
CHOROBY
GENETYCZNE U
CZŁOWIEKA

CHOROBY
GENETYCZNE
Choroby genetyczne są chorobami przekazywanymi z pokolenia
na pokolenie. Są to też choroby powstające na nowo na skutek
zmian i zaburzeń w mechanizmach przekazywania cech
dziedzicznych. Te nieprawidłowości mogą być przekazywane
potomstwu jako choroby dziedziczne. Jednym słowem, choroba
genetyczna może mieć swój początek - na skutek sprzężenia się
różnych czynników - w każdej chwili u każdego z nas.
Trzy procent dzieci rodzi się z wadami wrodzonymi, z tego 85%
defektów jest uwarunkowanych genetycznie.

ABERRACJE
CHROMOSOMOWE
Często spotykanymi aberracjami chromosomowymi
są trisomie polegające na występowaniu w komórce
trzech chromosomów homologicznych zamiast dwóch. Mogą one
powstać w wyniku nieprawidłowej segregacji chromosomów w
czasie podziału mejotycznego, prowadzącego do
powstania gamet, lub po zapłodnieniu, a także wskutek
nieprawidłowego rozdziału chromosomów
podczas mitotycznych podziałów bruzdkowania. Aberracje
chromosomów mogą wystąpić również jako skutek działania
promieniowania jonizującego - aberracje popromienne
chromosomów
.
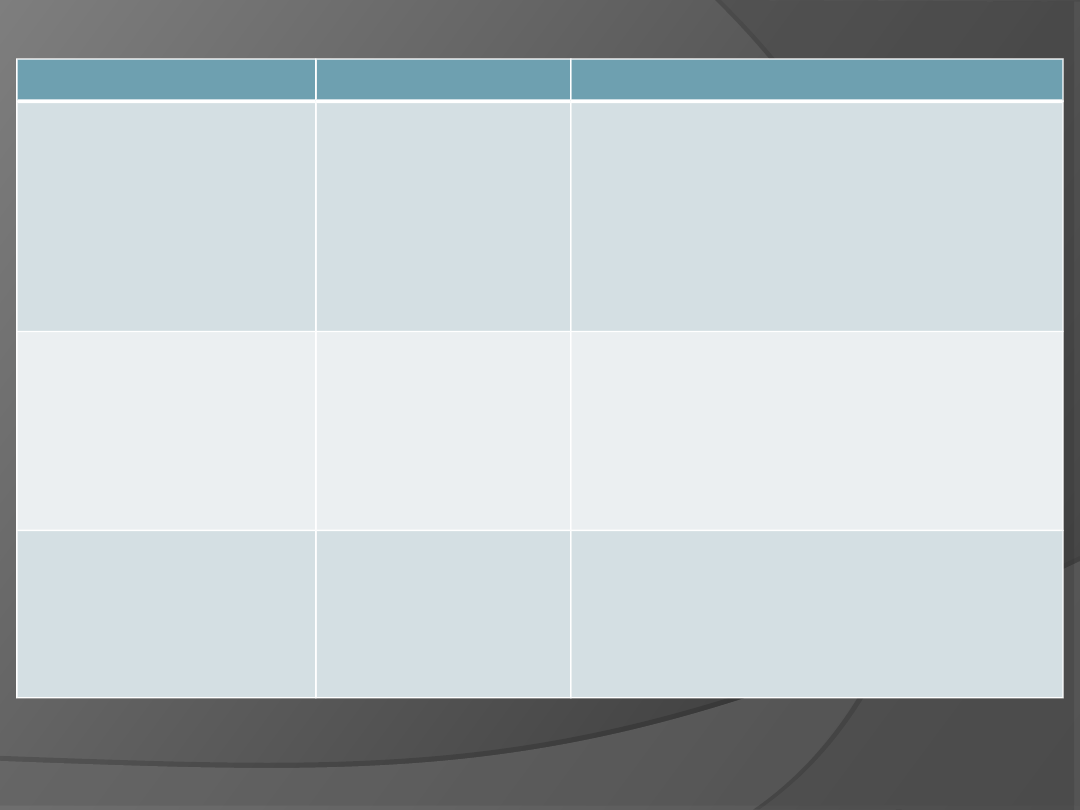
nazwa zespołu
genotyp
charakterystyka
z. Downa
47 XY
(trisomia 21)
Charakterystyczny wygląd i
skośny przebieg szpar ocznych,
płaski profil twarzy, uch
przechylone ku tyłowi, poprzeczna
bruzda na dłoni, duży język, wady
serca, upośledzenie umysłowe,
średnia długość życia 45 lat
z. Edwardsa
47 XY
(trisomia 18)
Liczne zniekształcenia,
niedorozwój serca, nisko
umieszczone małżowiny uszne,
zwiększone napięcie mięśni,
średnia długość życia 6 miesięcy
do 2 lat.
z. Patau
47 XY
(trisomia 13)
Rozszczep warg i podniebienia,
nieprawidłowy rozwój licznych
narządów (mózgu, serca),
dodatkowe palce, średnia długość
życia do 3 miesiecy

ZESPÓŁ DOWNA
Trisomia chromosomu 21. jest przyczyną
wystąpienia tzw. zespołu Downa, dawniej
zwanego pospolicie mongolizmem. Osoby z tą
wadą mają mongoidalne rysy twarzy, niski
wzrost, szczególny układ bruzd na dłoniach,
niezborność ruchów i niedorozwój umysłowy,
a przy tym pogodne usposobienie. Zespół
Downa wykrywa się metodami diagnostyki
prenatalnej. Zaobserwowano, że starsze
kobiety w większym stopniu są narażone na
ryzyko urodzenia dziecka z tą wadą niż
kobiety młodsze. U matek, które nie
przekroczyły 28. roku życia, z zespołem
Downa rodzi się jedno dziecko na tysiąc, u
matek czterdziestoletnich częstotliwość ta
wynosi jeden do stu, a u starszych nawet
jeden do pięćdziesięciu.

ZESPÓŁ EDWARDSA
Trisomia 18. pary
chromosomów.
Powoduje
niedorozwój
umysłowy. Prowadzi
do śmierci we
wczesnym
dzieciństwie z
powodu poważnych
nieprawidłowości w
budowie
wewnętrznej (m.in.
niezrośnięty otwór
międzyprzedsionkow
y).
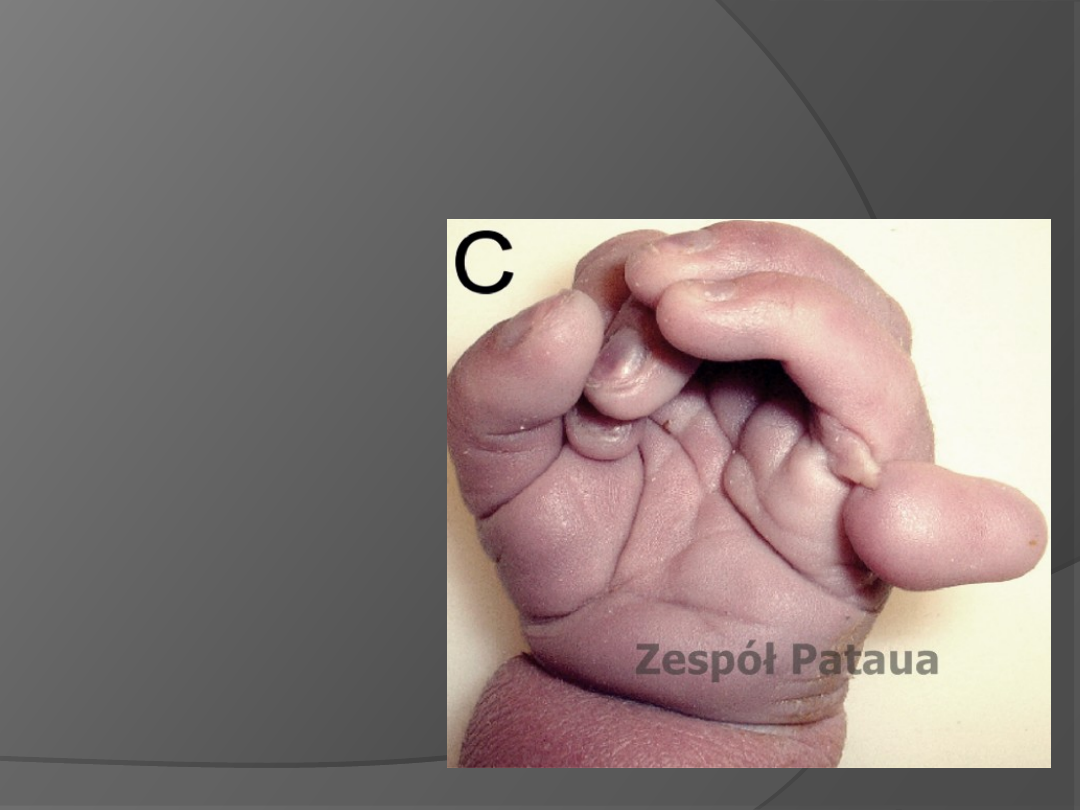
ZESPÓŁ PATAUA
Trisomia 13. pary
chromosomów. Przykładowe
skutki: niedorozwój
umysłowy, niezrośnięty
otwór
międzyprzedsionkowy w
sercu, wnętrostwo,
szczelina w
tęczówce, rozszczep
wargi(tzw. zajęcze usta).
Ok. 0,02% dzieci rodzi się z
tą chorobą. Takie dzieci
najczęściej umierają w
pierwszym roku życia,
jedynie kilka procent
dożywa 3 lat -
spowodowane jest to
wewnętrznymi wadami
wrodzonymi.

ZABURZENIA
W LICZBIE CHROMOSOMÓW
PŁCI
Wśród chorób genetycznych częste są również zaburzenia w liczbie
chromosomów płci. Niektóre z nich, np. dodatkowy chromosom Y u
mężczyzn (XYY), trisomia chromosomu X u kobiet (XXX), często ale
nie zawsze nie powodują istotniejszych nienormalności: kobiety
(jedna na 1000) mają uwydatnione cechy płciowe i niską
inteligencję, natomiast mężczyźni (również 1 na 1000)
charakteryzują się wysokim wzrostem oraz bywają nadpobudliwi i
infantylni. Badania przeprowadzone w więzieniach wykazały, że 1 na
50 skazanych mężczyzn posiada dodatkowy chromosom Y

nazwa zespołu
genotyp
charakterystyka
z. Turnera
45 X0
Płeć żeńska, ale całkowity brak
jajników, niski wzrost, płetwista
szyja, brak zaznaczonych
żeńskich cech płciowych; jeśli w
dzieciństwie podane zostaną
estrogeny, powyższych cech nie
ma, rozwija się zdrowa i aktywna
seksualnie, ale niepłodna kobieta
z. Klinefeltera
47 XXY
Mężczyzna, zwykle szczupły i
wysoki, brak owłosienia,
niedorozwój jąder i bezpłodność,
rozrost gruczołów sutkowych,
możliwe lekkie upośledzenie

ZESPÓŁ TURNERA
Jedną z przyczyn chorób genetycznych
jest monosomia, aberracja chromosomowa
polegająca na występowaniu jednego
chromosomu X, zw. zespołem Turnera. U
człowieka występuje 5% zygot, ale tylko
jedna na 40 rozwija się dalej. Zespół Turnera
dotyka 1 na 3000 kobiet - występuje częściej,
gdy matka jest w młodszym wieku. Osoby z
tą chorobą posiadają na ciele liczne
znamiona barwnikowe. Charakteryzują się
również niższym wzrostem, szerokim karkiem
i niedorozwojem drugorzędowych cech
płciowych (niewykształcone piersi, brak
owłosienia łonowego); są bezpłodne.

ZESPÓŁ
KLINEFELTERA
Inną aberracją chromosomową jest
występowanie u mężczyzn dodatkowego
chromosomu X (XXY); nie leczona powoduje
występowanie tzw. zespołu Klinefeltera,
który charakteryzuje nienormalne
wydłużenie członów, brak lub ograniczony
rozwój drugorzędowych cech płciowych
oraz niepłodność spowodowana brakiem
spermatogenezy. Występuje on w
przybliżeniu u jednego mężczyzny na 1000.

WAŻNIEJSZE CHOROBY
BĘDĄCE SKUTKIEM
MUTACJI GENOWYCH
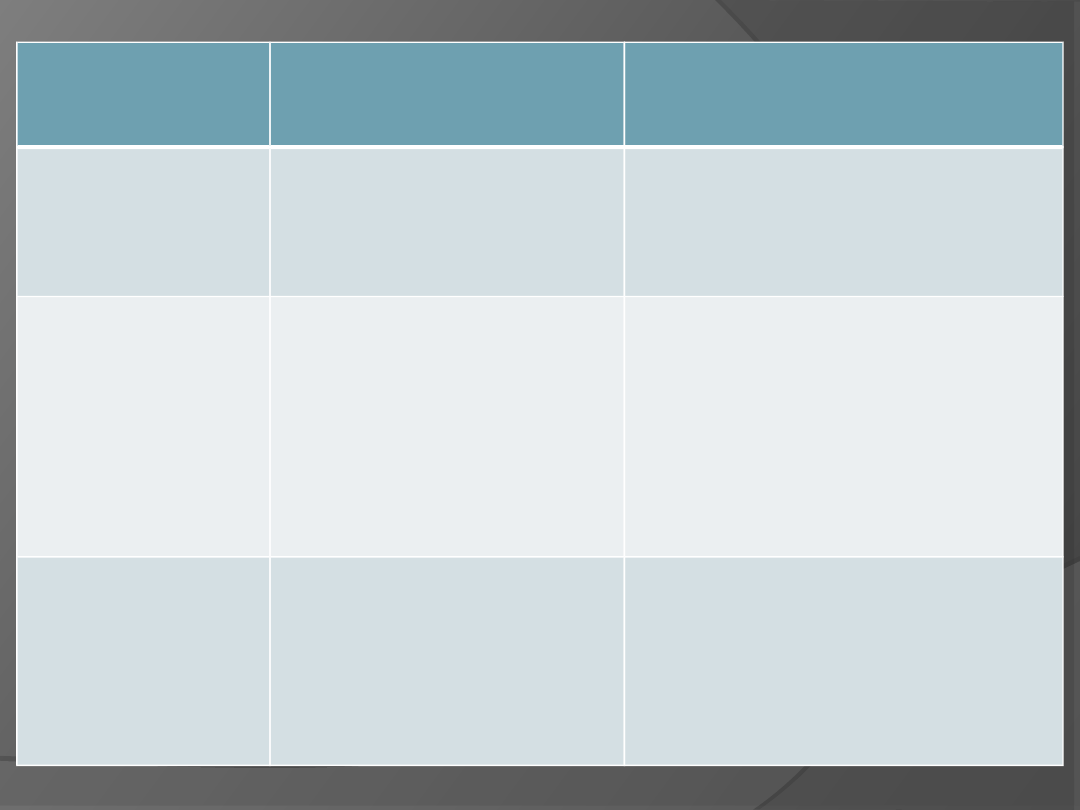
nazwa choroby
geny ulegające
mutacji i sposób
dziedziczenia
objawy
albinizm
Mutacja genu kodującego,
enzym konieczny do
syntezy melaniny,
cecha recesywna
Brak barwnika (melaniny) w
skórze, włosach i tęczówce oka –
bardzo jasna karnacja, białe
włosy, czerwone tęczówki
anemia sierpowata
Mutacja genu kodującego
hemoglobine (zamiana
jednego nukleotydu)
cecha recesywna
Nieprawidłowa hemoglobina ma
słabe powinowactwo tlenu.
Krwinki z taką hemoglobiną
przyjmują nieprawidłowy
''sierpowaty'' kształt, czopują
naczynia krwionośne, a narządy
nie otrzymują wymaganej liczby
tlenu. Często kończy się śmiercią.
fenyloketonuria
Mutacja genu kodującego
enzym konieczny przy
przemianach aminokwasów
(fenyloalaniny)
cecha recesywna
Niedorozwój umysłowy,
zaburzenia ruchowe (upośledzenie
układu nerwowego). Możliwość
zapobiegania ujawnieniu się
choroby poprzez stosowanie diety
ubogiej w fenyloalaninę.
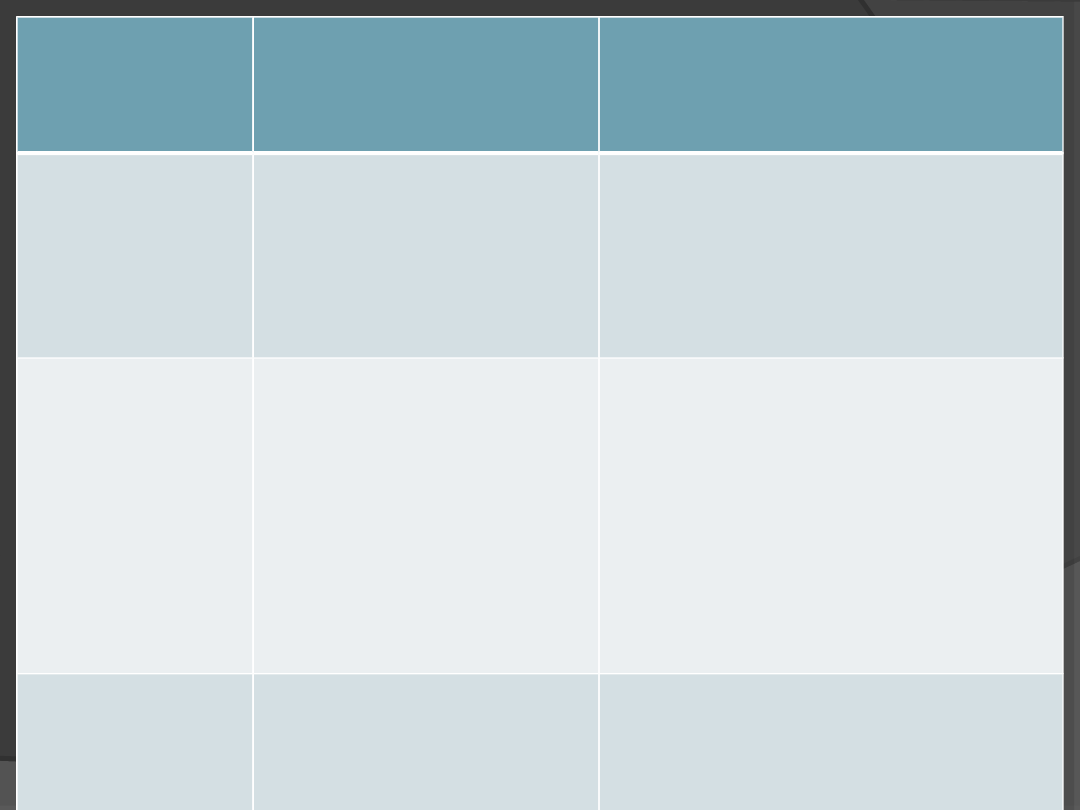
nazwa
choroby
geny ulegające
mutacji i sposób
dziedziczenia
objawy
hemofilia
Mutacja genu kodującego
jeden czynnik krzepliwości
krwi,
cecha sprzężona z płcią
(gen na chromosomie X),
recesywna
Nie zachodzi krzepniecie krwi –
zranienia i stłuczenia grożą silnymi
krwotokami, aż do śmierci przez
wykrwawienie. Współcześnie
leczona przez podawanie
brakującego czynnika.
mukowiscydoza
Mutacja genu
odpowiedzialnego za
białko transportujące w
błonach komórkowych,
cecha recesywna
Produkowanie nadmiernej ilości
śluzu, szczególnie dotyczy to układu
oddechowego – w którym utrudnia
wymianę gazową i zwiększa
podatność na infekcje; oraz
pokarmowego, w którym prowadzi
do upośledzenia funkcjonowania
trzustki a wątroby i w konsekwencji
do śmierci. Jedna z częstszych
chorób genetycznych.
daltonizm
Mutacja genu
odpowiedzialnego za
białko receptorowe –
odbierające bodźce
świetlne, cecha sprzężona
z płcią, recesywna
Upośledzone barwne widzenie,
najczęściej dotyczy czerwonego
koloru.

nazwa choroby
o chorobie
dystrofia mięśniowa
Mutacje w pojedynczym genie zlokalizowanym na
chromosomie X są przyczyną dystrofii mięśniowych
typu Duchenna (DMD) i Beckera (BMD). Szacuje się, że
DMD występuje u 1 na 3500 chłopców, ujawniając się w 2-3
roku życia postępującym zanikiem mięśni. BMD ma
przebieg łagodniejszy.
z. Retta
Mutacja genu MECP2 na chromosomie X, dziedziczona jako
cecha dominująca, w większości przypadków letalna dla
mężczyzn. Objawia się zaburzeniami rozwoju
psychoruchowego i wadami somatycznymi (m.in. skolioza).
alkaptonuria
Rzadka, genetycznie uwarunkowana choroba polegająca na
enzymatycznym defekcie metabolicznym w szlaku
przemian aminokwasów aromatycznych: tyrozyny i fenyloal
aniny. Alkaptonuria jest dziedziczona w sposób
autosomalny recesywny i charakteryzuje się wydalaniem z
moczem dużych ilości kwasu homogentyzynowego
(ciemniejącego na powietrzu), niebieskawo-czarnym
zabarwieniem tkanki łącznej (ochronoza) oraz zmianami
zwyrodnieniowymi stawów i kręgosłupa
.

DELECJE
Delecja to jeden z typów (najczęściej
spontanicznej) mutacji genowej dotyczącej
zmiany składu nukleotydowego DNA
.

nazwa zespołu
o zespole
Zespół kociego krzyku
wywołany delecją krótkiego ramienia
chromosomu 5. Objawy:
niepełnosprawność intelektualna (od
lekkiej do głębokiej), jasna karnacja,
płaczliwość, po porodzie specyficzny
płacz dziecka przypominający
miauczenie kota.
Zespół Wolfa-Hirschhorna
wywołany częściową delecją krótkiego
ramienia chromosomu 4. Objawia się
szczególnym wyglądem twarzy,
niedorozwojem żuchwy, zahamowaniem
wzrostu, zaburzeniami rozwoju
umysłowego oraz wrodzonymi wadami
serca.
Zespół Angelmana oraz Pradera –
Williego
wywołane mikrodelecją w chromosomie
15
.
Zespół Di George'a
wywołany mikrodelecją krótkiego
ramienia chromosomu 22.

Dziękujemy za uwagę
Paulina Ratzman
Marek Nosalski
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
Wyszukiwarka
Podobne podstrony:
4 Charakterystyka chorób genetycznych człowieka
Choroby genetyczne człowiekai ich diagnostyka Biologia
Choroby genetyczne czlowieka 2
Choroby genetyczne człowiekai ich diagnostyka Biologia
318 choroby genetyczne czlowiek Nieznany
Choroby genetyczne czlowieka
19101-choroby genetyczne człowieka(2), semestr IV, genetyka, Genetyka
Choroby Genetyczne człowieka
Choroby genetyczne u człowieka spowodowane są mutacjami
Choroby genetyczne człowieka, SZKOŁA, Biologia
BIOLOGIA Choroby genetyczne człowieka
Choroby genetyczne człowieka wynikajAce z mutacji chromosomowych strukturalnych
Choroby genetyczne człowieka związane ze zmianą liczby chromosomów
Choroby genetyczne człowieka
Wykł XIV genetyka człowieka choroby genetyczne
więcej podobnych podstron