
Zespół Pradera-Willego
oraz
zespół Angelmana

Zmiany DNA w tym samym
cytogenetycznym regionie 15q 11-13
są odpowiedzialne za wystepowanie
dwóch odmiennych zespoł
dymorficznych:
– zespołu Pradera-Willego
- zespołu Angelmana.

Przyczyny występowania zespołów Pradera-Willego i
Angelmana
Zespoł Pradera-Willego
Zespół Angelmana
Mikrodelecja w regionie
15q 11-13 pochodzenia
ojcowskiego
Mikrodelecja w regionie
15q 11-13 pochodzenia
ojcowskiego
Matczyna
jednorodzicielska disomia
(UPD)
Ojcowska
jednorodzicielska disomia
(UPD)
Mutacje „imprintigowe”
pochodzenia ojcowskiego
centrum imprintingu
Mutacje genu UBE3A
przekazane przez matke
Mutacje „imprintigowe”
pochodzenia matczynego
w centrum imprintingu

Zespół Pradera-Willego
Częstość -> 1:15000 – 1:30000 żywo
urodzonych dzieci.

Zespół Pradera-Willego
W około 65-75% przepadków przyczyną
jest delecja interstycjalna(de novo)
długiego ramienia chromosomu 15
(15q11-q13) pochodzenia
ojcowskiego (pat).
46,XX, del (15)(q11q13)pat
46, XY, del(15)(q11q13)pat

Zespół Pradera-Willego
¼ przypadków tego zespołu występuje
uniparentalna disomia pochodzenia
matczynego :
46, XY, upd(15)mat lub 46, upd(15)mat.
W regionie krytycznym (15q11-q13) ->
kilka genów podlegających zjawisku
rodzicielskiego piętnowania, m.in. loci
SNUFR-SNRPN, MKRN, MAGEL2, NDN.
U około 1-2% chorych z rozpoznanym
zespołem Pradera – Willego nie stwierdza
się zmian w obrębie regionu 15q11-q13.

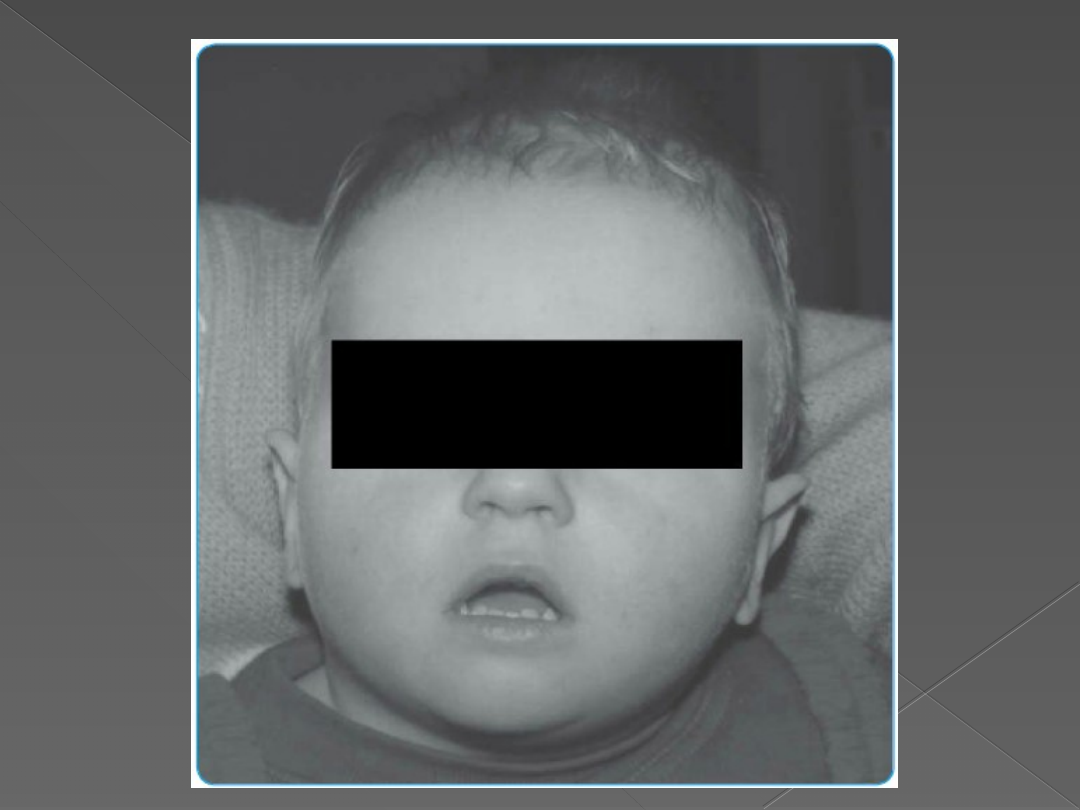
Zespół Pradera-Willego
charakterystyczne objawy:
- hipotonia mieśniowa(już w okresie przedurodzeniowym słabe ruchy płodu lub brak postępu
porodu)
- w okresie noworodkowym i wczesnoniemowlecym:
a) obniżone napiecie mięśniowe wiotkość)
b) słaby odruch ssania
c) opóźniony rozwój psychoruchowy.
- w wieku 2-3 lat
a) nadmierne łaknienie(hiperfagia)
b) dysmorfii twarzy: wąskie czoło, szpary powiekowe o migdałowatym kształcie ustawione
skośnie w góre, długie rzęsy, głęboko osadzone gałki oczne, zez rozbieżny, szeroka nasada
nosa, małe usta w kształcie trapezu, wąska czerwień wargi górnej, cofnięta czerwień wargi
dolnej, wysokie podniebienie, małe zęby.
c) hipopigmentacja skóry i włosów.
- W 3-5 r.ż.:
a) odbiegające od normy zachowanie
b)zmiane osobowości dziecka
c)opóźnienie rozwoju mowy.
d)kapryśność, kłótliwość, upartość.
Cała waga dziecka koncentruje się na zdobywaniu pokarmu i ciągłym zaspokajaniu
wzmożonego łaknienia.

Cechy nastoletnich pacjentów:
a) niski wzrost (średnio 155cm u
chłopców i 148cm u dziewcząt)
b) małe dłonie i stopy
c)delikatne zwężające się ku końcowi
palce
Także otyłość -> powikłania: cukrzyca,
nadciśnienie tętnicze, skrzywienia
kręgosłupa.
Zespół Pradera-Willego

hipogonadyzm hipogonadotropowy - Ekspresja zróżnicowana i
zmienia się wraz z wiekiem.
dysfunkcją podwzgórza + niewystarczającym wydzielaniem
gonadotropin -> otyłość, hipogenitalizm i hipogonadyzm.
♂ - cechy hipogonadyzmu hipogonadotropowego stwierdza się już u
noworodków:
- małe prącie
- małe jądra
- wnętrostwo
♀ - w pokwitaniu:
- pierwotny brak miesiączki, opóźnione miesiączkowanie lub wtórny
brak miesiączki.
Uważa się, że bezpłodność jest regułą u chorych z tym zespołem.
Ryzyko powtórzenia się zespołu w następnej ciąży jest małe (1%).
Zespół Pradera-Willego

Ostateczne rozpoznanie zespołu Pradera-
Willego stawia się na podstawie cech
fenotypowych i ukierunkowanych badań
genetycznych. W badaniach tych wykorzystuje
się techniki cytogenetyki klasycznej – techniki
prążkowe GTG, RBG o wysokiej rozdzielczości
(HRT), cytogenetyki molekularnej- techniki
FISH z zastosowaniem sondy dla regionu
15q11-q13 oraz badania molekularne: analize
wzorów metylacji DNA, badanie polimorfizmu
długości fragmentów restrykcyjnych (RFLP).
Zespół Pradera-Willego

Zespół Angelmana
Wystepuje z częstością 1:25000
urodzeń.

Zespół Angelmana
U około 70% pacjentów z objawawi klinicznymi zespołu
Angelmana stwierdza się delecje interstycjalną (de novo)
w regionie 15q11-q13 długiego ramienia matczynego
chromosomu 15.
46,XX, del (15)(q11q13)mat,
46,XX,del(15)(q11q13)mat
W około 1-2% przypadków tego zespołu wystepuje
uniparentalna disomia chromosomu 15 pochodzenia
ojcowskiego.
Około 3-5% przypadków tego zespołu jest wynikiem
defektu imprintingu genowego, którego powodem jest
mutacja w tzw. centrum imprintingu(IC).

Zespół Angelmana
Jako przyczynę wystąpienia zespołu Angelmana(5-
10%) wymienia się również mutacje pojedynczego
genu UBE3A, którego produkt białkowy w sprzężeniu
z ubikwityną działa proteolitycznie na szlaku
ubikwityno-proteosomowych. W tkance nerwowej
mózgu osoby zdrowej kopia kopia genu UBE3A
odziedziczona po ojcu jest prawie całkowicie
nieaktywna, stąd fizjologiczną rolę przejmuje
matczyna kopia tego genu. Należy zaznaczyć, że
przekazanie zmutowanego genu UBE3A przez matkę
powoduje rozwój zespołu Angelmana, natomiast
przekazywanie zmutowanego genu UBE3A przez ojca
nie powoduje u dziecka objawów chorobowych.

Zespół Angelmana
U około 70% noworodków :
- trudności w ssaniu
- wymioty po posiłkach
- mały przyrost masy ciała.
Cechy zewnetrzne:
- krótkogłowie.
- małogłowie
Kilkuletnie dzieci:
- cechy dysmorfii twarzy:
a) szeroko i głęboko oczny
b) duże usta z wąską wargą górną
c) szeroko rozstawione zęby
d) duża żuchwa z prognatyzmem
e) język wystający i ślinienie się
f) jasna karnacja skóry, blond włosy i jasne tęczówki.

Zespół Angelmana
Chore dzieci:
- opóźnieniem rozwoju psychoruchowego
- dziecko siada dopiero około 2 roku życia
- zaczyna chodzić w wieku 3-4 (ruchy tułowia i kończyn są
wzmożone i zamaszyste)
- drżenie kończyn lub zaburzenia równowagi.
Około 10% dzieci z zespołem Angelmana
- ciężkie zaburzenia mowy
- głęboka niesprawność intelektualna
U około 90% dzieci jest padaczka.
W 1/3 przypadków - niewielkie zmiany atroficzne w korze
mózgu.

Zespół Angelmana
W 2006 zweryfikowano kryteria
diagnostyczne zespołu Angelmana.
Weryfikacja rozpoznania klinicznego
opiera się na analizie stopnia metyzacji
regionu 15q11.2-q13, detekcji
mikrodelecji techniką FISH z sondą, czy
też techniką CGH.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Zespół Pradera-Willego
- Zespół Pradera-Willego
- Zespół Pradera-Willego
- Zespół Pradera-Willego
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Zespół Angelmana
- Zespół Angelmana
- Zespół Angelmana
- Zespół Angelmana
- Zespół Angelmana
- Zespół Angelmana
Wyszukiwarka
Podobne podstrony:
Zespół Pradera Willego i Angelmana
zespol Angelmana i zespol Pradera - Willego, VI rok, Genetyka, Genetyka, Egzamin
Zespół Pradera Williego
03 0000 037 02 Leczenie dzieci z zespolem Prader Willi hormonem wzrostu
Zespół Pradera Williego
Zespół Pradera
zespół pradera Williego
zespol angelmana, AM, rozne, genetyka, genetyka, GENETYKA, Genetyka ze strony
zespół angelmana
Zespół Angelmana
Zespół nerczycowy
9 RF ZEspól 0 Środki trwałe
więcej podobnych podstron