
Tadeusz Robak
Ostre białaczki i przewlekłe
zespoły
mieloproliferacyjne

Choroby mieloproliferacyjne
Klonalne choroby komórki pnia
przebiegające z proliferacją jednej lub wielu
linii mieloidalnych
•Przewlekła białaczka szpikowa
•Przewlekła białaczka neutrofilowa
•Czerwienica prawdziwa
•Nadpłytkowość samoistna
•Przewlekła mielofibroza idiopatyczna
•Przewlekła białaczka eozynofilowa/zespół
hipereozynofilowy

Przewlekła białaczka szpikowa
(PBS) –definicja
• PBS jest klonalną chorobą
rozrostową układu krwiotwórczego
spowodowaną nabytym defektem
genetycznym wielopotencjalnej
komórki macierzystej

John Hughes Bennett 1812-
1875
Edinburgh physician.
Credited with the first report of
leukaemia, in 1845
Virchow in Vienna published a
similar report 5 weeks later,
noting in 1847 the association
with an enlarged spleen
Henry Fuller (St Georges,
London) noted in 1850 that
there were different sorts of
blood cells in leukaemia.
Leukos =
White
Aima
= Blood
•www.cmlsupport.org.uk/ Templates/CMLSeminar.aspx?action=RC accessed June 06

John Hughes Bennett
1812-1875
Bennett later thought
that leukaemia was
due to a peculiar
infection, maybe in the
spleen.
Virchow argued that
leukaemia arose
because of disturbance
in marrow production
of blood cells, and
proposed ‘splenic’ and
‘lymphatic’ forms.
WRONG
RIGHT
•www.cmlsupport.org.uk/ Templates/CMLSeminar.aspx?action=RC accessed June 06

•
Mężczyzną między 35 a 70 rokiem życia, (rzadziej chorują kobiety, M:K=1,3:1,
wyjątkowo rzadko dzieci). Średni wiek w momencie zachorowania to 50 lat
•
•
Wśród 1 miliona mieszkańców można się spodziewać około 10 nowych chorych
w ciągu roku
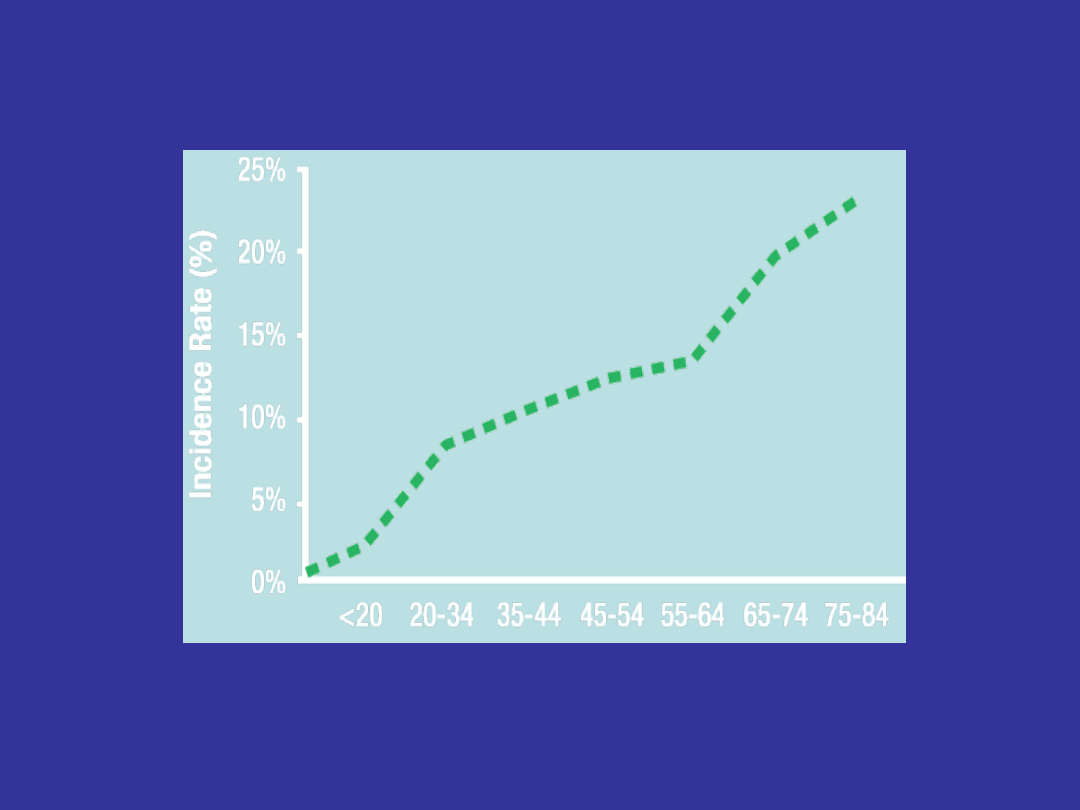
CML Incidence vs Age
•
• Overall incidence rate = 1 to 2 patients per 100,000 people per year
1
1. Faderl S, Talpaz M, Estrov Z, et al. N Engl J Med. 1999;341:164-172.
Incidence of CML Increases With Age
Age

Etiologia PBS
•W większości przypadków nieznana
•Narażenie na promieniowanie jonizujace
(po wybuchu bomby atomowej, po
radioterapii innego nowotworu)
Ref -http://info.cancerresearchuk.org/cancerandresearch/cancers/leukaemiachronic/ accessed
Aug 06

Patogeneza PBS
Obecność chromosomu Philadelphia i genu
BCR-ABL jest kluczowa dla rozwoju PBS
Ref -http://info.cancerresearchuk.org/cancerandresearch/cancers/leukaemiachronic/ accessed
Aug 06

History
History of the Philadelphia
Chromosom
Source: http://www.medscape.com/viewarticle/448490_1, Nowell, P. C.; Hungerford, D. A. :
A minute chromosome in human chronic granulocytic leukemia. Science 132: 1497, 1960.
In 1960, Nowell and Hungerford identified the Philadelphia chromosome
of chronic myeloid leukemia. It was called the Philadelphia chromosome
because it was thought to be useful to follow the practice of
hemoglobinologists and name anomalous chromosomes after the city of
discovery.

History
History of the Philadelphia
Chromosom
Source: http://www.medscape.com/viewarticle/448490_1, Nowell, P. C.; Hungerford, D. A. :
A minute chromosome in human chronic granulocytic leukemia. Science 132: 1497, 1960.
„These findings suggest a
causal relationship
between the chomosome
abnormality and chronic
granulocytic leukemia“
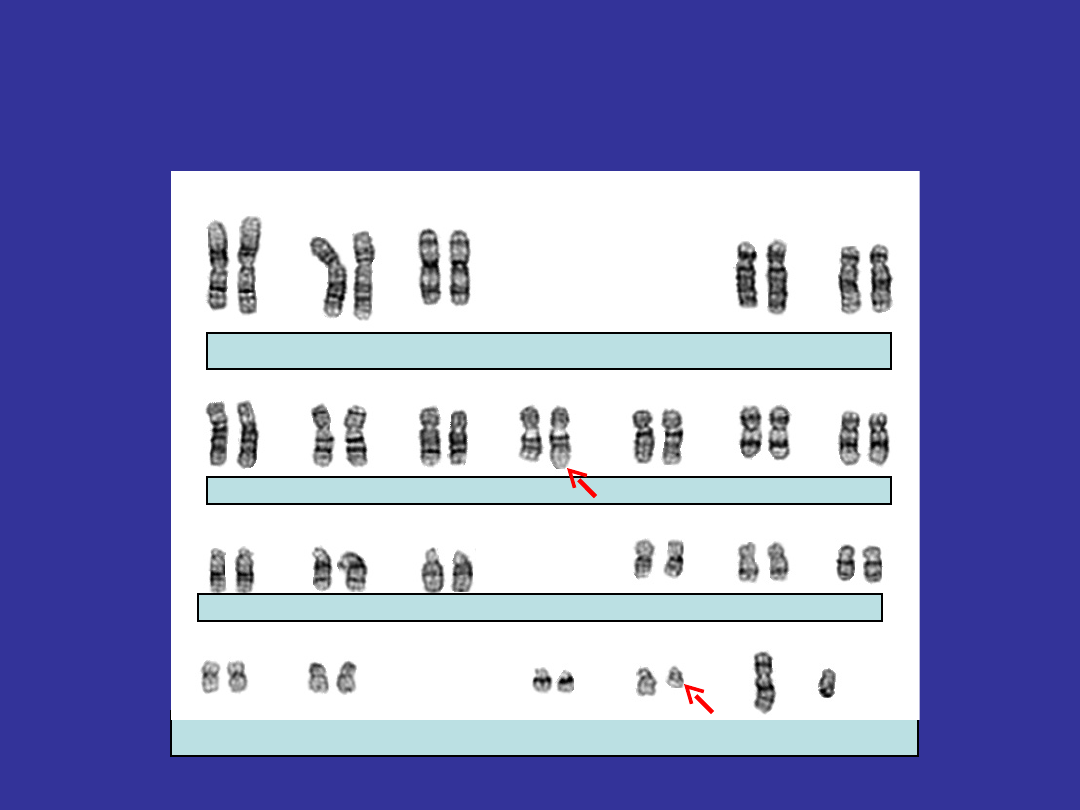
The Philadelphia Chromosome
2
3
4
5
1
7
8
9
1
0
11
12
6
13
14
15
16
17
18
20
21
22
X
Y
19
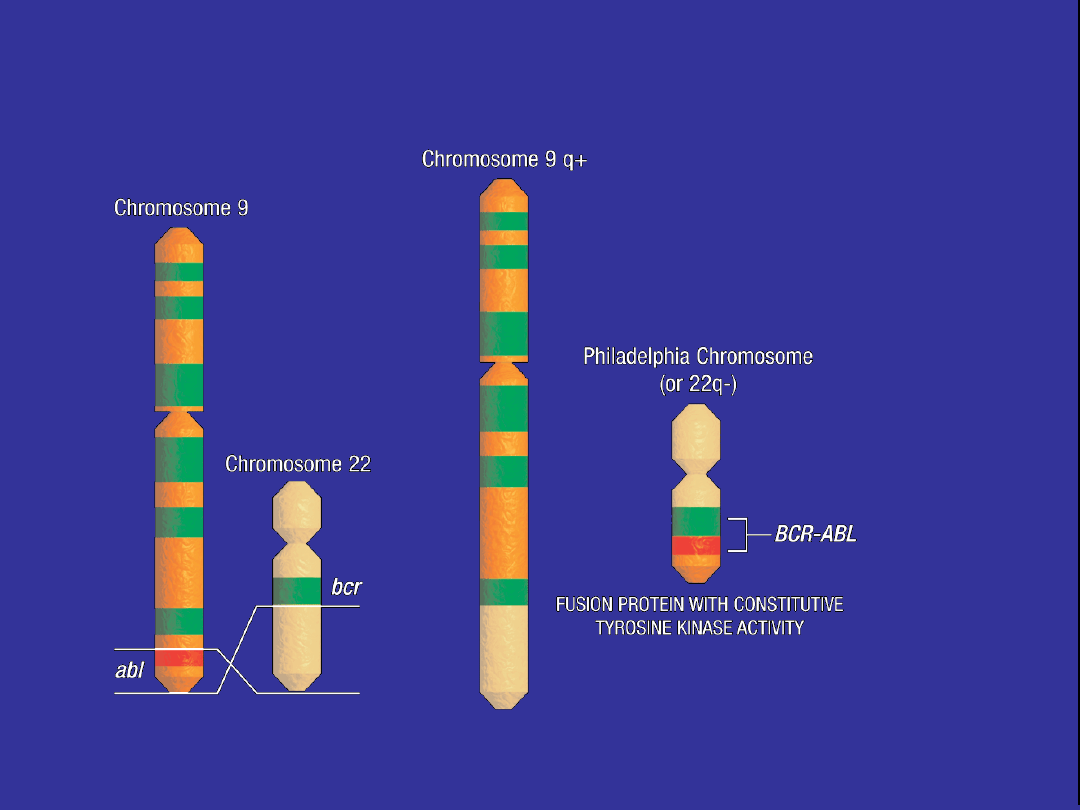
BCR-ABL Gene Structure
•
1. Faderl S, Talpaz M, Estrov Z, Kantarjian HM. Ann Intern Med. 1999;131:207-219.
•
2. Faderl S, Talpaz M, Estrov Z, et al N Engl J Med. 1999;341:164-172.

• Dokonuje jej kinaza tyrozynowa (TK) - 2 typy
enzymu:
– Receptorowa kinaza tyrozynowa (RTKs): c-
Kit, PDGF-R
– Niereceptorowa wewnątrzkomórkowa
kinaza tyrozynowa (TKs): Bcr-Abl, Abl
• Każda z TK jest wysoce specyficzna dla swych
substratów
• Dokonuje jej kinaza tyrozynowa (TK) - 2 typy
enzymu:
– Receptorowa kinaza tyrozynowa (RTKs): c-
Kit, PDGF-R
– Niereceptorowa wewnątrzkomórkowa
kinaza tyrozynowa (TKs): Bcr-Abl, Abl
• Każda z TK jest wysoce specyficzna dla swych
substratów
Fosforylacja Tyrozyny -2
Fosforylacja Tyrozyny -2
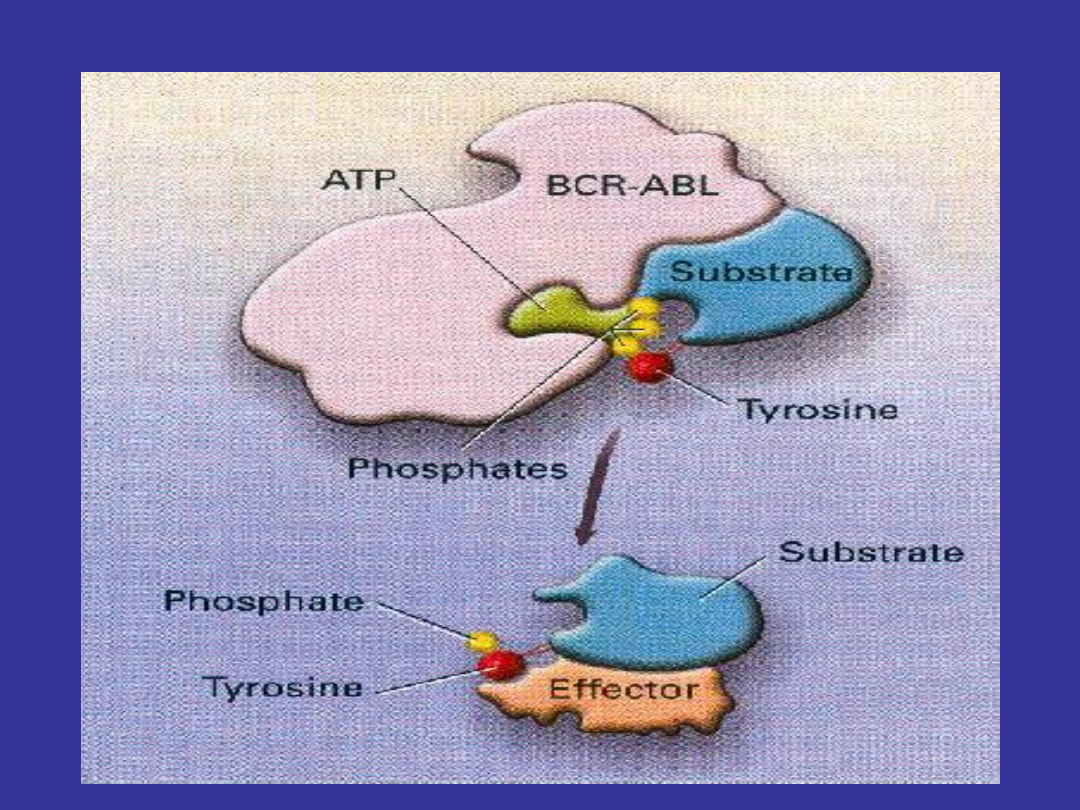
Kinazy tyrozynowe
Kinazy tyrozynowe

Badania laboratoryjne w PBS
• Morfologia krwi obwodowej
• Badanie cytologiczne szpiku kostnego
• Badanie cytogenetyczne
• Badanie techniką FISH (fluorescencyjnej hybrydyzacji in situ)
• Badania molekularne techniką PCR (reakcji łańcuchowej
polimerazy)
• Jakościowe
• Ilościowe
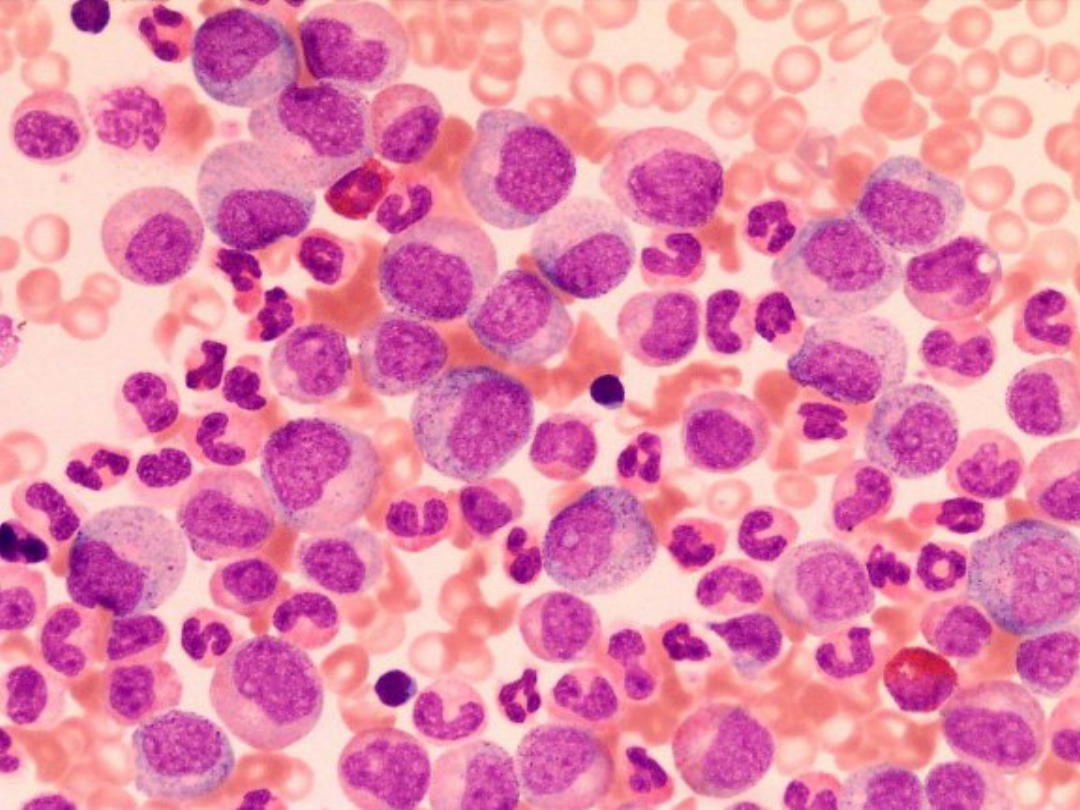
PBS KREW OBWODOWA
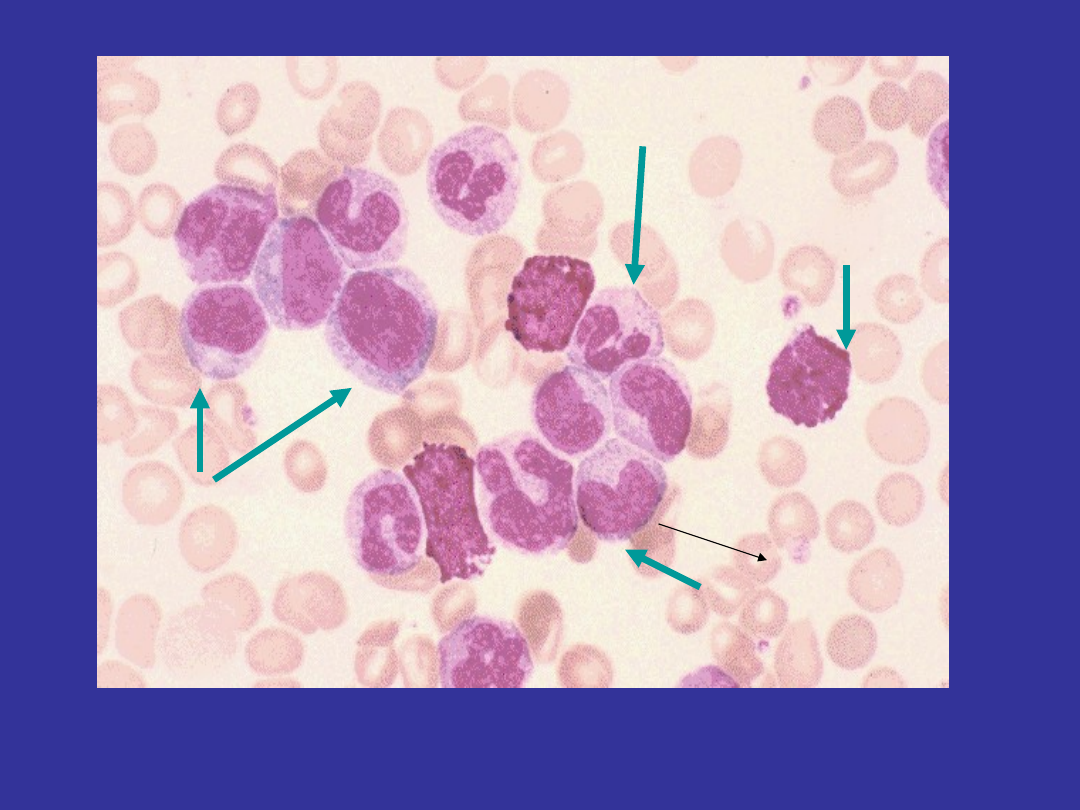
neutrophil
basophil
metamyeloc
yte
myelocytes
http://meds.queensu.ca/medicine/deptmed/hemonc/dload/cllcml.ppt
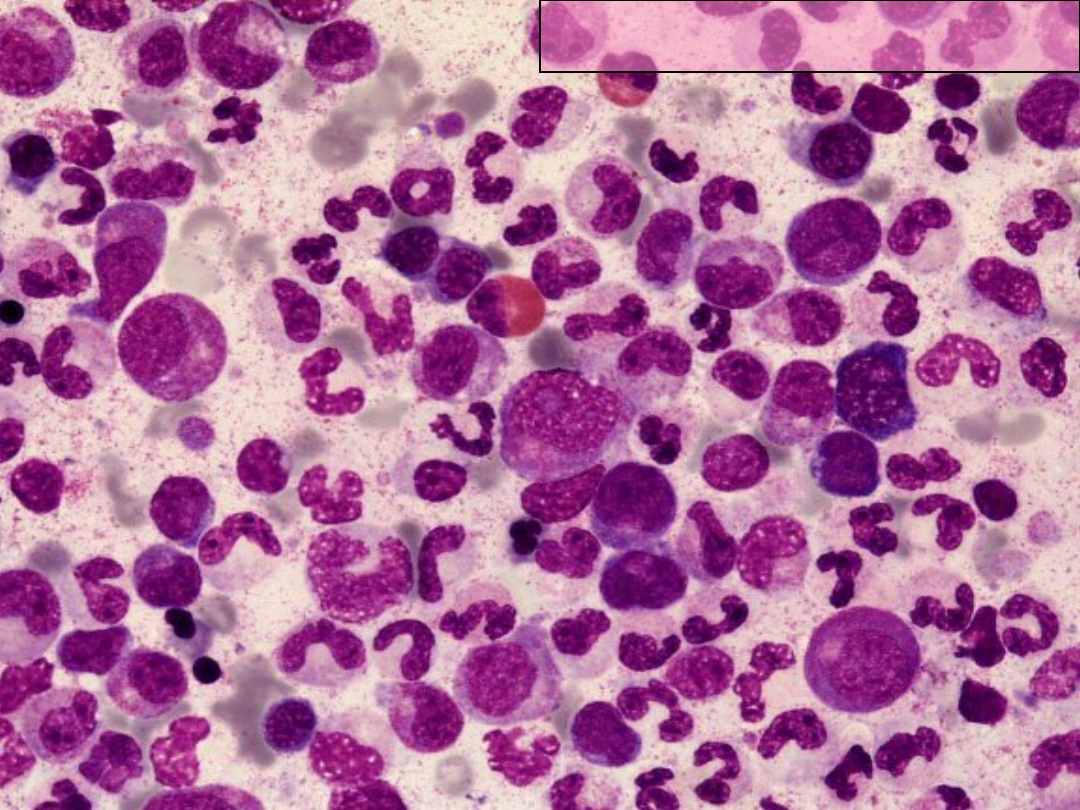
PBS SZPIK KOSTNY
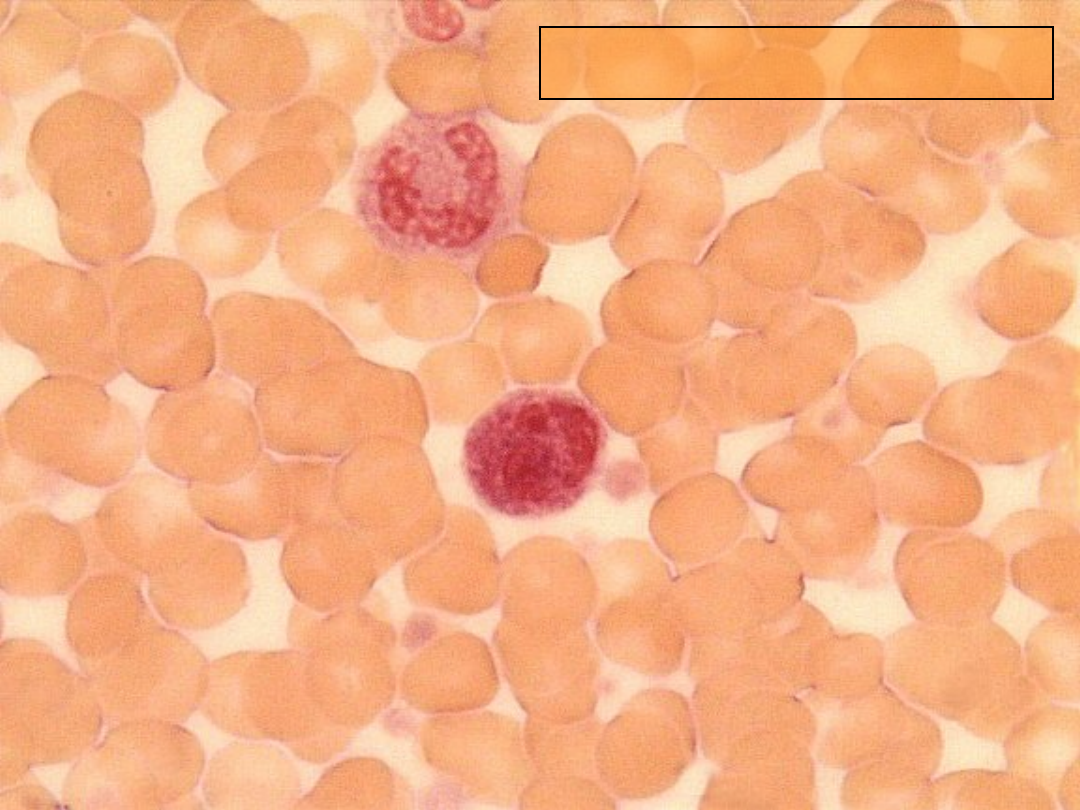
AKTYWNOŚĆ
FAG
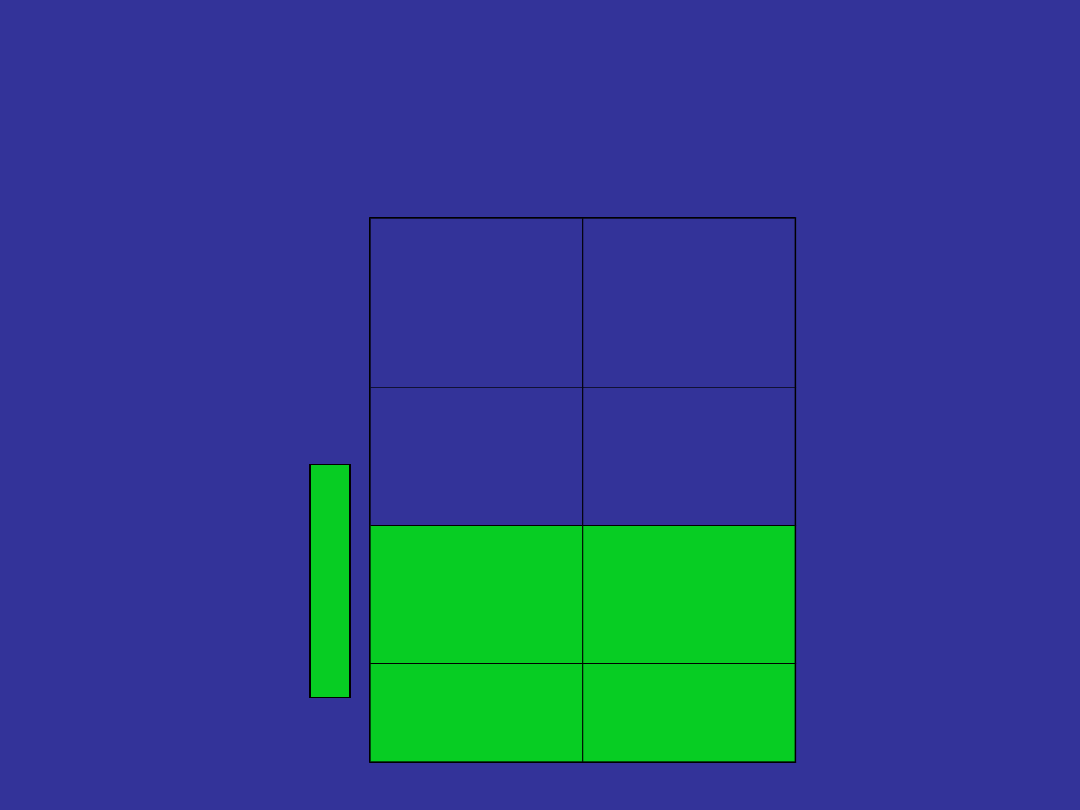
What’s a cytogenetic response and
why does it matter?
Type of
response
% of
Philadelphi
a-positive
cells
Minor/mini
mal
More than
35%
Partial
Less than
35%
Complete
0%
M
a
jo
r
re
sp
o
n
se
www.ncrn.org.uk/cml/Documents/ Patient%20forum/Pt_meeting_Milano.ppt –accessed June 06
Test performed on a sample of bone marrow every 6 months or so
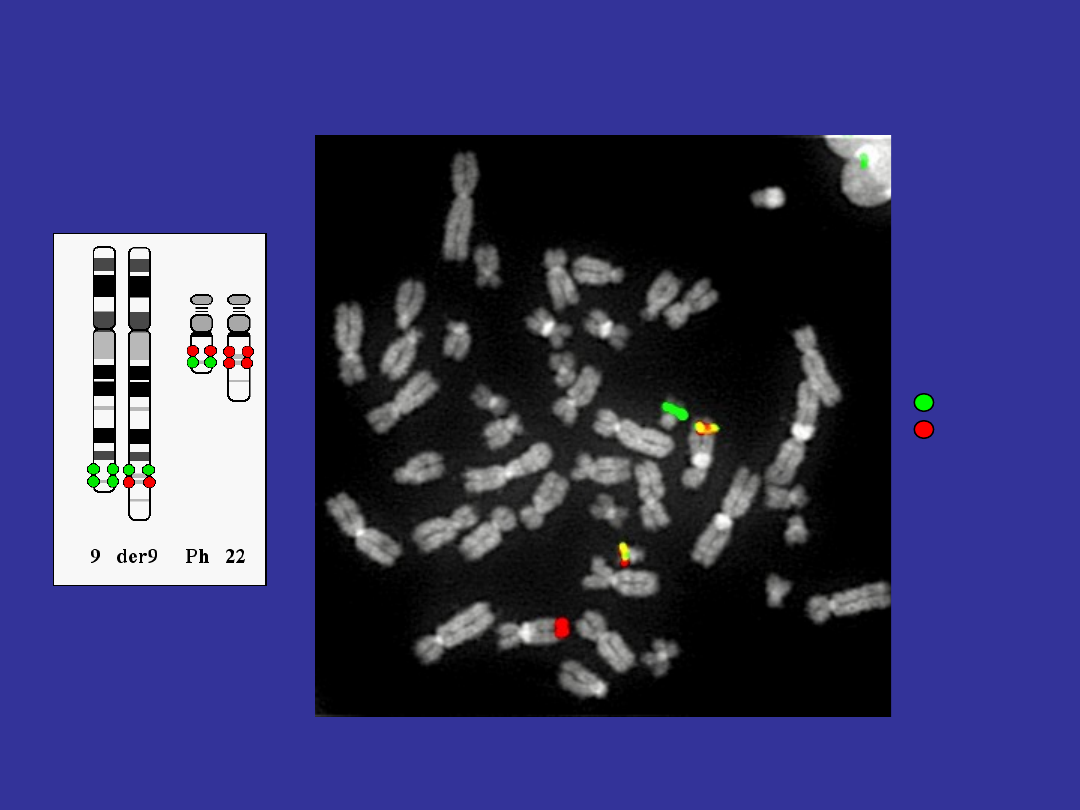
Ph
9
22 der(9)
BCR
ABL
Detection of BCR/ABL fusion using
dual-fusion (D-FISH) system
A kind gift from Dr Marin
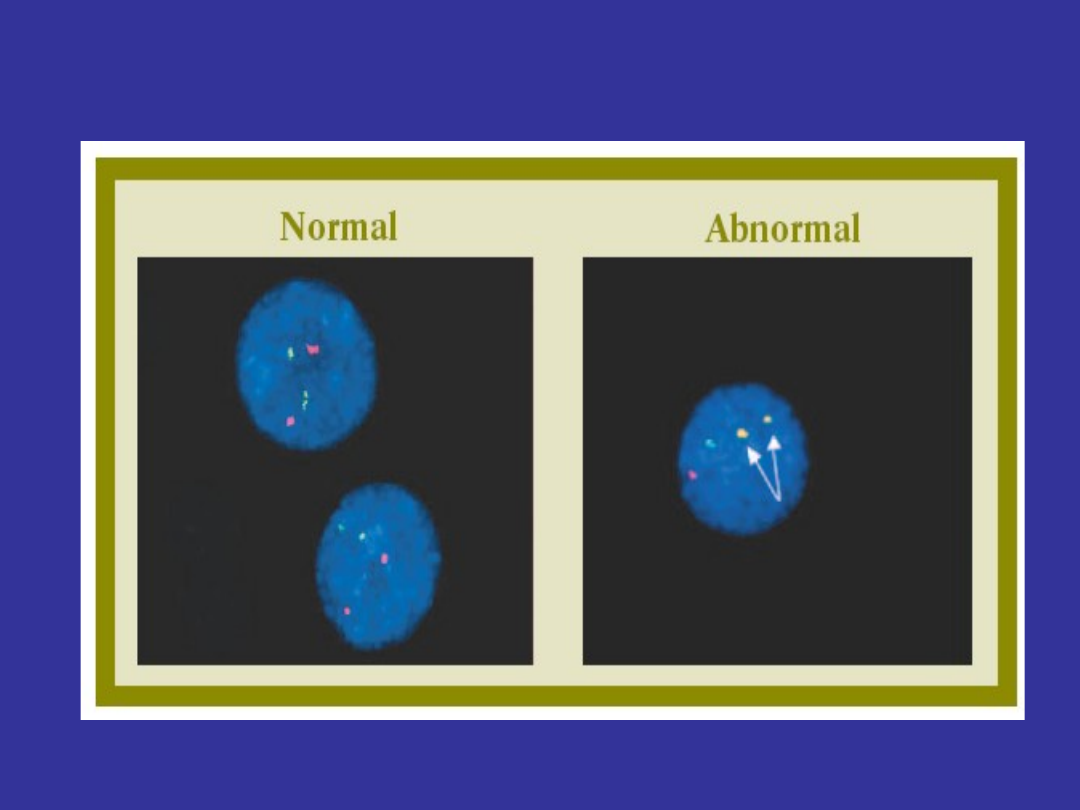
http://www.leukemia-lymphoma.org accessed June 06
FISH (fluorescence in situ
hybridization)

What are the
Different Phases of
CML?
• Chronic Phase (CP)
• Accelerated Phase (AP)
• Blast Phase (BP) / Blast Crisis (BC)
Faderl S, Kantarjian HM, Talpaz M. Chronic Myelogenous Leukaemia: Update on Biology and Treatment. Oncology. 1999;13:169-
184. accessed aug 06
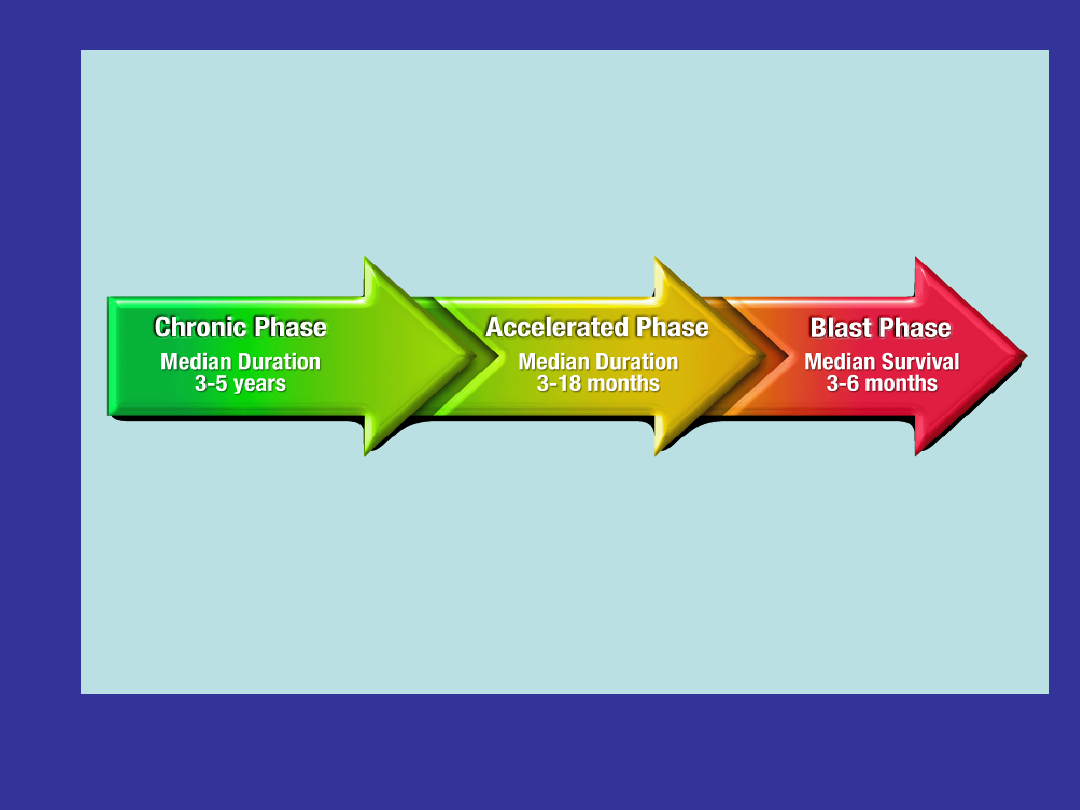
Clinical Course: Disease
Progression (untreated)
• 1. Faderl S, Talpaz M, Estrov Z, et al. N Engl J Med. 1999;341:164-172.
• 2. Faderl S, Talpaz M, Estrov Z, Kantarjian HM. Ann Intern Med. 1999;131:207-219.
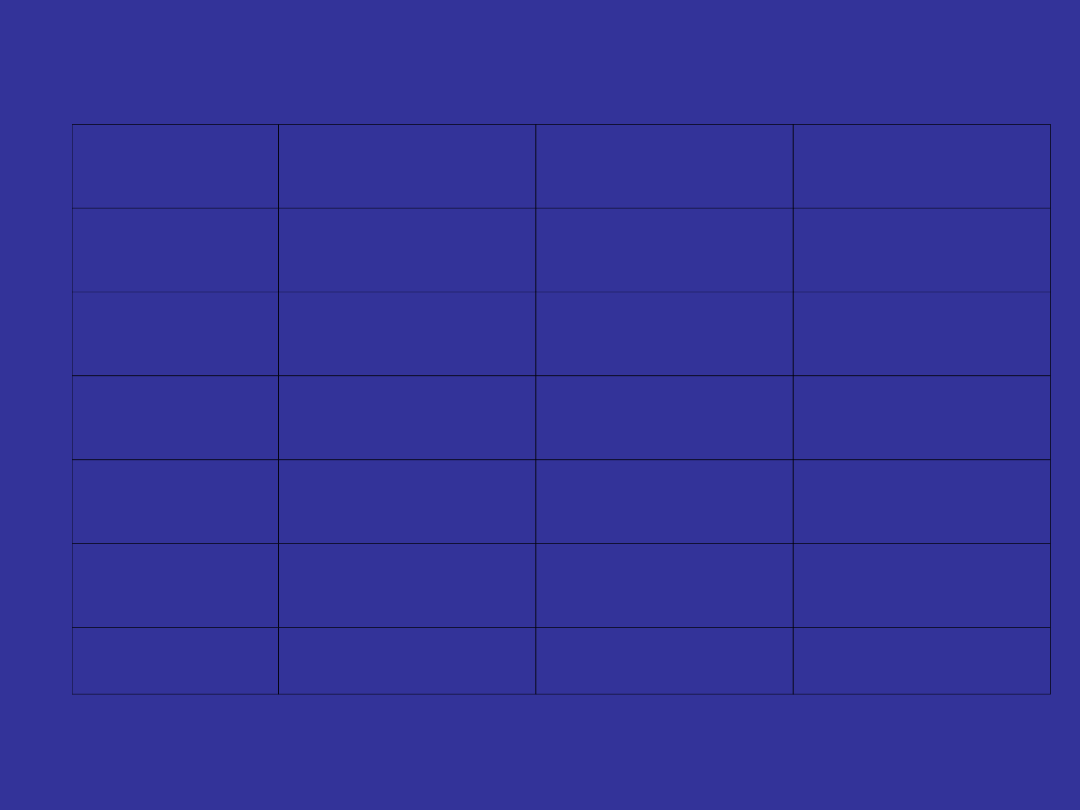
Clinical Criteria for the Phases of
CML
Chronic
Accelerated
Blast Crisis
WBC
≥ 20 x 10
9
/L
↑
↑
Blasts
<15%
15%-30%
>30%
Basophils
↑
≥ 20%
-
Platelets
Normal or ↑
↑ / ↓
↓
Marrow
cellularity
↑
↑
↑
Chromosome Ph+
Ph+
Ph+
www.glivec.com/content/target_cml/about_cml.jsp accessed
Aug 06

Czynniki prognostyczne PBS
Wskaźnik Sokala
(
Blood, 1984, 63:789-799)
-
dotyczy jedynie chorych w fazie przewlekłej
-opracowany dla chorych leczonych
busulfanem
Zróżnicowany czas przeżycia chorych
rozpoznanych w fazie przewlekłej
Wskaźnik Hasforda
(
J Natl Cancer Inst. 1998
3;90:850-8.)
-
dotyczy jedynie chorych w fazie przewlekłej
-opracowany dla chorych leczonych
interferonem
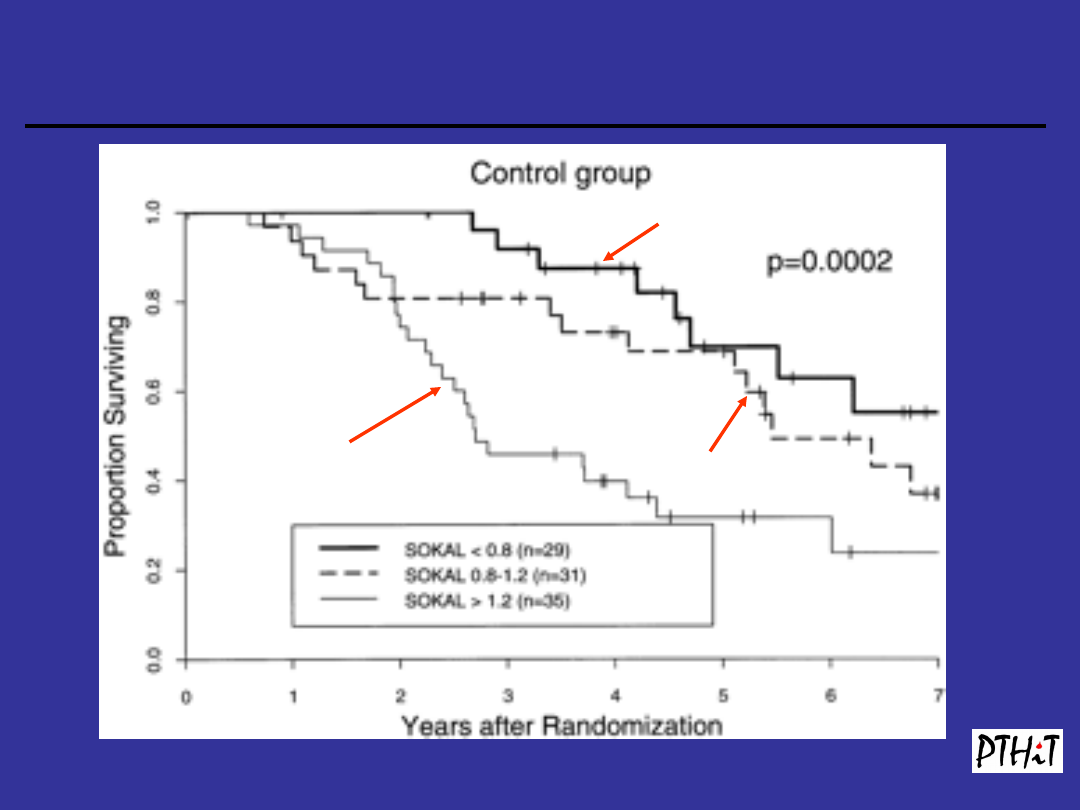
Wskaźnik Sokala
Sokal<0,8
Sokal 0,8-1,2
Sokal 1,2
Blood, Apr 1998; 91: 2713 - 2721.
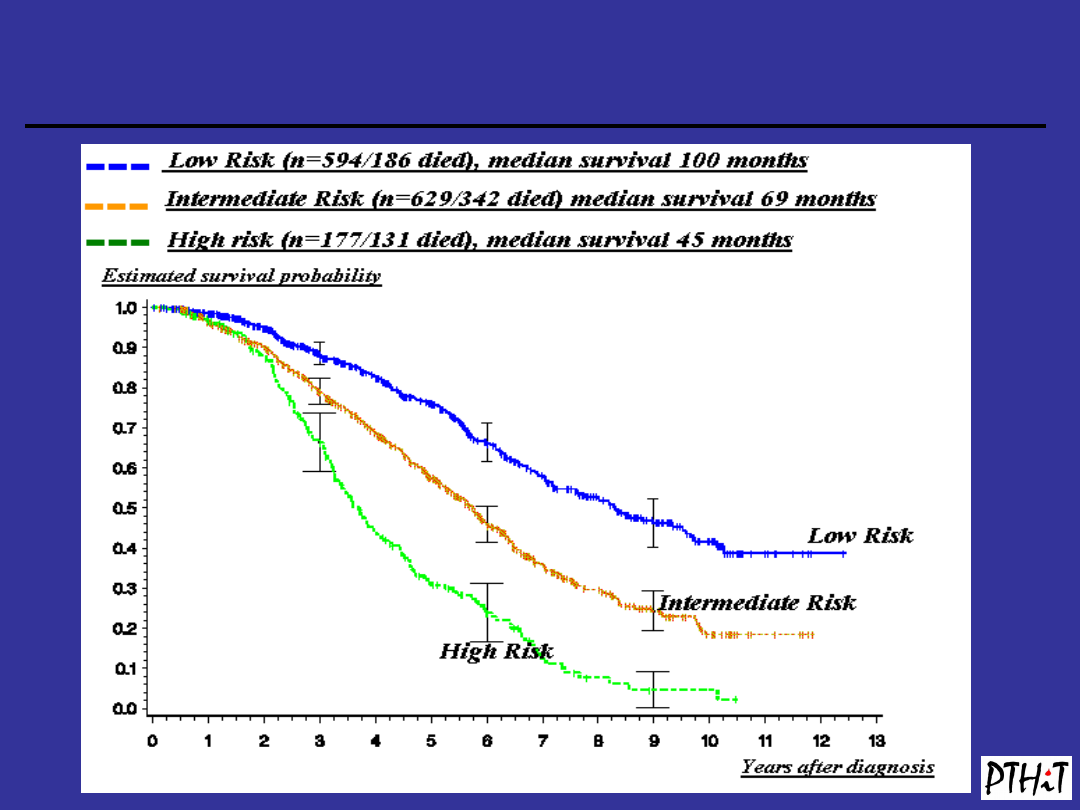
Wskaźnik Hasforda

Cel leczenia
przewlekłej białaczki
szpikowej
Zmniejszenie ilości
komórek
nowotworowych aż do
ich całkowitej
eliminacji

Cele pośrednie leczenia
PBS
Cytoredukcja
Remisja cytogenetyczna
Remisja molekularna
Wyleczenie

•
Typowa dawka: początkowo 2g/m
2
a
następnie 1-2 g/dobę
•
Inhibitor syntezy DNA (hamuje reduktazę
rybonukleotydów), działa na późne
prekursory hematopoezy
•
Nie wywołuje nieodwracalnej hipoplazji
szpiku - jest bezpieczniejszy niż wcześniej
stosowany busulfan, wydłuża 5 letnie
przeżycie chorych z 32 do 44 miesięcy
(p=0,008)
Hydroksymocznik
Hydroksymocznik

Dalsza eliminacja komórek
Ph+ po osiągnięciu
cytoredukcji
Tylko 3 dostępne metody
leczenia:
1. Allogeniczna transplantacja komórek
hematopoetycznych - od 1970
2. Zastosowanie interferonu alfa - od
1980
3. Zastosowanie imatinibu - od 1998

Medycyna oparta na faktach
ery „interferonowej”
1. Całkowita odpowiedź cytogenetyczna (CCR)
najlepszym wskaźnikiem wydłużenia
przeżycia
2. Ponowny wzrost ilości komórek Ph+ po
uzyskaniu odpowiedzi cytogenetycznej
rozwój oporności na stosowane leczenie +
zapowiedź fazy transformacji blastycznej
kwalifikacja do transplantacji lub innych
form leczenia
1. Najlepsza metoda leczenia- jak najwięcej chorych
uzyskuje MCR, a najlepiej CCR i utrzymuje ją jak
najdłużej
2. Konieczność jak najlepszego monitorowania leczenia
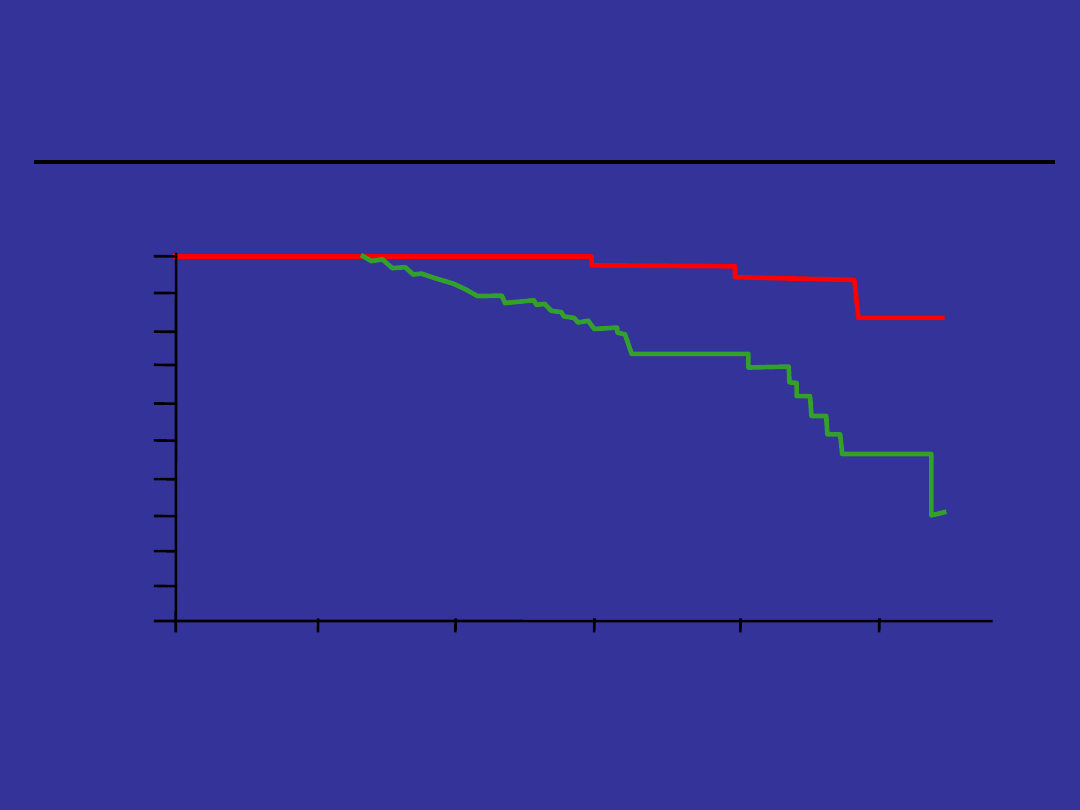
Odpowiedź
Odpowiedź
cytogenetyczna a przeżycie
cytogenetyczna a przeżycie
Guilhot F et al.. N Engl J Med.. 1997:337:223-229
Pr
a
w
d
o
p
o
d
o
b
ie
ń
st
w
o
Większa
odpowiedź
Brak lub mniejsza
odpowiedź
Miesiące po leczeniu
1.
0
0.
9
0.
8
0.
7
0.
6
0.
5
0.
4
0.
3
0.
2
0.
1
0.
0
P < .001
12
24
36
48
60

Brian J. Druker, M.D
.

28 May
2001
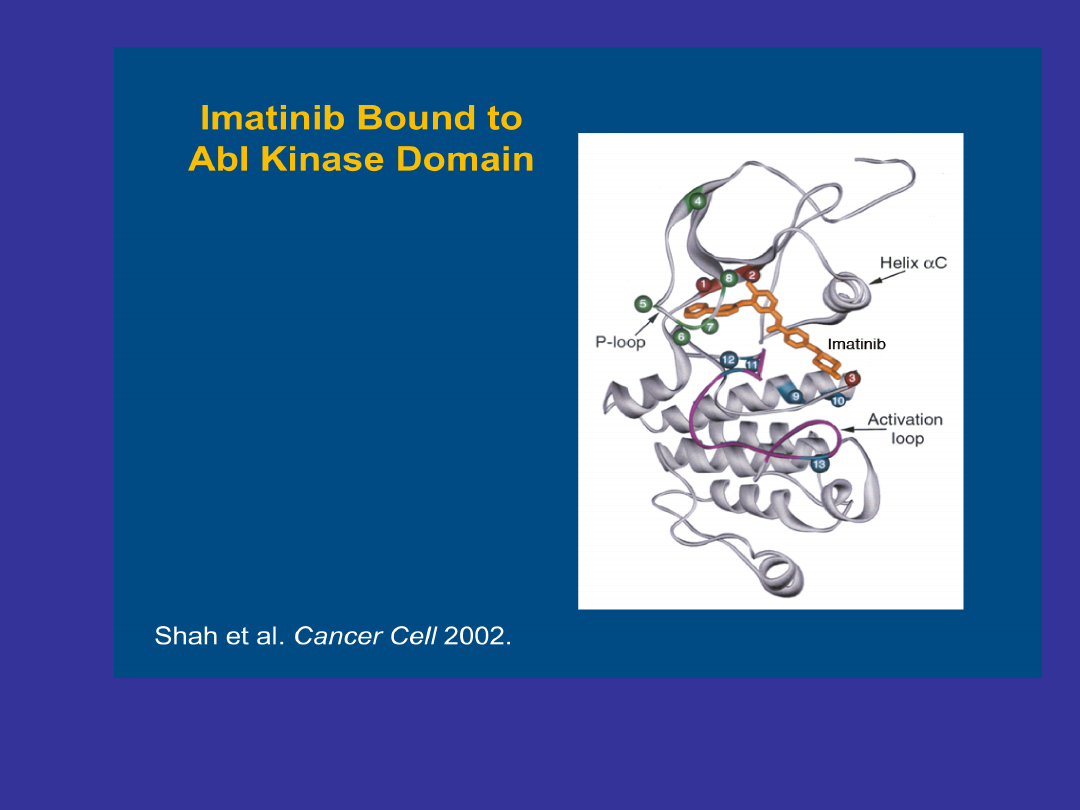

Wiązanie imatinibu do kinazy BCR-
ABL
• Kinaza BCR-ABL nieustannie zmienia konformację
(z nieaktywnej na aktywną i na odwrót)
•Imatinib ma zdolność wiązania się wyłącznie z
nieaktywną formą kinazy BCR-ABL.
• Od momentu przyłączenia się imatinibu, kinaza
BCR-ABL pozostaje w konformacji nieaktywnej, co
w konsekwencji zapobiega fosforylacji substratów i
prowadzi do zahamowania szlaków przewodzenia
sygnału (STI- signal transduction inhibitor)
• Imatinib powoduje również zmiany strukturalne
w pętli P (P loop), która ma wpływ na zdolność
wiązania leku i stabilność kinazy BCR-ABL w
konformacji nieaktywnej.
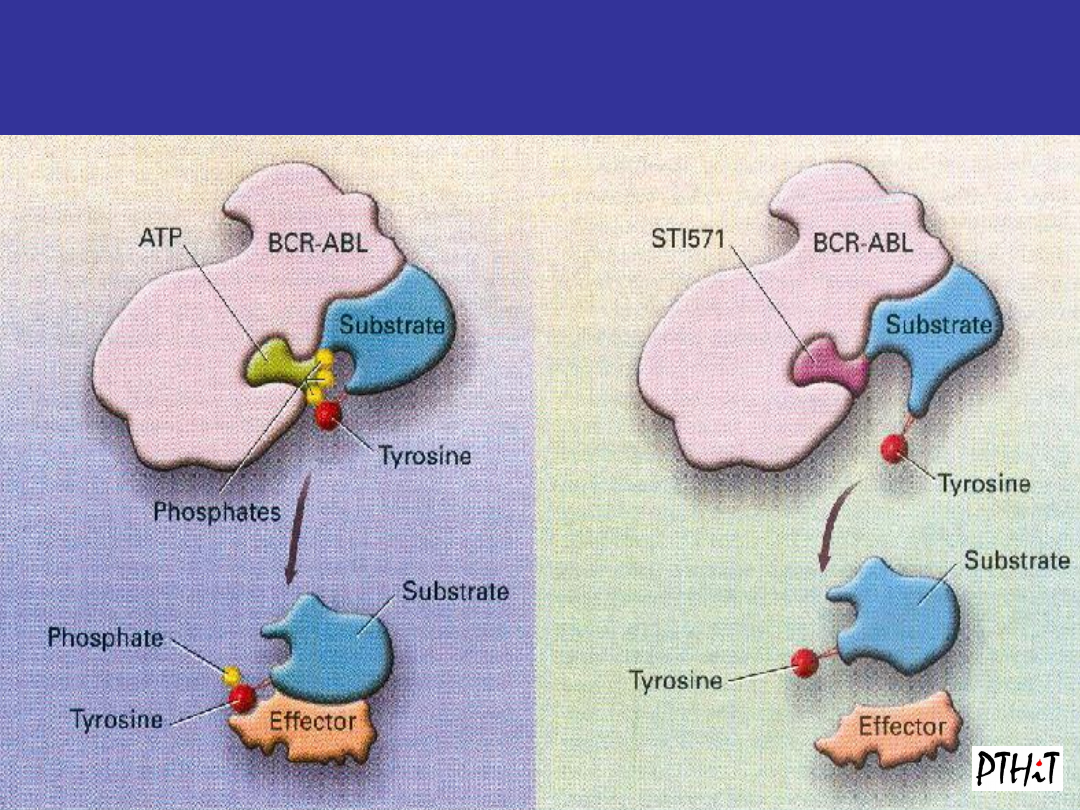
Leczenie celowane imatinibem
Goldman JM, Melo JV. N Engl J Med.. 344:1084-1086

Imatinib Mesylate:
Mechanism of Action
• Imatinib
mesylate
occupies the ATP
binding pocket of
the Abl kinase
domain
• This prevents
substrate
phosphorylation
and signaling
• A lack of
signaling inhibits
proliferation and
survival
P
P
P P
ATP
SIGNALING
Imatinib
mesylate
Garcia-Manero et al. Cancer. 2003;98:437- 457. Savage and Antman. N Engl J Med. 2002;346:683.
ABL Kinase domain
Glivec
®
is a trademark of Novartis
Europharm Ltd

Administration
•
Very dark yellow to brownish-orange film-coated
tablet in 100mg or 400mg strengths.
•
The prescribed dose should be administered
orally with a meal and a large glass of water to
minimise the risk of gastrointestinal irritations.
•
Doses of 400 mg or 600 mg should be
administered once daily, whereas a daily dose of
800 mg should be administered as 400 mg twice
a day, in the morning and in the evening.
Glivec SmPC; updated Feb 2006

Dosing
• The recommended dosage of Glivec
®
is 400
mg/day for patients in chronic phase CML
• The recommended dosage of Glivec
®
is 600
mg/day for patients in accelerated phase or blast
crisis
• Dose increases to 600mg or 800mg per day are
permitted in the absence of severe toxicity under
some circumstances…
Glivec SmPC; updated Feb 2006
Glivec
®
is a trademark of Novartis
Europharm Ltc

Wnioski (1)
• Imatinib powinien być stosowany jako
pierwsza linia terapii u pacjentów z
nowo rozpoznaną PBS
• Całkowite przeżycie 89%
(śmiertelność związana z PBS<5%) po
5 latach terapii imatinibem jest
wynikiem lepszym niż uzyskiwano za
pomocą wcześniej stosowanych
terapii

Wnioski (2)
• Ryzyko progresji ulega zmniejszeniu w
kolejnych latach terapii
• Osiągnięcie CCR po 12 miesiącach leczenia
jest istotnym czynnikiem prognostycznym
PFS (istotniejszym niż skala Sokala i
Hasforda)
• Osiągnięcie CCR i CMoR (bez względu na
czas i wyjściowy stopień zaawansowania)
jest najistotniejszym czynnikiem
prognostycznym

Badania przy rozpoznaniu
PBS
• Morfologia krwi obwodowej z
rozmazem
• Badanie cytogenetyczne (metodą
prążkową)
• Badanie FISH (del9q+)
• Zabezpieczenie materiału do badań
molekularnych (jakościowy PCR,
ilosciowy PCR, analiza mutacji)

Całkowita remisja
hematologiczna
• PLT < 450 X 10
9
/L
• WBC < 10 x 10
9
/L
• Rozmaz bez niedojrzałych form
granulocytów (mielocytów,
promielocytów, mieloblastów), < 5%
bazofilów
• Śledziona niepowiększona
* Morfologia co 2 tygodnie do uzyskania
remisji hematologicznej, potem co 1-3
miesiące

Odpowiedź cytogenetyczna
• Całkowita Ph+ 0
• Częściowa Ph+ 1-35%
• Mniejsza Ph+ 36-65%
• Minimalna Ph+ 66-95%
• Brak Ph+ > 95%
*Co 6 miesięcy, do osiągnięcia całkowitej remisji
cytogenetycznej, potem co 12 miesięcy
**FISH wyjściowy, potem tylko jeżeli nie można
uzyskać materiału do klasycznej cytogenetyki

Analiza mutacji
• Przed leczeniem – zabezpieczenie
materiału do badań
• Podczas leczenia:
- w przypadku niepowodzenia
leczenia lub odpowiedzi
suboptymalnej
- w przypadku wzrostu poziomu
transkryptu BCR-ABL (2 log?)

Odpowiedź optymalna
• 3 miesiące – całkowita odpowiedź
hematologiczna
• 6 miesięcy – częściowa odpowiedź
cytogenetyczna (Ph+ < 35%)
• 12 miesięcy – całkowita odpowiedź
cytogenetyczna (0 Ph+)
• 18 miesięcy – większa odpowiedź
molekularna

Dylemat przy niepowodzeniu
terpii Glivekiem
Czy rekomendować
transplantacje szpiku
czy leczenie
innymi inhibitorami
kinazy

•
szansę nie tylko całkowitego
przeżycia, ale również przeżycia
najbliższych lat
•
wyniki leczenia w danej grupie
wiekowej
•
czynniki rokownicze
•
preferencje i sytuacje chorego
Wybór optymalnej metody leczenia
Wybór optymalnej metody leczenia
Powinien uwzględniać:

Allogeniczna transplantacja komórek
Allogeniczna transplantacja komórek
hematopoetycznych
hematopoetycznych
•
Jedyna metoda leczenia pozwalająca na
trwałe wyleczenie u większości chorych
•
Najlepsze wyniki osiągane są przy
przeszczepieniu szpiku w przewlekłej
fazie choroby, w jak najkrótszym czasie
od rozpoznania PBS (< 1 roku) oraz u
chorych młodszych i mających dawców
rodzinnych lub w pełni zgodnych
(10/10) dawców niespokrewnionych
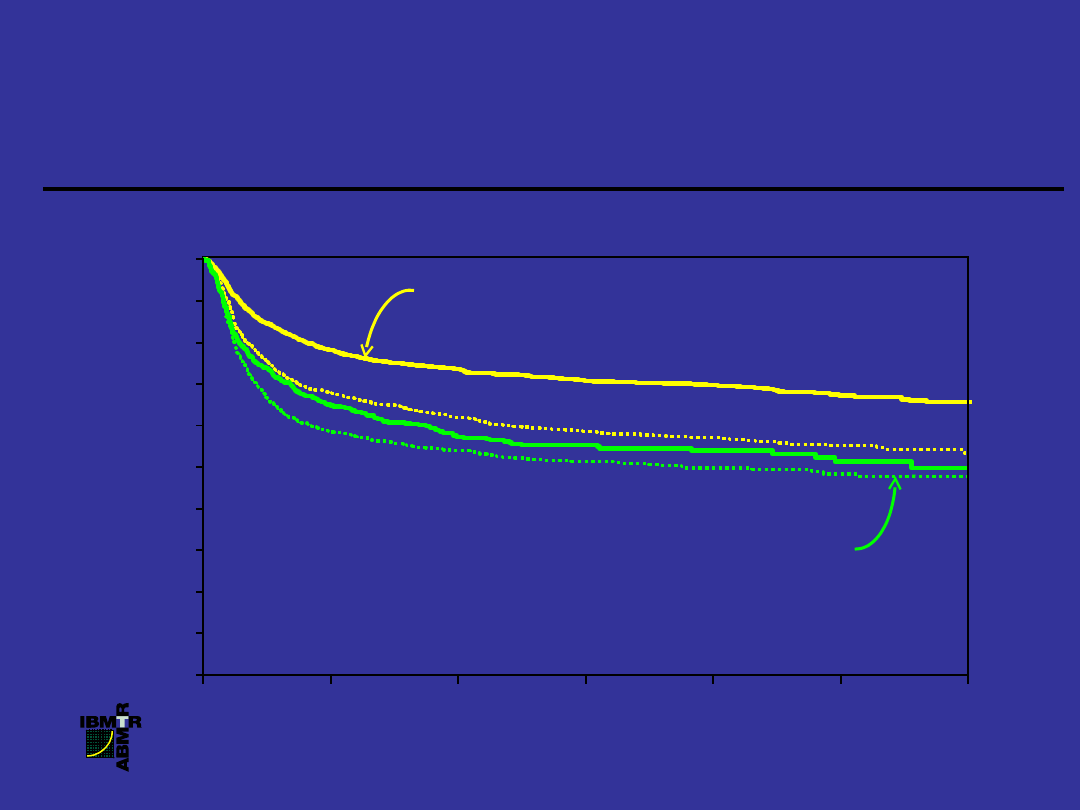
Prawdopodobieństwo przeżycia chorych z
PBS po allogenicznej transplantacji szpiku,
1996-2001
z uwzględnieniem typu dawcy i czasu trwania choroby
P
R
A
W
D
O
P
O
D
O
B
IE
Ń
S
T
W
O
,
%
100
0
20
40
60
80
LATA
HLA-zgodne rodzeństwo
, 1 rok (N
= 2,720)
HLA-zgodne rodzeństwo, 1 rok (N =
1,317)
Niespokrewnione
1 rok (N = 677
)
Niespokrewnione, 1 rok (N = 951
)
0
1
2
3
4
6
5
P = 0.0001
70%
50%

Allogeniczna transplantacja komórek
Allogeniczna transplantacja komórek
hematopoetycznych
hematopoetycznych
•
Większość niepowodzeń (80-90%)
występuje w ciągu pierwszego roku po
transplantacji.
•
Najczęściej (60-70%) wiążą się z chorobą
przeszczep przeciw gospodarzowi,
powikłaniami infekcyjnymi i rzadziej (10-
15%) toksycznością związaną z
postępowaniem przygotowawczym
•
Skala Gratwohla pozwala przewidzieć
ryzyko powikłań
okołotransplantacyjnych

FAZA ZAAWANSOWANA

FAZA AKCELERACJI PBS
FAZA AKCELERACJI PBS
Obowiązują te same zasady co w leczeniu fazy
przewlekłej
Należy jednak pamiętać:
1. Leczenie imatinibem zaczynamy od dawki co najmniej
600mg
2. Częściej występuje oporność pierwotna i nabyta
3. Po stwierdzeniu oporności chorych należy jak
najszybciej kwalifikować do innych dostępnych form
leczenia

Choroby mieloproliferacyjne -
patogeneza
Mutacja JAK2V6117F (Kinaza JAK majaca
zdolność aktywacji szlaku sygnałowego
JAK-STAT)
Czerwienica 90-95% pacjentów
Nadpłytkowość samoistna 50-70%
Przewlekła idiopatyczna mielofibroza 40-
50%

Nadpłytkowość samoistna
( Essential thrombocythemia
– ET)

Nadpłytkowość samoistna - objawy
•30% bezobjawowo
•Objawy naczynioruchowe (bóle
głowy, omdlenia, światłowstręt,
bóle w klatce piersiowej,
zaburzenia widzenia, parestezje,
erytromialgia, livedo reticularis
•Objawy krwotoczne ok. 25%
•Powikłania zakrzepowe ok. 20%
(częściej tętnicze: zawał serca,
incydenty naczyniowo-mózgowe)
•Splenomegalia 20%

Nadpłytkowość samoistna –badania
dodatkowe
•Liczba plt >600 x 10
9
/l
•Hb w normie
•Leukocytoza w normie
•W rozmazie krwi obwodowej:
nadpłytkowość, płytki róznego kształtu i
wielkości, płytki olbrzymie, zlepy płytek,
fragmenty megakariocytów, bazofilia
•Mielogram – nie jest podstawa
rozpoznania, nadpłytkowość, zlepy
płytek, megakariocyty atypowe
(mikromegakariocyty)
•Trepanobiopsja szpiku: zwiekszona
komórkowość, zwiekszona liczba
megakariocytów, z tworzeniem zlepów,
wielopostaciowością jader i nietypowa
ploidia jader
•Badanie cytogenertyczne:
niecharakterystyczne, u 5% wynik
nieprawidłowy

Nadpłytkowość samoistna – kryteria
rozpoznania
•Utrzymujące się zwiększenie
liczby plt >600 x 10
9
/l
•Brak przyczyny nadpłytkowości
odczynowej
•Hct<40%
•Prwidłowe zapasy żelaza w
szpiku (ferrytyna w normie,
prawidłowe MCV)
•Brak chromosomu Ph i genu BCR-
ABL
•Brak istotnego włóknienia szpiku
•Brak cytogenetycznych lub
morfologicznych cech zespołu
mielodysplastycznego

Nadpłytkowość samoistna –
różnicowanie
•Nadpłytkowość odczynowa
(wtórna)
•Czerwienica prawdziwa
•Przewlekła białaczka szpikowa
•Mielofibroza
•MDS z nadpłytkowościa (5q-)

Nadpłytkowość wtórna – przyczyny
•Infekcja
•Splenektomia
•Choroby nowotworowe
•Uraz
•Przewlekłe stany zapalne
•Kolagenozy
•Utrata krwi i niedobór żelaza

Nadpłytkowość samoistna –przebieg
i leczenie
Ryzyko transformacji w AML 5-
10%, w mielofibrozę 5%
•Leczenie: prewencja zakrzepów
(kwas acetylosalicylowy), leczenie
cytoredukcyjne
(Hydroksykarbamid), anagrelid,
interferon alfa

Czerwienica prawdziwa
(Polycythemia vera –PV)

Czerwienica prawdziwa – kryteria
rozpoznania
• A1 Zwiekszona masa
erytrocytów (> 25% normy)
lub Hct>60% u mężczyzn i
56% u kobiet
• A2 Brak przyczyny wtórnej
nadkrwistości
• A3 Splenomegalia
• A4 marker klonalnośći –
nieprawidłowy kariotyp
komórek szpiku
A1 + A2 + A3 lub A4 rozpoznanie
PV

Czerwienica prawdziwa – kryteria
rozpoznania cd
•B1 Nadpłytkowość > 400 x 10
9
/l
•B2 Leukocytoza neutrofilowa
•B3 Trepanobiopsja szpiku –
zwiększona komórkowość,
proliferacja układu
czerwonokrwinkowego i
płytkotwórczego
•Niski poziom erytropoetyny
A1 + A2 + 2 objawy B

Czerwienica prawdziwa – inne
badania dodatkowe
•FAG podwyższona
•B12 w surowicy – zwiększona
•Badanie cytogenetyczne –
30% aberracje: 20q-, 8+, 9+,
13+
•Tworzenie endogennych
kolonii erytroidalnych bez
dodania erytropoetyny

Czerwienica prawdziwa – objawy
kliniczne
•Bezobjawowo
•Bóle i zawroty głowy, szum w
uszach, zaburzenia widzenia
•Świąd skóry, erytromialgia
•Zaczerwienienie i pieczenie twarzy
•Splenomegalia
•Powikłania zakrzepowe

Czerwienica prawdziwa -
leczenie
•Krwioupusty
•Hydroksykarbamid
•Anagrelide
•Interferon alfa
•Kwas acetylosalicylowy

Czerwienica wtórna - przyczyny
Wynika ze zwiększenia syntezy
erytropoetyny endogennej
•Wady serca
•Przewlekła hypowentylacja
pęcherzyków płucnych (choroby płuc
i oskrzeli)
•Przebywanie na dużych
wysokościach
•Palenie tytoniu
•Choroby nerek
•Nowotwory wydzielające
erytropoetynę

Mielofibroza samoistna
(idiopatyczna)

Mielofibroza samoistna – objawy
kliniczne
•,20% bezobjawowo
•Objawy niedokrwistości
•Objawy hypermetabolizmu
(chudnięcie, poty, zmęczenie,
gorączka)
•Splenomegalia
•Hepatomegalia
•Dna moczanowa

Mielofibroza samoistna – badania
dodatkowe
Morfologia krwi obwodowej:
• Niedokrwistość
• Liczba leukocytów zmniejszona, w normie
lub zwiększona, odmłodzenie
• Liczba płytek zazwyczaj zmniejszona,
rzadko, zwykle początkowo – zwiększona
• Obraz leukoerytroblastyczny (erytroblasty)
• Wybitna poikilocytoza i polichromazja
erytrocytów, krople łez
• Płytki olbrzymie, fragmenty
megakariocytów,
• Nagie jądra megakariocytów

Mielofibroza samoistna –
badania dodatkowe cd
1. Biopsja szpiku – zazwyczaj nie uzyskuje się
treści, tzw sucha punkcja
2. Trepanobiopsja
• Włóknienie (włókna retikulinowe lub
kolagenowe)
• Zmniejszona komórkowość
• Rozplem układu megakariocytarnego i
dysplazja megakariocytów (nagie jądra,
jadra „cloud like”, „baloon like”
Włóknienie jest spowodowane cytokinami
wydzielanymi przez megakariocyty

Mielofibroza samoistna -
różnicowanie
•Inne zespoły
mieloproliferacyjne
•AML (M7) tzw ostra
mielofibroza
•MDS
•Białaczka
włochatokomórkowa
•Przerzuty nowotworów do
szpiku
•Gruźlica

Mielofibroza samoistna –
przebieg i leczenie
Czas przeżycia 4-5 lat, 5-10%
transformacja do AML
Leczenie:
•Objawowe
•Hydroksykarbamid
•Naświetlania śledziony
•U młodszych chorych allo-SCT

Ostre białaczki

OB - Klasyfikacja
• Ostra białaczka szpikowa (OBS)
• Ostra białaczka limfoblastyczna (OBL)

OB - epidemiologia
• Zachorowalność na OB - 4/100000
ludzi na rok (OBS-70%; OBL-30%)
OB- rozpoznanie
Charakterystyka OBS
OBL
Morfologia
komórki
blastycznej
pojawiają się
cechy
różnicowania
komórek do
granulocytów
i monocytów
brak
różnicowania

OB- rozpoznanie
Cytochemia
OBS
OBL
Myeloperoxidase (POX)
Sudan black
Non-specyfic esterase
(NSE)
Periodic acid-Schiff (PAS)
+
+
+ (M4,M5)
+ (M6)
-
-
-
+

OB- rozpoznanie
Markery immunologiczne
OBS
OBL
Szpikowe:
CD13, CD33
Glycophorin
antygeny płytkowe np.
CD41
MPO
Limfocytowe:
linia B CD19, CD 22, CD10
linia T CD7, CD3, CD2
+
+ M6
+ M7
+ M0
-
-
-
-
-
-
+ B-ALL
+ T-ALL

OBS - epidemiologia
• Zachorowalność 4-16 /100,000 ludzi
na rok
• średni wiek - 64 lata

OBS - etiologia
Czynniki prawdopodobnie związane z
występowaniem OBS :
• Wirusy (HTLV)
• promieniowanie jonizujące
• wcześniejsza chemioterapia
• benzen

OBS – objawy kliniczne
Objawy związane są z:
• Niedokrwistością
• Małopłytkowością
• Neutropenią

OBS – objawy kliniczne
Niedokrwistość
• bladość
• zmęczenie
• osłabienie
• kołatanie serca
• duszność wysiłkowa

OBS – objawy kliniczne
Małopłytkowość
• siniaki
• wybroczyny
• krwawienie z nosa
• krwawienie z dziąseł
• krwawienie dospojówkowe
• przedłużające się krwawienia
miesięczne lub po małych
skaleczeniach

OBS – objawy kliniczne
Neutropenia
• infekcje bakteryjne i grzybicze
• gorączka neutropeniczna FUO (fever
of unknown origin)

OBS – objawy kliniczne
Rzadsze objawy kliniczne
• Hepatomegalia i/lub splenomegalia (u
ok.1/3 chorych)
• Limfadenopatia
• Nacieki skórne i w dziąsłach (M4, M5)
• Zajęcie OUN (M4, M5)
• DIC (disseminated intravascular
coagulation) (M3)

OBS – diagnoza (wg WHO)
• Mielogram lub trepan (>20%
blastów)
• Badania morfologii krwi obwodowej
– Najczęściej niedokrwistość
normochromiczna, normocytarna
– Najczęściej małopłytkowość
– WBC może być podwyższona,
prawidłowa lub obniżona (przerwa
białaczkowa), neutropenia

OBS - diagnoza
• Cytochemia szpiku kostnego (POX, NSE)
• Imunofenotyp (CD13, CD33, MPO, GfA)
• Cytogenetyka
• Markery molekularne (FLT3, MLL, GPp)
• Biochemia (LDH, kwas moczowy)

OBS – klasyfikacja FAB
• M0 - niezróżnicowana
• M1 - bez cech dojrzewania
• M2 - z cechami dojrzewania blastów
• M3 - promielocytowa
• M4 - mielomonocytowa
• M5 - monocytowa
• M6 - erytroleukemia
• M7 - megakarioblastyczna

OBS – klasyfikacja WHO
• OBS z określonymi zmianami
cytogenetycznymi
– OBS z t(8;21)
– OBS z eozynofilią (inv 16) or t(16;16)
– Ostra białaczka promielocytowa (APL
t(15;17))
– OBS z 11q23

OBS – klasyfikacja WHO
• Ostra białaczka szpikowa z
wieloliniową
dysplazją
– poprzedzona MDS
– bez wcześniejszego MDS
• polekowe OBS i MDS
– po lekach alkilujących
– po inhibitorach Topoisomerazy II
– inne

OBS – klasyfikacja WHO
• Ostra białaczka szpikowa niesklasyfikowana
niezróżnicowana
– bez cech dojrzewania
– z cechami dojrzewania blastów
– promielocytowa
– mielomonocytowa
– monocytowa
– erytroleukemia
– megakarioblastyczna
– ostra białaczka bazofilowa
– mięsak szpikowy

Grupy ryzyka
cytogenetycznego
(klasyfikacja SWOG)
Grupa ryzyka
Anomalie kariotypu
Korzystne
inv(16)/del(16q)/t(16;16),
t(8;21)
Pośrednie
+8, -Y, +6, del(12p)
Kariotyp prawidłowy
Niekorzystne
-5/del(5q), -7/del(7q),
inv(3q), abn11q, 20q,
21q, del(9q), t(6;9),
t(9;22), abn 17p, kariotyp
złożony
nieznane
inne

Inne czynniki ryzyka
Związane z chorobą
Liczba krwinek białych w chwili rozpoznania (pośrednio
jest wykładnikiem masy guza)
Opóźnione uzyskanie remisji (>1 cykl indukujący)
(pośrednio jest wykładnikiem wrażliwości komórek
białaczkowych na CHT)
Związane z chorym
(ograniczające
intensywność CHT)
Wiek (>60 lat w OBS, >35 lat i >55 lat w OBL)
Obecność innych współistniejące choroby,
ograniczających intensywność CHT

OBS - leczenie
• Intensywna chemioterapia
• Leczenie wspomagające

OBS – leczenie
(chemioterapia)
• Indukcja
– Cel – uzyskanie remisji hematologicznej (brak
danych klinicznych i laboratoryjnych choroby)
– „złoty standard” 3+7 (Daunorubicyna + Cytozar)
– AM3L – all-trans retinoic acid (ATRA) +
antracyklina (DNR, IDA)
• Konsolidacja
– Cel – eliminacja „ukrytych” komórek
białaczkowych (minimal residual disease; MRD)
– I: HAM (Cytozar + Mitoksantron)
– II: HDAra-C (Cytozar)

Leczenie podtrzymujące
Polega na stosowaniu łagodniej
chemioterapii przez okres 2-3 lat od
uzyskania remisji.
Całkowita remisja hematologiczna to
•Ustąpienie subiektywnych dolegliwości
•Brak pozaszpikowych ognisk choroby
•Normalizacja krwi obwodowej (neut>1,5 G/l,
Plt>100 G/l, brak zależności od przetoczeń KKCz)
•<5% blastów w szpiku przy komórkowości > 20%

OBS – leczenie
(przeszczep
komórek krwiotwórczych)
• autologiczny SCT (intensyfikacja
remisji)
– zmniejsza odsetek nawrotów
– dodaje dodatkową toksyczność do
leczenia
• allogeniczny SCT
– 1CR - OBS z grupy złego rokowania
– 2 i kolejna CR – niezależnie od grupy
rokowniczej

OBS – leczenie
(leczenie
wspomagające)
• profilaktyka nudności i wymiotów
• przetaczanie produktów krwiopochodnych
(KKCz, KKP, FFP)
• allopurinol i płyny jako profilaktyka
zespołu lizy guza
• profilaktyka i leczenie infekcji
(bakteryjnych, grzybiczych, wirusowych)

OBL - epidemiologia
• choroba głównie dotyczy dzieci
• 75% przypadków pojawia się przed 6
rokiem życia
• zachorowalność w USA 3200 na rok

OBL - etiologia
• nieznana
• istnieją pewne dane sugerujące tło
genetyczne

OBL – objawy kliniczne
objawy związane z:
• niedokrwistością
• małopłytkowością
• neutropenią
• hepatomegalią i/lub splenomegalią,
• objawami z OUN (bóle głowy, nudności, wymioty,
zaburzenia widzenia)
rzadsze objawy:
• limfadenopatia
• nacieki skórne
• obrzęk jąder

OBL – diagnoza (wg WHO)
• Mielogram lub trepan (>20% blastów)
• Badania morfologii krwi obwodowej
– najczęściej niedokrwistość
normochromiczna, normocytarna
– najczęściej małopłytkowość
– WBC może być obniżona, prawidłowa lub
podwyższona (>200tysG/L), przerwa
białaczkowa, neutropenia

OBL - diagnoza
• Cytochemia szpiku kostnego (PAS+, POX-)
• Imunofenotyp
– linia B (CD 19, CD22, CD10)
– linia T (CD3, CD7)
• Cytogenetyka
• Punkcja lędźwiowa i ocena płynu mózgowo-
rdzeniowego (wzrost ciśnienia, blasty)
• Biochemia (LDH, kwas moczowy)
• ocena wydolności wątroby i nerek

OBL- klasyfikacja FAB
Charakterysty
ka komórki
L1
L2
L3
Rozmiar
Cytoplazma
Jądro
małe,
jednakowe
skąpa
cytoplazma
reguralny
kształt,
1-2 jąderka
duże,
niejednakowe
bardziej obfita
cytoplazma
nieregularny
kształt,
większe
jąderka
duże
jednakowe
obfita
cytoplazma,
z licznymi
wodniczkami
,
intensywnie
zasadochłon
na regularny
kształt

OBL- leczenie
Intensywna
chemioterapia
Leczenie wspomagające

OBL-leczenie
(chemoterapia)
• Indukcja
Cel – uzyskanie remisji hematologicznej (brak danych
klinicznych i laboratoryjnych choroby);
Prednizon, Epirubicyna, Winkrystyna, L-asparaginaza
• Konsolidacja
Cel – eliminacja „ukrytych” komórek białaczkowych
(minimal residual disease; MRD); Metrotreksat,
Cytozar, Etozpozyd, Cyklofosfamid, 6-MP
• Podtrzymywanie
Cel- utrzymanie remisji hematologicznej; Metokreksat,
6-MP
cc

OBL – Leczenie (przeszczep
komórek krwiowórczych)
• autologiczny SCT
-
zmniejsza odsetek nawrotów
- rekomendowany jest jako intensyfikacja remisji
po leczenie konsolidującym u chorych
młodszych
• allogeniczny SCT
- MRD (matched related donor)– rekomendowany u
wszystkich chorych z dawcą rodzinnym w 1CR
- MURD (matched unrelated donor)– chorzy z
niekorzystnej rokowniczo grupy 1CR, pozostali
w
2,3 CR

OBL - leczenie (leczenie
wspomagające)
• profilaktyka nudności i wymiotów
• przetaczanie produktów
krwiopochodnych (KKCz, KKP, FFP)
• Allopurinol i płyny jako profilaktyka
zespołu lizy guza
• Profilaktyka i leczenie infekcji
(bakteryjnych, grzybiczych, wirusowych)

OBL - rokowanie
• zależne od czynników
rokowniczych (wiek, cytogenetyka)
• Dzieci– 70-90% wyleczeń
• Dorośli - < 5 %
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
- Slide 99
- Slide 100
- Slide 101
- Slide 102
- Slide 103
- Slide 104
- Slide 105
- Slide 106
- Slide 107
- Slide 108
- Slide 109
- Slide 110
- Slide 111
- Slide 112
- Slide 113
- Slide 114
- Slide 115
- Slide 116
- Slide 117
Wyszukiwarka
Podobne podstrony:
Przewlekłe zespoły mieloproliferacyjne
przewlekle zespoly mieloprofileracyjne
ostre białaczki 24 11 2008 (kurs)
Biegunki przewlekle Zespol zlego wchlaniania, Biegunki przewlekłe
Patofizjologia, Nadkrzepliwość występuje w przewlekłych zespołach wewnąt, Nadkrzepliwość występuje
Ostre białaczki 2
ostre bialaczki, Medycyna, Interna, Hematologia
zespoły mieloproliferacyjne
OSTRE BIAŁACZKI semin
Ostre białaczki (2)
Przewlekle procesy mieloproliferacyjne
więcej podobnych podstron