fosfatydyloinozytolowi, kwasowi
fosfatydowemu
Nie wszystkie APL przedłużają in vitro testy krzepnięcia zależne
od fosfolipidów

Tylko przeciwciała antyfosfolipidowe należące do grupy ACL
powodują przedłużenie czasów krzepnięcia w obecności beta –2
– GP1
Inne przeciwciała nie potrzebują żadnego kofaktora do
blokowania kompleksu protrombinazy
Niektóre przeciwciała mają aktywność LA
Beta –2- glikoproteina 1 (apolipoproteina H)
białkowy
kofaktor niezbędny do wiązania ACL

LA w diagnostyce klinicznej
Toczeń rumieniowaty układowy – 20-60% niezależnie od stopnia
zaawansowania zmian
Sklerodermia
RZS
Periarteritis nodosa
Zespół Sneddona
Colitis ulcerosa
Polymyalgia rheumatica
Infekcje (AIDS)
Sarkoidoza
Nowotwory
Leki: chlorpromazyna, prokainamid
Osoby zdrowe

Inne przeciwciała w zespole antyfosfolipidowym
Przeciwciała przeciw protrombinie, przeciw białku C i S
Przeciwciała przeciw białkom powierzchni komórek śródbłonka:
przeciw trombomodulinie
Przeciwciała przeciwpłytkowe
Przeciwciała przeciw anneksynie V
Przeciwciała narządowo nieswoiste

SLE
Postępowanie diagnostyczne
Badania podstawowe: OB., CRP, proteinogram, morfologia z rozmazem
Badania serologiczne zawarte w pkt. 10 i 11 kryteriów ARA oraz
dodatmowo: ACL i/lub LAC, SS-A/Ro, SS-B/La
Chorzy gorączkujący: posiewy krwi i moczu
Potwierdzenie zmian narządowych
Skóra, błony śluzowe, oczy: LBT, badanie okulistyczne, laryngologiczne
Narząd ruchu: rtg stawów
Układ oddechowy: rtg płuc, ew: scyntygrafia/TK,
testy
czynnościowe, badanie płynu opłucnowego
Układ sercowo-naczyniowy: EKG, ECHO, przepływy
naczyniowe
Układ moczowy
Układ nerwowy

Leczenie tocznia rumieniowatego układowego
1.
Zmiany skórne, zapalenie stawów
Niestrydowe leki p/zapalne, leki antymalaryczne,
glikokortykosteroidy miejscowo, kremy z filtrem UVA/UVB
2.
Zapalenie błon surowiczych
Prednison 1mg/kg m.c. (4-6 tygodni) lub Metylprednisolon
iv.0.5-1.0g (pulsy – 3 dni) co 4-6 tygodni, w przerwie Prednison
20-30mg/dz
Brak poprawy: Azatiopryna (Imuran) 1-2mg/kg m.c./dobę lub
Cyklofosfamid (Endoksan) 1g/miesiąc (i.v lub p.o)
3.
Zajecie nerek – jak 2.; proteinuria >1 g/dobę Metylprednisolon
i.v 0.5-1.0g (pulsy 3 dni), co 4-6 tygodni, w przerwie Prednison
20-30mg/dz oraz Cyklofosfamid 1g/dobę (i.v. lub p.o) przez 6
miesięcy, potem 1g co 3 miesiące przez 2 lata; w razie
przeciwwskazań: Azatiopryna 1-2mg/kg mc /dobę
Brak poprawy:IVIG

Twardzina układowa
Systemic sclerosis (SSc)
Choroba układowa, występująca głownie u kobiet w średnim
wieku, charakteryzująca się stwardnieniami i zanikami skóry,
tkanki podskórnej i zajęciem narządów wewnętrznych.
Czynniki środowiskowe: związki krzemu, silikon, związki
toksyczne: chlorek winylu, trójchloroetylen i inne(bleomycyna,
tryptofan)
Dowodem na tło autoimmunizacyjne jest występowanie zmian
skórnych w przebiegu przewlekłej reakcji przeszczepu przeciwko
gospodarzowi (GVH) po przeszczepie szpiku

Twardzina układowa
Pierwotnym zjawiskiem w SSc wydaje się być uszkodzenie
(toksyczne, autoimmunologiczne) komórek śródbłonka
mikrokrążenia w następstwie czego powstają nacieki
okołonaczyniowe (CD4, makrofagi), wytwarzające cytokiny
prozapalne (IL-1, IL4, IL-6, TNF-alfa) oraz czynniki
uszkadzające naczynia, co prowadzi do powstawania
mikrozakrzepów i uwalniania z płytek PDGF i TGF-beta
Cytokiny:IL-1, IL-4, TGF-beta, TNF-alfa pobudzają
wytwarzanie kolagenu typu I, III , VI i VII przez fibroblasty
Odzwierciedleniem procesu włóknienia jest wzrost stężenia
aminopropeptydu kolagenu I i III w surowicy

Twardzina uogólniona
Autoprzeciwciała
Przeciw topoizomerazie I (Scl-70)
Przeciw centromerom (ACA)
Nieswoiste przeciwciała przeciw antygenom jąderkowym (przeciw
polimerazie RNA I i II, białku – fiblarynie)

Sklerodemia
Objawy kliniczne
Postaci kliniczne
rozlana (diffuse SSc) z uogólnionymi stwardnieniami w obrębie
całej skóry; Scl-70 Ab – 85%, ACA -0%, przebieg szybki, ciężki
ograniczona (limited SSc) – zwana tez akrosklerodermią – z
zajęciem twarzy i odsiebnych części kończyn górnych; Scl-70 Ab –
50%, ACA-20%, przebieg wolniejszy, łagodniejszy

Sclerodermia
Objawy kliniczne
Objaw Raynauda
Sklerodaktylia (przykurcze i stwardnienie palców) oraz nadżerki
Teleangiektazje
Zmiany narządowe: przełyk – rozszerzenie, atonia, dysfunkcja;
serce – arytmia, zapalenia wsierdzia, zaburzenia przewodzenia;
mięśnie – polymyositis; płuca – zwłóknienie śródmiąższowe,
zmniejszona pojemność życiowa; nerkii – zwłóknienie,
nadciśnienie złośliwe; kości – bóle stawowe, osteoliza, kalcynoza
Postać ISSc – zespół CREST (Calcinosis, Raynaud, Esophagus,
Sclerodactylia, Teleangiectsia)

Sclerodermia
leczenie
Penicylamina – 250-500 mg/dobę (hamowanie wiązań krzyżowych
w kolagenie)
IFN – gamma – hamuje syntezę kolagenu in vitro (ale zapalenie
naczyń!)
Kalcytriol (pochodna witaminy D3) ; UVA – próby
W stanie ciężkim: leki immunosupresyjne: metotrexat,
cyklosporyna, cyklofosfamid
Kortykosterydy – u chorych ze zwłóknieniem płuc i nasilonymi
objawami mięsniowymi

Zapalenie skórno-mięśniowe
Dermatomyositis - DM
Odmiana zapalenia wielomięśniowego (polymyositis) z
charakterystycznymi zmianami skórnymi; humoralnie
uwarunkowana mikroangiopatia
Immunopatologia
HLA-DR3, DR7, DRw52, DRw53, DQ1, DQ2
Zakażenie: wirusy Coxsackie, ECHO, retrowirusy,
paramyksowirusy
Uszkodzenie śródbłonka – przeciwciała + aktywne
składowe dopełniacza C5-C9 (tzw.MAC –membrane attack
complex) + komórki cytotoksyczne

Zapalenie skórno-mięśniowe
Objawy kliniczne
Dzieci, dorośli (5-24rż, 45-64 r.ż)
Zmiany skórne poprzedzają objawy zapalenia mięsni
Zmiany rumieniowo-naczyniowe i obrzękowe twarzy z zajęciem
powiek (rzekome okulary), na ramionach, szyi, dekolcie (tzw.znak
V)
Zmiany rumieniowo-plamiste (objaw Gottrona) i grudkowe (grudki
Gottrona) na grzbietowych stronach rąk, w okolicy stawów
śródręcznopalicznkowych i międzypaliczkowych.
Wybroczyny w obrębie wałów paznokciowych – tzw. pętle
drzewkowate w badaniu kapilaroskopowym

Zapalenie skórno-mięśniowe
Objawy kliniczne
Osłabienie siły mięśniowej (pas barkowy i biodrowy)
Zajęcie mięśni przełyku i mięśnia serca, zaniki mięśni
Czasem zmiany skórne przypominające twardzinę
Odmiana: dermatomyositis amyopathica – typowe zmiany
skórne bez zajęcia mięśni
Odzwierciedleniem zmian w mięśniach jest: wzrost
stężenia enzymów mięśniowych - aldolazy, izoenzymu MM
fosfokinazy kreatyny, badanie EMG; zmiany
zwyrodnieniowo-naciekowe w wyc. z mięśnia (CD4, B
wokół naczyń w perimysium, zanik okołopęczkowy)
Mi-2Ab – skierowane przeciw helikazie, enzymowi
biorącemu udział w procesie transkrypcji

Zapalenie wielomięśniowe – polymyositis
Idiopatyczne miopatie
Zapalenie wielomięsniowe
Zapalenie skórno-mięśniowe
Postać dziecięca zapalenia skórno-mięśniowego
Zapalenie mięśni w przebiegu układowych chorób tkanki łącznej
Zapalenie mięśni towarzyszące chorobie nowotworowej
Wtrętowe zapalenie mięśni
Inne postaci miopatii zapalnych
zapalenie mięśni z eozynofilią
ogniskowe zapalenie mięśni
zapalenie mięśni oczodołu

Zapalenie wielomięśniowe
Nie występuje u dzieci
Badanie elektrofizjologiczne nie pozwala na zróżnicowanie
zapalenia skórno-mięśniowego od wielomięśniowego; dla
obu miopatii charakterystyczna jest obecność potencjałów
drżenia włókienkowego i dodatnich ostrych fal,
wykrywanych u 93-100% chorych (w obrębie mięśni
przykręgosłupowych w odcinku piersiowym)
Badanie histopatologiczne: komórki zapalne (CD8 i
makrofagi) otaczają i naciekają niezmienione martwiczo
włókna mięśniowe
Zdrowe włókna mięśniowe (w miejscach odległych od
nacieku) wykazuję ekspresję HLA klasy 1
We wczesnych postaciach – brak zmian histopatologicznych

Overlap Syndromes
Scleromyositis – dominacja zmian skórnych
twardzinopodobnych, HLA-DQA1, -DRB1, Scl70/PM1
Zespół antysyntetazowy – śródmiąższowe zwłóknienie płuc,
zmiany twardzinopodobne, HLA-DRw52, Jo-1Ab
Mieszana choroba tkanki łącznej (MCTD – mixed connective tissue
disease) – cechy twardziny układowej, tocznia układowego i
polymyositis,
przeciwciałą przeciw różnym peptydom U1RNP

Zespoły z polymyositis
Leczenie
Kortykosteroidy – poprawa po 3-6 miesiącach (leki pierwszego
rzutu)
Leki immunosupresyjne (metotreksat, azatiopryna)
Immunoglobuliny G we wlewach dożylnych (3 miesięczna próba);
IgG blokują odkładanie się dopełniacza
Cyklosporyna, cyklofosfamid (leki 3.rzutu)
Mykofenolan mofetilu (w Polsce stosowany po przeszczepie
narządów)
Dieta i styl życia
Ograniczenie spożycia soli, cukrów prostych i węglowodanów
złożonych
Suplementacja wapnia (1g/24h) i wit. D (400-800jm/24H)
Ćwiczenia z obciążeniem celem zmniejszenia ryzyka osteopenii

Reumatoidalne zapalenie
stawów

Klasyfikacja chorób reumatycznych
Amerykańskiego Towarzystwa Reumatologicznego
z 1983 roku
(Arthritis and Rheumatism 1983,1030)
I.
Układowe choroby tkanki łącznej
II.
Zapalenie stawów z tow. zapaleniem kręgosłupa
(spondyloarthritis)
III.
Choroby zwyrodnieniowe stawów (osteoarthrosis)
IV.
Zespoły „reumatyczne” towarzyszące zakażeniu
V.
Choroby metaboliczne i gruczołów dokrewnych
VI.
Nowotwory
VII.
Zaburzenia nerwowo-naczyniowe
VIII.
Choroby kości i stawów
IX.
Zmiany pozastawowe
X.
Różne choroby, którym towarzyszą objawy stawowe

Reumatoidalne zapalenie stawów (RZS)
Patogeneza
Predyspozycja genetyczna
DR1 (DRB1*010101, *0102,*010404,*010401,*01040)
DR4 (DRB1*0401,*0404,*0405,*0408)
DR6 (DRB1*140 2)
DR10 (DRB1*1001)
Teoria wspólnego epitopu – trzeci hyperzmienny region łańcucha
beta cząsteczek HLA wykazuje identyczną identyczną sekwencję
aminokwasów w pozycji 67-74 (70-74)
Wspólny epitop występuje u 80% chorych na RZS i u 50%
zdrowych (rasa biała)
Postać agresywna RZS – DR4 (DRB1*0401/*0401 lub *0401/*0401
Postać łagodna RZS – DR1

RZS
Patogeneza
Przypuszczalna stymulacja antygenowa:
Reakcja krzyżowa (zjawisko mimikry molekularnej)
Cząsteczki pochodzenia bakteryjnego (np. białka szoku
cieplnego) lub wirusowego (npgp110 –EBV) – homologia
aminokwasów z cząsteczkami HLA aktywacja limfocytów
Autoantygeny :
glikoproteina chrząstki o ciężarze cząsteczkowym 39 kDa,
białko wiążące łańcuch ciężki, białko zawierające cytrulinę

RZS
Immunopatologia
Krew obwodowa
Obniżenie odsetka subpopulacji limfocytów T (aktywna faza
choroby) w stosunku do limfocytów B; zmienne wartości
CD4:CD8, wzrost odsetka limfocytów z receptorami γδ
Hyperplastyczna błona maziowa: CD4, CD45RO, makrofagi,
komórki plazmatyczne

RZS
Immunopatologia - cytokiny
Cytokiny prozapalne
TNF-α (czynnik martwicy nowotworów), IL-1, IL-6 – monocyty/makrofagi ; TNF-α, IL-1
synowiocyty typu A i B prostoglandyna E
2
(PGE
2
)
metaloproteinazy (kolagenaza, żelatynaza, stromielizyny)
IL-6 – wzmaga produkcję metalopeoteinaz i PGE
2
, indukuje produkcję TNFαR, IL-1RA
Induktory odpowiedzi ostrej fazy
IL-1, TNFα –produkcja (wątroba): składniki dopełniacza, białka układu krzepnięcia, inhibitory
proteinaz
IL-6 – CRP ( białko C-reaktywne), SAA (amyloid A surowicy),
α1-antytrypsyna, fibrynogen
Duże stężenie CRP w płynie stawowym zwiększa aktywację makrofagów i stymuluje
wytwarzanie IL-1β i TNFα
Fragmenty proteolitycznej degradacji SSA odkładają się w postaci amyloidu w nerkach,
sercu.

RZS
Cytokiny a łuszczka stawowa
Łuszczka – wzmożona proliferacja fibroblastów pod wpływem
uwolnionych z synowiocytów typu A, komórek śródbłonka,
czynników wzrostowych: GM-CSF (czynnik stymulujący kolonie
granulocytarno-makrofagowe), FGF (czynnik wzrostu
fibroblastów), TGFβ (transformujący czynnik wzrostu)
Podtrzymywanie procesu chorobowego:
IL-12, IL-15, IL-18
Wzrost IL-8, MIP-1 = wzrost migracji granulocytów
i komórek jednojądrowych do tkanek

RZS
Przeciwciała
•
RF – czynnik reumatoidalny (rheumatoid factor) – autoprzeciwciało
rozpoznające część stałą (Fc) własnej IgG jest prduktem genów, które
uległy somatycznym mutajom i należy zarówno do klasy M, G jak i A
immunoglobulin. Kompleksy IgG-RFIgM poprzez uruchomienie kaskady
dopełniacza jak i IgG-RFIgG poprzez uruchomienie produkcji cytokin
prozapalnych w makrofagach są zaangażowane w proces zapalny.
Ujawniane u 50-80% chorych, czułość 79%, specyficzność 63%
•
Przeciwciała przeciw zdenaturowanemu kolagenowi typu II (klasa IgG3)
prowadzą do uszkodzenia tkanki poprzez wiązanie z powierzchnią
chrząstki i aktywację kaskady dopełniacza
•
Przeciwciała przeciwkeratynowe (AKA) reagujące z filagryną nabłonka
płaskiego (czułość – 36-59%, specyficzność 88-99%)

RZS
Autoprzeciwciała
Czynnik okołojądrowy (AFP – perinuclear factor) ) – przeciwciała
przeciw filagrynie nabłonka śluzówki policzka ; ujawniane u 49-
91% chorych, specyficzność 73-99%
Przeciwciała przeciw cytrulinie (CCP – antibodies against cyclic
ctrullinated peptide) – specyficzność – 97%, czułość 80%; CCP-Ab
(+) – nasilone zmiany radiologiczne
Inne przeciwciała: ANCA, ANA, SMA, ACA i inne
przeciwfosfolipidowe

Reumatoidalne zapalenie stawów (RZS)
Kryteria diagnostyczne ARA zrewidowane w 1987 roku
Kryteria
Uwagi
1.
Sztywność poranna
Czs trwania >1 h
2.
Zapalenie 3 lub >stawów Jednoczasowy obrzęk lub wysięk 3 st.
3.
Zapalenie stawów ręki
1 staw śódręczno-palcowy (MCP) lub
międzypaliczkowy bliższy (PIP) oraz
staw nadgarstkowy
4.
Symetryczne zapalenie Jednoczasowość, obustronność
stawów
5.
Guzki reumatoidalne
Nad wyniosłościami kostnymi po stronie
wyprostnej lub w okolicach stawów
6.
RF (+)
Metoda – w kontroli nie więcej niż
5%
7.
Zmiany radiologiczne
Obecność nadżerek i osteoporozy
typowe dla RZS
okołostawowej (przednio-tylny rtg)

RZS
Cel postępowania z chorymi na RZS
Zapobieganie/kontrolowanie uszkodzenia stawów
Zapobieganie utracie funkcji
Ustąpienie lub zmniejszenie bólu
Dążenie do uzyskania całkowitej remisji
(kinezo-, fizyko, psychoterapia)
Wczesny etap postępowania
Szybkie ustalenie rozpoznania
Ocena stanu zdrowia i aktywności choroby
Ustalenie prognozy
Wczesne zastosowanie najskuteczniejszego leczenia

RZS
Postępowanie
Przed włączeniem leczenia należy ustalić:
stopień nasilenia bólu
czas trwania sztywności porannej
liczbę bolesnych i obrzękniętych stawów
ograniczenie funkcji

RZS
Kryteria prognostyczne aktywnej fazy choroby
Aktywne zapalenie stawów (>6 obrzękniętych i bolesnych stawów)
OB>30
CRP >8mg/l (metoda nefelometryczna)
RF
Kryteria złej prognozy
Młody wiek
Wysoki poziom RF
Obrzęk ponad 20 stawów
Jeden z objawów : guzki reumatoidalne
zespół Sjögrena
śródmiąższowe zapalenie płuc
zapalenie naczyń, osierdzia
zespół Felty’ego
Arthritis Reum. 2002
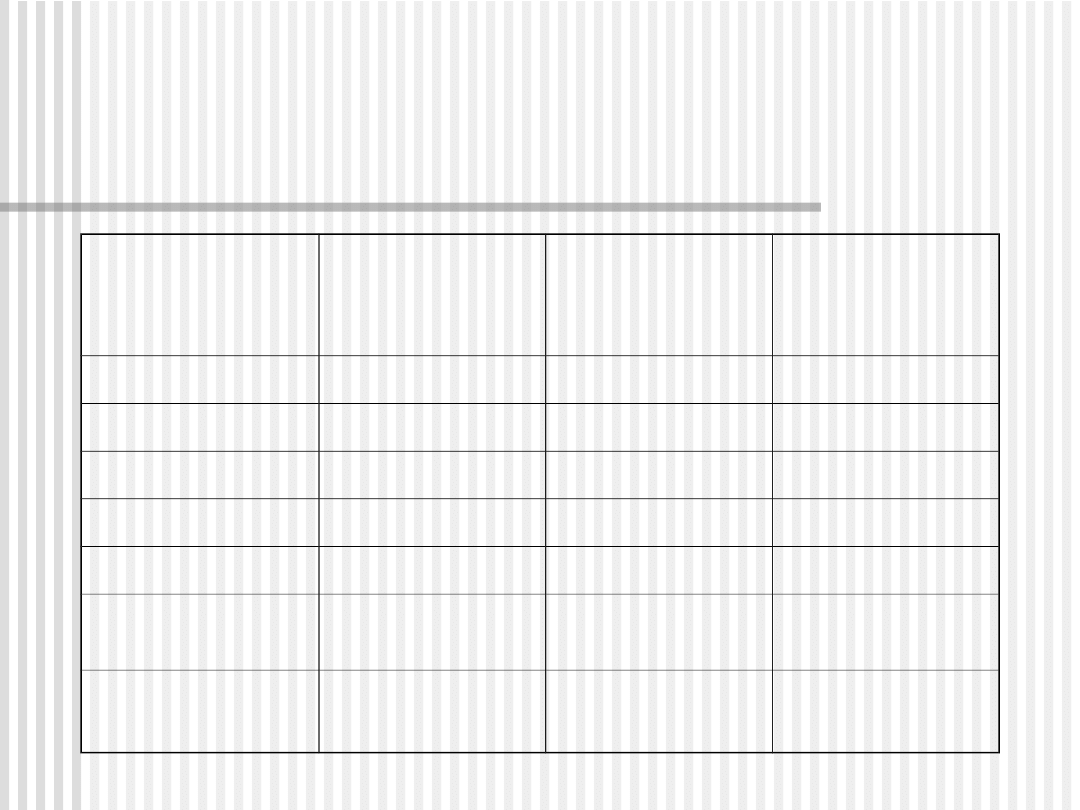
Leki modyfikujące przebieg choroby o
udokumentowanej skuteczności
Lek
Średni czas
poprawy
Opóźnienie/
hamowanie
nadżerek
Koszt
Sulfasalazyna
3 miesiące
spowalnia
Wysoki
Cyklosporyna
2-4 miesiące
spowalnia
Bardzo wysoki
Azatioprina
3 miesiące
spowalnia
Niski
Minocyklina
3 miesiące
brak danych
Wysoki
Sole złota
6 miesięcy
spowalnia
Niski
D-
penicylamina
6 miesięcy
spowalnia
Niski
Hydroksy-
chlorochina
6 miesięcy
nie
Niski

RZS
Leki modyfikujące przebieg choroby o najwyższej
skuteczności
Metotreksat – 7.5-25 mg/tydzień (śr.15mg), 1-2 miesiące, opóźnienie
powstawania nadżerek stawowych
Leflunomid (Arava) – 10-20 mg/dobę, 1-3 miesiące, związek izoksazolowy,
inhibitor pirymidyny o działaniu immunosupresyjnym – opóźnia
pobudzenie komórek docelowych, zmniejsza wytwarzanie cytokin, hamuje
pobudzenie osteoklastów do resorpcji tkanki kostnej i chondrocytów do
degradacji chrząstki (opóźnienie zmian)
Etanercept (Enbrel) – bloker TNFα (immunoglobulina powstała na drodze
inżynierii genetycznej poprzez fuzję fragmentu Fc IgG1 z receptorem p75
dla TNFα) – 25mg 2x w tygodniu, kilka dni do 3 miesięcy,redukuje
nadżerki (hamowanie zmian)
Infliximab (Remicade) – bloker TNFα (monoklonalne przeciwciało –
75%ludzkie, 25% mysie), 3-10/kg m.c. i.v. co 4-8 tygodni (hamowanie
zmian)

Udział badań immunologicznych
w diagnostyce zaburzeń rozrodu

Niepłodność
Niepłodność męska („male factor”) – 30-50% par
Niepłodność żeńska – 45% par
Niepłodność małżeńska
Niepłodność idiopatycznna
Niepłodność – nie dochodzi do poczęcia po 2 latach współżycia ( w Polsce) lub
po roku (w krajach wysokorozwiniętych) bez zabezpieczenia.
Podział niepłodności
Pierwotna (sterilitis primaria) – kobieta nigdy nie była w ciąży
Wtórna (sterilitis secundaria) – ciąża zakończona porodem lub poronieniem,
niemożność ponownego zajścia w ciązę
Niemożność donoszenia ciąży (infertilitas) – poronienia, porody przedwczesne

Przyczyny niepłodności
>100
„Nowy styl życia” – kobiety – wygasanie procesów rozrodczych
> 35 roku życia
mężczyźni – zdolność do spermatogenezy
> 90 roku życia
Sztuczne poronienia
Zaburzenia autoimmunologiczne
Zakażenia
Chlamydia trachomatis
Mycoplasma hominis
Wirus świnki – 25%

Przyczyny niepłodności
Wady wrodzone
Wnętrostwo
Zespół Klinefeltera (2 chromosomy X i jeden Y)
brak komórek plemnikotwórczych w kanalikach
nasiennych w jądrach
Wrodzony brak jąder
Spodziectwo
Syringomyelia

Przyczyny niepłodności
Leki
cymetydyna
metotreksat
metadon
kolchicyna
spironolakton
sulfasalazyna
fenytoina
tiorydazyna
cytostatyki (cyklofosfamid, busulfan, chlorambucil)

Przyczyny niepłodności
Narkotyki
Kokaina
zmniejszenie ilości i jakości
Marihuana plemników
Alkohol
Czynniki genetyczne
Mikrodelecje chromosomu Y
Nieprawidłowości genu dla apolipoprroteiny B

Przyczyny niepłodności
Środki chemiczne
DDT
Aldrin
działanie na zarodki męskie w czasie
Dioksyny
ciąży; efekt estrogenopodobny
Furany
Dieldrin
Metale ciężkie
Ołów
Kadm
hamowanie enzymów akrosomalnyc
Arszenik
(toksyczny wpływ na spermatogenezę)

Niepłodność małżeńska
Przeciwciała przeciwplemnikowe (Antisperm antibodies – ASA)
Występowanie: u kobiet - 5%, u niepłodnych – 7-17%, u niepłodnych
o
niewyjaśnionej etiologii – 14-40%
Czynniki predysponujące:
brak aktywacji przez składniki nasienia supresorowych limfocytów
T w drogach rozrodczych, spadek ilości CD8 w szyjce macicy,
ASA u partnera
przełamanie stanu tolerancji: infekcja, zmiany w nabłonku
pochwy, żołądkowo-jelitowa ekspozycja plemników, stany
nowotworowe
Obecność unikalnych antygenów plemnika, wykazujących
zdolność odpowiedzi immunologicznej

ASA u mężczyzn
Czynniki zabezpieczające: bariera krew-jądro, supresja
odpowiedzi na antygeny plemników (przewaga CD8 w warstwie
podstawnej nabłonka jądra, w nabłonku najądrza, przewodach
odprowadzających, gruczole sterczowym)
Mechanizmy powstawania ASA
Spadek aktywności i liczebności CD8 w męskich narządach
rozrodczych
Brak lub niedobór czynników rekrutujących CD8 do męskich
narządów rozrodczych
Zaburzona antygenowość plemników
Wnętrostwo

Efekt działania ASA
Zahamowanie ruchu postępowego plemników (aglutynacja; >
70% unieruchomiona + niepłodność)
Zahamowanie dojrzewania i/lub uszkodzenie rozwijających się
plemników
Upośledzenie penetracji śluzu szyjkowego przez plemniki
Wywołanie przedwczesnej reakcji akrosomalnej
Zahamowanie łączenia się plemników z osłonką przejrzystą
Zahamowanie zdolności przylegania plemnika do błony
komórkowej oocyta (oolemny) i upośledzenie penetracji do
komórek jajowych
Poronienia nawykowe - zachowanie antygenów plemników na
powierzchni zarodka, zwiększona ekspresja antygenów MHC ojca
na trofoblaście w następstwie stymulacji interferonu gamma
przez ASA

Metody oznaczania ASA
Test kontaktowy nasienie-śluz szyjkowy (zjawisko „bicia witki” –
unieruchomionego plemnika)
Pośrednia, mieszana reakcja antyglobulinowa
Test immunokuleczek

Leczenie niepłodności spowodowanej przeciwciałami
przeciwplemnikowymi
Terapia kortykosteroidami
Inseminacja wewnątrzmaciczna
Różne formy zaplodnienia pozamacicznego: pobieranie plemników z
najądrzy (MESA) lub jądra (TESA) i dokonanie zapłodnienia in vitro
techniką ICSI (intracytoplasmic sperm injaction) poprzez połączenie z
pobraną komórką jajową; dojajowodowe przeniesienie gamet,
zapłodnienie pozaustrojowe i transfer zarodka (IVF-ET – in vitro
fertilization with embrion transfer)
Oddzielenie plemników związanych z przeciwciałami od plemników
wolnych od przeciwciał

Przeciwciała antyjajnikowe
Skierowane przeciw różnym determinantom antygenowym jajnika:
Komórkom tekalnym
Komórkom tekaluteinowym
Komórkom ziarnistym
Komórkom śródmiąższowym
Oocytom
Zaburzenie procesu owulacji?
Występowanie u chorych z: SLE, chorobą Addisona, przedwczesnym
wygasaniem funkcji jajnika, endometriozą

Przeciwciała przeciwfosfolipidowe a
niepłodność
Fosfolipidy (Fs) – molekuły uczestniczące w tworzeniu
syncytiotrofoblastu
Odsłonięcie Fs (fosfatydyloseryny, fosfatydyloetanoaminy) może
indukować powstawanie przeciwciał (APA)
Efekt działania APA
Zaburzenia krzepnięcia (spadek stężenia białka S, mutacja
czynnika V, nieprawidłowa funkcja bialka C, adhezja płytek do
śródbłonka i ich agregacja)
Modulacja metabolizmu kwasu arachidonowego (skurcz naczyń i
zakrzepica)
Zwiększona ekspresja cząsteczek adhezyjnych
Zwiększona sekrecja cytokin prozapalnych
Zaburzenie procesu powstawania syncytiotrofoblastu
Zmniejszenie ekspresjii aneksyny V na kosmkach łożyska

Efekt działania APA
Przyczyna 5-15% poronień na podłożu immunologicznym
Zaburzenie procesu inplantacji
Poród przedwczesny
Przedwczesne pęknięcie błon płodowych
Przenoszenie ciąży
Wewnątrzmaciczne zahamowanie wzrostu płodu
Stan przedrzucawkowy
Rzucawka

Leczenie chorych z APA
Kwas acetylosalicylowy (75-100mg) / heparyna
Glikokortykosteroidy
IVIG

Inne autoprzeciwciała
TA (przeciw tyreoglobulinie i peroksydazie) – zaburzenia owulacji,
poronienia nawykowe (IVIG, IVIG/heparyna/kwas acetylosalicylowy)
ANA – zaburzenia procesu implantacji; przyczyna niepowodzeń
zapłodnień in vitro, poronień wczesnych (glikokortykosteroidy, IVIG)
Anty-DNA - ?
RF – niewydolność fazy luteinowej, ASA, przeciwciała antyjajnikowe
SMA – związek z zaburzeniami owulacji, zmiana perystaltyki
jajowodów

Immunologiczne mechanizmy utrzymania ciąży
Płód – alloprzeszczep w organizmie matki
Dobór osób odmiennych antygenowo
Płód – antygeny zgodności tkankowej pochodzące od ojca i matki
Antygeny płodu rozpoznawane przez limfocyty T i B matki
Dlaczego allogenowo niezgodny płód przeżywa?
Peter Medawar – lata 50-te XX wieku
„Immunodewiacja” – przejściowe (na czas ciąży) dostosowanie się
układu odpornościowego matki dla prawidłowego rozwoju płodu

Immunoregulacyjna rola łożyska
Komórki trofoblastu – granica między krwią matki a krążeniem płodowym
- słaba antygenowość trofoblastu
- brak antygenów klasy I i II MHC
- obecność HLA – G, HLA –E, HLA – F
- znaczenie ekspresji HLA - G:
ochrona trofoblastu przed działaniem cytotoksycznych komórek NK i
cytotoksycznych limfocytów T, indukcja indukcja komórek supresorowych w
doczesnej o fenotypie CD8, CD16, CD56, indukcja ab blokujących, stymulacja
limfocytów matki do wytwarzania cytokin warunkujących zagnieżdżenie i wczesne
stadia rozwoju jaja płodowego, udział w zwalczaniu infekcji – prezentacja
endogennych białek limfocytom matki
Trofoblast jako bariera fizyczna – utrudnione przechodzenie limfocytów T od matki
do płodu
Mechanizmy warunkujące szczelność bariery łożyskowej: ujemny ładunek warstwy
sialomucynowej (utrudnienie kontaktu z ujemną błoną naczyniową błoną
limfocytów matki), właściwości immunoabsorpcyjne komórek syncytiotrofoblastu i
komórek Hofbauera (wychwyt, fagocytoza i degradacja licznych komórek, KI, Abs
matki skierowanych przeciw antygenom płodu, immunoregulacyjne działanie
białkowych i lipidowych składowych błony komórkowej syncytiotrofoblastu,
oporność na działania uszkadzające wywołane przez komórki układu
odpornościowego, Ab cytotoksyczne, KI, składowe dopełniacza

Czynniki immunosupresyjne pochodzenia łożyskowego
Transformujący czynnik wzrostu β-2 (TGFβ), IL-10
Łożyskowy czynnik supresorowy, czynnik pochodzący z trofoblastu,
czynnik ciepłostabilny, białko osoczowe towarzyszące ciąży, ludzka
gonadotropina kosmówkowa, ludzki laktogen łożyskowy, białko łozyskowe
14, izoferrytyna łożyskowa, estrogeny
Progesteron indukujący powstanie immunoregulacyjnego białka – PIBF –
(progesteron induced immunoregulatory protein), które:
hamuje aktywację limfocytów indukowaną alloantygenami, komórek NK
produkujących czynnik martwicy nowotworów
indukuje powstanie limfocytów supresorowych hormonozależnych w
doczesnej
hamuje produkcję IL-2, a nasila IL10
Podanie ciężarnym kobietom inhibitora receptora progesteronu = poronienie,
wzrost aktywności cytotoksycznej limfocytów i komórek NK

Immunologia oka
Alicja Bąkowska
Zakład Immunopatologii
Akademii Medycznej w Gdańsku

Penetracja czynników zakaźnych
i potencjalnie szkodliwych
Spojówka pokrywająca gałkę oczną i wyścielająca
wewnętrzną powierzchnię powiek
Gruczoł łzowy

Tkanka limfatyczna narządu wzroku
Tkanka limfatyczna spojówek (conjunctiva-associated
lymphoid tissue – CALT);
Exp. Eye Res. 1997, 64, 905-912
Tkanka limfatyczna spojówek i gruczołów łzowych (lacrimal
drainage-associated lymphoid tissue – LDALT) jako
integralna składowa tkanki limfatycznej związanej z
błonami śluzowymi (mucosal-associated lymphoid tissue –
MALT);
Invest. Ophtalmol.
Vis.
Sci. 2001. 42, 566- 574
Tkanka limfatyczna oka (eye-associated lymphoid tissue –
EALT);
Ophtalmologe 2003, listopad, 100(11); 929-942

Tkanka limfatyczna oka jako składowa
wspólnego układu limfatycznego błon
śluzowych
( MALT - koncepcja sformułowana w końcu lat 70. przez Bienenstocka i
współpracowników)
Nabłonek – CD3
+
(189+/-27 komórek/mm2), CD8
+
/CD45Ro
+
/HML-1
+
,
komórki, nieliczne CD4
+
( I CD4:CD8 =0.75), komórki Langerhansa (CD1+
a nie jak w skórze CD6
+
; 85+/-16 komórek/mm
2
); nieliczne komórki
nabłonka wykazują ekspresję HLA-DR . W warunkach fizjologicznych brak
mastocytów, granulocytów kwasochłonnych i zasadochłonnych.
Blaszka właściwa - pierwotne i wtórne grudki chłonne (60% badanych
dawców narządów); typowe żyłki z wysokim śródbłonkiem (HEV), przez
które limfocyty przedostają się do węzłów chłonnych; rozproszone
limfocyty, makrofagi, mastocyty, komórki plazmatyczne.

Podstawowe funkcje układu limfatycznego
oka
Wytwarzanie przeciwciał we wszystkich klasach immunoglobulin (A, M, G, E)
Przeciwciała w klasie A immunoglobulin przedostają się do łez w w formie
wydzielniczych IgA (S-IgA), stanowią 80% całkowitego stężenia
immunoglobulin i pełnią znaczącą rolę ochronną poprzez zdolność do:
Opłaszczania i aglutynacji mikroorganizmów
Zapobiegania adhezji mikroorganizmów do nabłonka, a tym samym
wnikania w głąb spojówki
Działania bakteriostatycznego
Neutralizacji toksyn bakteryjnych
Przeciwciała w klasie G immunoglobulin, produkowane w spojówce,
ujawniane są również w rogówce i twardówce.

Film łzowy
Funkcje filmu łzowego
Tworzy gładką, wilgotną powierzchnię gałki ocznej
Dostarcza tlen i substancje odżywcze do powierzchownych warstw
rogówki, pozbawionej własnych naczyń
Usuwa ciała obce, resztki tkankowe z powierzchni oka przez drogi łzowe
Aktywnie uczestniczy w ochronie narządu wzroku
lizozym
beta lizyna
naturalne inhibitory enzymów proteolitycznych
laktoferyna
transferyna
ceruloplazmina
immunoglobulinu w klasie A (S-IgA), G, E
składniki dopełniacza
substancja P

Oko jako narząd „immunologicznie
uprzywilejowany”
1873 rok – van Dooremaal i 1884 rok Zahn po raz pierwszy opisali
wydłużone przeżycie ludzkich komórek nowotworowych wprowadzonych do
przedniej komory oka królika oraz przeżycie przeszczepu ksenogenicznego
(królik/rogówka płodowa człowieka)
1944 rok – doświadczenia Greene’a i Lunda – przeszczep ludzkich komórek
nowotworowych do oka królika
1945 rok - doświadczenia Medawara potwierdziły możliwość wydłużonego
przeżycia tkanek przeszczepionych do oka i mózgu
1948 rok – wprowadzenie przez Medawara określenia „miejsce
immunologicznie uprzywilejowane” w odniesieniu do narządów, w których
przeszczepy allogeniczne i ksenogeniczne nie podlegają odrzuceniu bądź
przeżycie ich jest wydłużone
1953 rok – Billingham – potwierdzenie wyników badań
(oko, wątroba, mózg, nadnercza, łożysko, jądra, chrząstka, jajnik, gruczoł
krokowy).

Oko jako miejsce „immunologicznie
uprzywilejowane
”
„Immunologiczne uprzywilejowanie” w narządzie wzroku dotyczy:
Zrębu rogówki
Tęczówki
Soczewki
Komory przedniej
Ciała szklistego
Siatkówki
Jest to następstwo szczególnej struktury anatomicznej jak i aktywnej
modyfikacji reakcji immunologicznych przez mikrośrodowisko gałki ocznej
zawierające cząsteczki o działaniu przeciwzapalnym i
immunosupresyjnym, hamujące reakcje immunologiczne nieswoiste
(wrodzone) i swoiste (wtórne).

Czynniki warunkujące uznanie oka za miejsce
„immunologicznie uprzywilejowane”
Uwarunkowania strukturalne
Brak unaczynienia – soczewka, zrąb rogówki
Bariera krew-tkanka – tęczówka, ciało szkliste, nabłonek barwnikowy siatkówki
Brak naczyń limfatycznych odprowadzających
Brak lub zmieniona funkcja APC – rogówka , komora przednia – brak APC, zrąb tęczówki
i ciało szkliste – zmieniona funkcja
Brak, obniżona ekspresja antygenów zgodności tkankowej klasy I i II - keratocyty,
śródbłonek rogówki, nabłonek tęczówki, ciało szkliste, siatkówka – brak ekspresji HLA-
DR
Ekspresja cząsteczek MHC Ib - HLA –G – na keratocytach, komórkach śródbłonka i
nabłonka rogówki (Human Immunology 2003, 64, 1039-1044) są rozpoznawane przez
komórki NK przekazując im sygnał hamujący ich aktywację.
Stała obecność liganda białka Apo-1/Fas na komórkach: śródblonka rogówki, nabłonka
tęczówki i ciała rzęskowego, nabłonka barwnikowego siatkówki i na włóknach
nerwowych siatkówki

Czynniki warunkujące uznanie oka za miejsce
„immunologicznie uprzywilejowane”
Czynniki rozpuszczalne w cieczy wodnistej
Transformujący czynnik wzrostu β-2 (TGF- β-2) - hamuje aktywację
limfocytów T (proliferację jak i produkcję cytokin), komórek NK, APC
Hormon pobudzajacy melanocyty α (α-MSH) – hamuje produkcję cytokin
(IFN-γ) promując wydzielanie TGF β-2 przez limfocyty T
Peptyd zbliżony do kalcytoniny (CGRP) – hamuje efekt aktywnych
makrofagów
Naczynioaktywny polipeptyd jelitowy (VIP) – potencjalny inhibitor
aktywacji limfocytów T
Czynnik zahamowania migracji makrofagów (MIF) – obecny w wysokim
stężeniu, głęboki inhibitor lizy komórek NK
Kortizol
Białko hamujące aktywację składowych dopełniacza
Somatostatyna – wzrost produkcji czynników immunomodulujących w
mikrośrodowisku oka (TGF-β, α-MSH)
IL-10 – hamuje odpowiedź typu komórkowego i zapalną

Nadzór produkcji IgE przez:
Antygeny ukladu MHC – allel HLA-DRB1*1501
(pyłki ambrozji)
allel HLA-DRB3*0101
(pyłki brzozy)
HLA-B8 –
wysokie miano swoistych IgE
wysokie stężenie całkowitych IgE
Geny „atopii” - chromosom 5 – region 5q31 (gen zespołu IL4, IL-13, IL-5)
chromosom 11 – region 11q13
(gen dla łańcucha β receptora FcεRI – odpowiedzialnego
za przekazywanie sygnału do wnętrza komórki)

Alergia
(Johansson S.G.O i wsp.,Allergy 2001, 56, 813)
Nadwrażliwość – objawy, wywołane przez ekspozycję na patogen, w dawce tolerowanej przez
osoby zdrowe
Alergia – reakcje nadwrażliwości zainicjowane przez mechanizmy immunologiczne; z udziałem
przeciwciał (IgE, rzadziej IgG) lub komórek zapalnych.
IgE – zależna – ostre alergiczne zapalenie spojówek i tkanek wokół oczu
wiosenne zapalenie spojówek i rogówki (50%)
atopowe zapalenie spojówek i rogówki (33%)
IgE – niezależna (CD3, KI, mastocyty, eozynofile)
olbrzymiokomórkowe zapalenie spojówek
całoroczne zapalenie spojówek (40%)
nietolerancja leków, kosmetyków
atopowe zapalenie spojówek i rogówki (67%)
wiosenne zapalenie spojówek i rogówki (50%)

Typ II – cytotoksyczność zależna od przeciwciał
Reakcja cytotoksyczna zależna od przeciwciał IgG/IgM z udziałem dopełniacza
Reakcja ADCC – cytotoksyczność zależna od przeciwciał
Alergia na leki (penicylina,
tyreostatyki)
Zapalenie naczyń z udziałem ANCA

Typ
III – reakcje antygen - przeciwciało
Przewlekłe zapalenie naczyń
Zapalenie siatkówki
Zapalenie twardówki

Typ
IV – nadwrażliwość typu opóźnionego
>
12 h, reakcja typu komórkowego
Olbrzymiobrodawkowe zapalenie spojówek
(typ I i IV)
Kontaktowe zapalenie skóry powiek
i spojówek
(hapten+białko)
o
Gruźlica
o
Sarkoidoza
o
Choroba Crohna

Toczeń rumieniowaty układowy
(Kryteria diagnostyczne wg Amerykańskiego Towarzystwa Reumatologicznego
z 1982 roku, zaktualizowane w 1997 roku)
Rumień twarzy
Stały rumień płaski lub lekko uniesiony
Rumień krążkowy Zmiany rumieniowate, lekko ubiesione
Nadwrażliwość na światło słoneczne
Osutka - jako wynik nietypowej
reakcji
Owrzodzenia jamy ustnej Zwykle zmiany niebolesne
Zapalenia stawów bez nadżerek
2 stawy obwodowe – ból, obrzęk
Zapalenie opłucnej i osierdzia
Tarcie, wysięk – zmiany udokumentowane
badaniami dodatkowymi
Zmiany w nerkach
Białkomocz 0.5g/d, wałeczki wmoczu
Zmiany w układzie nerwowym
Napady drgawkowe, zaburzenia psychiczne
Zaburzenia hematologiczne
Niedokrwistość hemolityczna, limfopenia,
małopłytkowość
Zaburzenia immunologiczne
Przeciwciała przeciw: nDNA, Sm,
kardiolipinie w
klasie G lub M immunoglobulin. Obecność
fałszywie dodatniego testu VDRL
Obecność przeciwciał
Wysokie miano
(lupus 1999, 8,8, 586)

Toczeń rumieniowaty układowy a
zmiany oczne
Suche zapalenie spojówek i rogówki – 10-25% chorych
Zapalenie nadtwardówki
Zapalenie twardówki
Choroidopatia centralna surowicza
Neuropatia niedokrwienna nerwu wzrokowego
Pozagałkowe zapalenie nerwu II
Retinopatia – 10 % chorych
Znamienna korelacja
Przeciwciała przeciwkardiolipinowe (p<0.05)
Postać neurologiczno- psychiatryczna (p<0.01)
Stężenie kreatyniny (p<0.01)
Aktywność SLE (p<0.03)
Annals of the Rheumatic Diseases 2000, 59, 9,
705)
Toczeń rumieniowaty układowy
(Kryteria diagnostyczne wg Amerykańskiego Towarzystwa Reumatologicznego
z 1982 roku, zaktualizowane w 1997 roku)
Rumień twarzy
Stały rumień płaski lub lekko uniesiony
Rumień krążkowy
Zmiany rumieniowate, lekko ubiesione
Nadwrażliwość na światło słoneczne
Osutka - jako wynik nietypowej
reakcji
Owrzodzenia jamy ustnej
Zwykle zmiany niebolesne
Zapalenia stawów bez nadżerek
2 stawy obwodowe – ból, obrzęk
Zapalenie opłucnej i osierdzia
Tarcie, wysięk – zmiany udokumentowane
badaniami dodatkowymi
Zmiany w nerkach
Białkomocz 0.5g/d, wałeczki wmoczu
Zmiany w układzie nerwowym
Napady drgawkowe, zaburzenia psychiczne
Zaburzenia hematologiczne
Niedokrwistość hemolityczna, limfopenia,
małopłytkowość
Zaburzenia immunologiczne
Przeciwciała przeciw: nDNA, Sm, kardiolipinie w
klasie G lub M immunoglobulin. Obecność
fałszywie dodatniego testu VDRL
Obecność przeciwciał
Wysokie miano
(lupus 1999, 8,8, 586)

Pierwotny zespół
antyfosfolipidowy
Przeciwciała przeciwkardiolipinowe (ACA) ujawniane są u 5 – 33%
chorych z zakrzepicą naczyń oka
Wysokie stężenie ACA w klasie G i M immunoglobulin to
potencjalne ryzyko zaostrzenia choroby oczu i choroby układowej.
(Surv. Ophthalmol. 2002, 47, 3)
Przeciwciała pezeciwkardiolipinowe mogą być znacznikiem
toczącego się procesu chorobowego u chorych z zapaleniem
naczyniówki a zwłaszcza u pacjentów z zapaleniem naczyń
siatkówki.
(Am. J. Ophthalmol. 2001, 131,451)

Przewlekłe zapalenie naczyń
Choroba Takayasu
Zmiany oczne u 60% chorych
Zapalenie błony naczyniowej
Retinopatia
(
APLAR J. Rheumathol. 2002, 5, 1, 25)
Korelacja z aktywnością choroby – przeciwciała
przeciwmonocytarne
(J. Rheumathol. 2003,30, 9)

Przewlekłe zapalenie naczyń
Ziarniniak Wegenera – kryteria diagnostyczne
Zmiany w obrębie nosa lub jamy ustnej
Bolesne lub niebolesne zmiany wrzodziejące lub ropne, krwista wydzielina z nosa
Zmiany w obrazie radiologicznym płuc
Cienie okrągłe, nacieki, jamy
Patologiczny obraz osadu moczu
Erytrocyturia >5wpw lub wałeczki erytrocytów
Ziarniniaki w badaniu histopatologicznym biopsji
Ziarniniaki w ścianach lub przestrzeniach okołonaczyniowych
małych i średnich naczyń

Ziarniniak Wegenera a zmiany oczne
>58% chorych
Wyniki badań przeprowadzonych w latach 1988- 1998
57.4% choroba układowa ze zmianami ocznymi
6.3% zmiany oczne wyprzedzały rozwój zmian układowych
30% ! – tylko zmiany oczne bez objawów choroby układowej
(Am. J. Ophthal. 2001, 132, 3)

Pierwotny niedobór immunologiczny
a zmiany w narządzie wzroku
Niedobór IgA (IgA deficiency)
– zatrzymanie różnicowania limfocytów B IgM do komórek
plazmatycznych
Alergia
Choroby autoimmunizacyjne
Pospolity zmienny niedobór odporności (Common variable
immunodeficiency, CVID)
- defekt syntezy przeciwciał
Zapalenie naczyń siatkówki
Infekcje oportunistyczne
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
Wyszukiwarka
Podobne podstrony:
socjologia konspekt 2006, II rok II semestr, BWC, socjologia
kolo 2 stoma 2006, 3 rok stoma, patomorfa, 2 kolo, 2 kolo, testy
anatomia egzamin 2006, I rok, Anatomia, cxrtsjxcgvhbjnkmugjyfghkjl, Anatomia egzamin, Egzaminy
Udar - Książka 2006, V rok, Neurologia, Sem. V rok, Udar mózgu
konspekty 2006 2007, 1
ZUS W wa PN pismo 400 10 06 2006 rok
rok IV se. zimowa, Stary Testamen 2006 rok IV sem I(2), Stary Testament
atf konspekt II, I rok Filozofia, Analiza tekstów filozoficznych
ZWIERCIADŁO - konspekt, POLONISTYKA, rok 1, Różne
bugental - konspekt, Psychologia rok I, HMP
Pytania Rat 2006, Rok II, Med. ratunkowa wieku dziecięcego
Budżet Gminy Karpacz na 2006 rok - Skąd mamy pieniądze i na co je wydajemy
konspekt 3 i 4, Filozofia, Rok IV, Filozofia człowieka
2006, 5 ROK, INTERNA, # EGZAMIN, KOLOKWIA, GIEŁDY
Chirurgia z LEPow 2004-2006, V rok, Chirurgia
Chemia 2006, I rok, I rok, gieldy, od Karoliny, medycyna, 2 semestr, Chemia, z jagiellonskiego, Chem
atf konspekt III, I rok Filozofia, Analiza tekstów filozoficznych
więcej podobnych podstron