
14-17.12.2009
Paweł Domagała
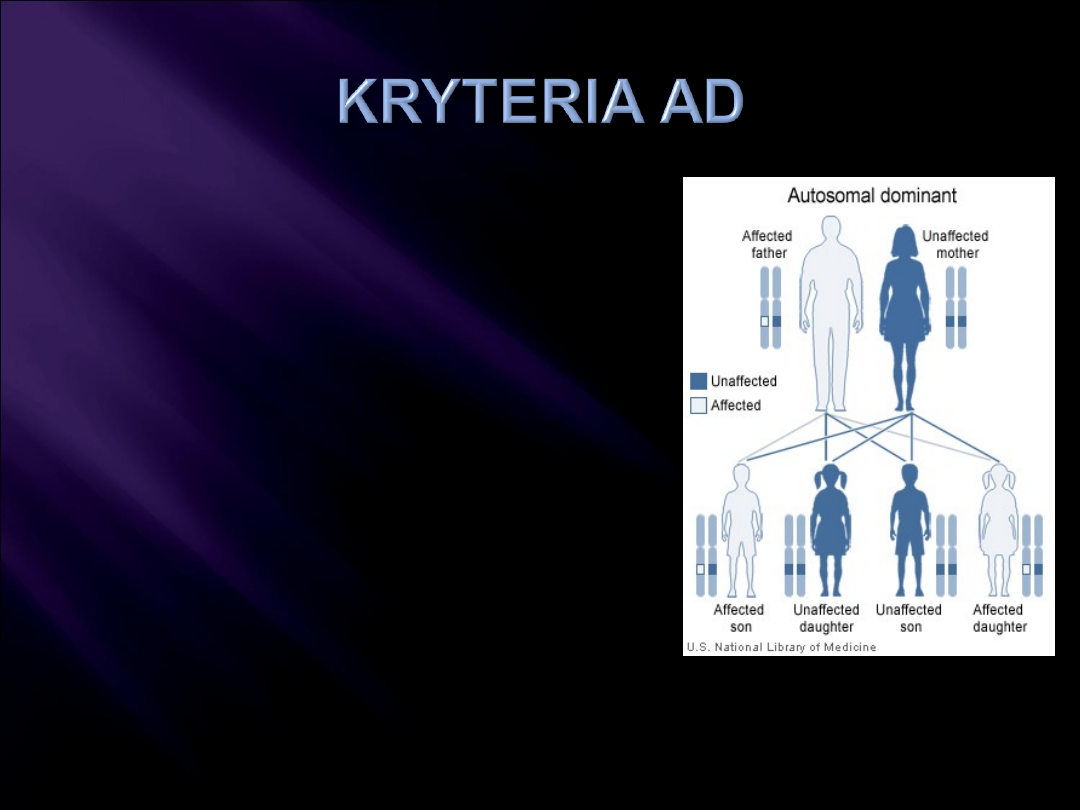
Pionowy wzór rodowodu
Chorują głównie
heterozygoty, b. rzadko
homozygoty
Jednakowa częstość
objawów K/M
Ryzyko odziedziczenia
zmiany – 50%
75% (gdy dwoje chorych
rodziców)

FH
pląsawica Huntingtona
wielotorbielowatość nerek
zespół Marfana
achondroplazja
nerwiakowłókniakowatość
osteogenesis imperfecta
HNPCC

ZMIENNA EKSPRESJA
ZMNIEJSZONA PENETRACJA
PENETRACJA = CHORZY / NOSICIELE
MOZAIKOWATOŚĆ GERMINALNA =
zmiany tylko w części komórek gonad
=>
zdrowi rodzice => chore dzieci

Mutacja genu receptora
LDL
,
apoB-100
(FDB =familial defective APOB-100) i
PCSK9
Podwyższony poziom LDL, TG, CH
Częstość występowania:
Postać hetero - 1:500
Postać homo – 1: 1 000 000
TC mg/dl
LDL-Ch mg/dl
norma
< 200 - 250
< 135 - 155
heterozygoty
250 – 500
200 - 400
homozygoty
> 700
> 500
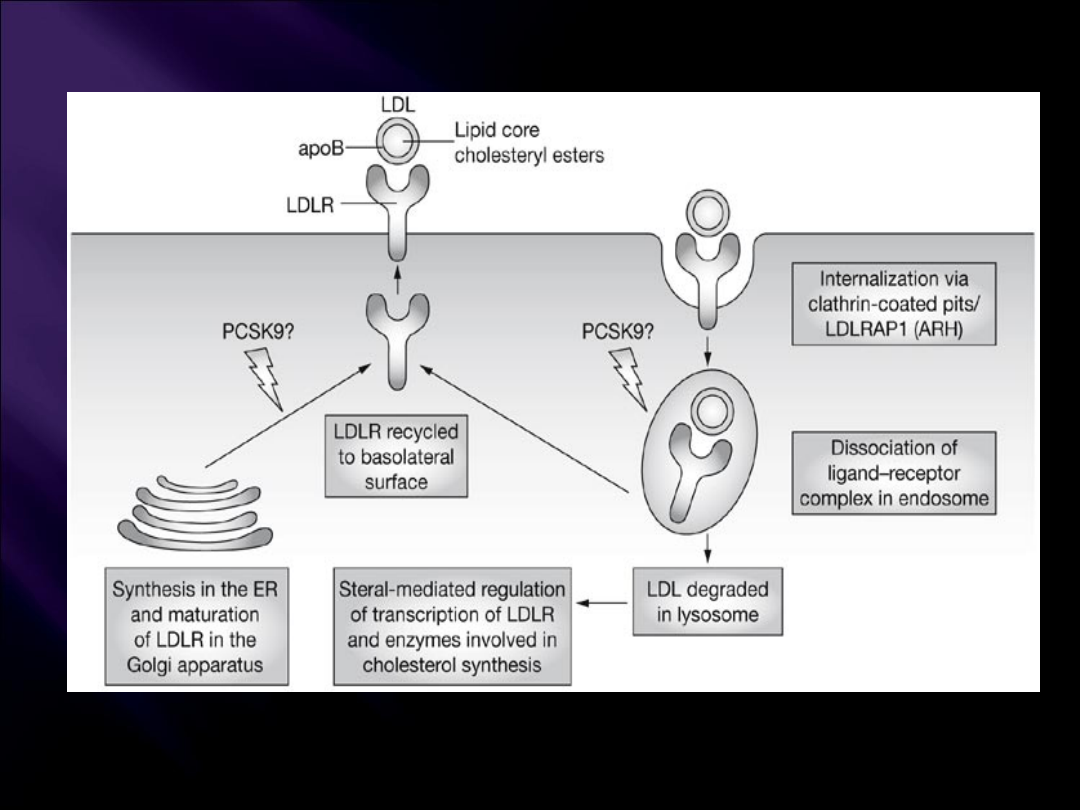
Soutar AK and Naoumova RP (2007) Mechanisms of Disease: genetic causes of familial
hypercholesterolemia
Nat Clin Pract Cardiovasc Med 4: 214–225

> 1000
10x częściej niż mutacje apoB-100
Występują w każdym exonie,
ale najwięcej w 4
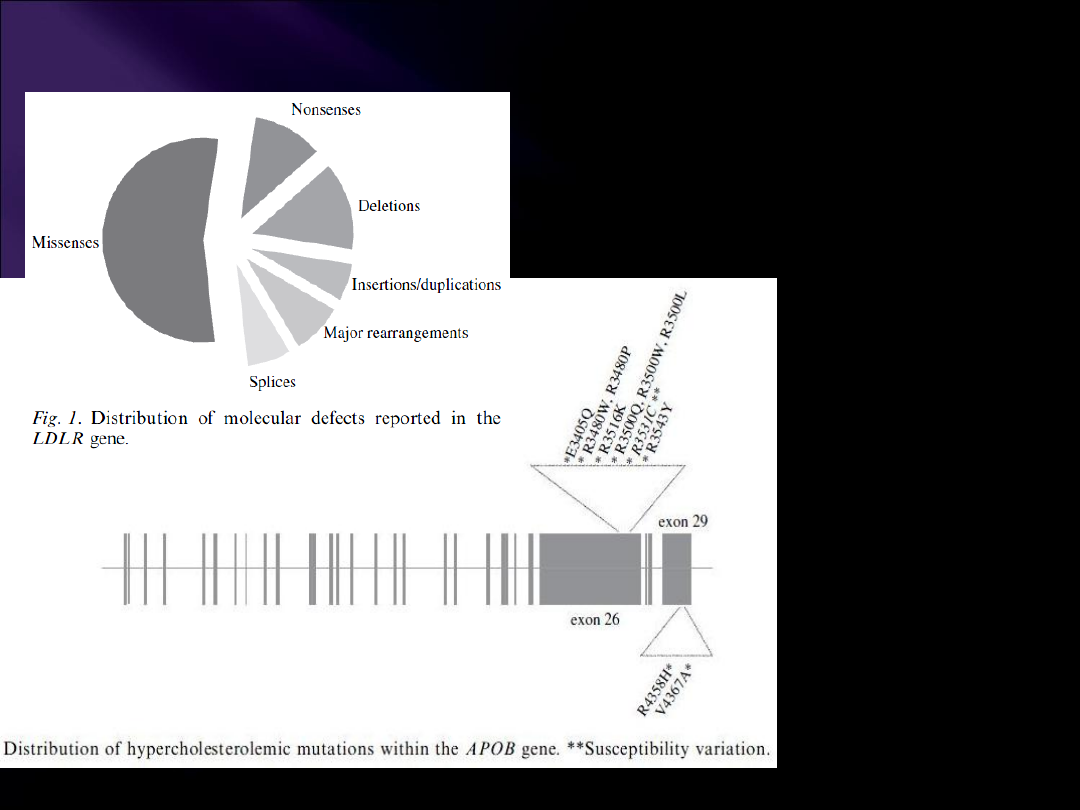
Genetic heterogeneity of
autosomal dominant
hypercholesterolemia. Clin
Genet 2008; 73: 1-13.

HETEROZYGOTY
żółtaki ścięgien (xanthoma)
s. Achillesa, prostowniki ręki , stóp
kępki żółte na powiekach (xanthelasma) i rąbek
rogówki
nasilona miażdżyca po 30 roku życia
zgrubienie zastawek aorty i zgrubienie pnia
aorty – niegomykalność aorty lub stenoza aorty
IHD
ryzyko zgonu wieńcowego jest zwiększone 100x

HOMOZYGOTY
żółtaki w skórze (cutaneous xanthomas) od 3
r.ż.
miażdżyca jest b. ciężka, rozsiana i
obejmuje tętnice wieńcowe, szyjne, biodrowe,
udowe oraz początkowy odcinek aorty.
miażdżyca n. wieńcowych, i zawał mięśnia
sercowego, zwykle przed 20 r.ż..
Rzadko dożywają 20 r.ż.

DANE RODOWODOWO-KLINICZNE
WYNIKI TESTÓW BIOCHEMICZNYCH
TESTY DNA (LDL-R, ApoB-100, PCSK9)

u wszystkich -
zmiana stylu życia
(dieta, ruch)
rozpocząć stosowanie leków
zmniejszających [LDL]
statyny
stanowią leki pierwszego wyboru
żywice jonowymienne
(w skojarzeniu ze
statynami, jeśli to konieczne)
jeśli to konieczne, rozważyć
leczenie
trójlekowe
(statyna + żywica
jonowymienna + kwas nikotynowy)

leczenie za pomocą diety nieskuteczne
żywice jonowymienne nieskuteczne
kwas nikotynowy mało skuteczny
statyny mogą być umiarkowanie skuteczne
u niektórych chorych
zespolenie omijające jelito kręte
nieskuteczne
przeszczep wątroby skuteczny, ale
niepraktyczny
obecnie stosuje się LDL-aferezę
(usuwa
VLDL i LDL)
W przyszłości – terapia genowa ?


POSTAĆ DZIECIĘCA – ARPKD 1:20 000
POSTAĆ DOROSŁYCH – ADPKD
1:1 000
o bardzo znacznej penetracji
MOŻE WYJĄTKOWO UJAWNIĆ SIĘ U DZIECI !
W bardzo rzadkich sytuacjach wykrywana w
badaniach prenatalnych.

Mutacje w genach PKD1 i PKD2
Najczęstsza spośród dziedzicznych
nefropatii
4 przyczyna PNN 7-15%
Zwykle Late-onset
Zaburzenie wielonarządowe

Dwa geny związane są z ADPKD:
PKD1, dotyczy 85% chorych – przebieg
szybki
PKD2, dotyczy 15% chorych – przebieg
wolny
PKD1 Policystyna 1
PKD2 Policystyna 2
mnóstwo pojedynczych mutacji
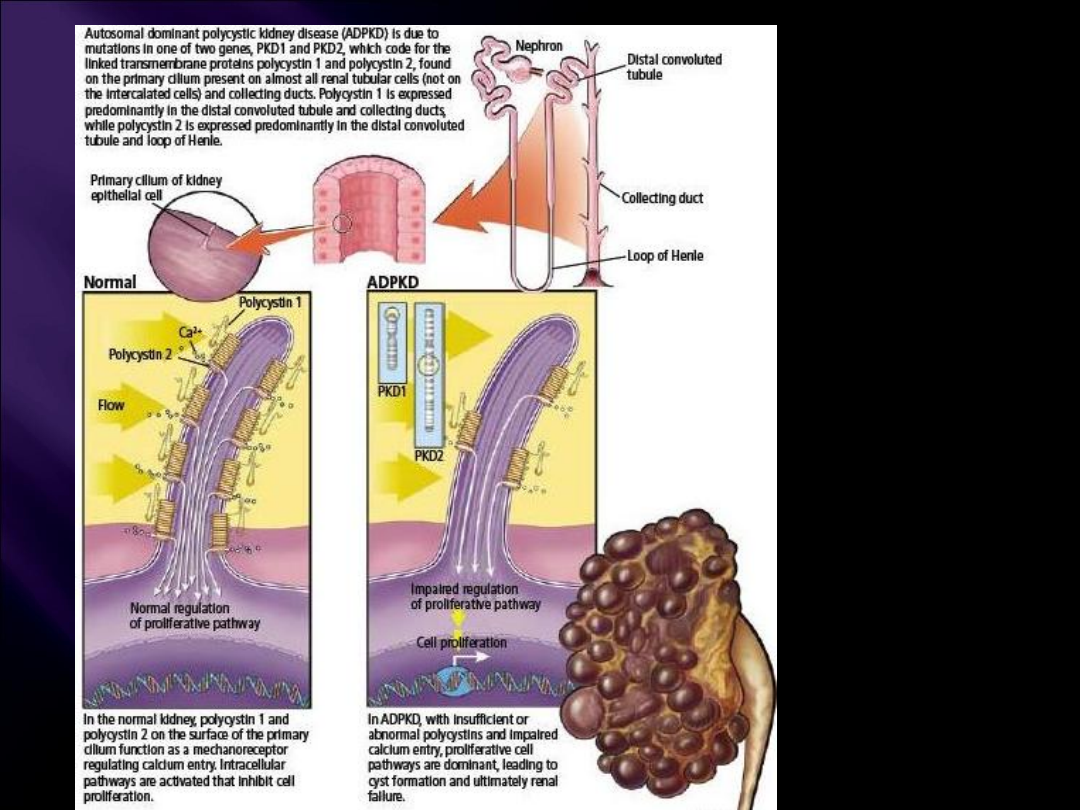
Autosomal dominant polycystic
kidney disease: emerging
concepts of pathogenesis and
new treatments. Cleve Clin J
Med. 2009 Feb;76(2):97-104.
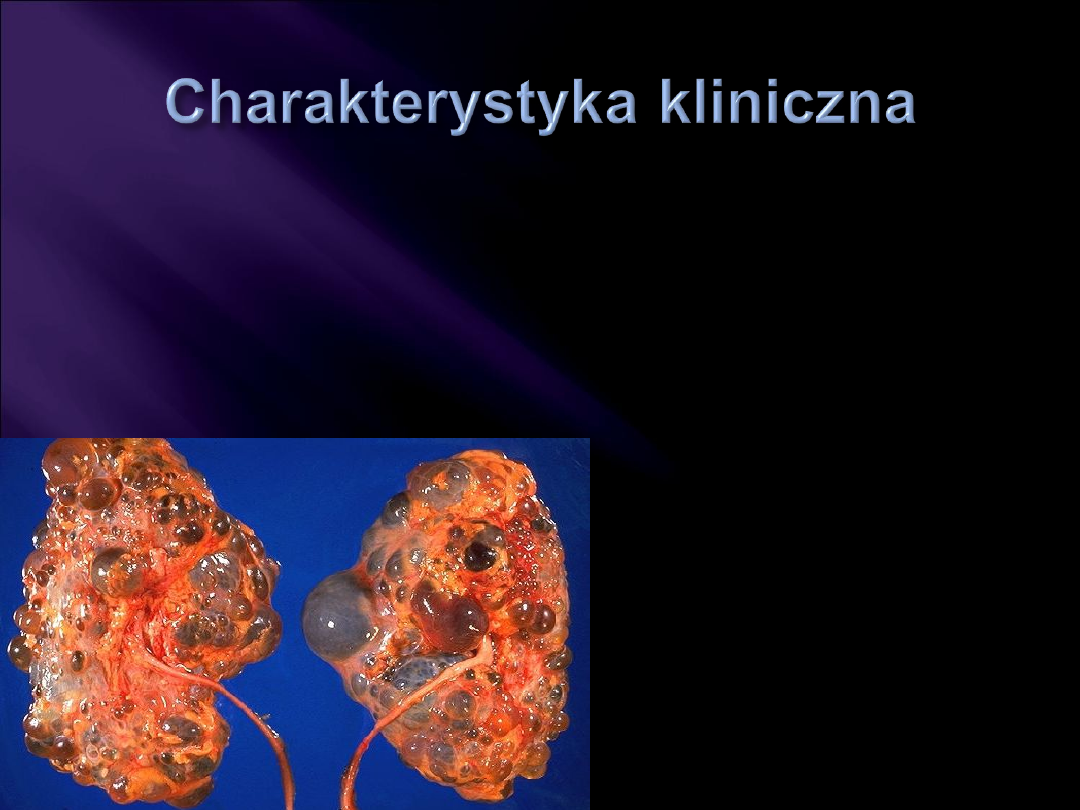
we wczesnym okresie brak objawów, później mogą się
pojawić: bóle brzucha, nadciśnienie, krwiomocz
zmiany torbielowate
zazwyczaj w obu nerkach
zmiany torbielowate w innych narządach:
wątroba (90%), trzustka (9%), śledziona, płuca
pęcherzyki nasienne,
pajęczynówka;

zaburzenia naczyniowe
:
tętniaki wewnątrzczaszkowe- tt.podstawy
mózgu – 12%
poszerzenie pnia aorty
rozwarstwienie aorty piersiowej
wypadanie płatka zastawki mitralnej
(mitral valve prolapse) – najczęstsza
wada zastawkowa (25% chorych)
przepukliny brzuszne, uchyłkowatość
jelita grubego

Manifestacja nerkowa ADPKD zawiera:
nadciśnienie
ból nerek
niewydolność
Zmiany policystyczne wątroby to najcz.
pozanerkowa manifestacja.
20% w III dekadzie do 75% po 60 r.ż.
Średnio 50% chorych z ADPKD -
schyłkowa NN do 60 r.ż.
Zmienność narządowa, ciężkość zmian
różni się u członków tej samej rodziny.
Wywiad rodzinny 50%
Testy laboratoryjne
Zmiany w obrazowaniu nerek – USG/CT
RR, ECHO, angiografia OUN
Kryteria dgn u osób z dodatnim wywiadem
rodzinnym :
Co najmniej 2 jedno- lub obustronne torbiele u
młodszych niż 30 lat; [5 bez wywiadu rodzinnego]
2 torbiele w każdej nerce u 30-59 latków; [5]
4 torbiele w każdej nerce ≥ 60 r.ż. [8]

objawowe
Celem leczenia jest spowolnienie tempa
rozwoju NN oraz minimalizowanie
objawów:
nadciśnienia
bólu
ZUM
7-10% chorych leczonych nerko-zastępczo
z powodu NN to chorzy z ADPKD

Nadciśnienie
Nie ustalono stałego schematu leczenia
(USA)
Z uwagi na rolę układu R-A w patogenezie
nadciśnienia w ADPKD, zaleca się
stosowanie:
IKA
Antagonistów receptora angiotensyny II.
zwiększają przepływ nerkowy,
mało efektów ubocznych,
redukują proliferację mięśniówki naczyń

Ból
Leki nienarkotyczne – często stosowane.
Ostrożnie – efekt nefrotoksyczny
TLPD- pomocne w leczeniu długotrwałych
zespołów bólowych
Leki narkotyczne – zarezerwowane dla
ostrych epizodów
Blokada zwojów trzewnych - czasowa
terapia
Odnerwienie nerek – u 1 chorego
[
].

Krwawienia do torbieli i ich nadmierny
wzrost
Zakażenia torbieli (50%)
Kamica nerkowa (20%)
Krwiomocz (30-50%)
Nadciśnienie tętnicze (70%)
Niewydolność nerek

Średni czas przeżycia
od 1 objawów
NN wynosi ok.
10 lat
Czynniki przyspieszające NN
Nawracające ZUM
Źle kontrolowane HA
Główna przyczyna zgonu obok
niewydolności narządowej –
pęknięcie
tętniaka

częstość 1:15 000

Pełna, zależna od wieku penetracja genu
Wiek pene
tracj
a
30
10%
40
30%
50
60%
60
85%
70
95%

Przebieg choroby postępujący
1 objawy w wieku 3-80 lat (średnio 40
rż)
Nieuchronnie prowadzi do śmierci
P. typowa – 17 lat
P. młodzieńcza – 7 lat

Postać młodzieńcza (Westphala)
10%
(przed 20 rokiem życia)
Od początku choroby
dominuje sztywność
Pojawiają się
napady padaczkowe
Objawia się ona początkowo trudnościami w
nauce, czasami drobnymi zmianami pisma czy
pewną ociężałością ruchów.

Gen huntingtyny –
IT-15
Mutacja polega na wystąpieniu nadmiernej
liczby powtórzeń trójnukleotydowej sekwencji
CAG
(glutamina) na końcu 5’ genu
(...CAGCAGCAG...) >36
Zdrowi średnio 10-29 powtórzeń
Chorzy od
36
(średnio 45-55)
W zakresie 36-39 penetracja jest niepełna
Wiek zachorowania koreluje z liczbą powtórzeń
Zmiana od matki – ta sama liczba CAG
Zmiana od ojca –
WZROST LICZBY
POWTÓRZEŃ !!!, POSTAĆ MŁODZIEŃCZA (83%)
Antycypacja!

Mimowolne ruchy pląsawicze
występujące
w różnych częściach ciała
Pląsawiczy chód
– taneczny ale przerywany
nagłym zatrzymaniem i prostowaniem ciała
Wzmożenie ruchów mimowolnych przy
wykonuwaniu ruchów celowych a redukcja
nasilenia ruchów w czasie snu
Postępujący
zespół otępienny
Zaburzenia mowy, połykania
(zachłystowe
zapalenie płuc 85% † )
Zaburzenia psychiczne - niewytłumaczalne
zmiany nastroju, apatia, aspołeczność,
drażliwość
Zaburzenie skokowych ruchów gałek ocznych
W końcowej fazie – sztywność pozapiramidowa

trudność w utrzymywaniu wysuniętego
języka
zaburzenie rozpoznawania grymasu
obrzydzenia na prezentowanej twarzy
test Luria-Nebraska

CT , MRI, PET
Znacznego stopnia zanik jądra
ogoniastego i skorupy z wyraźnym
poszerzeniem układu komór i zanikiem
kory
Zmiany neuropatologiczne pojawiają się
z opóźnieniem w stosunku do objawów
klinicznych

Jeśli penetracja rodzinnej polidaktylii
(choroba dziedziczona AD) wynosi 80%
to jakie jest prawdopodobieństwo, że
kobieta z rodzinną polidaktylią będzie
miała fenotypowo zdrowe dziecko?

Całkowite prawdopodobieństwo posiadania
zdrowego dziecka będzie wynosiło:
prawdopodobieństwo że dziecko jest
zdrowe +
prawdopodobieństwo że dziecko jest
nosicielem fenotypowo zdrowym
50%
+ (20% z 50%)
=
½ + (2/10 x 5/10) =
3/5 = 60%
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
Wyszukiwarka
Podobne podstrony:
Wyk ad 5 6(1)
uk ad pokarmowy
Wyk ad II
Tkanki wyk ad 1
Ekonomika Transportu wyk+ad 1
Wyk ad Fizyka 2
Wyk ad 04
AD Order Of Battle
freeRadius AD tutorial
Na wyk ad id 312279 Nieznany
!BSI, wyk ad 4
Birthright - zasady, Pozostałe rpg, Ad&d
więcej podobnych podstron