
WPROWADZENIE
DO INŻYNIERII
BIAŁEK
dr Paweł Wysocki

Podczas ostatniej dekady, określono sekwencję genomów różnych
organizmów. Ogrom informacji zakodowanej w genomach
dramatycznie zmienił naukę.
Jeśli możesz zidentyfikować gen w strukturze genomowego DNA,
możesz poznać sekwencję aminokwasów białka oraz sekwencje
regulatorowe kontrolujące ekspresję genu. Nie znasz jednak
strukturalnej organizacji białka oraz jakiej podlega regulacji.
Jeżeli nie ma genomowej bazy danych albo nie możesz
zidentyfikować regionu kodującego w genomie, musisz oczyścić
białko aby można było wyizolować gen.

Jeśli nie udało się wyizolować genu kodującego
białko, możesz być zmuszony do izolacji białka
z jednego z poniższych powodów:
- oczyszczone białko może zostać wykorzystane
do określenia sekwencji aminokwasowej.
Sekwencja może posłużyć jako próba
pomagająca w izolacji genu.
- oczyszczone białko może posłużyć do
wytworzenia przeciwciał mogących pomóc w
izolacji genu kodującego białko.
- oczyszczone białko może być poddane analizie
metodą spektrometrii masowej.

Jeśli wyizolowałeś gen kodujący białko, możesz
izolować
białko z jednego z następujących powodów:
-
czyste białko jest niezbędne do analiz
strukturalnych
- czyste białko jest niezbędne do badania funkcji
enzymów.
- oczyszczone białka są konieczne do badania
interakcji
z innymi białkami lub kwasami nukleinowymi
- oczyszczone białka są potrzebne do badań
regulacji
aktywności enzymatycznej (czy posiadają
podjednostki
regulacyjne, czy są regulowane na drodze
modyfikacji
potranslacyjnych, czy podlegają regulacji na
skutek
interakcji z innymi białkami).

Genom i transkryptom nie są wystarczające do
konstruowania modeli układów biologicznych i
przewidywania ich funkcjonowania.
Aktywność biologiczna białek jest bardzo często
regulowana
na
etapie
modyfikacji
potranslacyjnych
(np.
po
proteolizie
ograniczonej czy fosforylacji).
Zawartość białek w komórce nie zawsze
pozostaje
w
bezpośrednim
związku
z
szybkością transkrypcji mRNA. Bardzo istotna
funkcję
regulacyjną
odgrywają
procesy
translacji i degradacji białek.
Dlaczego badać białka ?

Genom człowieka:
• ~3.2 x 10
9
bp (trzydzieści razy większy od genomu
D.melongaster)
• sekwencje kodujące stanowią tylko 5% genomu
• sekwencje powtórzone ponad 50%
• „Tylko” ~32.000 genów
• eksony są relatywnie małe w porównaniu do innych
genomów eukariotycznych
• introny są relatywnie długie
• średnia długość genu ~ 8,000 bp
• średnio 5-6 eksonów/gen
• średnia długość eksonu ~200 bp
• średnia długość intronu ~2,000 bp
• ~8% genów zawiera jeden ekson
• niektóre eksony mogą być złożone z 1 do 3 bp

PROTEOMIKA
If the genome is a list of the instruments in an
orchestra, the proteome is the orchestra playing
a symphony.
R.Simpson

Taki sam GENOM
Inny PROTEOM
Zdarzenia mające miejsce ponad genomem
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
1997
1998
1999
2000
2001
2002
2003
2004
Papers
Reviews
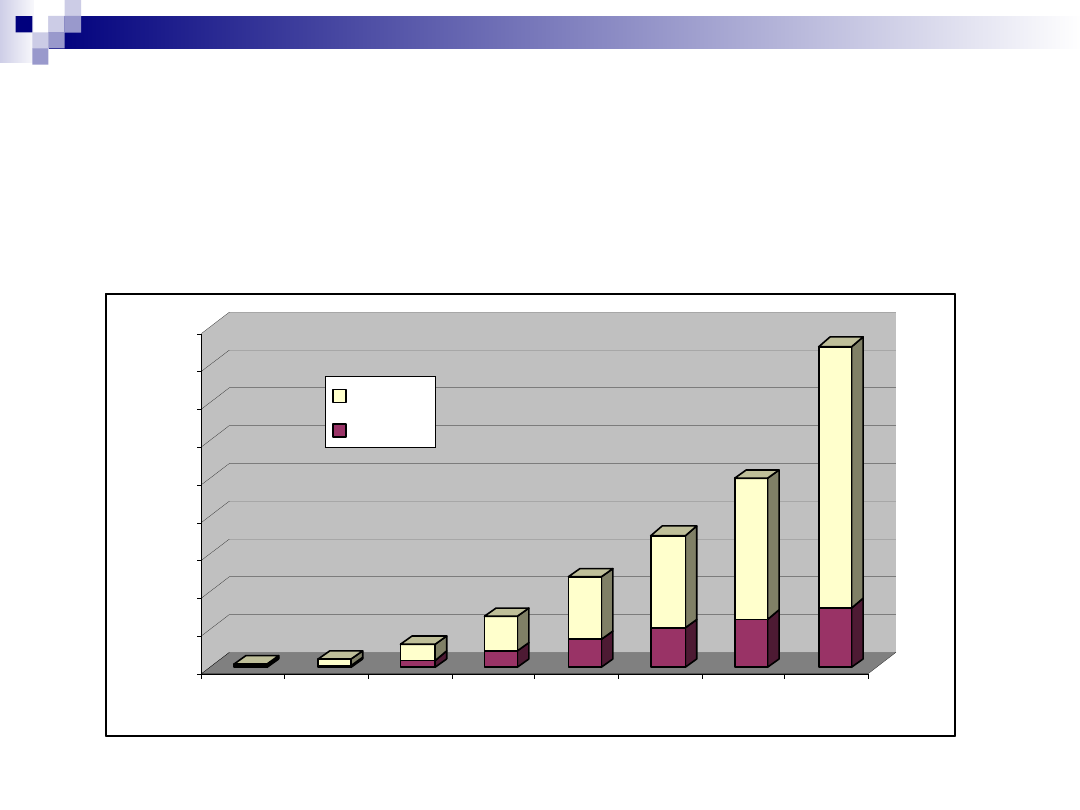
Publikacje dotyczące proteomiki według Medline
Od
220
publikacji w minionym wieku (‘94-’99)
do
21,350
(!!!) publikacji w pierwszych latach tego wieku
(‘00-’05)

• około 1x10
14
komórek
• 250 typów komórek
• szacowana ilość białek 25 000 - 30 000 (120
000)
• 8 000 - 10 000 różnych białek w komórce
• od kilku do kilku milionów kopii danego białka
w komórce (większość białek w komórce to
białka konstytutywne występujące w ponad 10
000 kopii)
• kilkadziesiąt-kilkaset białek specyficznych dla
danego typu komórki lub tkanki
• brak liniowej zależności pomiędzy ilością
mRNA a proteomem
Organizacja proteomu człowieka
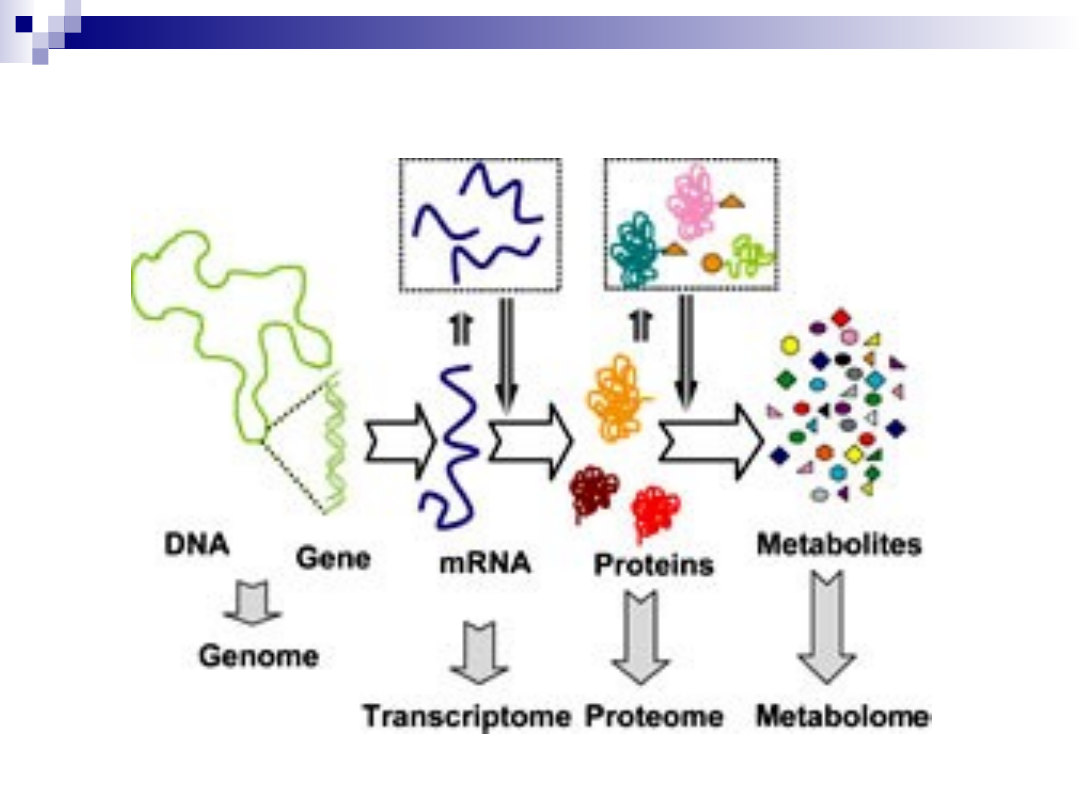
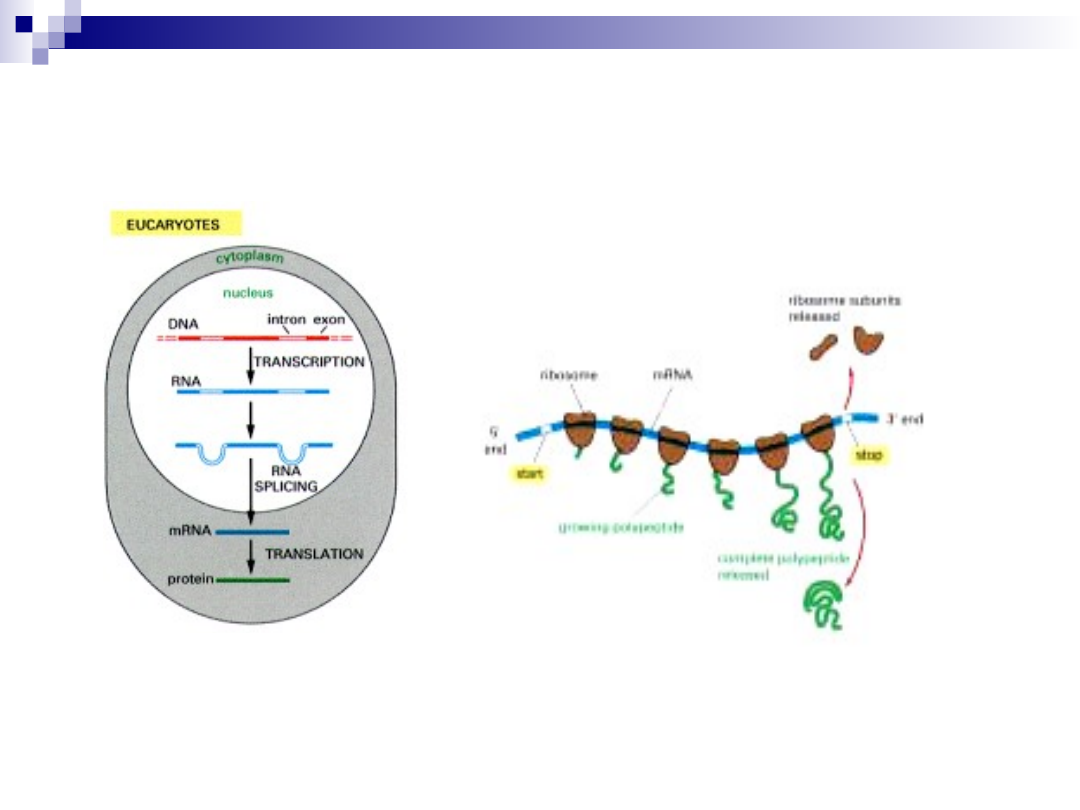
DNA
RNA
mRNA
Białko
Transkrypcja
Kontrola
transkrypcji
Dojrzewanie
Splicing
Redagowanie
Alternatywna
poliadenylacja
Translacja
Kontrola
translacji
oraz
degradacji
>200
potranslacyjnyc
h modyfikacji
•aktywność
katalityczna
•oddziaływani
e
•stabilność
•czas życia
•lokalizacja
•aktywność
Genomika
Transkryptomika Proteomika Metabolomika
Zależność pomiędzy genomem oraz proteomem

Dynamika proteomu
Całkowita ilość białek w organizmie 14kg
Dzienny obrót białek 350g
Połowiczne czasy degradacji
Dekarboksylaza ornitynowa
11 min
Oksygenaza tryptofanowa
2 h
Glukokinaza
12 h
Transaminaza alaninowa
1 dzień
Albumina surowicza
3,5 dnia
Dehydrogenaza mleczanowa
16 dni
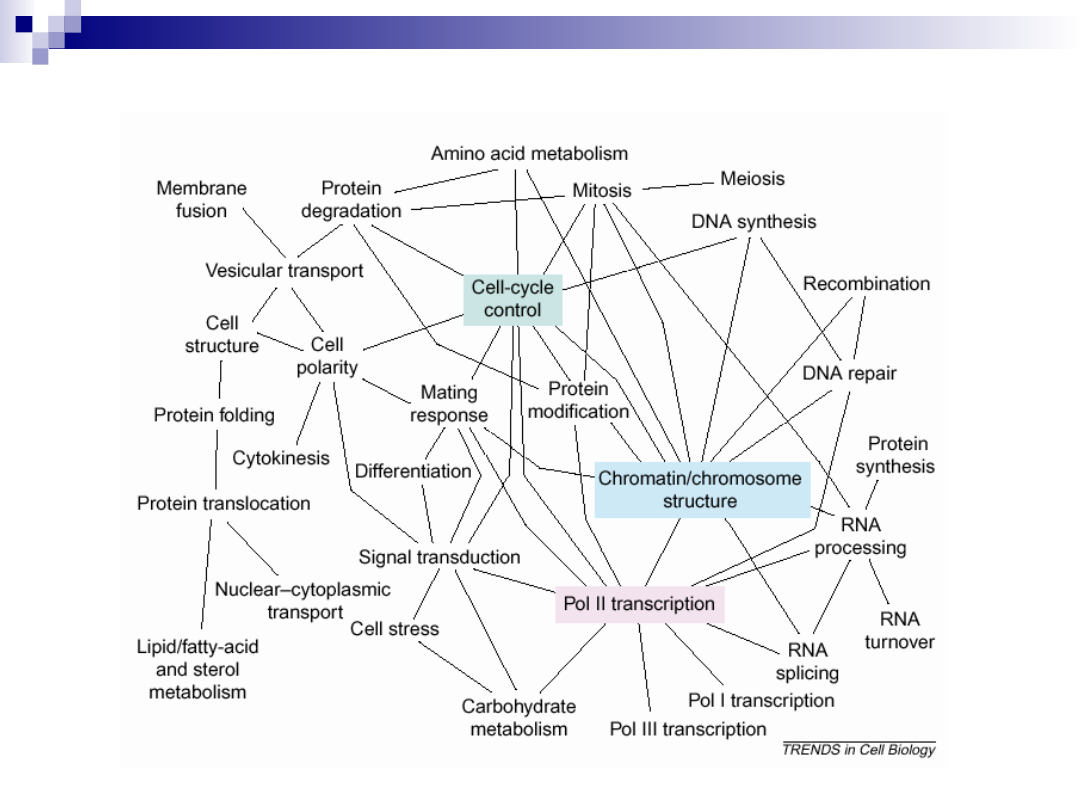
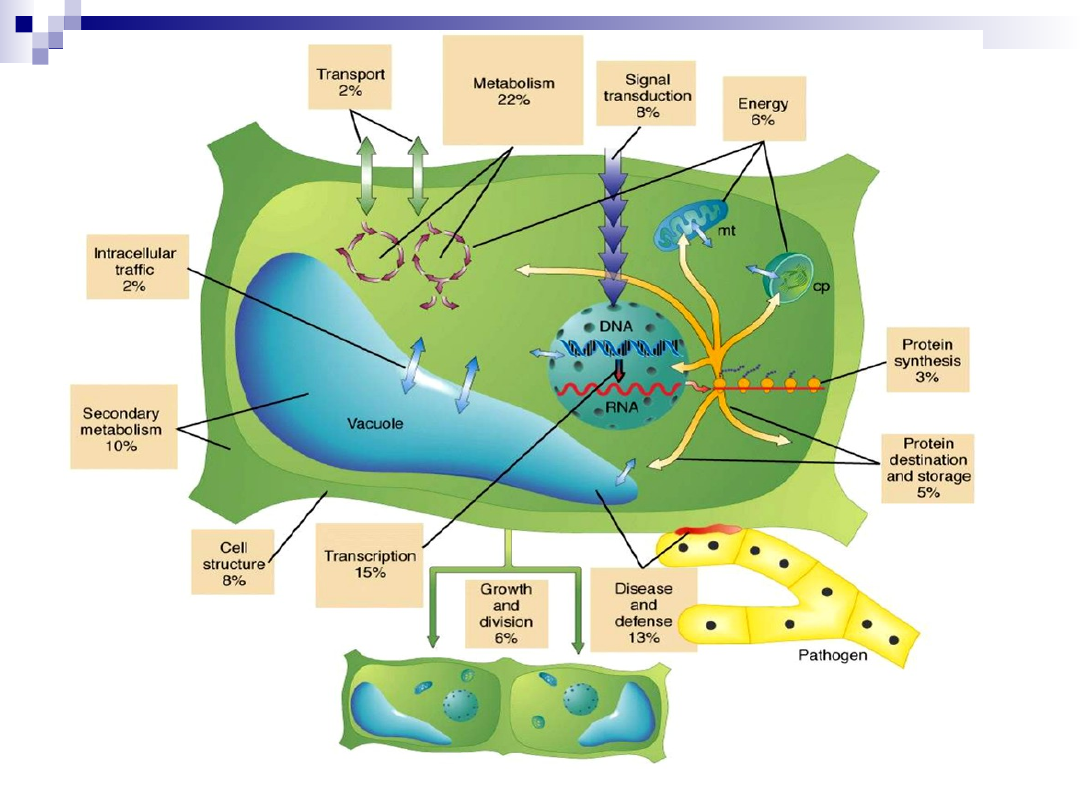
Mapa połączeń pomiędzy funkcjonalnymi grupami białek

Sygnalizacja komórkowa z udziałem
czynnika wzrostu naskórka (EGF)
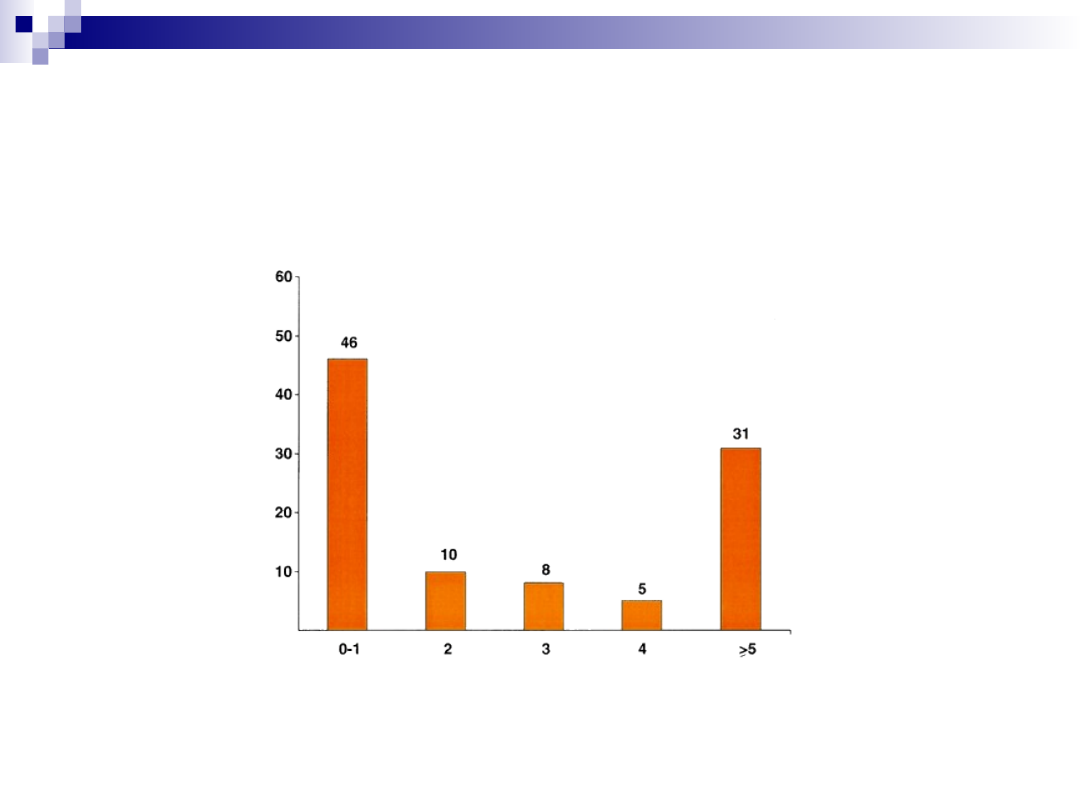
Średnia ilość oddziaływań pomiędzy białkami
Człowiek 7 – 10
x 30 000 białek 210 000 - 300 000
Drożdże 5
x 6 000 białek 30 000
Baza danych białek ludzkich HPRD
Ilość oddziaływań
P
ro
ce
n
t
b
ia
łe
k
w
H
P
R
D

Proteomika 886,000 hits
(2004)
4,700,000 hits
(2005)
Genomika 2,070,000 hits
(2004)
16,000,000 hits
(2005)
Proteomika – Google

Olbrzymi zakres ilości
Olbrzymi zakres ilości
analizowanych molekuł
analizowanych molekuł
(
(
od
od
1
1
do
do
10
10
8
8
)
)
Szeroki zakres mas
Szeroki zakres mas
cząsteczkowych
cząsteczkowych
(
(
od
od
750 kDa
750 kDa
do
do
350 Da)
350 Da)
zakres
zakres
pI (3
pI (3
do
do
12)
12)
zakres hydrofobowości
Duże zróżnicowanie stabilności
białek
Proteomika: wyzwania
Proteomika: wyzwania

Kompletny zestaw białek, których
biosynteza i modyfikacje mają miejsce w
określonym momencie funkcjonowania
komórki, narządu lub organizmu.
Białka są „fizycznym” wyrażeniem
interakcji między genomem i środowiskiem.
PROTEOM
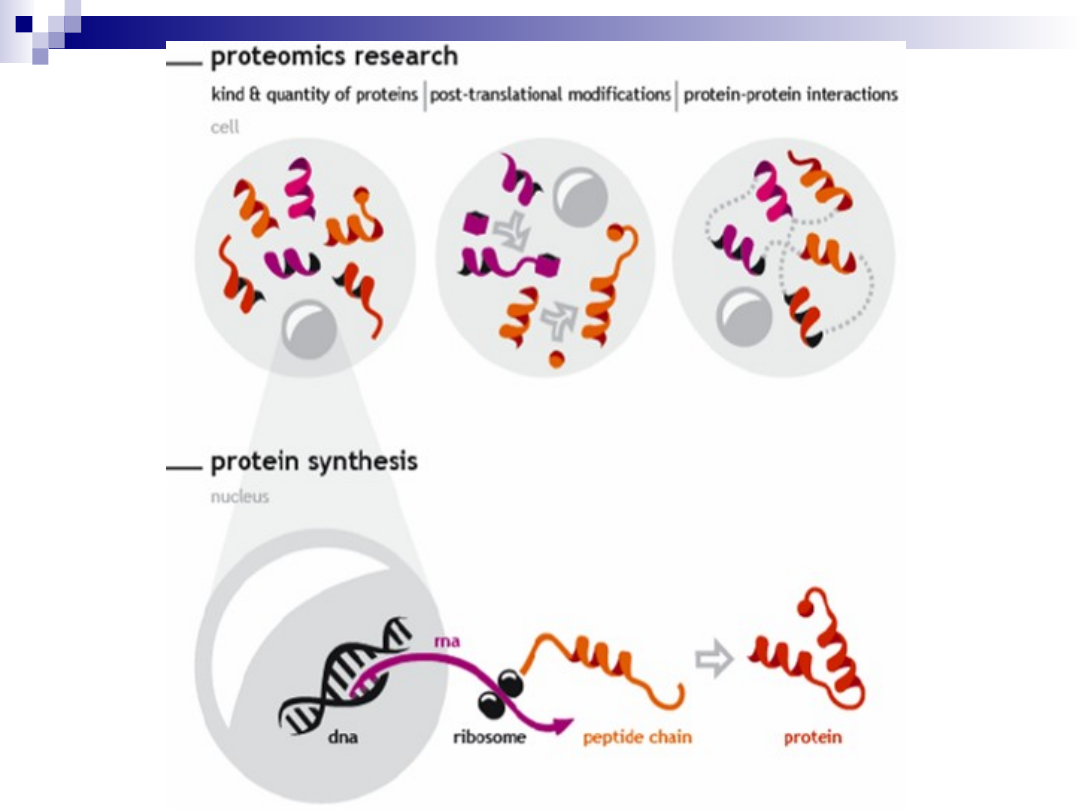
Gen A
Gen B
mRNA A
mRNA B
Białko A
Białko B
Białk
a
interakcje
lokalizacja
obieg białek
modyfikacje potranslacyjne
FUNKCJE
SCHEMAT ZALEŻNOŚCI MIĘDZY GENOMEM,
SCHEMAT ZALEŻNOŚCI MIĘDZY GENOMEM,
TRANSKRYPTOMEM I PROTEOMEM
TRANSKRYPTOMEM I PROTEOMEM
GENOM
TRANSKRYPTOM
PROTEOM

GŁÓWNE ZASTOSOWANIA PROTEOMIKI
IDENTYFIKACJA BIAŁEK PROTEOMU
(wykorzystanie metod pozwalających na
identyfikację białek na dużą skalę)
PROTEOMIKA PORÓWNAWCZA LUB
JAKOŚCIOWA
PROTEOMIKA FUNKCJONALNA (opisywanie
interakcji między cząsteczkami białek)

CZYM JESZCZE ZAJMUJE SIĘ
PROTEOMIKA?
-pomiarami ekspresji białek w komórkach – expression
proteomics
- badaniem składu białkowego organelli komórkowych
– cell
map proteomics
- analizą modyfikacji potranslacyjnych
- analizą interakcji białko-białko
- analizą interakcji białko-lek
- określaniem struktury białek (NMR, X-ray, in silico
etc.)
Badania te w konsekwencji prowadzić mają do
określenia funkcji białka
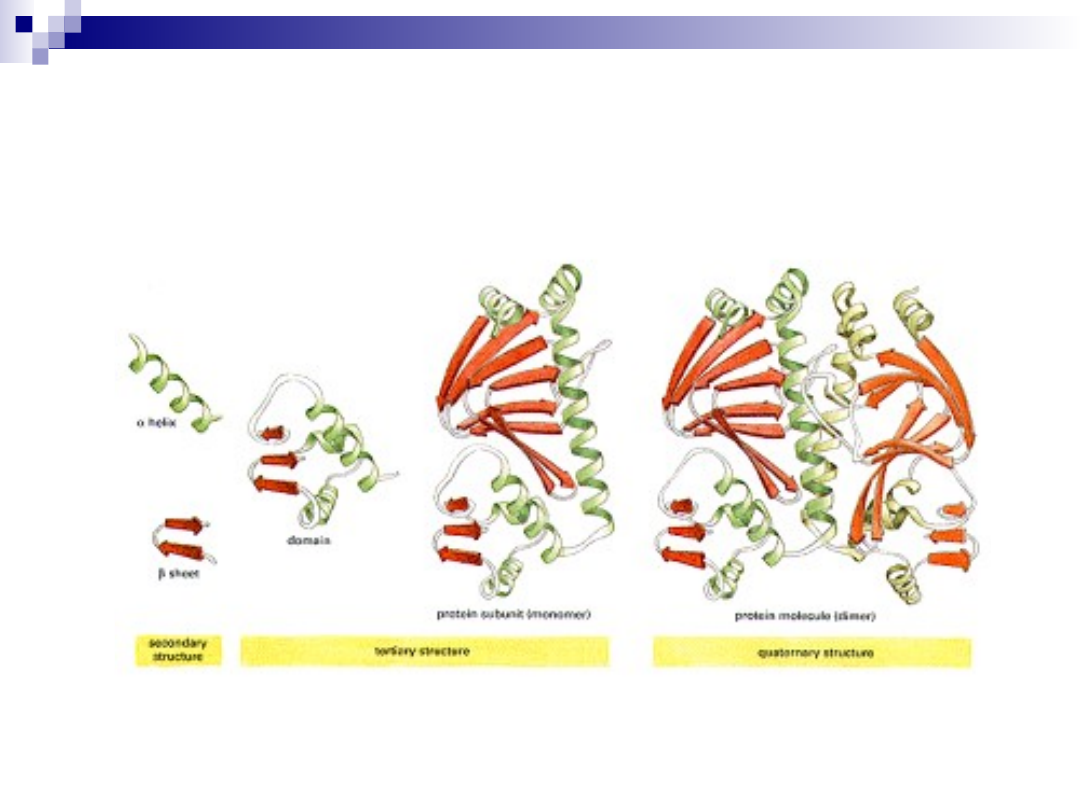
STRUKTURA BIAŁEK

DNA/RNA
Białka
Cząsteczki stabilne
Przechowywanie i procedury
postępowania tanie i proste
Minimalne modyfikacje cząsteczek
Dobrze opanowane procedury
izolacji
Cząsteczki labilne, podatne
na denaturację
Przechowywanie i procedury
zależne od konkretnego białka
Liczne modyfikacje cząsteczki
Interakcje międzycząsteczkowe
zależne od lokalizacji
Dobrze opanowane
sekwencjonowanie
Problemy z
sekwencjonowaniem
Testy DNA / genotypowanie/
ekspresja/
Protein Chip (nie
opanowane)
Antibodies array (nie
opanowane)

Funkcje białek są zależne od
kontekstu, w jakim są rozpatrywane
Określono całkowitą sekwencję białka.
Nadal nie wiemy nic o:
Cechach struktury ????????
Lokalizacji ????????
Interakcjach z małymi cząsteczkami ?????????
Interakcjach z innymi białkami ?????????
Typach/dynamice modyfikacji ?????????
Czasie funkcjonowania cząsteczki ?????????


Schemat żelu
elektroforetycznego
ilustrujący procedurę
izolacji białka
Procedura
Objętość
frakcji
(ml)
Białko
całkowite
(mg)
Aktywnoś
ć
(jednostki
)
Aktywnoś
ć
właściwa
jedn/mg
Ekstrakt
komórkowy
1400
10000
100000
10
Sączenie
molekularne
90
400
80000
200
Chromatografia
jonowymienna
80
100
60,000
600
Tabela oczyszczania białka enzymatycznego
Współczynn
ik
oczyszczeni
a
Odzysk
100%
20
80%
60
60%
1
Rodzaj i kolejność etapów jest opracowywana
indywidualnie dla każdego białka. Efektywna
procedura izolacji daje wysoki odzysk (minimalną
stratę aktywności enzymatycznej) i wysoki
współczynnik oczyszczenia (duży wzrost
aktywności właściwej).
IZOLACJA BIAŁKA
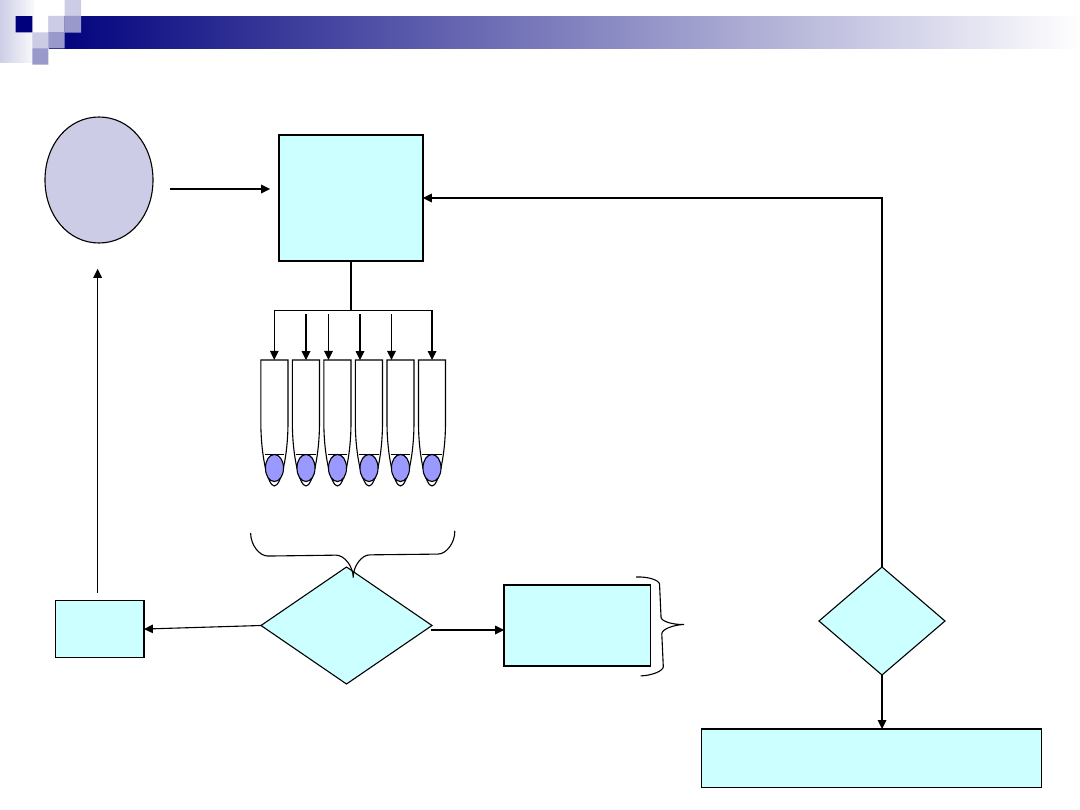
Próba
Technika
separacyjna
Frakcjonowanie
OCZYSZCZANIE BIAŁKA JEST PROCEDURĄ WIELOETAPOWĄ
Czy jest aktywność?
Start od
początku
NI
E
Połączenie frakcji
TAK
Sprawdzenie czystości
Pomiar zawartości białka
Pomiar aktywności
enzymu
Czyste?
Przygotowanie do technik analitycznych
Tak
Nie
Przeprowadzaj
rozdziały
innymi
technikami
separacyjnymi
do uzyskania
czystości
preparatu

Pierwsze etapy
1. Źródło materiału. Dobre źródło materiału jest tanie i łatwo dostępne. Wiele
białek występuje w zwiększonych ilościach w specyficznych tkankach np.
hemoglobina we krwi. Z tego powodu, tkanki te mogą być doskonałym źródłem
naszego białka
2. Pomiar zawartości: Większość pomiarów wykorzystuje reakcje chemiczne
katalizowane przez określone enzymy. Białka nie wykazujące aktywności
enzymatycznej mierzy się najczęściej z wykorzystaniem elektroforezy SDS-
PAGE.

33
Przygotowanie próby
Główne wskazania:
Wybierz procedurę ekstrakcji kierując się
źródłem i lokalizacją białka
Używaj, w miarę możliwości łagodnych
procedur aby minimalizować ryzyko zmiany pH
i uwolnienia enzymów proteolitycznych
Pracuj szybko w obniżonej temperaturze
Używaj buforów aby zachować wartość pH i
siły jonowej
CEL: stabilizacja białka
34
Czystość
Etap
Wstępna
Faza(y) pośrednia
Końcowa
Izolacja,
koncentracja, stabilizacja
Usunięcie głównych
zanieczyszczeń
Osiągnięcie końcowej
czystości, usunięcie
zanieczyszczeń
śladowych, wariantów
strukturalnych
białka,agregatów,
wirusów itd.
Trzy fazy oczyszczania białek:
35
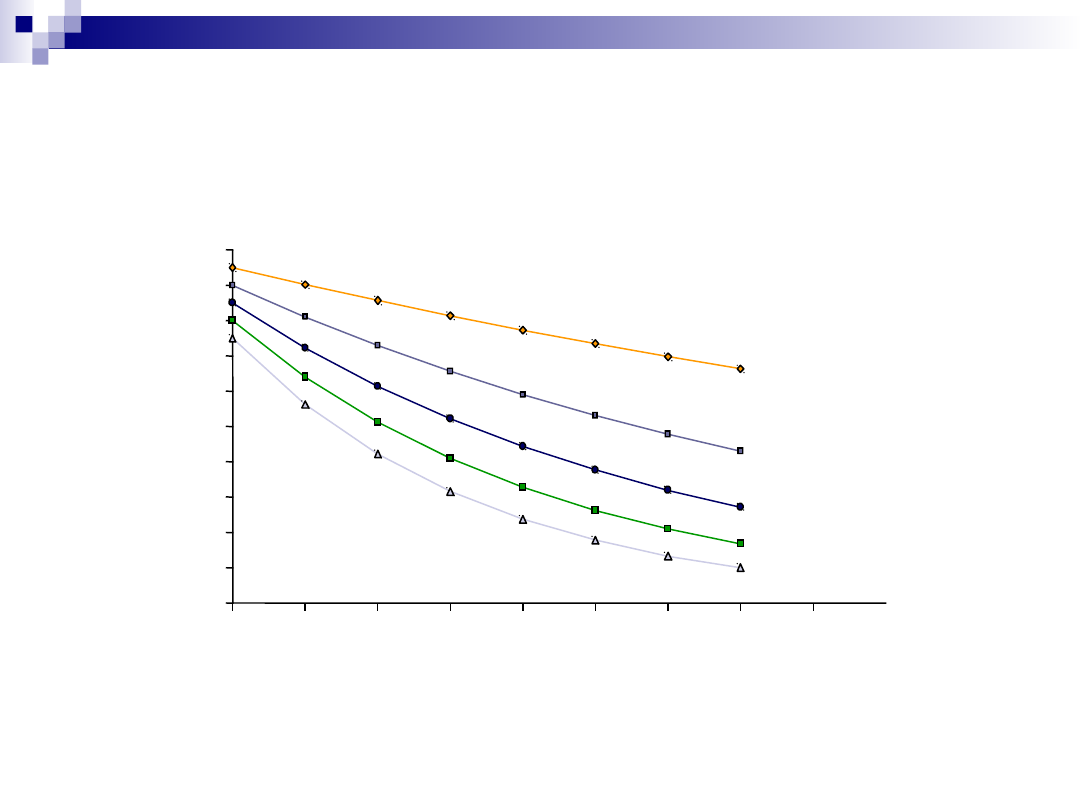
Zawsze ograniczaj ilość etapów izolacji
Utrzymuj wysoki odzysk na każdym z
etapów
Ilość etapów izolacji
Odzysk (%)
95% / step
90% / step
85% / step
80% / step
75% / step
0
20
40
60
80
100
1
2
3
4
5
6
7
8
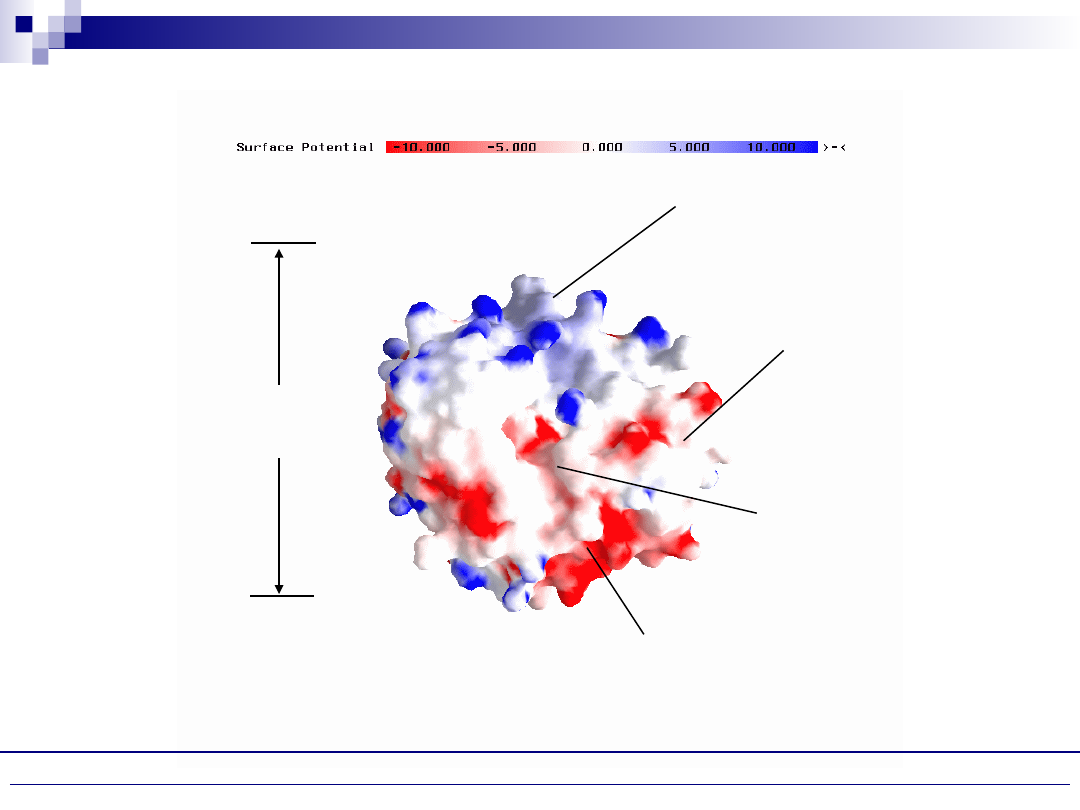
Dodatnio naładowane domeny zasadowe
Ujemnie naładowane domeny kwaśne
Region hydrofobowy
Miejsce wiążące ligand
ok. 40 Å
Rozmiar cząsteczki:
Białka są cząsteczkami amfipatycznymi
Obecność ładunków, regionów hydrofobowych, wielkość oraz rozpuszczalność wpływają
na właściwości biofizyczne białka i determinują strategię oczyszczania
37
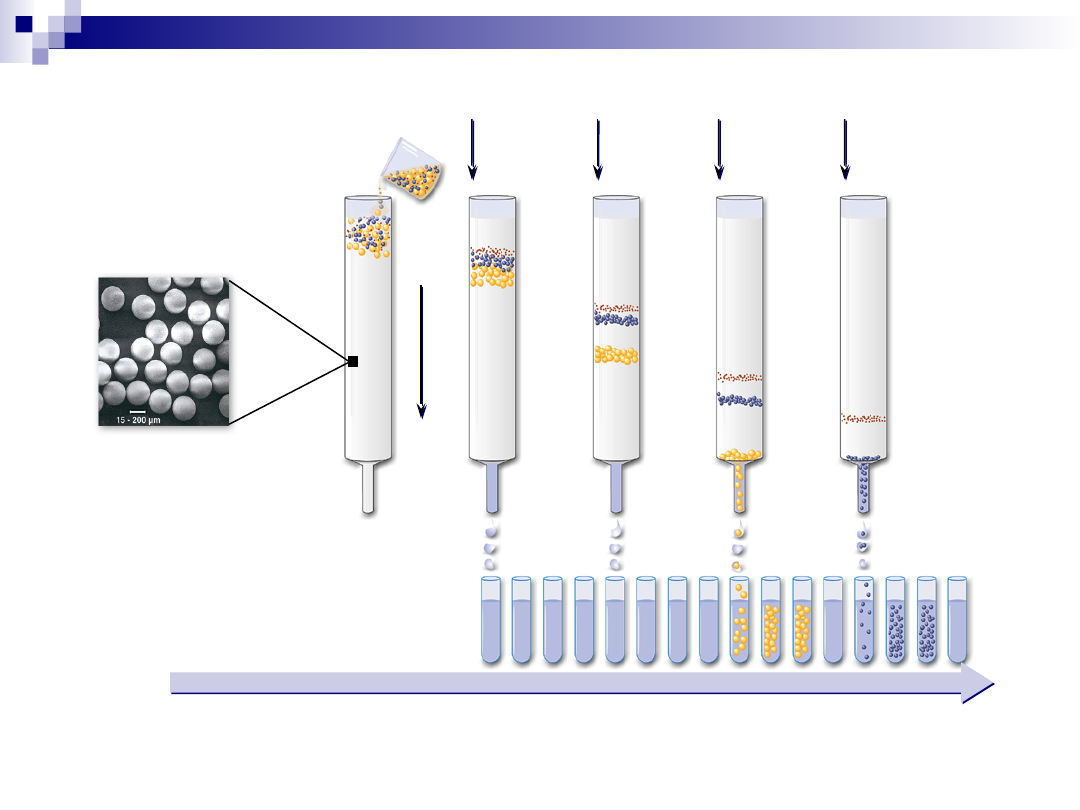
Chromatografia
Przepływ cieczy
Przepływ
cieczy
Czas
1
2
3
4
5
Rozdział ze względu na:
-masę cząsteczkową
-ładunek
-hydrofobowość
-powinowactwo
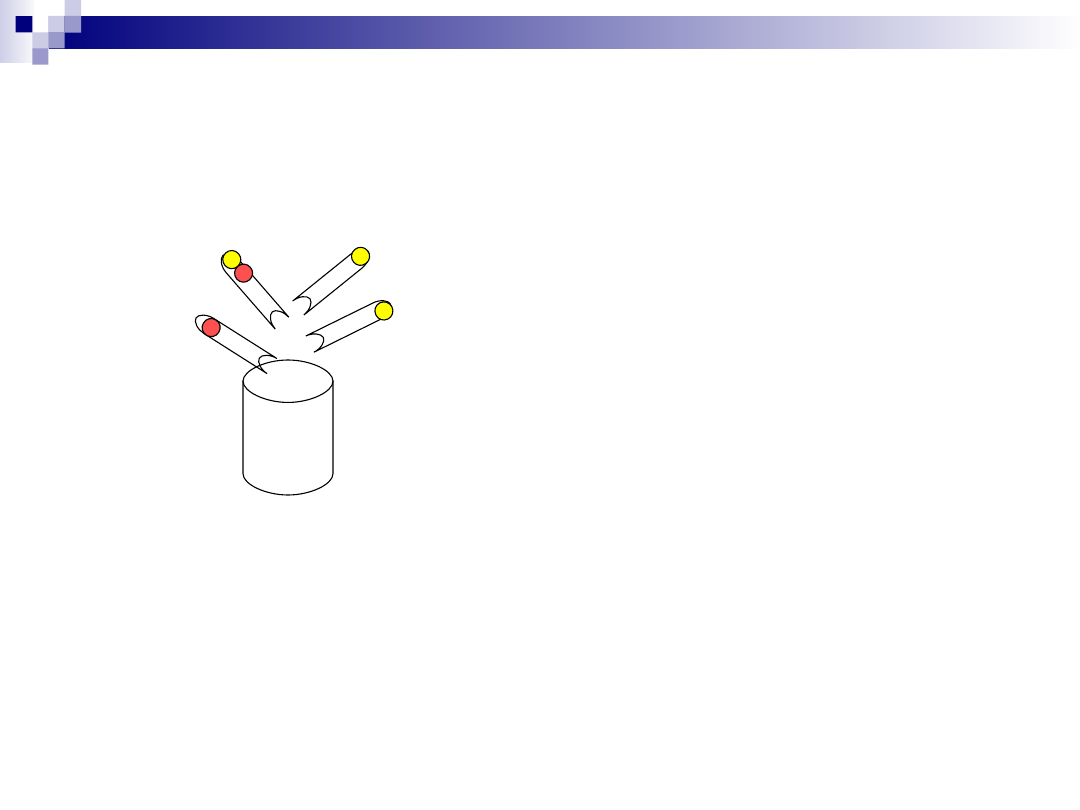
Próba zawierająca
białka lub peptydy

Schemat kolumny
chromatograficznej z mieszaniną
białek naniesioną na powierzchni
Ekstrakt jest nanoszony na
powierzchnię żelu w kolumnie
(w tym przypadku jest to
mieszanina 4 różnokolorowych
białek)
Białka przesuwają się w dół
kolumny z różnymi
prędkościami w zależności od
właściwości białka i złoża
chromatograficznego.
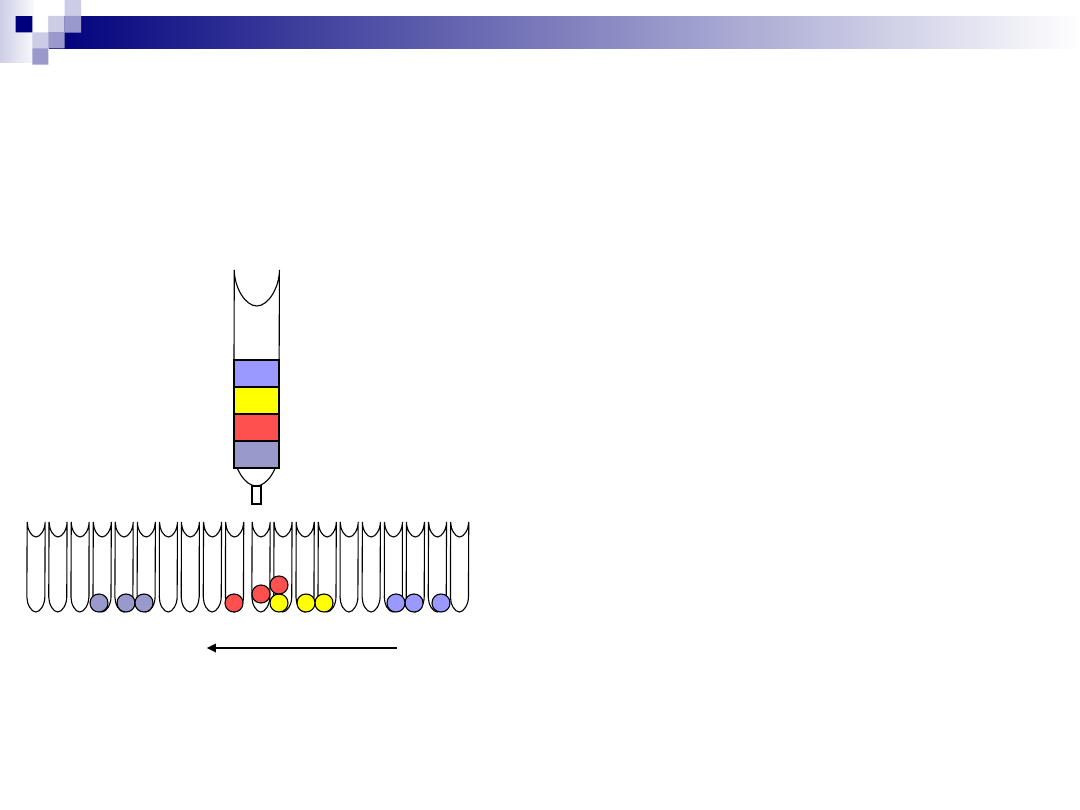
Frakcjonowanie
Podczas separacji na kolumnie
bufor wypływający z kolumny
jest zbierany do kolejnych
probówek , które przesuwane
są z odpowiednią prędkością.
Zawartość probówek
nazywana jest frakcjami
chromatograficznymi.

W rzeczywistości, białka
nie są kolorowe a więc
musimy znaleźć
odpowiedź na 3 pytania:
1.
Skąd mamy wiedzieć
która z frakcji zawiera
białka?
2.
Która z frakcji
białkowych zawiera to
białko, które chcemy
izolować?
3.
Jak zmierzyć czystość
frakcji?
???

Całkowita zawartość białka może
zostać oszacowana po pomiarze
absorbancji roztworu przy 280 nm
z użyciem spektrofotometru.
Może to być wykonane podczas
trwania rozdziału
chromatograficznego.
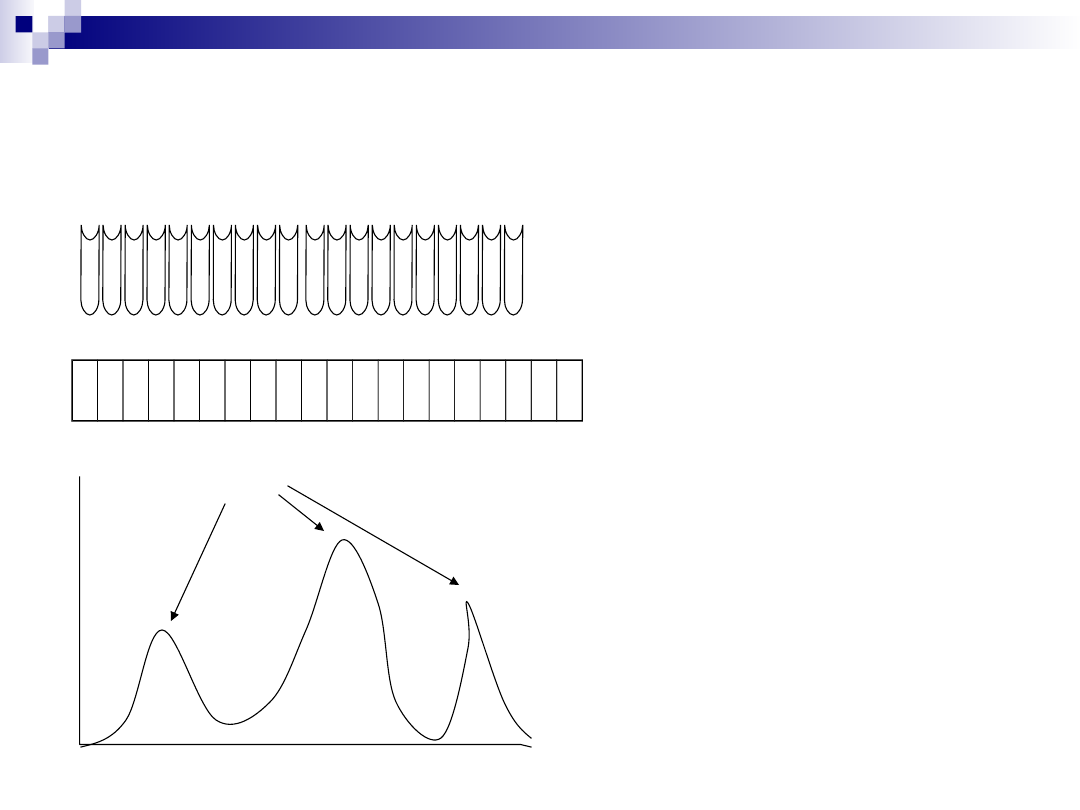
Pytanie 1. Skąd mamy wiedzieć która z frakcji zawiera białka
?
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20

Na podstawie uzyskanych
wartości sporządza się
tzw. Profil elucyjny
A
280
0 0 0 2 5 2 0 0 0 2 5 8 5 2 0 0 2 5 2 0
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Fraction
#
A
280
0 0 0 2 5 2 0 0 0 2 5 8 5 2 0 0 2 5 2 0
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Fraction
#
A
280
Kolejne frakcje
„Piki białkowe”
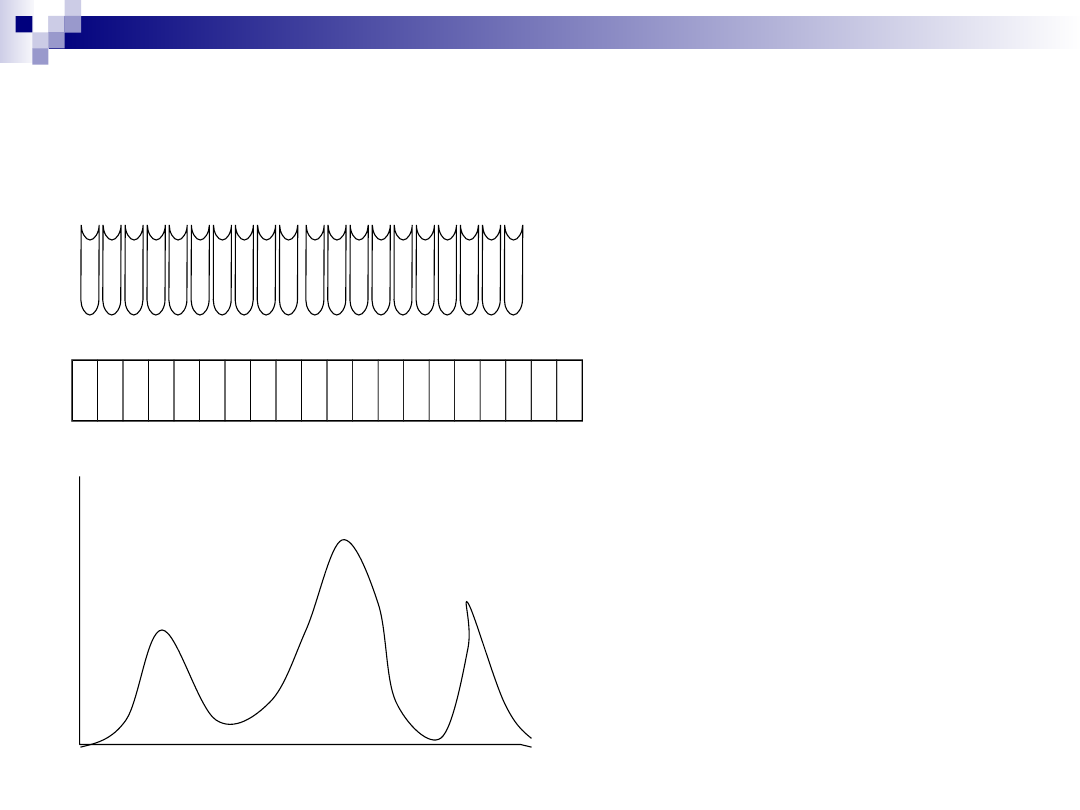
Aktywność enzymu może
zostać określona przez
przeprowadzenie oznaczania
aktywności enzymu w każdej
z frakcji zawierających białko.
Pytanie 2. Która frakcja zawiera interesujące nas białko?
A
280
0 0 0 2 5 2 0 0 0 2 5 8 5 2 0 0 2 5 2 0
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Fraction
#
A
280
Fraction #
Fraction
#
A
280
0 0 0 2 5 2 0 0 0 2 5 8 5 2 0 0 2 5 2 0
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Fraction
#
A
280
Fraction #
EnzAssa
y
Results
A
280
0 0 0 2 5 2 0 0 0 2 5 8 5 2 0 0 2 5 2 0
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Fraction
#
A
280
Fraction #
EnzAssa
y
Results
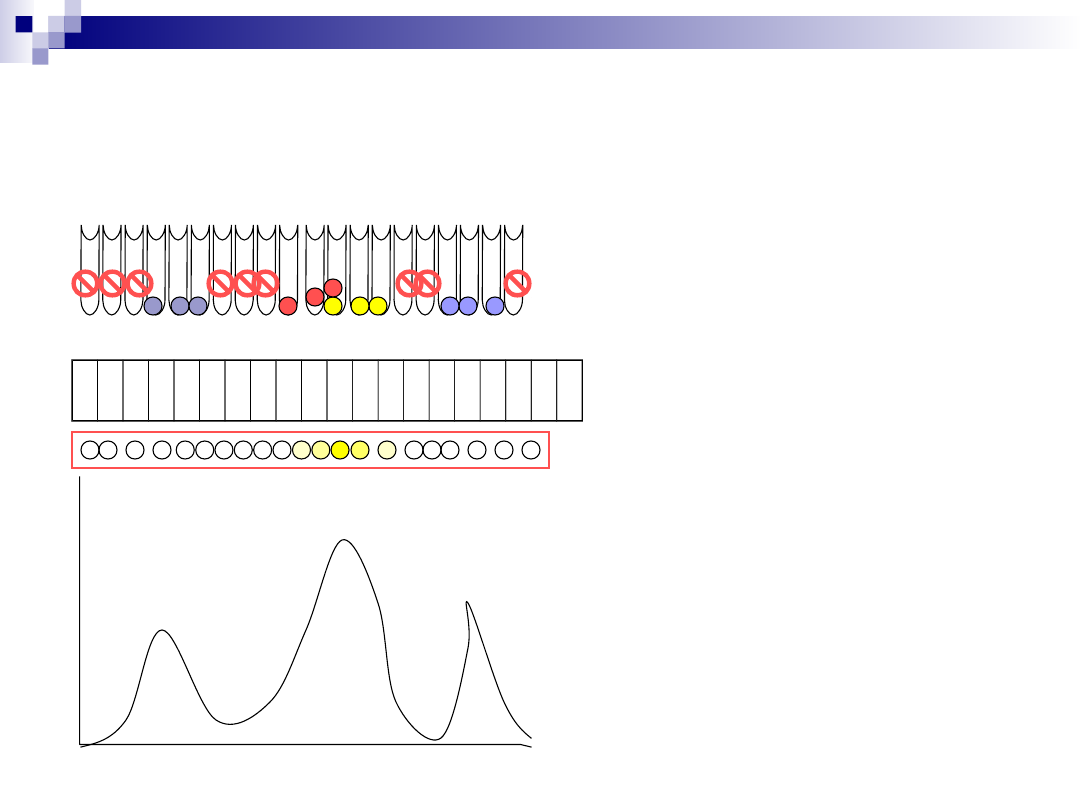
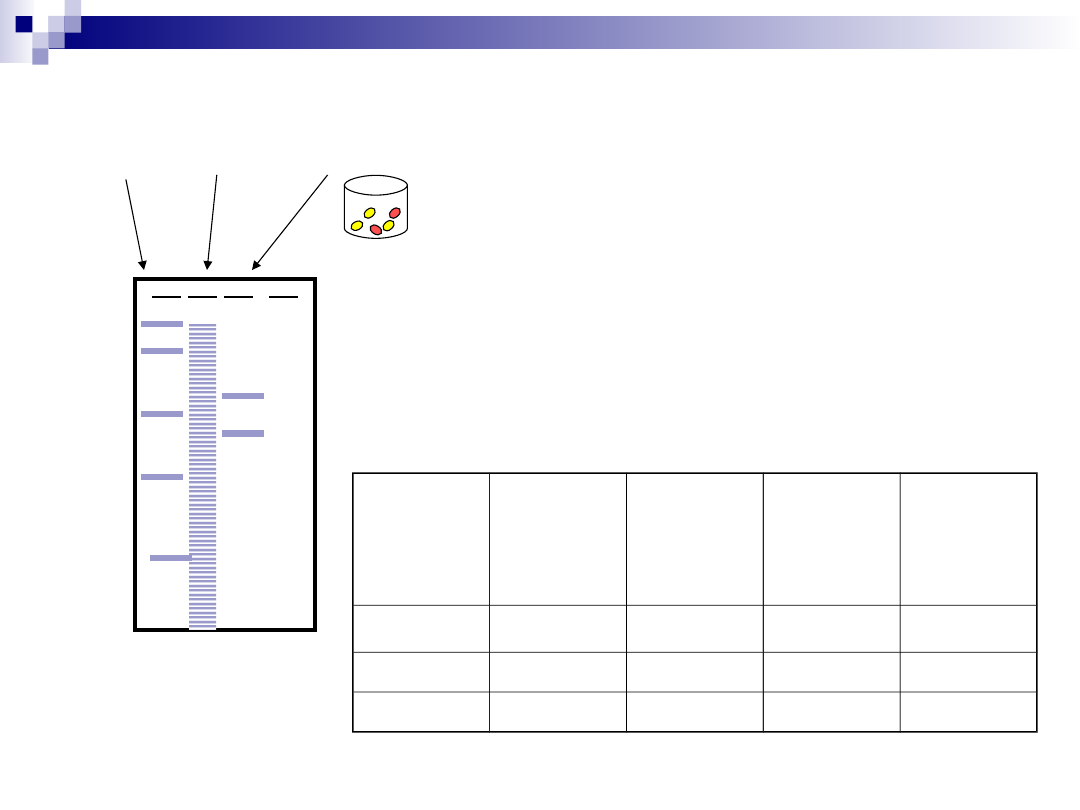
Zbierz frakcje z zawartością białka i aktywnością enzymatyczną
1.
Frakcje zostają zebrane.
Popatrz na żel. Nawet jeżeli naniesione jest zbyt
dużo materiału, jeżeli widoczna jest jedna
frakcja, prawdopodobnie białko jest czyste, i jest
monomerem, homo-dimerem lub homo-
multimerem. Jeżeli to nie jest jeden prążek,
dochodzi prawdopodobnie do równoczesnej
izolacji innego białka. Jest taka możliwość, że to
podjednostka izolowanego białka.
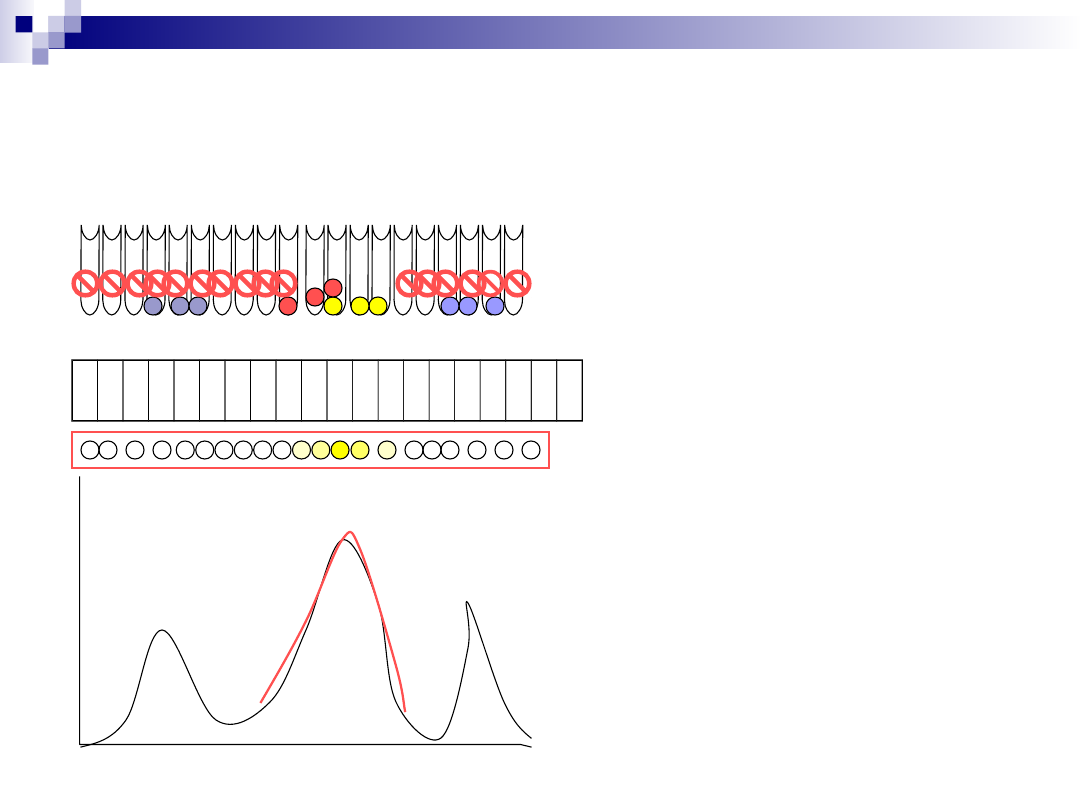
Oblicz aktywność właściwą przez
przeprowadzenie dokładnego jakościowego
pomiaru aktywności enzymu i ilości białka.
Czy zebrana próba jest czysta? Jak obserwować postęp izolacji?
Standardy | Ekstrakt | zebrane frakcje
Procedur
e
Obj.
frakcji
(ml)
Zawartoś
ć białka
(mg)
Aktywnoś
ć
(U)
Aktywno
ść
właściwa
Units/mg
Ekstrakt
wyjściowy
1400
10000
100000
10
Etap 1
90
400
80000
200
Tabela oczyszczania
Wyniki:
1.
Na żelu widoczna więcej niż jedna frakcja.
Ponieważ próba nie jest czysta potrzeba dodatkowej techniki separacyjnej
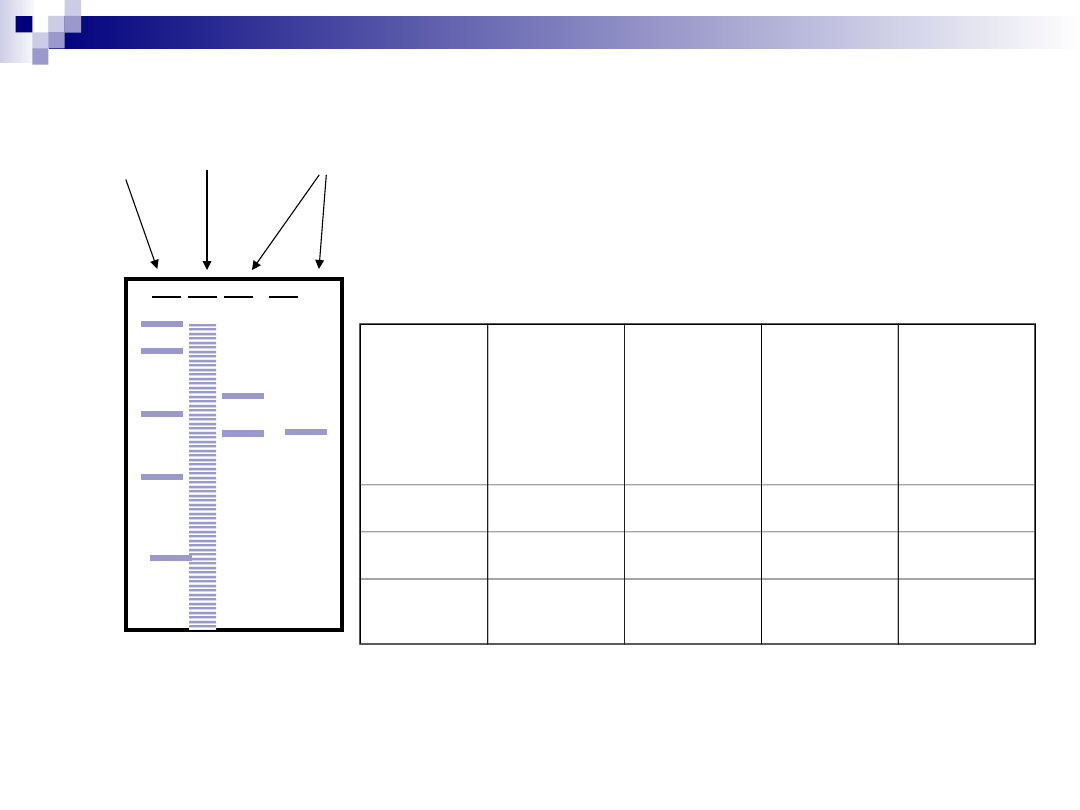
Czy próba jest czysta? Jak oceniasz postęp izolacji?
Standard | Ekstrakt | Zebrane frakcje
Procedura
Objętość
próby
(ml)
Całkowit
a
zawartoś
ć białka
(mg)
Aktywnoś
ć
(units)
Aktywno
ść
właściwa
Units/mg
Ekstrakt
komórkowy
1400
10000
100000
10
Metoda
separacyjna 1
90
400
80000
200
Metoda
separacyjna
2
8
4
60000
15000
Tabela oczyszczania
Wyniki:
1.
Na żelu widoczna 1 frakcja
2.
Aktywność właściwa wynosi 15 000

Kryteria czystości
preparatu białkowego:
Jednorodności
– obserwujemy
jedną frakcję po elektroforezie
PAGE i SDS – PAGE. Im bardziej
czułą metodę barwienia
zastosujemy, tym większą mamy
pewność, że białko jest czyste.
Inne wyniki można uzyskać po
rozdziale białek enzymatycznych
zbudowanych z heterogennych
podjednostek np.
PAGE
SDS-PAGE

Molekularne – podczas
analizy sekwencji N-
końcowej wykazywana jest
obecność jednego rodzaju
aminokwasu. Podczas analiz
stwierdza się jeden rodzaj
aminokwasu N-końcowego i
C-końcowego.
Katalityczne (enzymy) – nie
wykazywana jest żadna inna
aktywność enzymatyczna
Immunologiczna – po
immunizacji uzyskuje się
jednorodną frakcję
przeciwciał, dających jedną
linię precypitacyjną w
testach immunodyfuzji.

Rozbicie komórek:
Jest konieczne z wyjątkiem przypadków, gdy źródłem
białka jest płyn fizjologiczny (surowica krwi, mocz,
płyn mózgowo-rdzeniowy, pożywka hodowlana itd..)
Jeżeli białko występuje w jakimś oraganellum
komórkowym (mitochondrium, błonach, jądrze itd.)
najlepiej najpierw izolować określoną część komórki,
potem dopiero poddawać ją rozbiciu.
Najczęściej stosowane metody rozbijania komórek:
-
Szok osmotyczny – komórki umieszcza się w
środowisku hipo- lub hipertonicznym, co powoduje
uszkodzenie błon komórkowych (np. liza erytrocytów)
-
Zamrażanie-rozmrażanie – tworzące się kryształy lodu
powodują uszkodzenia błon komórkowych
-
Rozbijanie mechaniczne z zastosowaniem różnego
typu homogenizatorów mechanicznych, młynków,
mikserów itp.

-
Rozcieranie tkanek w ciekłym azocie.
-
Rozbicie ścian i (lub) błon komórkowych odpowiednimi
enzymami (np. lizozym podczas rozbicia komórek
bakterii).
-
Traktowanie tkanek odczynnikami silnie denaturującymi
białka (8M mocznik, 6 M chlorowodorek guanidyny, SDS,
NaOH itd.) Możliwe tylko wtedy, gdy interesujące nas
białko nie podlega denaturacji. Jest to tzw. denaturacja
wybiórcza.
Podczas rozbicia komórek dochodzi do uwolnienia
proteinaz z lizosomów, które mogą hydrolizować białka.
Hydrolizie enzymatycznej można zapobiec przez:
-
Pracę w obniżonej temperaturze np. w chłodni lub z
wykorzystaniem lodu
-
Dodatek inhibitorów proteinaz serynowych (PMSF,
benzamidyna, inne), metaloproteinaz (EDTA)

Pierwsze etapy
1. Źródło materiału. Dobre źródło materiału jest tanie i łatwo dostępne. Wiele
białek występuje w zwiększonych ilościach w specyficznych tkankach np.
hemoglobina we krwi. Z tego powodu, tkanki te mogą być doskonałym
źródłem naszego białka.
2. Pomiar zawartości: Większość pomiarów wykorzystuje reakcje chemiczne
katalizowane przez określone enzymy. Białka nie wykazujące aktywności
enzymatycznej mierzy się najczęściej z wykorzystaniem elektroforezy SDS-
PAGE.

DETERGENTY:
Niejonowe: Tween 80, Triton X-100, Lubrol, Triton X-114
Jonowe :SDS, deoxycholan sodu, CTAB
Obojętne dwujonowe (zwitterionic): CHAPS, CHAPSO, lizolecytyna
Używane są podczas izolacji białek błonowych, gdyż powodują dyspersję
lipidów i fosfolipidów. Każdy detergent ma określoną koncentrację
(CMC; critical micelle concentration), powyżej której cząsteczki tworzą
struktury zwane micellami. Są one trudne do usunięcia podczas
dalszych etapów izolacji.
Niskie stężenia detergentów mogą skutecznie chronić białka podczas
przechowywania.

Pierwsze etapy – opracowanie metody pomiaru
Pomiar enzymu jest metodą określającą jego
aktywność enzymatyczną .
Ponieważ pomiary będą przeprowadzane
wielokrotnie istotne jest, aby procedura była
prosta. Aktywność enzymatyczna jest mierzona
zwykle na podstawie zmian absorbancji
mierzonych przy pomocy spektrofotometru. Na
przykład pomiar aktywności rybonukleazy mierzy
zmianę absorbancji roztworu RNA spowodowaną
jego hydrolizą do rybonukleozydów.

Po co izolować enzymy?
Aby zrozumieć i opisać strukturę enzymu, kinetykę
aktywności, mechanizm działania, regulację
aktywności i jego udział w funkcjonowaniu złożonych
systemów biologicznych niezbędne jest badanie
enzymów w prostym, ściśle zdefiniowanym systemie,
złożonym wyłącznie z małych jonów, soli
buforujących, kofaktorów.
Izolacja czystych enzymów jest szczególnie istotna
w przypadku ich wykorzystania do celów
przemysłowych i medycznych.

Objectives of enzyme
purification
Objectives : maximum possible
yield + maximum catalytic activity
+ maximum possible purity

Metody homogenizacji
Metody mechaniczne
Homogenizatory wysokociśnieniowe* (55 MPa) :
istotne jest chłodzenie układu
Rozcieranie „na mokro” z wykorzystaniem
różnorodnych młynków lub szklanych kulek
Inne metody
Suszenie (odwadnianie) komórek
Liza z wykorzystaniem szoku osmotycznego,
detergentów lub enzymów
Ultradzwięki* (istotne chłodzenie próby)

Metody wstępnej separacji
enzymów
3. Solubility
Change in pH
Enzymes are least soluble at pI because there is no
repulsive force between enzymes
Enzyme must not be inactivated in a range of pH
Change in ionic strength
Large charged molecules are only slightly soluble
in pure water; Addition of ion promotes solubility
(Salting in)
Beyond a certain ionic strength, the charged
molecules
are quickly precipitated (Salting out)
Ammonium sulfate is popularly used 10-fold
increase in purity
Fructose-bisphosphate aldolase from rabbit muscle
can
be purified in high purity by ammonium sulfate

2.6 Methods of separation
3. Solubility
Decrease in dielectric constant
Addition of water-miscible organic solvent
(ethanol or
acetone)
Decrease dielectric constant
Sometimes deactivate the enzyme
Work at low temperature
PEG (poly ethylene glycol) ~ Mr 4000 to 6000 is
commonly used

2.6 Methods of separation
5. Choice of method
Time/Large scale -> Precipitation by ethanol or
ammonium sulfate or purification based on
solubility
Small scale/high purity -> Column
chromatography or electrophoresis
FPLC or HPLC -> Fast and high purity,
expensive

2.7 How to know the success of purification
Tests for catalytic activity
- By enzyme assay
- Check cofactors and inhibitors
Stabilizing factors
- Neutral pH, storage in 50% glycerol may help
- 2-mercaptoethanol or DTT(Dithiothreitol)*
- Protease inhibitor PMSF (Phenylmethylsulfonyl
flouride)
Active site titrations
Checking the proportion of active enzyme in
the purified enzyme
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
Wyszukiwarka
Podobne podstrony:
wykład 2 - preparaty galenowe, materiały farmacja, Materiały 3 rok, Od Ani, TPL, TPL
WYKLAD 3 Zastosowanie ziol i preparatow ziolowych w chorobach ukladu moczowego i nerek
WYKŁAD 3 Zastosowanie ziół i preparatów ziołowych w chorobach układu moczowego i nerek
preparatyka wyklady
wszystkie wykłady biologia, Lekarski, I, PIERWSZY ROK MEDYCYNA MATERIAŁY, BIOLOGIA, Bio preparaty 2
Napęd Elektryczny wykład
wykład5
Psychologia wykład 1 Stres i radzenie sobie z nim zjazd B
Wykład 04
geriatria p pokarmowy wyklad materialy
ostre stany w alergologii wyklad 2003
WYKŁAD VII
Wykład 1, WPŁYW ŻYWIENIA NA ZDROWIE W RÓŻNYCH ETAPACH ŻYCIA CZŁOWIEKA
Zaburzenia nerwicowe wyklad
Szkol Wykład do Or
Strategie marketingowe prezentacje wykład
Wykład 6 2009 Użytkowanie obiektu
więcej podobnych podstron