
CHOROBY DZIEDZICZONE
MITOCHONDRIALNIE
Gabriela Wójcik
Pedagogika specjalna, II rok

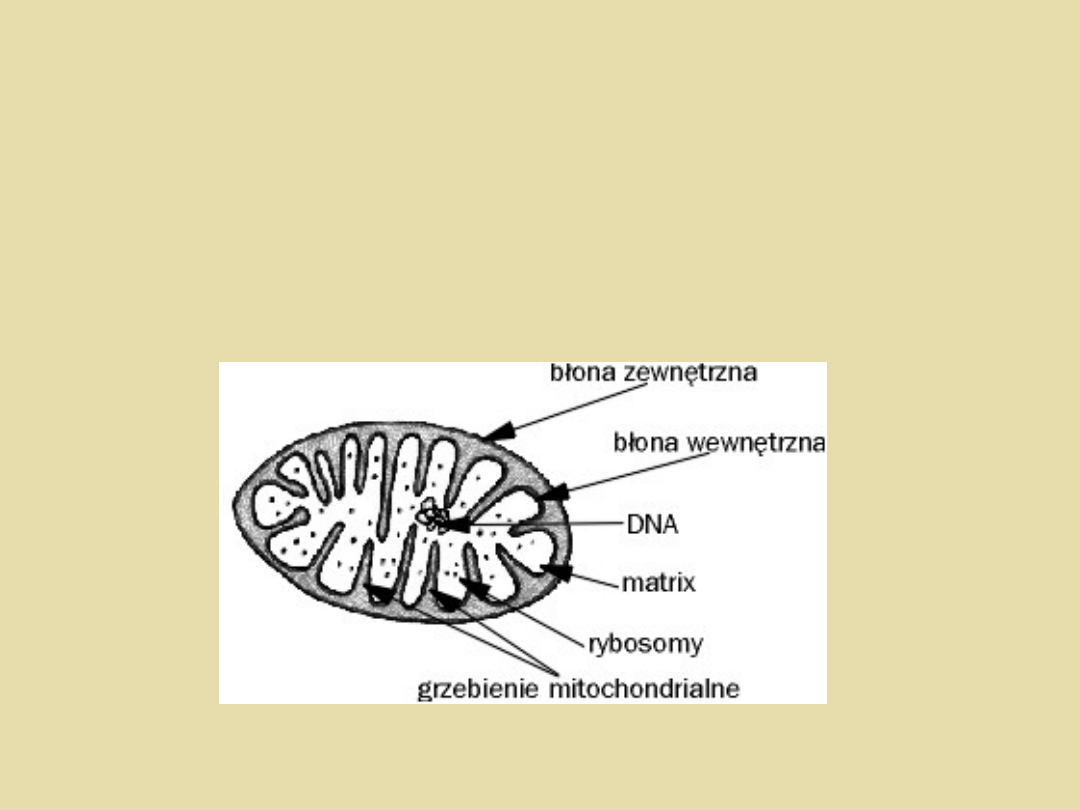
Choroby mitochondrialne są chorobami
genetycznymi, które wynikają z zaburzeń w
funkcjonowaniu jak i w strukturze mitochondriów.
Szacunkowo ok. 1 na 15 000 osób zapada na choroby
mitochondrialne.
Objawy najczęściej dotyczą mięśni szkieletowych lub
układu nerwowego, ponieważ są to tkanki o wysokim
zapotrzebowaniu energetycznym. Zazwyczaj
obejmują miopatie, encefalopatie oraz neuropatie.
Choroby mitochondrialne są wywoływane przez:
• mutacje w genomie mitochondrialnym (mtDNA)
• mutacje w jądrowym DNA kodującym białka
specyficzne dla mitochondriów oraz związane z
regulacją ich funkcjonowania.
Dziedziczenie mitochondriów
Mitochondria z reguły dziedziczone są tylko w linii matczynej,
ponieważ wszystkie pochodzą z oocytu. Bardzo niewiele
znajduje się ich w plemniku; w zygocie i tak są niszczone te,
które pochodzą od ojca. Dziedzicząc więc chorobę
mitochondrialną po matce mogą zapaść na nią obie z płci.

To obecność prawidłowego lub zmutowanego genu w
różnych organellach tej samej komórki. W
heteroplazmii obecny jest więcej niż jeden rodzaj
DNA pozajądrowego.
Cząsteczki mtDNA znajdujące się w mitochondrium
nie zawsze są identyczne. W czasie podziału komórek
mitochondria są rozdzielane do komórek potomnych
losowo.
HETEROPLAZMIA

Miopatie
Objawiają się osłabieniem mięśni, ponieważ
znajdujące się w nich włókna mięśniowe z różnych
przyczyn nie działają prawidłowo. Główna
przyczyna tych chorób znajduje się właśnie w
mięśniach. Pozostałe objawy miopatii to sztywność
lub częste skurcze mięśni.
Przykładowe miopatie to np.:
• zapalenie skórno-mięśniowe
• zapalenie wielomięśniowe
• postępujące kostniejące zapalenie mięśni
• tężyczka

Encefalopatia
To uszkodzenia mózgu spowodowane przez czynniki różnego
pochodzenia, w skutek czego dochodzi do tzw. charakteropatii, czyli
zaburzeń zachowania.
Encefalopatie dzielimy na:
WRODZONE:
• po urazie okołoporodowym
• po infekcjach płodowych
• po zatruciach
NABYTE:
• metaboliczna
• pourazowa
• miażdżycowa
• pozapalna
• poszczepienna
• nadciśnieniowa

Neuropatia
To stan chorobowy dotyczący nerwów – nazywany także
neuropatią obwodową lub zapaleniem nerwów obwodowych.
Objawami neuropatii są:
• drętwienie
• mrowienie
• palenie kończyn
• wrażenie osłabienia
• ból.
Występują one, ponieważ uszkodzenie bądź też stan zapalny
zaburza przewodzenie informacji ruchowych i czuciowych
wzdłuż włókien nerwowych.
Urazu nerwu możemy doznać podczas wypadku, zabiegu
chirurgicznego. Inne częste przyczyny to cukrzyca czy
alkoholizm.
Objawy uszkodzenia możemy dostrzec także w:
• miażdżycy tętnic
• nowotworach
• boreliozie
• AIDS

ZESPÓŁ LEIGHA
- miopatia mitochondrialna
Zespół Leigha to choroba dziedziczona na wiele sposobów,
zatem jest określana jako heterogenna etiologicznie choroba
mitochondrialna, uwarunkowana mutacjami w DNA
znajdującym się w mitochondrium lub jądrze komórkowym.
To defekt oksydazy cytochromu C – czyli IV kompleksu
oddechowego.
Objawy zwiastujące zespół, rozpoczynają się bardzo
wcześnie, bo nawet u dzieci, które nie ukończyły pierwszego
roku życia. Obejmują zasięgiem układ nerwowy, mięśnie, a
także oczy i rozwój umysłowy.
Rozpowszechnienie: <1 / 1 000 000

Około 10-30% osób ze zdiagnozowanym
zespołem Leigha ma mutację
mitochondrialną. Mówimy wtedy o zespole
Leigha dziedziczonym w linii matczynej
(maternally inherited Leigh syndrome –
MILS).
Przypadki MILS stanowią większą część –
powyżej 95% – mutacji mitochondrialnego
DNA. Mniejsza część tych mutacji,
związana jest z łagodniejszym fenotypem,
jak zespół NARP – NARP syndrome
(Neurogenic Ataxia and Retinitis
Pigmentosa – ataksja neurogenna i
barwnikowe zwyrodnienie siatkówki).

Objawy i rozpoznanie
W zespole Leigha pierwsze objawy możemy zaobserwować już
między 8, a 12 miesiącem życia dziecka. Zdarza się jednak, że
choroba może ujawnić się dopiero w okresie dojrzewania lub
wczesnej dorosłości. Zauważamy wtedy:
• cofanie się rozwoju psychoruchowego,
• zaburzenia połykania,
• trudności z utrzymaniem głowy (słabe mięśnie),
• anoreksja – znaczne niedożywienie i wychudzenie,
• objawy móżdżkowe (bezład, drżenie zamierzone – przy
podejmowaniu czynności, oczopląs – drżenia gałek ocznych,
drżenia drobne lub większe w kończynach górnych),
• deficyty umysłowe,
• ptozę – opadanie powiek
• delikatną skórę z tendencją do pękania naczynek
(powstawanie tzw. "pajączków"),
• Napady padaczkowe (nie są charakterystyczne, ale mogą się
pojawić)

Możliwości terapeutyczne
Choroba jest niewyleczalna i jak dotąd brak jest skutecznych
metod leczenia przyczynowego.
Zaleca się jednak suplementację witamin lub kofaktorów, w
tym B1, czyli tiaminy oraz witaminy B2 – ryboflawiny i
koenzymu Q10. Ich skuteczność zależy od wady podstawowej.
Obiektywne objawy poprawy stanu neurologicznego i mięśni
zaobserwowano po terapii preparatem EPI743, który odracza
postęp choroby.
Jeśli chodzi o kwasicę mleczanową (za duży poziom kwasu
mlekowego w organizmie), wyrównujemy ją wodorowęglanem
sodu lub cytrynianem sodu.
Chorzy z niedoborem dehydrogenazy pirogronianowej,
powinni mieć zapewnioną dietę bogatotłuszczową z niską
podażą węglowodanów, czyli dietę ketogenną
(ketogeniczną).
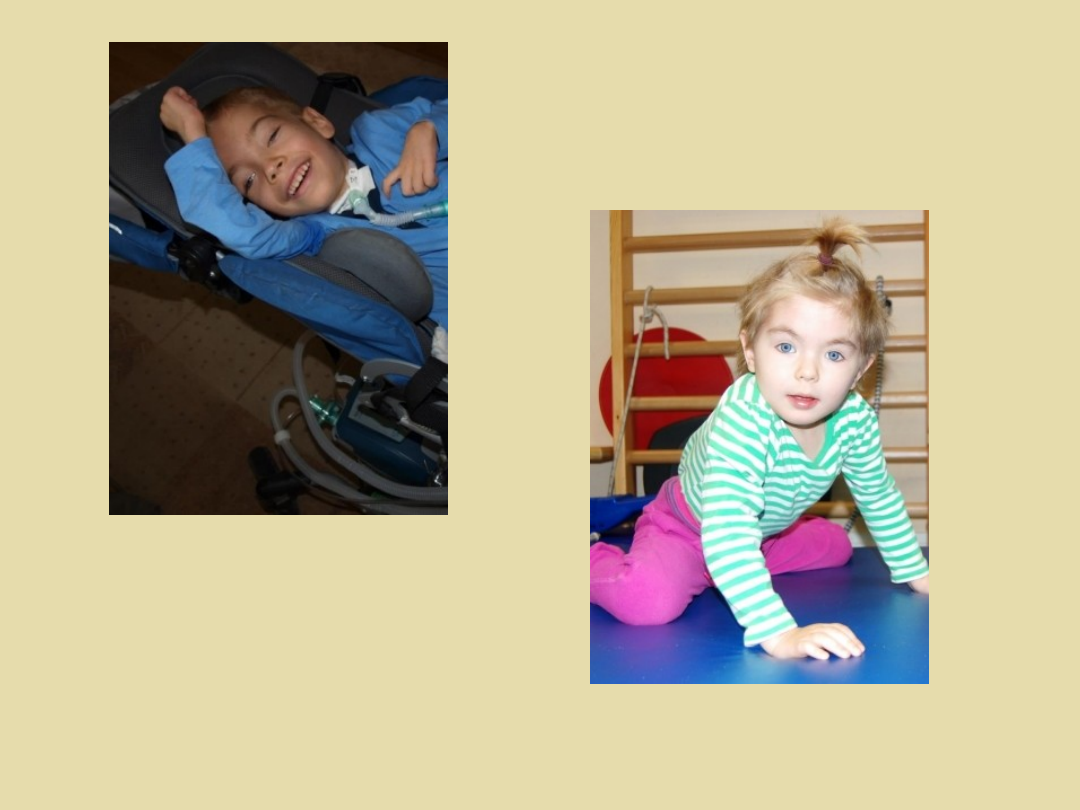
Dziewczynka z zespołem Leigha
leczona preparatem EPI-743
10-cio letni chłopiec z zespołem
Leigha

ZESPÓŁ MELAS,
czyli mitochondrialna encefalomiopatia
z kwasicą mleczanową oraz epizodami
udaropodobnymi.
Jest to wieloukładowe schorzenie mitochondrialne, należące
do najczęstszych chorób mitochondrialnych uwarunkowanych
mutacjami mitochondrialnego DNA. Jest on dziedziczony w
linii matczynej.
Mutacje te prowadzą do braku lub niedoborów białkowych
podjednostek łańcucha oddechowego. W wyniku tego
pojawiają się niedobory energetyczne w komórkach i
tkankach zawierających mutację mnDNA, co prowadzi do
upośledzenia ich funkcji, a następnie śmierci komórek.

W późniejszych etapach może dochodzić do martwicy
większych obszarów tkanki nerwowej, co uwidacznia się w
postaci dużych ognisk udarowych obejmujących zarówno korę,
jak i przylegającą istotę białą, oraz do postępującego zaniku
mózgu i móżdżku.
Zespół MELAS rozwija się w 80% przypadków w dzieciństwie.
Kliniczny obraz choroby ze względu na niezmiernie
skomplikowaną relację genotyp-fenotyp, jest zmienny w czasie
u każdego pacjenta, a zarazem wysoce indywidualny.
Charakterystycznymi, stałymi cechami klinicznymi zespołu
MELAS są:
• niedowidzenie połowiczne lub ślepota korowa
• encefalopatia z napadami padaczkowymi i/lub postępującym
otępieniem
• nawracające migrenopodobne bóle głowy
• epizody udaropodobne występujące przed 40 r.ż.
• miopatia z obecnością w tkance mięśniowej postrzępionych
włókien czerwonych

Pozostałe objawy zespołu MELAS:
• niedoczynność przytarczyc,
• cukrzyca typu II,
• głuchota korowa,
• niski wzrost,
• neuropatia czuciowa typu aksonalnego,
• ciężkie zaparcia.
Diagnozowanie zespołu
MELAS:
• badania biochemiczne: w surowicy i płynie mózgowo-rdzeniowym
stwierdza się podwyższone stężenie kwasu mlekowego,
• MR mózgu,
• TK głowy,
• badanie dna oka,
• biopsja mięśnia,
• badanie pośmiertne: ogniskowe zmiany martwicze tkanek mózgu
(duże ogniska udarowe) z zachowaniem niekiedy pojedynczych
neuronów, zgąbczenie i glejoza istoty białej oraz poszerzenie naczyń
krwionośnych.

DZIEDZICZNA NEUROPATIA
NERWÓW WZROKOWYCH LEBERA
To rzadka choroba wywołana mutacją w obrębie mitochondrialnego
DNA, która jest dziedziczona po matce. Chora kobieta przekazuje
zmutowany gen całemu swemu potomstwu, niezależnie od płci.
Chorują jednak głównie synowie, a córki stają się nosicielkami
nieprawidłowego zmutowanego DNA. W bardzo rzadkich
przypadkach również one mogą chorować. Stosunek chorujących
mężczyzn do kobiet wynosi 8:1.
Początek choroby ma miejsce zazwyczaj między 15. a 35. rokiem
życia, choć zdarzały się przypadki rozpoznania jej u osób między 2.
a 80. rokiem życia. Zmiany polegają na powolnej, bezbolesnej
utracie widzenia, początkowo w jednym oku. W ciągu następnych
kilku tygodni lub miesięcy dochodzi do rozwoju objawów choroby
w drugim oku. Spadek ostrości wzroku jest zazwyczaj znaczny.
Obok upośledzenia widzenia stwierdza się też nieprawidłowe
rozpoznawanie barw oraz zaburzoną reakcję źrenic na światło.

Niestety nie ma skutecznego leczenia w chorobie
Lebera. Neuropatia nerwów wzrokowych Lebera jest
chorobą nieuleczalną. Mimo prób stosowania takich
preparatów, jak kortykosteroidy czy
hydroksykobalamina (pochodna witaminy B
12
),
dochodzi zazwyczaj do postępującej utraty widzenia
i zaniku nerwów wzrokowych.
Jedyną skuteczną metodą mogłaby być terapia
genowa, która, jak na razie, jest przedmiotem badań.
Na rozwój choroby negatywny wpływ ma palenie
tytoniu i picie alkoholu, a także niedobory
pokarmowe, dlatego osobom z tym typem neuropatii
zaleca się prowadzenie zdrowego trybu życia.

ZESPÓŁ NARP,
czyli neurogenna miopatia z ataksją i
zwyrodnieniem barwnikowym siatkówki
To klinicznie heterogenny (niejednorodny) stan chorobowy
charakteryzujący się często połączeniem neuropatii czuciowo-
ruchowej, ataksji móżdżkowej oraz barwnikowej retinopatii
(zwyrodnienie siatkówki).
Zespół NARP najczęściej uaktywnia się u adolescentów i
młodych dorosłych, rzadko u dzieci i zaliczany jest
do zaburzeń mitochondrialnych.
Rozpowszechnienie: 1-9:100 000

NARP dziedziczy się w linii matczynej i prowadzi od
poważnych zaburzeń syntezowania ATP
mitochondrialnego, zmniejszenia produkcji energii
komórkowej i w efekcie śmierci komórki, szczególnie
w tkankach wysoce zależnych od fosforylacji
oksydacyjnej, jak mózg i siatkówka oka.
Mutacja ta występuje również u 8-10% pacjentów z
zespołem Leigha i są to najcięższe postaci zespołu
NARP. Zwykle przejawiają się w kolejnych
pokoleniach osób dotkniętych wadą.
Dziedziczenie i występowanie

Objawy występujące w zespole NARP
• wczesna retinopatia o charakterze „ziaren soli i pieprzu”;
• retinopatia barwnikowa siatkówki;
• oczopląs (drgania gałek ocznych);
• ślepota;
• opóźnienie rozwoju;
• demencja (otępienie);
• uszkodzenie słuchu;
• problemy z uczeniem się;
• napady drgawek;
• ataksja (niezborność ruchowa);
• neuropatia czuciowa;
• atrofia drogi piramidowej;
• zaburzenia pnia mózgu;
• proksymalne neurogenne osłabienie mięśni.
BIBLIOGRAFIA:
1. R. Michałowicz, J. Ślenzak, Choroby układu
nerwowego u dzieci i młodzieży, PWN, Warszawa,
1982
2. Podstawy genetyki dla studentów i lekarzy, pod.
red. G. Drewy, wydanie I, Wydawnictwo Volumed,
Wrocław, 1995
chorobyrzadkie.blogspot.com/2012/12/zespo-leigha-
miopatia-mitochondrialna.html
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
Wyszukiwarka
Podobne podstrony:
Choroby dziedziczne, Szkoła, przydatne w szkole
Choroby dziedziczne
Mechanizmy chorób dziedzicznych 1
Wykłady z genetyki (Choroby, dziedziczność) by Kusy
2006 hemochromatoza dziedziczna najczestsza choroba dziedz c
X-meni albo choroba dziedziczna, Naja Snake
Mechanizmy chorób dziedzicznych 2
CYSTYNURIA choroba dziedziczna
Zwyrodnienia siatkówki Choroby dziedziczne, Okulistyka-Optometria, Choroby siatkówki (umkc)
Choroby dziedziczne, Szkoła, przydatne w szkole
Dziedziczenie mitochondrialne 2(1)
choroby dziedziczone autosomalnie recesywnie
biologia choroby dziedziczne
diagnostyka chorób dziedzinych ppt
Ludzie mogą dziedziczyć mitochondrialne DNA po ojcu
więcej podobnych podstron