
ZESPÓŁ KLINEFELTERA
Etiologia
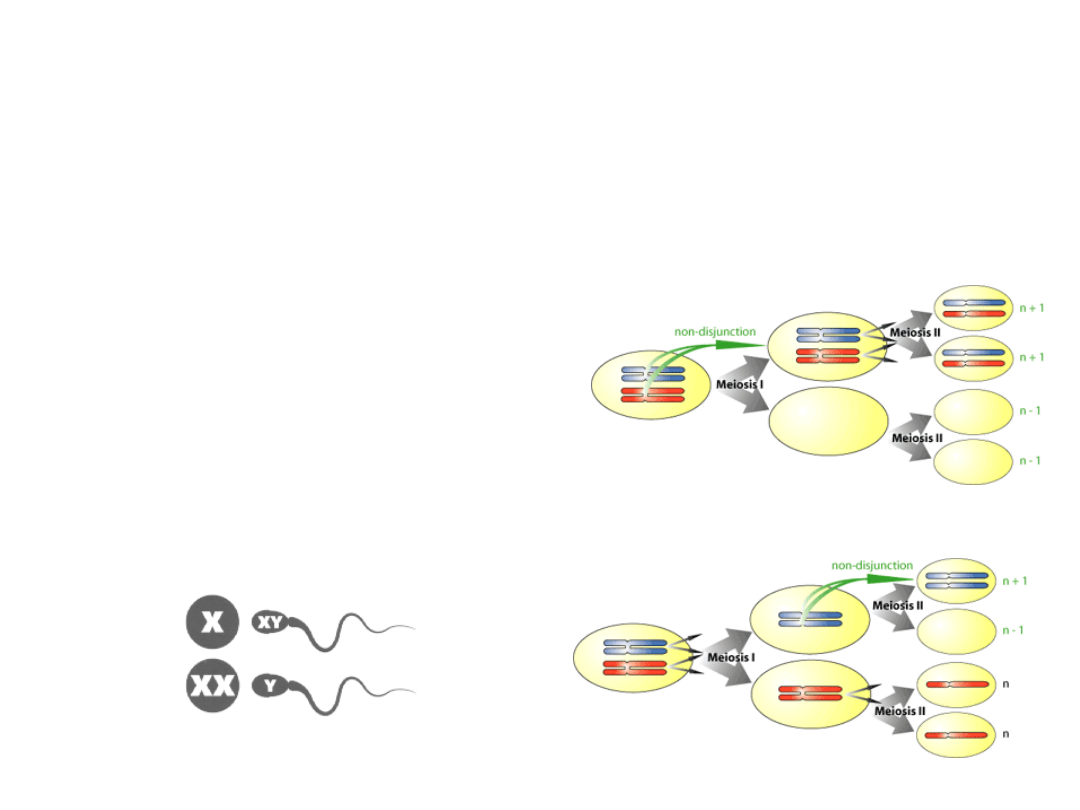
• nondysjunkcja chromosomu X
- I podział mejotyczny
- II podział mejotyczny
- podział mitotyczny
zygoty

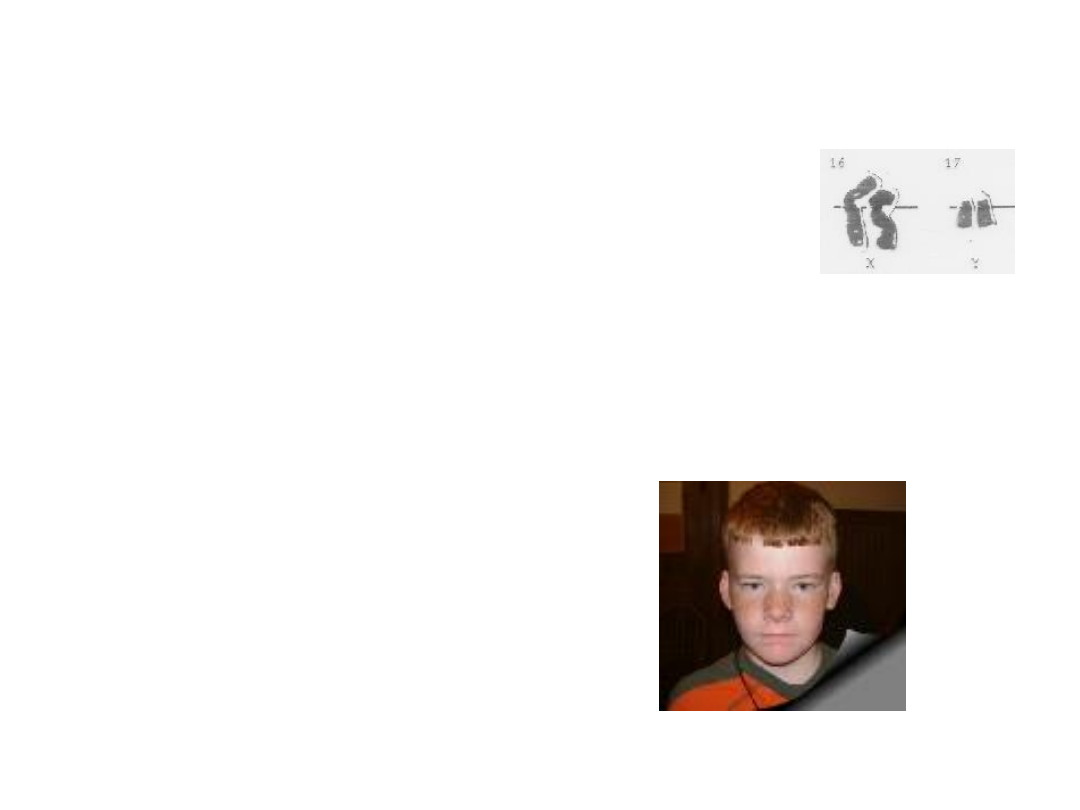
Epidemiologia
• 47,XXY -1:500 męskiej populacji
• Prawdopodobieństwo wzrasta z
wiekiem matki
• Rozpoznanie na
podstawie kariotypu

Objawy-dojrzewanie
• chłopcy nie różnią się od rówieśników z
kariotypem 46,XY
• długie ręce i nogi
• taurodontyzm
• częsta klinodaktylia
• dojrzewanie zaczyna się prawidłowo
• drugorzędowe cechy płciowe nie
wykształcają się prawidłowo
• rzadko występuje mutacja głosu
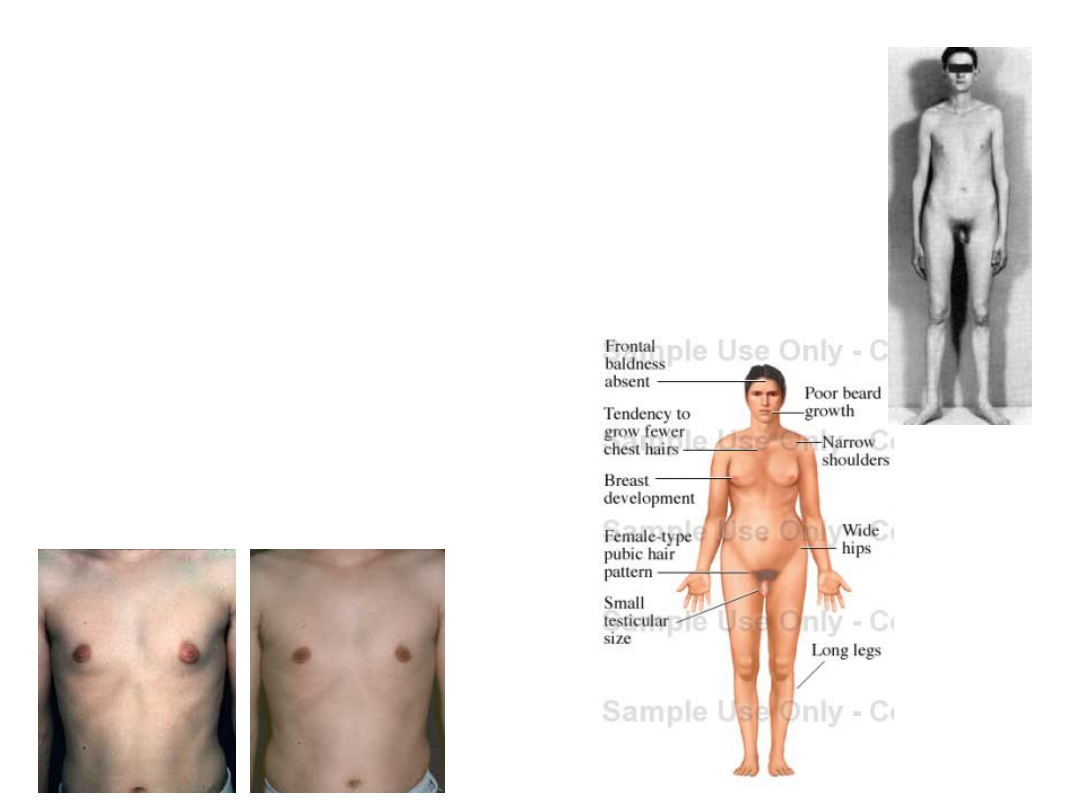
Objawy-dorośli
• Wysoki wzrost - 179,2 ± 6,2 cm
• Długie kończyny
• Kobieca sylwetka
• Kobiecy typ owłosienia
• Zmiany w obrębie kości
czaszki
• Ginekomastia
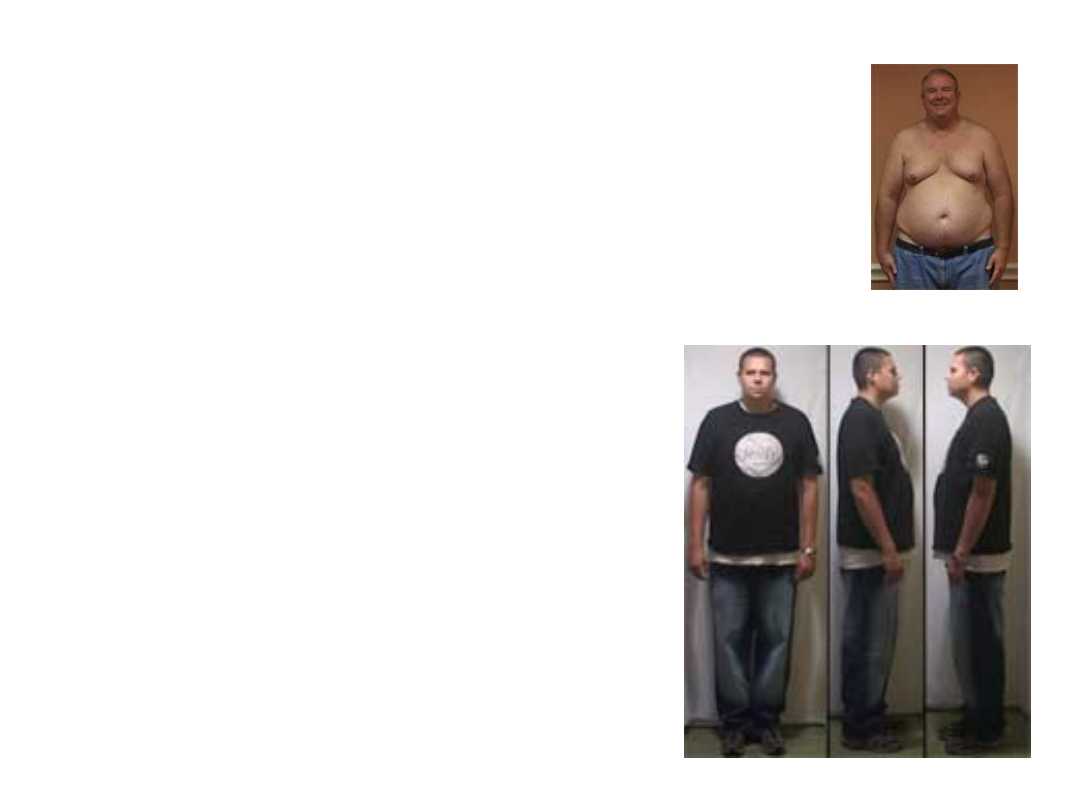
Objawy-dorośli
• małe jądra
• brak spermatogenezy
• obniżone libido
• tendencja do otyłości
• niższy poziom inteligencji
werbalnej
• nieprawidłowy poziom
hormonów
48,XXYY - 1:17,000
- nasilone zaburzenia psychiczne
- chorzy są zwykle nieśmiali, ale
mogą być kompulsywni i agresywni

48,XXXY - 1:50,000
• cechy dysmorficzne twarzy:
hiperteloryzm oczny, płaska nasada
nosa
• synostoza promieniowo-łokciowa
• klinodaktylia V palca
• małe jądra i penis

49,XXXXY - 1:85,000 -
1:100,000
zespół Fraccaro
• niski wzrost
• nasilone cechy dysmorficzne (hiperteloryzm,
płaską nasadę nosa, mongoidalne
ustawienie szpar powiekowych)
• mogą urodzić się z rozszczepem
podniebienia i języczka
• nierzadkie wady wrodzone serca (najczęściej
przetrwały przewód Botalla)
• synostoza promieniowo-łokciowa, stopa
wydrążona (pes cavus), kolana koślawe
(genu valgum), hipotonia i wiotkość w
stawach
• zaburzenia psychiczne

•
Streszczenie:
•
Charakteryzuje się dodatnią chromatyną płciową oraz kariotypem z obecnością dodatkowego chromoso mu X.
•
Zespół Klinefeltera (ang. Klinefelter syndrome) – grupa chorób spowodowanych aberracją chromosomalną polegającą
na obecności przynajmniej jednego dodatkowego chromosomu X w części lub we wszystkich komórkach organizmu
mężczyzny.
•
Najczęściej stwierdza, się kariotyp 47,XXY. Rzekomy zespół Klinefeltera powstaje prawdopodobnie pod wpływem
czynników uszkadzających jądra w życiu pozapłodowym. W przypadkach tych stwierdza się ujemną chromatynę
płciową. Prawdopodobieństwo zespołu Klinefeltera u dziecka wzrasta z wiekiem matki,
•
Rozpoznanie zespołu Klinefeltera można postawić jedynie na podstawie kariotypu. Prawidłowy kariotyp zmusza do
dalszej diagnostyki - istnieje możliwość mozaicyzmu komórek somatycznych (np. 46,XY/47,XXY) albo zespołu
mężczyzny XX. Inne jednostki chorobowe jakie należy uwzględnić w różnicowaniu to związane z nadmiernym
wzrostem zespół łamliwego chromosomu X i zespół Marfana, oraz inna przyczyna hipogonadyzmu
hipergonadotropowego - zespół Kallmanna.
•
Klasyczny zespół Klinefeltera występuje u mężczyzn o kariotypie 47,XXY i jest najczęstszą aneuploidią człowieka,
której częstość ocenia się na 1:500 noworodków płci męskiej .
•
Pacjenci z kariotypem 47,XXY nie mają w zasadzie charakterystycznych cech dysmorficznych, które ułatwiałyby
rozpoznanie w okresie przedpokwitaniowym. U około 40% mężczyzn XXY stwierdza się taurodontyzm (powiększone
zęby trzonowe); dosyć częsta jest klinodaktylia.
•
Dojrzewanie zaczyna się prawidłowo, ale poziom testosteronu stopniowo maleje i drugorzędowe cechy płciowe nie
wykształcają się prawidłowo. Normalnie rozwija się prącie. - często (56-88% [2]) rozwija się ginekomastia i rzadko
występuje mutacja głosu.
•
Mężczyźni 47,XXY
•
-wysokiego wzrostu (średni ostateczny wzrost chorych to 179,2 ± 6,2 cm [1]),
•
- słabiej umięśnionymi
•
- o bardziej kobiecej (gynoidalnej) sylwetce, wynikającej z charakterystycznej budowy klatki piersiowej i miednicy.
•
- Skóra jest cienka, często blada, słabo owłosiona
•
-Dłuższe niż przeciętnie są kończyny górne i dolne.
•
-Rozmieszczenie owłosienia może być typowo męskiego typu, zwykle jednak jest skąpe.
•
-Obserwuje się zmniejszenie rozmiaru jąder, które dodatkowo są nieprawidłowo twarde; ich objętość po ukończeniu
pokwitania zwykle nie przekracza 10 ml [2]. jądra ma łe, o zwiększonej konsystencji, najądrze z re guły jest
powiększone Badanie histologiczne wycinków z jąder stwierdza zeszkliwienie kanalików krętych oraz brak
spermatogenezy. Komórki Leydiga są liczne, ułożone w skupiskach, nieraz tworzących mikrogruczolaki; z biegiem lat
dochodzi jednakże do stopniowego zanikania komórek Leydiga

•
-W gonadach nie zachodzi spermatogeneza, czego konsekwencją jest niepłodność, dotycząca blisko 100%
pacjentów z zespołem Klinefeltera. Libido jest obniżone, częste są problemy z erekcją.
•
Inteligencja pacjentów na ogół nie odbiega od normy; odnotowano zarówno dużo niższe, jak i dużo wyższe od
przeciętnej wartości IQ. W teście inteligencji Wechslera stwierdza sie niższy poziom inteligencji werbalnej.
•
W badaniach laboratoryjnych stwierdza się obniżony poziom testosteronu <3,5 nmol/l (100 ng/dl),
podwyższone poziomy gonadotropin (FSH znacznie i LH > 15 j.m./l), SHBG, poziom estradiolu w osoczu jest
powyżej normy dla mężczyzn.
•
Zespół Klinefeltera często przebiega z nadwagą z tendencją do otyłości; wiąże się też ze zwiększonym
ryzykiem cukrzycy. Ryzyko raka sutka jest zwiększone i podobne do ryzyka u kobiet. U około 25% pacjentów
rozwinie się osteoporoza [1].
•
Objawy u pacjentów 48,XXYY, 48,XXXY i 49,XXXXY
•
Objawy i przebieg u pacjentów z większą liczbą chromosomów X są dużo cięższe niż w klasycznym zespole
Klinefeltera. Tak jak w klasycznym wariancie 47,XXY występują u nich gynoidalna sylwetka, nieprawidłowo
rozwinięte drugorzędowe cechy płciowe, hipogonadyzm hipergonadotropowy, ginekomastia.
•
U pacjentów z kariotypem 48,XXYY nasilone są zaburzenia psychiczne, chorzy są zwykle nieśmiali, ale mogą
być kompulsywni i agresywni [3]. Pacjenci z kariotypem 48,XXXY mają cechy dysmorficzne twarzy:
hiperteloryzm oczny, płaską nasadę nosa, synostozę promieniowo-łokciową, klinodaktylię V palca, ich jądra i
penis są małe.
•
W przeciwieństwie do chorych z kariotypem 48,XXYY i 48,XXXY, pacjenci 49,XXXXY są niscy; mają nasilone
cechy dysmorficzne (hiperteloryzm, płaską nasadę nosa, mongoidalne ustawienie szpar powiekowych), mogą
urodzić się z rozszczepem podniebienia i języczka, nierzadkie są wady wrodzone serca (najczęściej przetrwały
przewód Botalla), synostoza promieniowo-łokciowa, stopa wydrążona (pes cavus), kolana koślawe (genu
valgum), hipotonia i wiotkość w stawach. Również występują zaburzenia psychiczne.
•
----Wszyscy pacjenci z większą niż jeden dodatkową liczbą chromosomów X prezentują umiarkowane do
ciężkiego upośledzenie umysłowe: obliczono, że każdy dodatkowy chromosom X wiąże się z obniżeniem IQ o
przeciętnie 15-16 punktów [4].
•
Leczenie
•
Leczenie zespołu Klinefeltera polega na dożywotniej substytucji testosteronu w postaci enantatu lub cypionatu
testosteronu, począwszy od 12 roku życia zwiększając stopniowo dawkę, co naśladuje naturalny proces
pokwitania. Nie jest możliwe pobudzenie czynności kanalików plemnikotwórczych.
Document Outline
Wyszukiwarka
Podobne podstrony:
cw4 Zespół Klinefeltera
Zespół Klinefeltera, VI rok, Genetyka, Genetyka, Egzamin
Zespół Klinefeltera, oligofrenopedagogika - różne materiały i teksty
Zespół Klinefeltera
zespol klinefeltera przypadki
cw4 Zespół Klinefeltera
zespół Klinefeltera
Klinefeltera zespół, studia, I ROK, Biomedyka
Zespół nerczycowy
9 RF ZEspól 0 Środki trwałe
Zespół kanału łokciowego i nerw pachowy (tryb edytowalny)
Zespoly paranowotworowe
Zespoly interdyscyplinarne
więcej podobnych podstron