
Modele zwierzęce chorób
autoimmunizacyjnych

Eksperymentalne autoimmunologiczne
zapalenie mózgu i rdzenia kręgowego
(experimental autoimmune
encephalomyelitis, EAE)

• Eksperymentalne autoimmunologiczne zapalenie
mózgu i rdzenia kręgowego (EAE) - choroba zapalna
ośrodkowego układu nerwowego powszechnie
wykorzystywana
jako
model
stwardnienia
rozsianego (multiple sclerosi, MS).
• Cechuje się okresami pogorszenia oraz remisji,
utratą przewodnictwa nerwowego oraz stałym
postępem niepełnosprawności.
• Makrofagi oraz limfocyty T pośredniczą w destrukcji
osłonki mielinowej otaczającej nerwy, prowadząc do
zakłócenia przewodnictwa impulsów nerwowych.

Główną metodą indukcji EAE u zwierząt jest
immunizacja przy pomocy białka substancji białej
OUN, takiego jak:
• podstawowe białko mieliny (BMP),
• białko proteolipidowe (PLP),
• mielinowa glikoproteina oligodendrocytów (MOG),
co skutkuje powstaniem ostrej postaci choroby o
cechach MS.
Poprzez użycie różnych antygenów i adjuwantów,
można uzyskać przewlekłą, nawracającą postać EAE.

• Wyróżniamy 4 typy EAE (ze względu na
kliniczny przebieg choroby):
- ostre śmiertelne EAE;
- przewlekłe postępujące EAE;
- przewlekłe nawracające EAE;
- przewlekłe EAE z opóźnionym rozwojem.

• Ostre śmiertelne EAE - gwałtowny spadek masy ciała, osłabienie
tylnych kończyn, zaburzony chód zwierzęcia, gwałtownie
przekształcający się w paraliż zaatakowanych kończyn,
nietrzymanie moczu i osłabione oddychanie, śmierć zwierzęcia
krótko po immunizacji.
• Przewlekłe postępujące EAE – powolny rozwój choroby, ale
postępujący w przeciągu dwóch tygodni.
• Przewlekłe nawracające EAE - rozwój ostrej postaci choroby z
różnorodną intensywnością. Objawy choroby łagodne takie, jak
osłabienie kończyn tylnych, zaburzony chód oraz nietrzymanie
moczu lub ostra paraplegia tylnych kończyn. Po całkowitym
wyzdrowieniu, które najczęściej ma miejsce po ok. miesiącu,
obserwuje się nawrót choroby.
• Przewlekłe EAE z opóźnionym rozwojem - utrata wagi i ogólne
osłabienie w dwa tygodnie po immunizacji, z objawami
neurologicznymi rozpoczynającymi się miesiąc lub dwa miesiące
później.
•
EAE jest chorobą wielogenową, więc podatność
oraz przebieg kliniczny mogą się różnić w
zależności od użytego antygenu (np. MBP lub PLP)
oraz
od
szczepu/gatunku
zwierzęcia
wykorzystanego do badań. Tak więc EAE nie jest
pojedynczym modelem choroby, ale wieloma
modelami, które wykazują różnorodny poziom
podobieństwa do MS.

Aktywny model EAE
• W aktywnym modelu EAE podatne szczepy
myszy
immunizowane
są
odpowiednim
antygenem
mielinowym
lub
białkiem
emulgowanym w kompletnym adjuwancie
Freund’a (CFA).
• Myszom zwykle podaje się również toksynę
pałeczki krztuśca (PT) w dniu immunizacji oraz
48h później. Uważa się, że PT odgrywa istotną
rolę w przełamaniu bariery krew-mózg.
• W zależności od wykorzystanego szczepu myszy
oraz rodzaju antygenu, EAE rozwija się 10 do
15 dnia po pierwszej immunizacji.

• Po aktywnej immunizacji zachorowalność wynosi
75% do 80%.
• Bardzo często po aktywnej immunizacji przebieg
choroby jest monofazowy. Istotnym wyjątkiem
jest aktywny model EAE indukowany w myszach
szczepu SJL lub (SJL × SWR) F1 z
wykorzystaniem PLP 139-151 emulgowanego w
CFA. W tym przypadku myszy rozwijają
nawracająco-ustępującą postać choroby, która
rozwija się bez użycia PT.
• Dawka i rodzaj immunizacji oraz szczep
wykorzystany do badań decydują o ciężkości
przebiegu indukowanej choroby, który może
wahać się od łagodnego do śmiertelnego.

• Nie wszystkie szczepy gryzoni są jednakowo podatne na
indukcję EAE. Klasycznymi szczepami podatnymi na
immunizację MBP oraz PLP są szczury Lewisa i DA oraz
myszy SJL/J i PL/J. Immunizacja MOG wywołuje EAE u
kilku dodatkowych szczepów, przede wszystkim u
szczurów BN i u myszy C57BL/6 i BALB/c, które są
odporne na immunizację MBP czy PLP.
• Aktywnie indukowane EAE nie dotyczy wyłącznie
gryzoni, można je wywoływać u naczelnych, np. u rezusa
czy małpy szerokonosej (Callithrix jacchus). Tak jak u
gryzoni, dawka, rodzaj autoantygenu i adjuwant
wykorzystane do immunizacji decydują o charakterze
choroby. Obecnie większość badań nad EAE u
naczelnych
opartych
jest
na
immunizacji
rekombinowanym ludzkim MOG w CFA, co skutkuje
indukcją choroby neurologicznie i histologicznie
podobnej do ludzkiego MS.

Bierny model EAE
• W biernym modelu EAE jest indukowane poprzez wstrzyknięcie
do myszy aktywowanych komórek T mielinospecyficznych. Te
aktywowane komórki T są generowane poprzez immunizację
myszy (donorów) za pomocą białka mieliny lub peptydu
emulgowanego w CFA. Węzły chłonne są usuwane, a komórki T
aktywowane ponownie in vitro przy pomocy odpowiedniego
antygenu. Tak aktywowane komórki T są przenoszone do myszy
(biorców), które następnie rozwijają EAE.
• Najczęściej w biernym modelu EAE stosuje się myszy SJL lub
(PL x SJL) F1 oraz białko MBP jako antygen indukujący. W tym
modelu EAE myszy rozwijają nawracająco-ustępującą formę
choroby.

• Po
uzyskaniu
linii
komórkowych
monospecyficznych
limfocytów T ze szczurów Lewis, a następnie z myszy, okazało
się, że autoimmunologicznymi komórkami efektorowymi
przenoszącymi EAE są limfocyty T pomocnicze (CD4+), które
rozpoznają autoantygenne epitopy białka w kontekście
determinant MHC klasy II.
• Wiele z tych komórek odpowiada na aktywację wydzielając
interferon-γ i TNF-α, ale nie IL-4 czy IL-5, i dlatego zostały
sklasyfikowane jako komórki Th-1.
• Niedawno odkryto jednak, że ta populacja zawiera szczególny
podzbiór komórek T CD4+, które wydzielały IL-17 zamiast
interferonu-γ , były to komórki Th17. Obecnie komórki T Th17
uznaje się za „prawdziwe” patogenne mediatory.

• Bierny model EAE był również wywoływany u naczelnych. Z krwi
małej grupy rezusów wyizolowano MBPspecyficzne komórki T, z
których stworzono linię komórkową. Te komórki były ponownie
aktywowane in vitro i wstrzyknięte do donora. W jednym przypadku
na trzy komórki T spowodowały łagodne zapalenie OUN.
• Transfer komórek T pomiędzy osobnikami nie hodowanymi wsobnie
nie przyniesie spodziewanych rezultatów z powodu niezgodności
tkankowej.
• Inaczej jest w przypadku małp szerokonosych, które rodzą się jako
bliźnięta dzielące to samo łożysko. Rodzeństwo posiada
immunologiczną tolerancję na siebie nawzajem, co pozwala na
wzajemne transplantacje tkanek. Kiedy linia komórek T
mielinospecyficznych wyprowadzona od jednego z bliźniąt była
wprowadzana do drugiego, wywoływało to EAE.

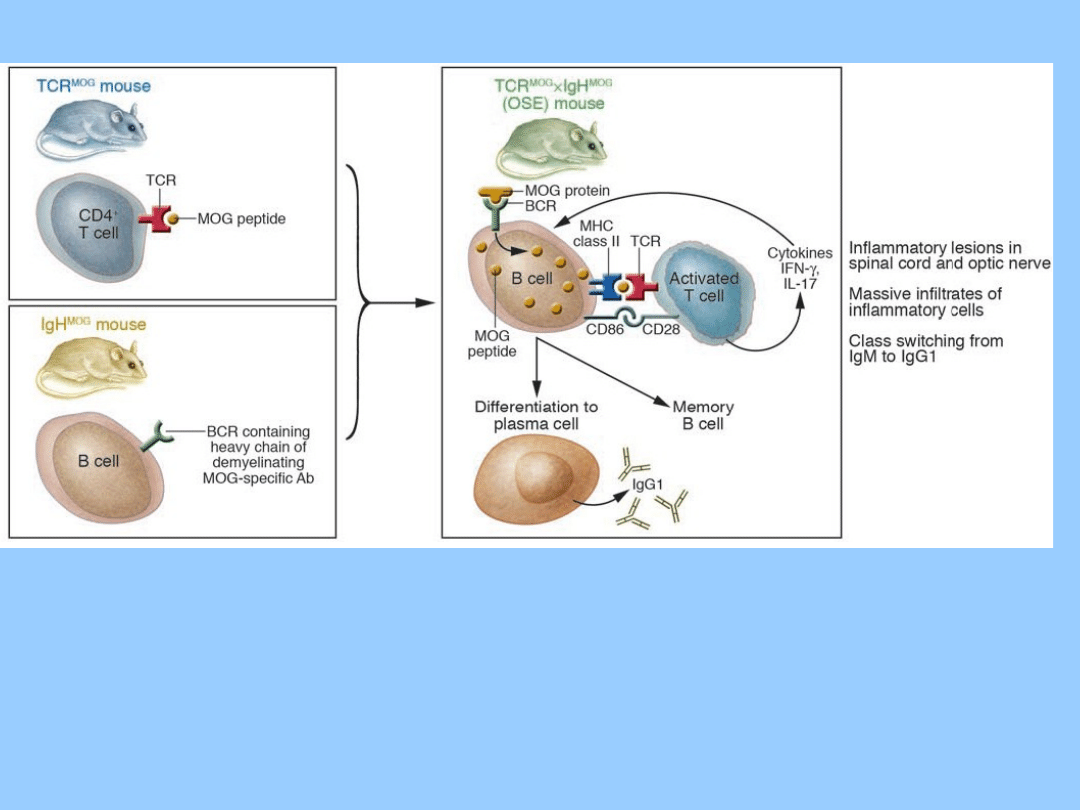
Spontaniczny model EAE
• Dwa zespoły badawcze (Krishnamoorthy i wsp. oraz Bettelli i
wsp.) wyhodowały podwójnie transgeniczne myszy u których
ekspresji ulegają receptory komórek B i T rozpoznające to samo
białko mielinowe. Myszy te powstały ze skrzyżowania dwóch linii
transgenicznych.
• Pierwsza linia myszy transgenicznych została wygenerowana za
pomocą
technologii
knock-in,
poprzez
wstawienie
rekombinowanego
łańcucha
ciężkiego
demielinizacyjnego
przeciwciała anty-MOG w region J immunoglobuliny. Dzięki temu
uzyskano myszy których 20-30% krążących komórek B wytwarza
przeciwciała anty-MOG oraz wykazuje ekspresję receptora
(BCR) rozpoznającego białko MOG. Myszy te nazwano IgH
MOG
.
• Druga linia, nazwana TCR
MOG
, została stworzona poprzez
ekspresję
TCR,
który
rozpoznaje
immunodominującą
determinantę białka MOG. Myszy te spontanicznie rozwijały
zapalenie nerwu wzrokowego, typowy wczesny objaw MS.

• Ponad połowa z tych myszy w wieku 8 tygodni
spontanicznie rozwinęła autoimmunizacyjną
demielinizację w ich rdzeniu kręgowym i nerwach
wzrokowych, co jest symptomem przypominającym
ludzkie stwardnienie rozsiane.
• Krishnamoorthy i wsp. nazwali ten model OSE. W
rdzeniu kręgowym uszkodzenia znajdowały się przede
wszystkim pod oponą miekką, tak jak w aktywnym
modelu EAE. W przeciwieństwie do OSE, spontaniczne
EAE występuje w większości powszechnie
wykorzystywanych modeli tylko w niewielkim zakresie,
lub po ekspozycji na drobnoustroje ewentualnie poprzez
eliminację komórek regulatorowych.
• Badania nad OSE wykazały, że ostry przebieg EAE oraz
zachorowalność nie są zakłócane przez takie czynniki,
jak płeć czy warunki hodowli.
(„A mighty mouse: building a better model of multiple sclerosis”
Richard M. Ransohof
)
Patogeneza EAE
• Komórki T odgrywają istotną rolę w przełamaniu
bariery krew-mózg oraz w patogenezie EAE. Za
indukcję procesu autoimmunizacji w EAE
odpowiadają
aktywowane
autoreaktywne
limfocyty T CD4+ swoiste względem składników
mieliny, wydzielające IFN-γ, IL-2, TNF-β. W
patogenezie EAE biorą udział również limfocyty
Th2, limfocyty T CD8+, makrofagi, limfocyty B,
limfocyty Th17 i prawdopodobnie komórki NK.

• Eksperymentalne autoimmunologiczne zapalenie mózgu i
rdzenia rozwija się wskutek infiltracji OUN przez limfocyty
Th1 uwalniające cytokiny prozapalne (IFN-γ, IL-2, TNF-β)
oraz makrofagi.
• Autoreaktywne komórki T rozpoznają antygeny mieliny,
które mogą być prezentowane przez okołonaczyniowe
makrofagi bariery krew-mózg. Po aktywacji limfocyty T
uwalniają cytokiny i chemokiny, które zmieniają ekspresję
molekuł adhezyjnych na otaczających komórkach, zwiększa
się przepuszczalność bariery krew-mózg i dochodzi do
rekrutacji komórek z krążenia do mózgu.
• Następstwem
autoimmunizacyjnego
zapalenia
jest
uszkodzenie mieliny i oligodendrocytów, jak również
aksonów, co prowadzi do utraty ich funkcji.

Podobieństwa i różnice między MS i
EAE
• Główną różnicą między MS i EAE jest to, że MS jest chorobą
spontaniczną, podczas gdy EAE jest indukowana poprzez
immunizację antygenami tkanki mózgowej.
• Ponadto, w większości protokołów, silny adjuwant jest
wykorzystywany do indukcji choroby. Nie wydaje się, że
podobnie silny immunologiczny czynnik występuje w
fizjologicznych warunkach, nawet w chorobach zakaźnych.
• Dla zapewnienia powtarzalności uzyskanych wyników, EAE
jest badane u osobników wsobnych lub homogennych
genetycznie. W ten sposób, genetyczna heterogenność,
która jest bardzo ważna w populacji osób ze stwardnieniem
rozsianym, jest odzwierciedlana tylko, kiedy różnorodne
modele EAE są badane równolegle.
• Podobnie jak w MS, EAE jest związane z wczesnym
naruszeniem bariery krew-mózg i ogniskowym przenikaniem
okołonaczyniowych jednojądrzastych komórek.

Model zwierzęcy
Podobieństwa do MS
Różnice z MS
Szczur Lewis
Aktywny model EAE
udział komórek T w
zapaleniu,słaba odpowiedź
przeciwciał
choroba jednofazowa, niewielka
demielinizacja
Bierny model EAE
udział komórek T w zapaleniu,
topografia choroby
choroba jednofazowa, niewielka
demielinizacja
Aktywny lub bierny model
EAE + wzajemny transfer
przeciwciał anty-MOG
udział komórek T w zapaleniu,
demielinizacja
tylko krótkotrwała demielinizacja
Szczur kongeniczny
Lewis, DA, BN
Aktywny model EAE
choroba nawracająco-ustępująca,
może naśladować histopatologię
SM
nie jest chorobą spontaniczną
Mysie EAE
Aktywny model EAE
choroba nawracająco-ustępująca
lub przewlekła postępująca,
demielinizacja i uszkodzenie
aksonów
nie jest chorobą spontaniczną
Myszy transgeniczne lub
typu knockout
szczegółowe określenie roli
badanych molekuł
immunologicznych/ cytokin
neurotroficznych/ szlaków
neuroanatomicznych
sztucznie stworzone zwierzęta
transgeniczne lub knock out

Eksperymentalna
autoimmunologiczna miastenia
rzekomoporaźna (experimental
autoimmune myasthenia gravis,
EAMG)

• Miastenia
rzekomoporaźna
(MG)
jest
chorobą
autoimmunologiczną nerwowo-mięśniową, w której uczestniczą
przeciwciała skierowane przeciwko nikotynowym receptorom
acetylocholinowym (AChR). Przeciwciała po połączeniu się z
AChR aktywują kaskadę dopełniacza.
• Główny mechanizm zaangażowany w destrukcję AChR jest
związany z aktywnością przeciwciał i dopełniacza.
• Również wiązanie krzyżowe AChR przez przeciwciała prowadzi
do antygenowej modulacji i może przyczyniać się do
zredukowania liczby tych receptorów w płytce nerwowo-
mięśniowej (neuromuscular junction, NMJ).
• Niewielka liczba przeciwciał anty-AChR może również wiązać
się z AChR i w ten sposób może zakłócać przewodnictwo
nerwowo-mięśniowe.
• Powyższe zjawiska patologiczne mogą prowadzić do defektów
w przekaźnictwie nerwowo-mięśniowym, objawiających się
osłabieniem i zmęczeniem mięśni szkieletowych u pacjentów z
MG.

• Eksperymentalna
autoimmunologiczna
miastenia
rzekomoporaźna zazwyczaj jest indukowana poprzez
immunizację
zwierząt
za
pomocą
AChR
drętwy
kalifornijskiej lub węgorza elektrycznego z adjuwantem.
EAMG może być również indukowana u szczurów i myszy
poprzez podanie surowicy od gryzoni z EAMG lub od
pacjentów,
lub
poprzez
podanie
przeciwciał
monoklonalnych lub poliklonalnych przeciwko AChR.

Model aktywny EAMG
• W modelu aktywnym EAMG króliki, szczury i myszy
immunizowane są rybim, szczurzym, mysim lub ludzkim
AChR emulgowanym w CFA i rozwijają przewlekłą
postać EAMG, która w dużym stopniu przypomina ludzką
MG.
• W przewlekłej EAMG, typowe cechy patologiczne są
bardzo podobne do tych obserwowanych w MG i
obejmują redukcję
NMJ, znaczny spadek zawartości
AChR na błonie postsynaptycznej oraz odkładanie się
IgG i dopełniacza w NMJ.
• W tej formie EAMG nie obserwuje się żadnej znacznej
infiltracji komórkowej w lub blisko NMJ.

• Immunizacja szczurów Lewis za pomocą AChR w CFA
razem z toksyną pałeczki krztuśca (PT) prowadzi do
indukcji ostrej postaci EAMG, 7-11 dni po immunizacji,
charakteryzującej
się
odkładaniem
przeciwciał
i
dopełniacza oraz inwazją komórek fagocytarnych do NMJ.
• Małpy również rozwijają EAMG poprzez immunizację
AChR. Co jest ważne, małpy z EAMG wykazują
charakterystyczne opadanie powiek często obserwowane
w ludzkiej MG, natomiast nieobecne u gryzoni.

Model bierny EAMG
• EAMG może być również efektywnie indukowane u
gryzoni poprzez bierny transfer patogennych przeciwciał
(np. podanie przeciwciał monoklonalnych skierowanych
przeciwko głównemu regionowi immunogennemu (MIR)
AChR) lub poprzez podanie surowicy zwierząt z EAMG.
• Bierny transfer EAMG z użyciem przeciwciał może
powodować osłabienie mięśniowe w ciągu 1 dnia. Próbki
mięśni
wykazały
infiltrację
NMJ
przez
komórki
jednojądrzaste, podobnie jak w ostrej postaci EAMG
indukowanej przez szczepienie receptorem ACh i PT.
Podobnie do modelu ostrej EAMG, bierny model nie jest
dokładnym
odzwierciedleniem
ludzkiej
miastenii
i
reprezentuje tylko efektorowy (czynny) etap choroby.

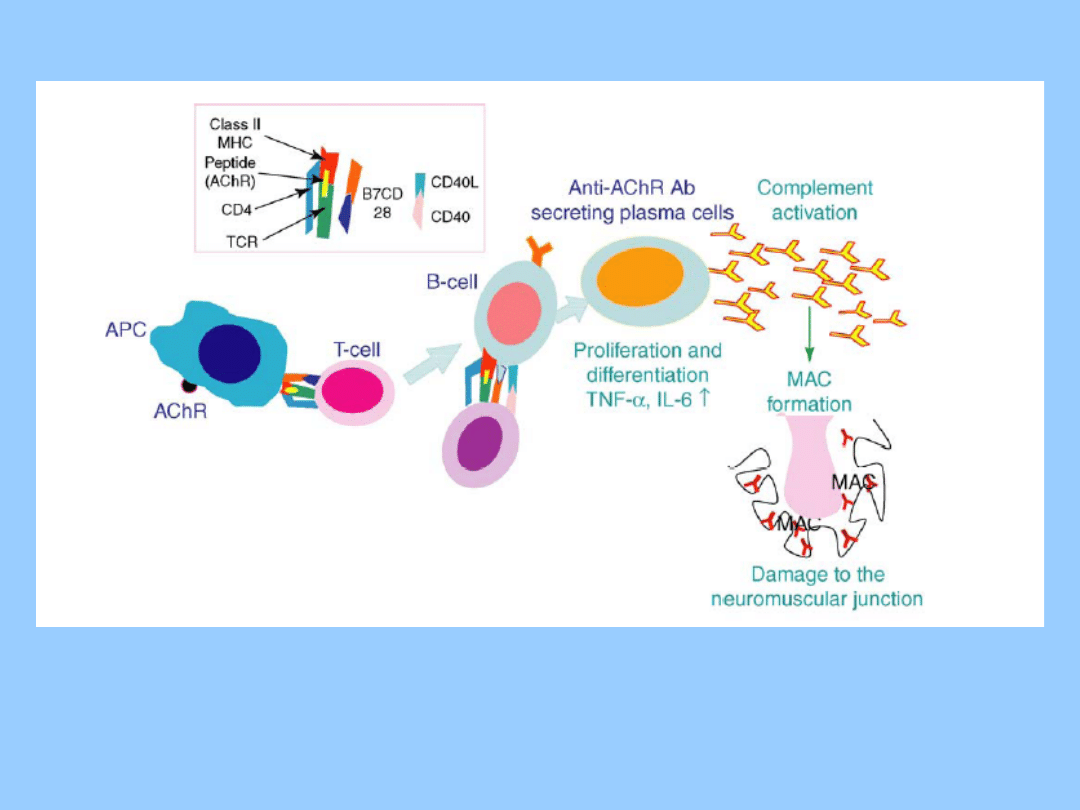
Patogeneza EAMG
• W EAMG i MG, wytwarzanie przeciwciał przeciwko AChR
jest zależne od komórek T i B.
• Różnorodne cytokiny wspomagają rozwój odpowiedzi
immunologicznej przeciwko AChR poprzez indukcję
ekspresji molekuł kostymulujących, dojrzewania komórek
T i B oraz produkcji przeciwciał i dopełniacza.
• Co ciekawe, zarówno cytokiny typu Th1, jak i Th2
odgrywają ważną rolę w indukcji EAMG i nieprawidłowe
funkcjonowanie jednego z tych systemów sprawia, że
myszy są bardzo odporne na indukcję choroby.
• Przeciwciała anty-AChR aktywują kaskadę dopełniacza i
formowanie
kompleksu
atakującego
błonę
(MAC),
wzmagając destrukcję NMJ pierwotnie dokonywaną przez
przeciwciała anty-AChR.

• Pomimo, że komórkowa infiltracja tkanki mięśniowej
wydaje się być istotnym czynnikiem przyczyniającym się do
rozwoju osłabienia mięśniowego w ostrej postaci EAMG u
gryzoni, niewielkie ilości komórek są obserwowane w
tkance mięśniowej przewlekłej postaci EAMG u gryzoni czy
ludzkiej MG. Prawdopodobnie, w przewlekłej postaci
EAMG, komórki jednojądrzaste wspomagają indukcję
osłabienia
mięśniowego
nie
poprzez
bezpośrednie
uszkodzenie komórek mięśniowych czy NMJ, ale poprzez
pomoc w wytworzeniu silnej odpowiedzi komórek T i B
przeciwko AChR.

Przeciwciała anty-AChR odgrywają istotną rolę w
patogenezie EAMG i MG poprzez blokowanie
receptora ACh, przez zwiększanie jego endocytozy lub
przez aktywowanie przebiegającego przy udziale
dopełniacza zapalnego niszczenia NMJ. Szczególnie
przeciwciała wiążące się do MIR na podjednostce α
cząsteczki AChR (ponad 50% wszystkich przeciwciał
anty-AChR w EAMG i MG) mają znaczenie
chorobotwórcze.
(„Unraveling myasthenia gravis immunopathogenesis using
animal models”
E. Tüzün, P. Christadoss
)

Podobieństwa i różnice między
EAMG i MG
• Podobieństwa:
- uogólnione osłabienie mięśniowe i męczliwość
zmieniające się w ciągu dnia
- predyspozycja genetyczna przynajmniej częściowo
kontrolowana przez cząsteczki MHC klasy II
-
zmniejszenie
amplitudy,
ale
nie
częstości
miniaturowych potencjałów płytki nerwowo-mięśniowej
- słabnąca odpowiedź na powtarzające się stymulacje
- zmniejszająca się wrażliwość na acetylocholinę
- przeciwciała anty-AChR, dopełniacz C3, MAC, IgG w
NMJ
- utrata AChR w płytce nerwowo-mięśniowej
- choroba immunologiczna związana z komórkami T i B

• Różnice:
- mięśnie gałki ocznej nie zmienione chorobowo, brak
podwójnego widzenia, brak opadania powiek
- brak zmian chorobowych w obrębie grasicy
- choroba monofazowa
- nie jest chorobą spontaniczną
- węzły chłonne, a nie grasica, są miejscem
autouczulania
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
Wyszukiwarka
Podobne podstrony:
11 Konspekt choroby baktereyjneid 12206 ppt
Choroby układu nerwowego ppt
11 CWICZENIE 1 SEMESTR LETNIid 12747 ppt
11 wyklad sys o przid 12675 ppt
Choroba niedokrwienna serca ppt 2009 ppt
choroby psychogenne dietetyka ppt
Choroby z autoimmunizacji
03 11 2013 Choroba wysokościowa
11 urazy i choroby czaszkiid 12652
Wykład 4 Choroby autoimmunizacyjne
Limfocyty regulatorowe w chorobach autoimmunologicznych, Limfocyty regulatorowe w chorobach autoimmu
Choroby zakaźne układu nerwowego świń 26.11.2009 (1), CHOROBY ZAKAŹNE UKŁADU NERWOWEGO ŚWIŃ
11 Charakteryzowanie chorób oczu, leczenie i profilaktyka
11 ZABURZENIA UKŁADU HEMOSTAZYid 12267 ppt
więcej podobnych podstron