
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
MINISTERSTWO EDUKACJI
NARODOWEJ
Alicja Królak
Wykonywanie instrumentalnej analizy żywności
321[09].Z4.03
Poradnik dla ucznia
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy
Radom 2006

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
1
Recenzenci:
mgr inż. Teresa Kubiak
mgr inż. Aleksandra Ptak
Opracowanie redakcyjne:
Konsultacja:
mgr inż. Maria Majewska
Korekta:
Poradnik stanowi obudowę dydaktyczną programu jednostki modułowej 321[09].Z4.03
„Wykonywanie instrumentalnej analizy żywności” zawartego w modułowym programie
nauczania dla zawodu technik technologii żywności
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy, Radom 2006

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
2
SPIS TREŚCI
1. Wprowadzenie
3
2. Wymagania wstępne
5
3. Cele kształcenia
6
4. Materiał nauczania
7
4.1. Areometryczny, piknometryczny hydrostatyczny i pomiar gęstości
7
4.1.1. Materiał nauczania
7
4.1.2. Pytania sprawdzające
10
4.1.3. Ćwiczenia
11
4.1.4. Sprawdzian postępów
14
4.2. Pomiary refraktometryczne, polarymetryczne i kolorymetryczne
15
4.2.1. Materiał nauczania
15
4.2.2. Pytania sprawdzające
25
4.2.3. Ćwiczenia
25
4.2.4. Sprawdzian postępów
28
4.3. Oznaczenia potencjometryczne i chromatograficzne
29
4.3.1. Materiał nauczania
29
4.3.2. Pytania sprawdzające
34
4.3.3. Ćwiczenia
34
4.3.4. Sprawdzian postępów
38
5. Sprawdzian osiągnięć
39
6. Literatura
44

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
3
1. WPROWADZENIE
Poradnik będzie Ci pomocny w przyswajaniu wiedzy dotyczącej stosowania różnych
metod oznaczania zawartości składników w produkowanej żywności. W poradniku
zamieszczono:
−
wymagania wstępne, w których określono co powinieneś umieć przystępując do realizacji
tej jednostki modułowej,
−
cele kształcenia, które określają umiejętności jakie powinieneś opanować w wyniku
procesu kształcenia,
−
materiał nauczania, który pomoże Ci samodzielne przygotować się do wykonania ćwiczeń
i zaliczenia sprawdzianów. Wykorzystaj do poszerzenia wiedzy wskazaną literaturę oraz
inne źródła informacji. Obejmuje on również ćwiczenia zawierające polecenie, sposób
wykonania oraz wyposażenie stanowiska pracy.
−
sprawdzian postępów, który umożliwi Ci sprawdzenie poziomu wiedzy po wykonaniu
ćwiczeń,
−
wykaz literatury.
Sprawdzian osiągnięć opracowany jest w formie testu zawierającego:
−
instrukcję,
−
zestaw zadań testowych,
−
punktację zadań,
−
kartę odpowiedzi.
Bezpieczeństwo i higiena pracy
Przebywając w laboratorium analizy żywności musisz przestrzegać regulaminu pracowni,
przepisów bezpieczeństwa i higieny pracy oraz przepisów przeciwpożarowych. Przy
wykonywaniu ćwiczeń zachowaj ostrożność podczas ogrzewania roztworów. Szyjkę kolby lub
probówki trzymaj otworem od siebie. Postępuj ostrożnie z roztworami kwasów i zasad
szczególnie stężonych. Kwasy do pipety naciągaj za pomocą pompki a nie ustami.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
4
Schemat układu jednostek modułowych
321[09].04
Analiza żywności w przetwórstwie spożywczym
321[09].04.01
Wykonywanie wagowej analizy żywności
321[09].04.02
Wykonywanie objętościowej analizy żywności
321[09].04.05
Wykonywanie towaroznawczych badań żywności
321[09].04.03
Wykonywanie
instrumentalnej
analizy żywności
321[09].04.04
Wykonywanie
mikrobiologicznych
badań żywności

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
5
2. WYMAGANIA WSTĘPNE
Przystępując do realizacji programu jednostki modułowej powinieneś umieć:
−
korzystać z różnych źródeł informacji,
−
posługiwać się sprzętem laboratoryjnym i łączyć go w zestawy zgodnie z instrukcją,
−
wskazywać zagrożenia związane z pracą w laboratorium oraz podczas wykonywania
ćwiczenia,
−
przeliczać stężenia,
−
korzystać z wag technicznych i analitycznych,
−
interpretować wyniki badań laboratoryjnych,
−
stosować metody Dobrej Praktyki Laboratoryjnej,
−
korzystać z dostępnych programów komputerowych.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
6
3. CELE KSZTAŁCENIA
W wyniku realizacji programu jednostki modułowej powinieneś umieć:
−
określić znaczenie analizy instrumentalnej w przetwórstwie spożywczym,
−
rozróżnić i scharakteryzować analizy fizykochemiczne,
−
określić budowę i zasadę działania aparatów stosowanych w analizie instrumentalnej,
−
obsłużyć aparaty do analiz instrumentalnych żywności,
−
przeprowadzić próby badawcze analiz instrumentalnych,
−
obliczyć ilość badanego składnika w próbie laboratoryjnej,
−
opracować wyniki badań laboratoryjnych z zastosowaniem programów komputerowych,
−
zinterpretować wyniki badań,
−
przeprowadzać wybrane analizy instrumentalne w zakładach przetwórstwa spożywczego
monitorujące procesy produkcyjne w celu otrzymania bezpiecznej żywności,
−
zastosować zasady Dobrej Praktyki Laboratoryjnej,
−
zastosować przepisy bhp, wymagania ergonomii, zasady ochrony przeciwpożarowej
i ochrony środowiska,
−
skorzystać z dokumentacji technicznej i technologicznej przy wykonywaniu analiz
instrumentalnych żywności.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
7
4. MATERIAŁ NAUCZANIA
4.1. Areometryczny, piknometryczny , hydrostatyczny i pomiar
gęstości
4.1.1. Materiał nauczania
Gęstość ciała d jest to masa jego jednostki objętości, gęstość określa się zatem stosunkiem
masy ciała m do jego objętości v. Rozróżnia się dwa rodzaje gęstości ciał: bezwzględną i
względną. Gęstość bezwzględna ciała określana jest więc wzorem:
3
/ dm
g
V
m
d
=
Gęstość względna jest ilorazem gęstości bezwzględnej danego ciała i gęstości
bezwzględnej innego ciała, uznawanego za wzorcowe, np. wody.
Gęstość ciała jest wielkością stałą, zależną od temperatury i ciśnienia. W praktyce gęstość
oznaczana jest przez porównanie gęstości ciała badanego z gęstością wody, którą przyjęto za
wzorzec. Gęstość wody w temperaturze 4°C jest równa 1 g/cm
3
. Oznaczenie gęstości
roztworu polega na porównaniu masy pewnej jego objętości w temperaturze t°C, z masą takiej
samej objętości, w tej samej temperaturze:
t
w
t
x
t
t
m
m
d
=
gdzie: m
x
t
– masa danego ciała w temperaturze t°C,
m
w
t
– masa wody w temperaturze t°C,
d
t
t
– gęstość ciała temperaturze t°C, w stosunku do wody w tej samej temperaturze.
Otrzymana wartość stanowi gęstość względną. Gęstość bezwzględną można obliczyć ze
wzoru:
4
4
w
t
x
t
m
m
d
=
aby przeliczyć gęstość względną na bezwzględną należy pomnożyć wartość liczbową gęstości
względnej danego ciała przez gęstość wody w temperaturze t°C.
Gęstość ciał w analizie produktów spożywczych wyznaczyć można trzema metodami:
−
areometrycznie,
−
hydrostatycznie,
−
piknometrycznie.
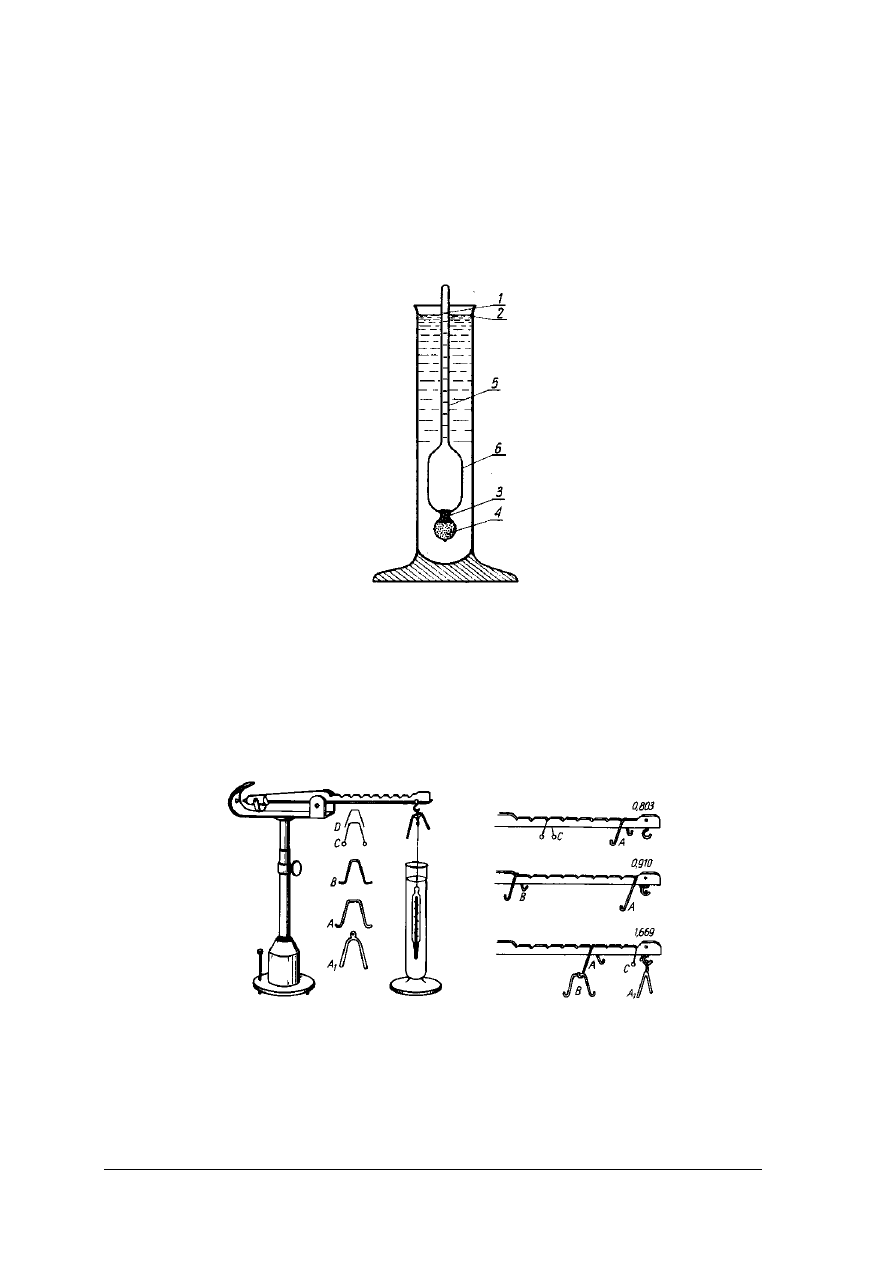
W analizie żywności do mierzenia gęstości cieczy używa się areometrów. Areometr (rys.1)
składa się z rurki szklanej, w której trzpieniu (część wydłużona rurki) umieszczona jest skala.
W dolnej części rurki wydętej w postaci bańki znajduje się balast, którym jest śrut lub rtęć.
Areometr może zawierać także termometr. Ponieważ gęstość jest zależna od temperatury
areometry są kalibrowane w określonej temperaturze, w której należy dokonać pomiaru. Przy
zastosowaniu innej temperatury pomiaru należy wyniki korygować. Areometr utrzymuje się
w cieczy w pozycji pionowej i zanurza się w niej tylko częściowo. Oznacza to, że jego ciężar
powinien być mniejszy od ciężaru wypartej cieczy. Wynika z tego, że im gęstość cieczy jest
większa tym zanurzenie areometru jest mniejsze i odwrotnie. Działanie areometru opiera się na
prawie Archimedesa. Areometr ma stałą masę, a objętość wypieranej cieczy jest zmienna
i zależna od gęstości. Dokładność pomiaru areometrem jest dość duża, może wynosić ±
0,0005. Dokładność areometru wiąże się z błędem skalowania, błędem skali termometrycznej,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
8
błędem popełnionym przy odczytywaniu wyniku. Błąd zależy od szerokości podziałki skali co
wiąże się z budową areometru. W przybliżeniu można przyjąć, że ciężar areometru jest równy
ciężarowi wypartej przez niego cieczy co można zapisać równaniem:
(
)
d
s
h
V
m
⋅
⋅
+
=
gdzie: m – masa areometru,
V – objętość bańki areometru,
h – wysokość zanurzonej w cieczy części trzpienia,
d – gęstość cieczy.
Miernikiem czułości areometru jest jego zanurzenie
d
h
∆
∆
.
Biorąc pod uwagę sposób skalowania areometry dzieli się na dwie grupy:
−
wyskalowane w wartościach liczbowych gęstości,
−
wyskalowane w stopniach umownych.
Najczęściej stosowane są areometry: Gay-Lussaca, Oechsle, Baumé, Ballinga, Briexa, Trallesa
i laktodensymetr. Laktodensymetr służy do oznaczania gęstości mleka. Zakres skali
laktodensymetru obejmuje 20÷40°Ld. Rozwodnienie mleka obniża w pewnym stopniu gęstość,
a odtłuszczenie powoduje pewne podwyższenie gęstości. Temperatura normalna wskazań
laktodensymetrów wynosi 20°C. W przypadku dokonywania pomiarów w innych
temperaturach niż 20°C koryguje się wskazania laktodensymetrów o odpowiednie poprawki,
które oblicza się przyjmując 0,2° Ld na każdy 1°C rozbieżności temperatury od 20°C lub
odczytuje z odpowiednich tabel. Przy pomiarach w temperaturach niższych niż 20°C poprawki
dodaje się, przy wyższych odejmuje.
Areometr Gay-Lussaca obejmuje zakres 0,6÷1,9 i wskazuje bezpośrednio gęstość. Aby
odczyt był dokładniejszy zakres ten dzieli się na 0,6÷1,0 i 1,0÷1,9 lub jeszcze mniejsze.
Temperatura skalowania wynosi 15°C, a w nowszych typach 20°C.
Areometr Baumé (°Bé) stosowany jest do pomiaru gęstości syropu ziemniaczanego,
melasy, zasady sodowej, kwasu siarkowego itp. Stopnie Baumé odpowiadają w przybliżeniu
procentowej zawartości NaCl w roztworach wodnych przy czym pomiędzy wskazaniami
areometru i gęstością istnieje zależność liczbowa:
1000
1
15
15
Oe
d
°
+
=
Areometr Ballinga zwany cukromierzem wskazuje w roztworach sacharozy procentową
zawartość tego cukru w temperaturze normalnej 20°C. W przypadku roztworów
zawierających inne substancje np. białka, kwasy, barwniki itp. wskazania areometru
odpowiadają zawartości tzw. ekstraktu. Składniki te wpływają na podwyższenie gęstości
roztworów wodnych w stosunku do gęstości wody.
Występujący w niektórych produktach alkohol etylowy obniża gęstość. W takich
przypadkach wskazania areometru Ballinga odpowiadają zawartości tzw. ekstraktu pozornego,
w odróżnieniu od rzeczywistego, którego zawartość jest oznaczana po odparowaniu alkoholu.
Zakres skali areometru Ballinga obejmuje 0÷70 °Blg. Jeżeli roztwór zawiera kilkanaście
procent alkoholu i niewielkie ilości ekstraktu możliwe są stopnie ujemne.
Cukromierze są stosowane w różnych przemysłach np. spirytusowym, gdzie używa się
nazwy areometr Brixa.
Areometr Oschle stosowany jest do pomiaru gęstości moszczu gronowego. Określa ile
gramów alkoholu może wytworzyć się w 1000 cm
3
wina po całkowitym odfermentowaniu
zawartego w moszczu cukru. Temperatura normalna dla wskazań tego areometru wynosi
15°C.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
9
Aerometr Trallesa wskazuje procenty objętościowe alkoholu etylowego w roztworach
wodnych. 100 cm
3
alkoholu etylowego o x °Tr zawiera w 100 cm
3
wodnego roztworu x cm
3
absolutnego alkoholu etylowego i nieco więcej niż (100 – x) cm
3
wody. Temperatura normalna
wskazań termometru Trallesa wynosi 15°C. Pomiar jest możliwy tylko w czystych, wodnych
roztworach alkoholu etylowego. Zakres skali termometru wynosi 0÷100°Tr. Pomiar powinien
być wykonywany w temperaturze normalnej. W przypadku innej wynik należy sprowadzić do
temperatury normalnej za pomocą odpowiednich tabel.
Rys. 1. Aerometr [2, s. 205]
Pomiar hydrostatyczny
Zasada pomiaru gęstości metodą hydrostatyczną (rys. 2) opiera się na prawie
Archimedesa. Do pomiarów stosowana jest waga Mohra-Westphala, która pozwala oznaczyć
gęstość z dokładnością do czterech miejsc po przecinku.
Rys. 2. Waga hydrostatyczna Mohra-Westphala . Koniki o zróżnicowanych masach: A, A
1
– po 5g;
B 2,–
0,5g; C – 0,005g i D – 0,005g [2, s. 221]
Waga jest bezszalkowa, składa się z podstawy i przymocowanego do niej statywu, na
którym zawieszona jest belka. Belka ma z jednej strony przeciwwagę, a z drugiej jest
wyskalowana, od zera w miejscu zawieszenia do 10 w miejscu nakładania nurnika. Waga jest
wyposażona w nurnik o masie 15 g i objętości 5 cm
3
oraz zestaw koników o masach: 5; 0,5;
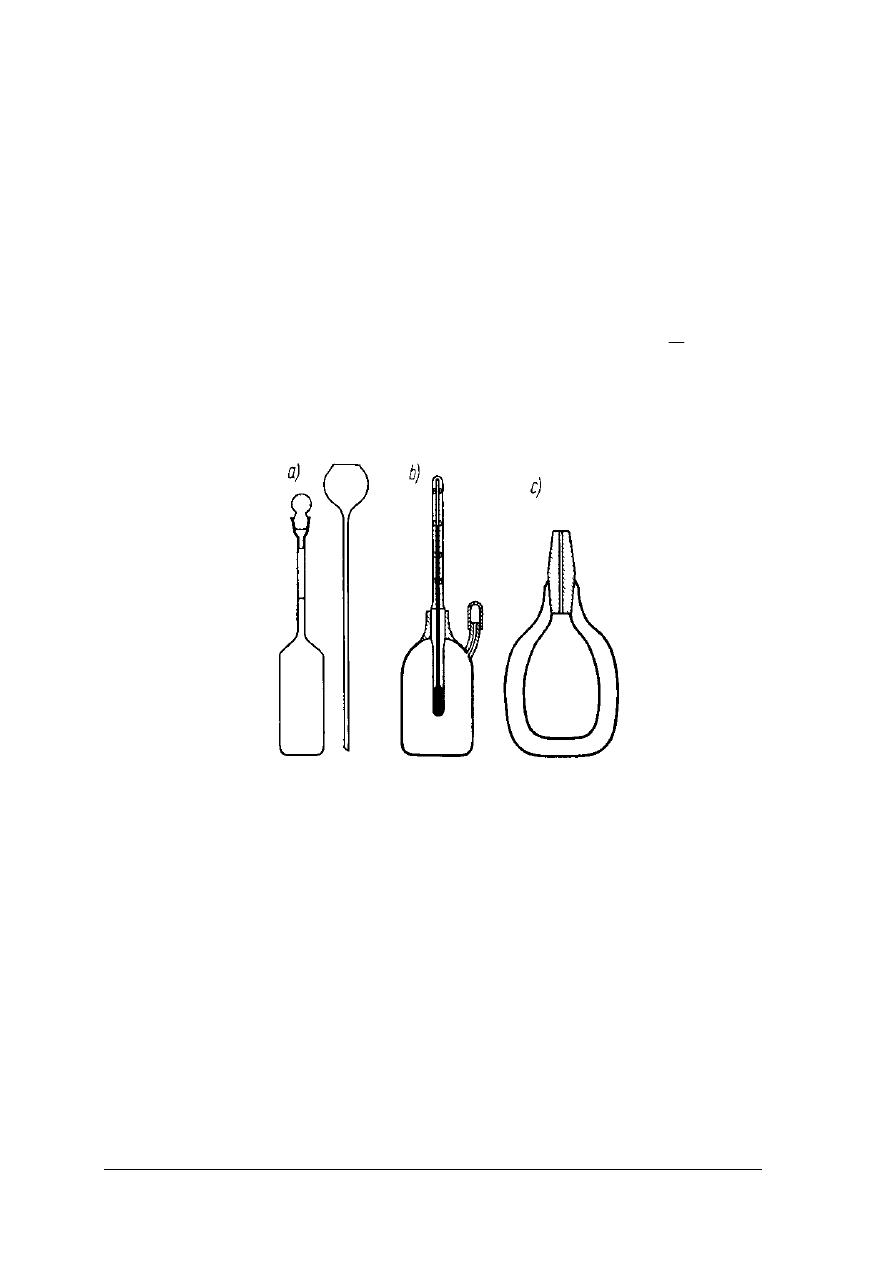
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
10
0,05 i 0,005 g. Nurnik zawieszony w pozycji 10 jest równoważony przeciwciężarem. Jeżeli
nurnik zanurzony w badanym roztworze wyprze 5 cm
3
roztworu, dla ponownego
zrównoważenia wagi należy na wyskalowanej belce zawiesić koniki. Konik 5-gramowy
zawieszony w pozycji 10 wskazuje gęstość 1 g/cm
3
, w pozycji 9 wskazuje 0,9 g/cm
3
itd.
Koniki o masach 0,5; 0,05 i 0,005 g wskazują kolejne miejsca po przecinku. W przypadku
gęstości roztworu 1,2834 g/cm
3
położenie koników będzie następujące:
5-gramowy w pozycji 10, drugi 5- gramowy w pozycji 2, 0,5-gramowy w pozycji 8, 0,005-
gramowy w pozycji 4. Waga skalowana jest w temperaturze 15°C i wskazuje gęstość d
15
15
.
Wagi skalowane są również na gęstość bezwzględną d
20
4
Pomiar piknometryczny
Zasada oznaczania piknometrycznego opiera się na definicji gęstości
3
/ dm
g
V
m
d
=
. Masa
pewnej objętości roztworu badanego porównywana jest z masą tej samej objętości wody o tej
samej temperaturze. W użyciu znajdują się piknometry: Reischauera, Geisslera i próżniowy
przedstawione na rysunku 3.
Rys. 3. Typy piknometrów spotykanych w pracowniach analitycznych a) Reischauera, b) Geisslera, c)
próżniowy [2, s. 222]
Wykonując starannie oznaczenie można uzyskać dokładność do 5 miejsca dziesiętnego.
Piknometr powinien być dostatecznie duży o pojemności 10 – 30cm
3
. Im objętość piknometru
jest większa tym oznaczenie jest dokładniejsze. Dokładność zależy także od temperatury, która
powinna być stała. Piknometr należy utrzymywać w stanie bardzo czystym.
4.1.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1. Jakie definicje i wzory potrzebne są do obliczeń gęstości ciała?
2. Jakich przyrządów używa się do mierzenia gęstości cieczy w analizie żywności?
3. Jak zbudowany jest areometr?
4. Od czego zależy dokładność pomiaru areometrem?
5. Jakie znasz typy areometrów i jaka jest ich zasada działania?
6. Jakie prawo wykorzystane jest w metodzie pomiaru gęstości wagą hydrostatyczna?
7. Jak działa waga hydrostatyczna?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
11
8. Jak wykonać pomiar gęstości za pomocą piknometru?
4.1.3. Ćwiczenia
Ćwiczenie 1
Porównaj
wskazania
areometrów:
Ballinga,
Trallesa,
Gay-Lussaca,
Baumé
i laktodensymetru.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) napełnić czyste i suche cylindry otrzymanymi od nauczyciela płynami,
2) wlewać po ściance cylindra, aby uniknąć ich spienienia,
3) opisać zachowanie się przyrządów,
4) zaproponować tabelę do zapisania obserwacji,
5) sporządzić zestawienie tabelaryczne wyników obserwacji.
Wyposażenie stanowiska pracy:
−
areometr Ballinga o zakresie skali 0÷15°Blg,
−
areometr Trallesa o zakresie skali 5÷10°Tr,
−
areometr Gay-Lussaca o zakresie gęstości 1,0÷1,2 g/dm
3
,
−
areometr Baumé o zakresie skali 0÷15°Bé,
−
laktodensymetr o zakresie skali 20÷40°Ld.
−
cylindry miarowe o pojemności 250 cm
3
– 5 sztuk,
−
roztwór alkoholu ok.10-procentowy,
−
roztwór cukru ok. 10-procentowy,
−
mleko,
−
woda destylowana.
Ćwiczenie 2
Oznacz ilość sacharozy w próbce za pomocą areometru.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przenieść ilościowo otrzymaną (ok.20g) próbkę sacharozy do zlewki i zadać 150cm
3
wody
destylowanej,
2) rozpuścić cukier mieszając zawartość bagietką,
3) przenieść roztwór ilościowo za pomocą lejka i bagietki do kolby miarowej o pojemności
250 cm
3
popłukując zlewkę wodą destylowaną,
4) uzupełnić zawartość kolby wodą destylowaną do kreski, wymieszać,
5) przelać zawartość kolby miarowej do cylindra, unikając spienienia,
6) zanurzyć ostrożnie areometr Ballinga,
7) odczytać wskazania areometru według górnej linii menisku,
8) odczytać temperaturę,
9) sprowadzić wskazania areometru Ballinga do temperatury normalnej (20°C), a następnie
obliczyć masę próbki sacharozy otrzymanej do analizy.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
12
Wyposażenie stanowiska pracy:
−
areometr Ballinga o zakresie skali 0÷15 Blg,
−
termometr o zakresie temperatury 0÷150°C,
−
zlewka o pojemności 250cm
3
,
−
lejek szklany,
−
bagietka szklana,
−
kolba miarowa o pojemności 250cm
3,
−
cylinder miarowy o pojemności 250cm
3
,
−
próbka sacharozy ok. 20g.
Ćwiczenie 3
Oznacz stężenie alkoholu za pomocą areometru Trallesa.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) popłukać pipetę o pojemności 20cm
3
niewielką ilością alkoholu otrzymanej próbki
i odmierzyć nią 20cm
3
badanej próbki,
2) przenieść próbkę do kolby miarowej o pojemności 200cm
3
,
3) uzupełnić kolbę miarową wodą destylowaną do kreski i wymieszać,
4) przelać roztwór do cylindra, po uprzednim popłukaniu go niewielką ilością tego roztworu,
5) zanurzyć w roztworze areometr Trallesa,
6) odczytać po ustaleniu się stanu równowagi wskazania areometru według dolnej linii
menisku,
7) odczytać temperaturę roztworu,
8) sprowadzić wskazania areometru do temperatury 15°C,
9) obliczyć stężenie alkoholu w próbce pierwotnej,
10) podać wynik w procentach objętościowych.
Wyposażenie stanowiska pracy:
−
areometr Trallesa o zakresie skali 5÷10°Tr,
−
termometr o zakresie skali 0÷150°C,
−
pipeta jednomiarowa o pojemności 20cm
3
,
−
kolba miarowa o pojemności 200cm
3
,
−
cylinder miarowy o pojemności 200cm
3
,
−
próbka alkoholu.
Ćwiczenie 4
Oznacz za pomocą piknometru gęstość syropu wysokosłodzonego.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) umieś w szafce wagi piknometr na 30 minut przed ważeniem w celu uzyskania
temperatury otoczenia,
2) zważyć piknometr z dokładnością 0,001g i zanotować jego masę jako m,
3) napełnić piknometr wodą destylowaną przy użyciu specjalnego lejka, nadmiar wody
zebrać bibułą,
4) zanotować temperaturę wody w piknometrze,
5) zważyć piknometr z wodą i zanotować jego masę jako m
1
,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
13
6) wylać wodę z piknometru i przepłukać go kilkakrotnie badaną cieczą,
7) napełnić piknometr badaną cieczą zwracając uwagę, by badany płyn nie zawierał
pęcherzyków powietrza,
8) doprowadzić ciecz do temperatury badania wody,
9) zważyć i zanotować masę piknometru z badaną cieczą jako m
2
,
10) obliczyć gęstość względną cieczy badanej ( w temperaturze badania) według wzoru:
11) umyć piknometr.
m
m
m
m
d
t
t
−
−
=
1
2
Wyposażenie stanowiska pracy:
–
piknometr,
–
waga analityczna,
–
lejek,
–
termometr,
–
ciecz do badania.
Ćwiczenie 5
Oznacz gęstość płynu przy użyciu wagi hydrostatycznej.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) sprawdzić punkt zerowy wagi wodą destylowaną w temperaturze 15°C i osuszyć pływak,
2) wlać ostrożnie wymieszany płyn do cylindra,
3) zebrać pianę bibułą,
4) zanurzyć pływak w badanym płynie do takiej głębokości do jakiej był zanurzony w wodzie
5) odczytać gęstość z położenia koników na odpowiednich podziałkach ramienia wagi.
Wyposażenie stanowiska pracy:
–
waga hydrostatyczna,
–
woda destylowana,
–
płyn do badania,
–
bibuła.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
14
4.1.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) zdefiniować pojęcia gęstości ciał?
2) dokonać pomiaru gęstości różnymi typami areometrów?
3) obliczyć stężenia roztworów na podstawie pomiarów gęstości/?
4) porównać wskazania areometrów różnych typów?
5) wyjaśnić działanie wagi hydrostatycznej?
6) określić zasadę działania piknometru?
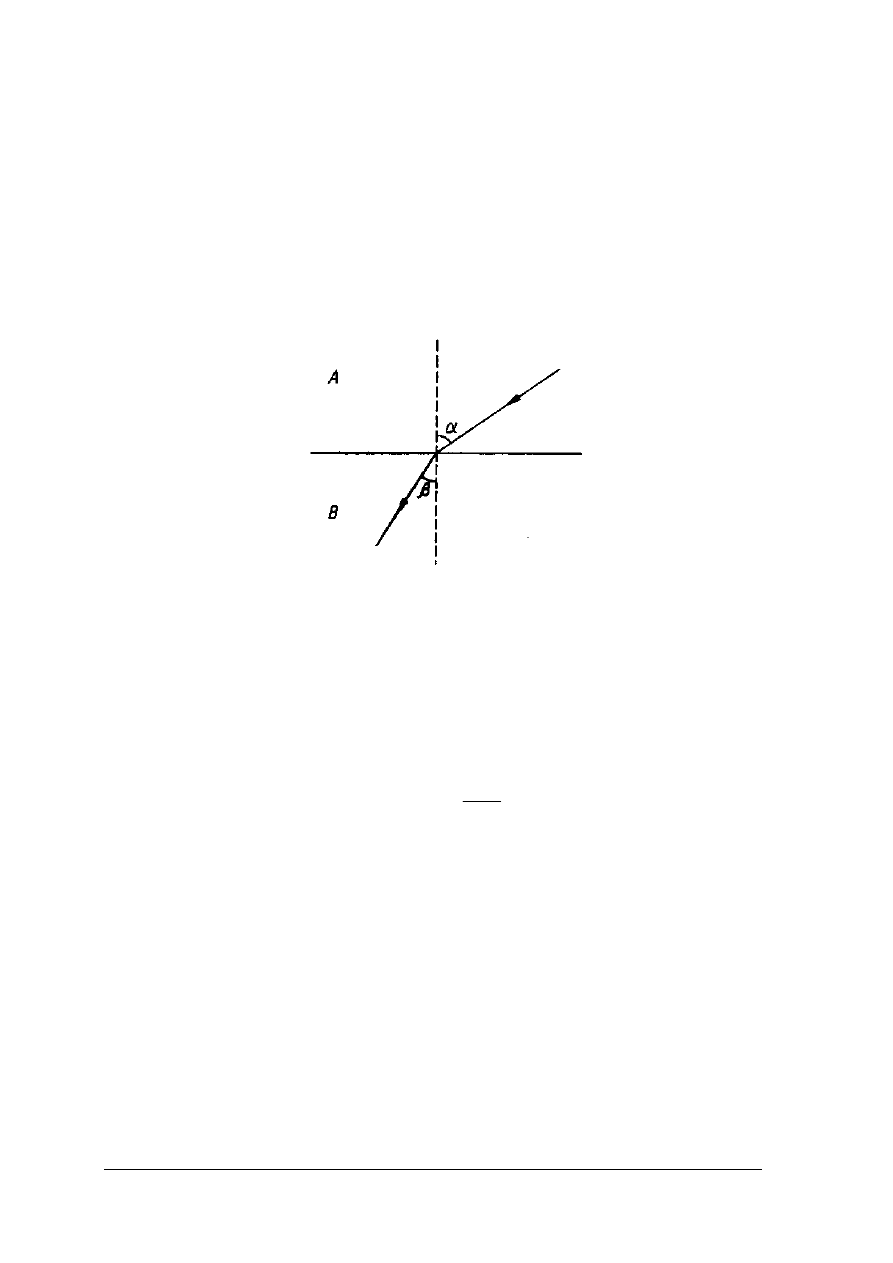
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
15
4.2. Pomiary
refraktometryczne
,polarymetryczne
i kolorymetryczne
4.2.1. Materiał nauczania
Pomiary refraktometryczne
Metody refraktometryczne opierają się na zjawisku załamania światła. Promień świetlny
przechodząc z ośrodka optycznie rzadszego (A) do ośrodka optycznie gęstszego (B) zostaje
częściowo odbity, a częściowo załamany (rys. 4)
Rys. 4. Załamanie promienia świetlnego na granicy dwóch ośrodków
α
- kąt padania,
β
- kąt załamania, A – środowisko optycznie rzadsze, B – środowisko optycznie gęstsze
[2, s.257]
Stosunek sinusa kąta padania (
α
) do sinusa kata załamania (
β
) nosi nazwę współczynnika
załamania światła, a jego wartość zależy od rodzaju padającego światła oraz od rodzaju
substancji i jej stężenia w badanym środowisku. Współczynnik refrakcji odnosi się do
temperatury 20°C oraz jednobarwnego światła żółtego które odpowiada linii widma
słonecznego, o długości fali 589,3 nm.
β
α
sin
sin
20
=
D
n
W analizie żywności częściej dokonuje się pomiarów stężeń roztworów cukru niż wartości
współczynników refrakcji. Wykorzystywany jest fakt, że z wartościami współczynników
refrakcji są skorelowane stężenia roztworów cukru. Wartość współczynnika refrakcji dla wody
wynosi n
D
20
= 1,333, natomiast dla kryształu cukru sacharozy n
D
20
= 1,57. Wynika stąd, że
różnym stężeniom roztworów wodnych sacharozy odpowiadają wartości liczbowe
współczynników refrakcji mieszczące się w zakresie 1,333 ÷ 1,57. Wodny roztwór cukru nie
wykazuje wartości współczynnika refrakcji, która byłaby sumą wartości współczynników
refrakcji obydwu składników wody i cukru lecz ich wypadkową. Do pomiarów stężeń
wodnych roztworów cukru oraz ich współczynników refrakcji służy refraktometr, którego
zasada działania oparta jest na kącie granicznym załamania.
Refraktometr Abbego składa się z dwóch pryzmatów szklanych o jednakowych
współczynnikach załamania, między którymi umieszcza się cienką warstewkę badanej cieczy.
Światło odbite od lusterka przechodzi przez pierwszy pryzmat i ulega rozproszeniu na jego
matowej powierzchni. Dalej przechodzi przez warstewkę badanej cieczy i przenika przez
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
16
pryzmat (jeżeli pada pod kątem mniejszym niż 90°C) oświetlając jasną część pola widzenia
w lunetce. Linia rozgraniczająca pole widzenia na jasną i ciemną część odpowiada kątowi
granicznego załamania. Pomiar sprowadza się do sprawdzenia, za pomocą dźwigni,
skrzyżowania nici pajęczych lub niekiedy trzech ruchomych kresek na granicy pól jasnego
i ciemnego oraz odczytania na skali wartości współczynnika refrakcji oraz stężenia cukru
w roztworze. Refraktometry wyposażone są w pryzmaty kompensacyjne, dzięki czemu
odczyty mogą być dokonywane przy użyciu światła dziennego lub sztucznego. Odczyty są
prawidłowe jeśli odczyty są wykonywane w temperaturze skalowania przyrządu (20°),
w przeciwnym wypadku należy je korygować odpowiednimi poprawkami. Dla skali cukrowej
poprawka wynosi 0,065 na każdy 1°C, a dla skali współczynnika refrakcji 0,00013 na 1°C
temperatury różnej od 20°C. Przy odczytach w temperaturach wyższych od 20°C poprawki się
dodaje, a przy niższych odejmuje.
Refraktometr zanurzeniowy
Składa się z jednego pryzmatu, podlegającego zanurzeniu w badanej cieczy. Zasada
działania jest podobna do refraktometru Abbego. Skalowane są w zakresie – 5 do 105
podziałek, a na noniuszu odczytuje się dziesiąte części. Odczytane wartości podziałek przelicza
się na wartości współczynników refrakcji lub procentową zawartość składników w badanych
środowiskach. W takim przypadku należy korzystać z tabel przeliczeniowych.
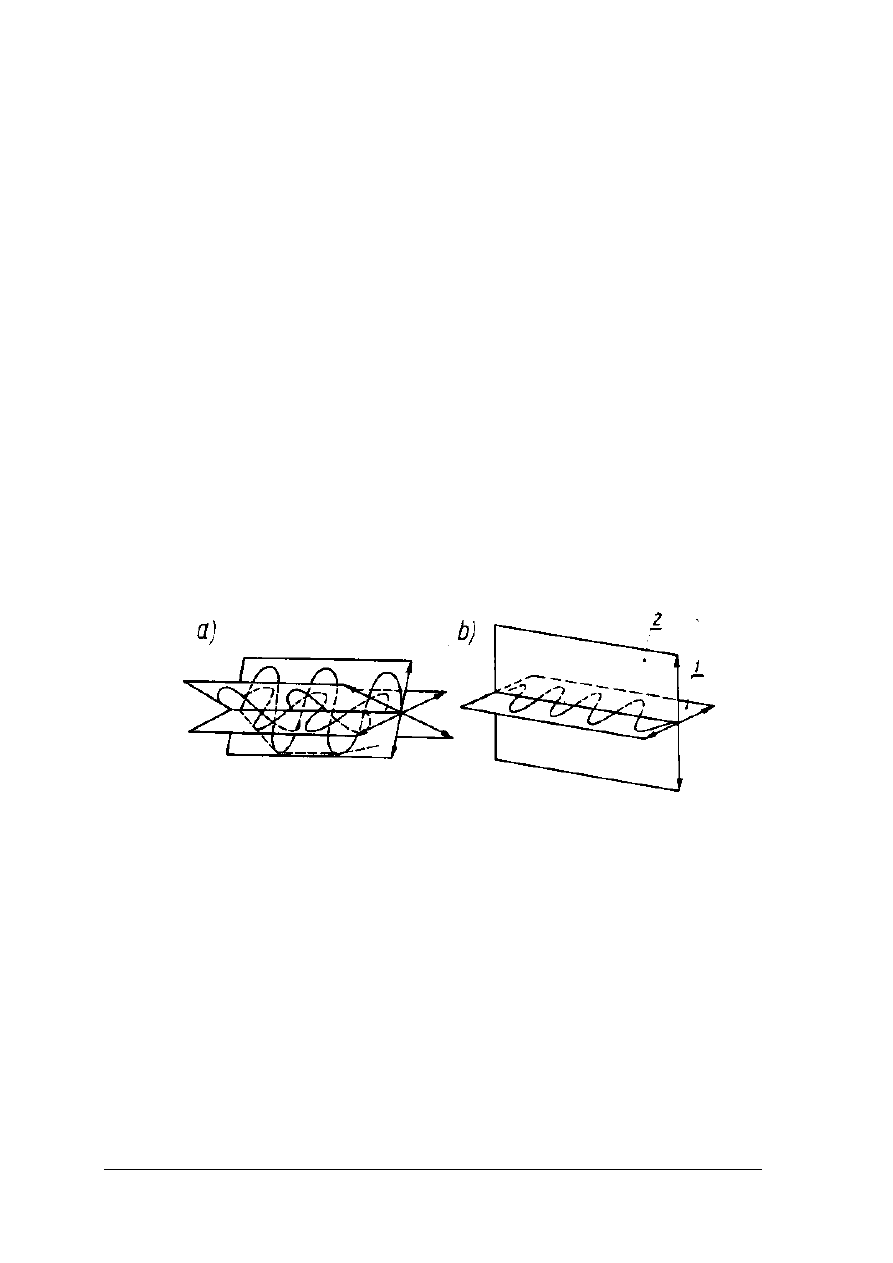
Metody polarymetryczne
W metodach polarymetrycznych wykorzystywana jest zdolność cukrowców do skręcania
płaszczyzny światła spolaryzowanego. Drgania światła spolaryzowanego zachodzą tylko
w jednej płaszczyźnie (rys. 5)
Rys. 5. Zasada rozchodzenia się światła [2,s.262]
a) zwykłego, b) spolaryzowanego 1- płaszczyzna drgań, 2 – płaszczyzna polaryzacji
Zdolność skręcania płaszczyzny światła spolaryzowanego mają związki zawierające tzw.
atomy asymetryczne. Wielkością charakterystyczną dla związków optycznie czynnych jest
skręcalność właściwa. Jest to kąt o jaki skręcona zostaje płaszczyzna światła spolaryzowanego
przez roztwór substancji optycznie czynnej, o stężeniu 1 g w 1cm
3
, przy grubości warstwy
1dm, w określonej temperaturze i przy określonej długości fali światła. Skręcalność odnosi się
do linii D widma słonecznego, o długości fali 589,3 nm. Skręcenie może być w prawą stronę,
czyli dodatnie i w lewą czyli ujemne. Pomiary polarymetryczne wykonywane są
w urządzeniach zwanych polarymetrami.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
17
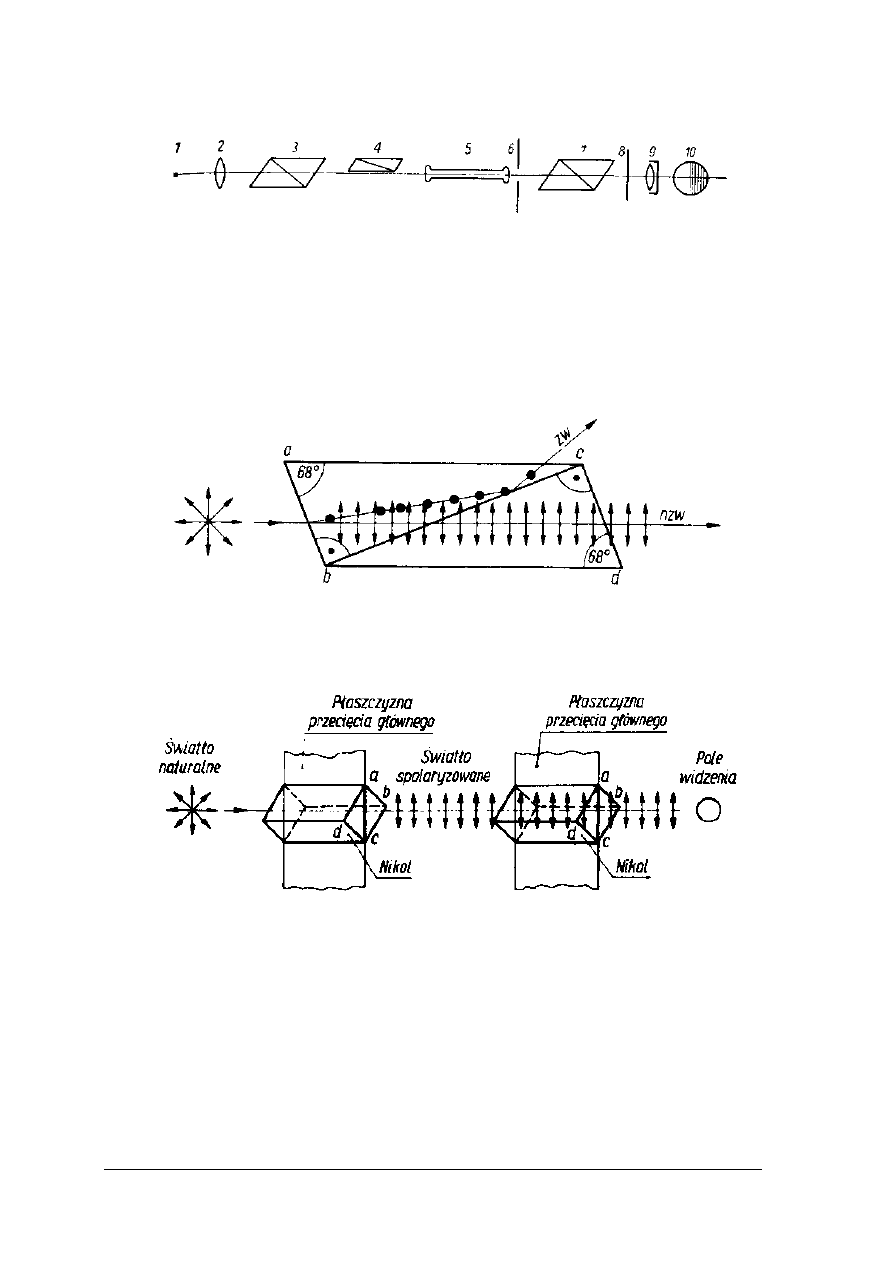
Rys. 6. Schemat budowy polarymetru [2, s.265]
W polarymetrach wykorzystywane jest jednobarwne światło monochromatyczne. Światło
podlega polaryzacji w pryzmacie Nicola. Pryzmat Nicola to specjalnie przycięty szpat islandzki
sklejony balsamem kanadyjskim. Promień świetlny, padając na pryzmat Nicola ulega
rozszczepieniu na dwie frakcje: promień zwyczajny i nadzwyczajny. Pierwszy ulega
całkowitemu wewnętrznemu odbiciu, a drugi przechodzi przez pryzmat jako promień
spolaryzowany (rys. 7).
Rys. 7. Podział promienia świetlnego na zwyczajny (zw) i nadzwyczajny (nzw) w pryzmatach Nicola
[2, s. 265]
Rys. 8. Równoległe ustawienie nicoli [2, s. 266]
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
18
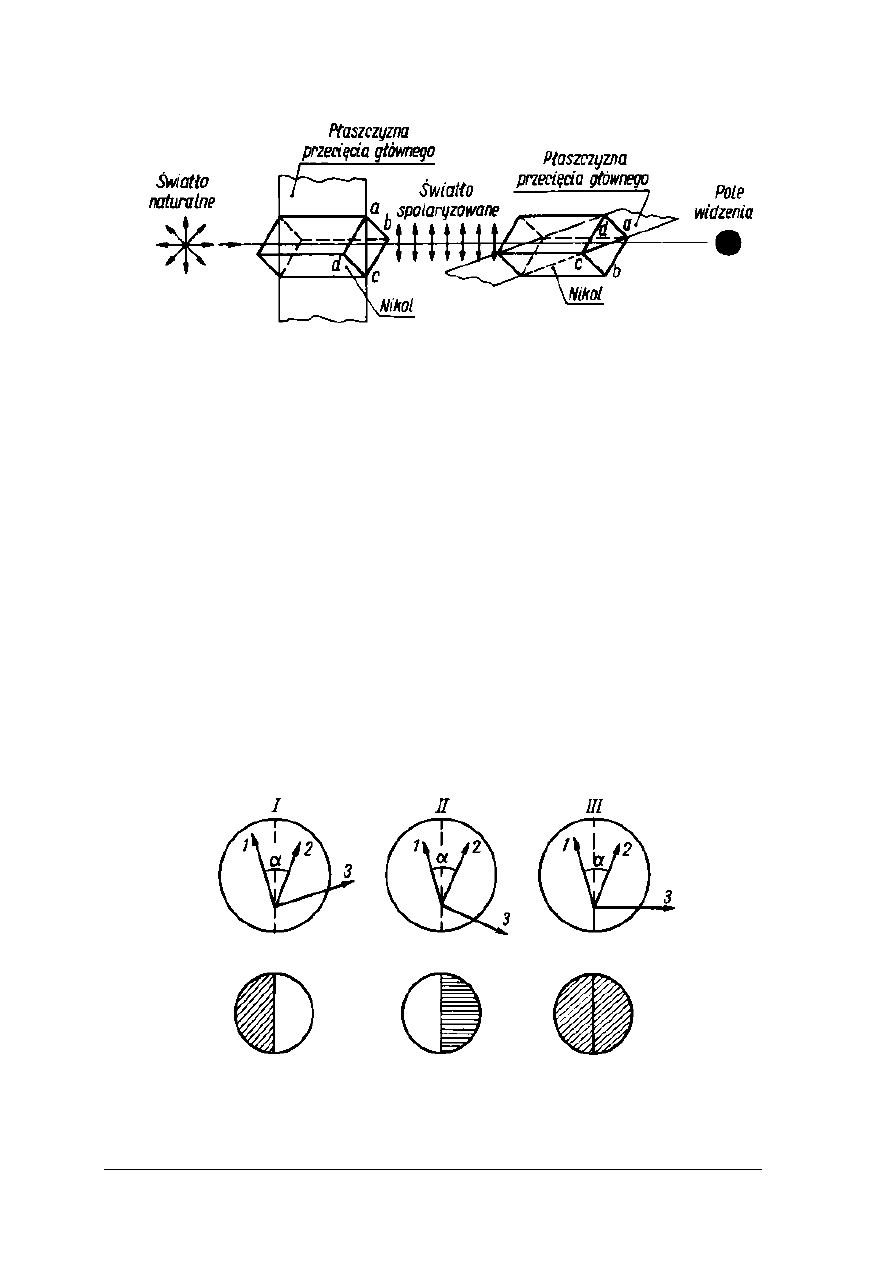
Rys. 9. Nikole skrzyżowane [2, s. 266]
Polarymetr składa się z dwóch nicoli (dwa pryzmaty Nicola). Jeden jest nieruchomy,
a drugi obraca się wokół własnej osi w analizatorze. W zależności od kierunku ustawienia
płaszczyzn dwóch nikoli względem siebie w okularze otrzymuje się ciemne (rys. 9) lub
rozjaśnione pole widzenia (rys. 8). Skrzyżowanie płaszczyzn nikoli względem siebie powoduje
całkowite wygaszenie. Pole widzenia jest ciemne, a pole jasne pojawia się tylko w przypadku
pokrycia się płaszczyzn obydwu nikoli. Jeżeli pole widzenia w okularze zostanie ustawione na
pewien stan oświetlenia lub zaciemnienia, a pomiędzy nikole wstawi się roztwór substancji
optycznie czynnej, nastąpi skręcenie światła spolaryzowanego. W celu przywrócenia
poprzedniego stanu oświetlenia pola widzenia potrzebna jest zmiana położenia nikola
analizatora. Kąt o jaki zostanie obrócony analizator odpowiada kątowi skręcenia płaszczyzny
polaryzacji.
Zasada półcienia wykorzystywana jest w polarymetrach nowszych typów. Obok nikola
polaryzatora umieszcza się mały polaryzator dodatkowy, którego płaszczyzna jest skręcona
o 10 ÷ 20° w stosunku do płaszczyzny nikola polaryzatora. Pole widzenia zostaje podzielone
na część jasna i ciemną. Kąt skręcenia płaszczyzny światła spolaryzowanego przez badany
roztwór substancji czynnej należy odczytać doprowadzając nikol do położenia, w którym obie
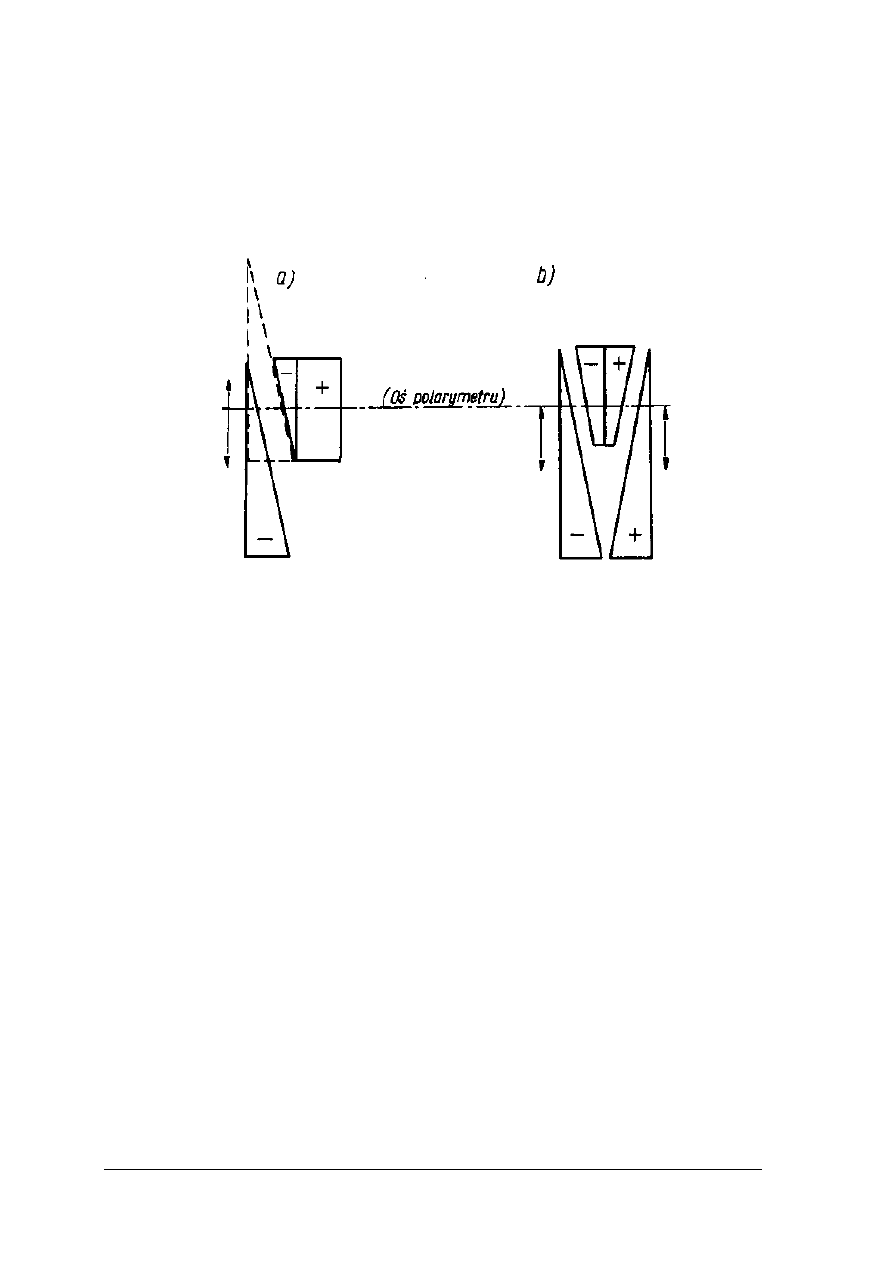
połówki pola widzenia są jednakowo oświetlone. Sytuację powyższą obrazuje rysunek 10.
Rys. 10. Powstawanie półcieni w polarymetrze Lippicha [2, s. 267]
1- oś krystalizacji nikola polaryzatora, 2 – oś krystalizacji małego nikola, 3 – oś
krystalizacji nikola analizatora.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
19
W polarymetrach klasycznych odczyt dokonywany jest w stopniach kątowych
Sacharymetry, które posiadają kompensację klinową (rys. 11) są wyskalowane w stopniach
Ventzkego. Skala Ventzkego wywodzi się stąd, że roztwór sacharozy, który zawiera 26 g tego
cukru w 100 cm
3
, przy grubości warstwy roztworu równej 2 dm wykazuje skręcenie
płaszczyzny polaryzacji o 100° Ventzkego.
Rys. 11. Kompensacja klinowa stosowana w sacharymetrach [2, s. 268]
a) kompensacja pojedyńcza, b) kompensacja podwójna
Metody polarymetryczne oznaczania zawartości cukrowców mogą być stosowne tylko
w przypadku rozpuszczenia ich w wodzie oraz gdy dają one roztwory klarowne i możliwie
bezbarwne.
Oznaczenia kolorymetryczne
Metody kolorymetryczne oparte są na działaniu promieniowania elektromagnetycznego na
badane substancje a głównie na absorpcji promieniowania w zakresie widzialnego światła
białego o zakresie długości fali 380 ÷ 780 nm. Światło padając na jakiś układ absorpcyjny
zostaje częściowo rozproszone, częściowo pochłonięte i częściowo przepuszczone. Zdolność
absorpcji pewnych frakcji promieniowania elektromagnetycznego przez różne ciała zależy od
ich budowy cząsteczkowej. Strumień światła I
0
padając ulega częściowemu odbiciu (I
odb
),
częściowemu rozproszeniu (I
r
) i częściowej absorpcji (I
a
) i tylko w części przechodzi przez ten
ośrodek (I
t
) jak pokazuje rys. 12
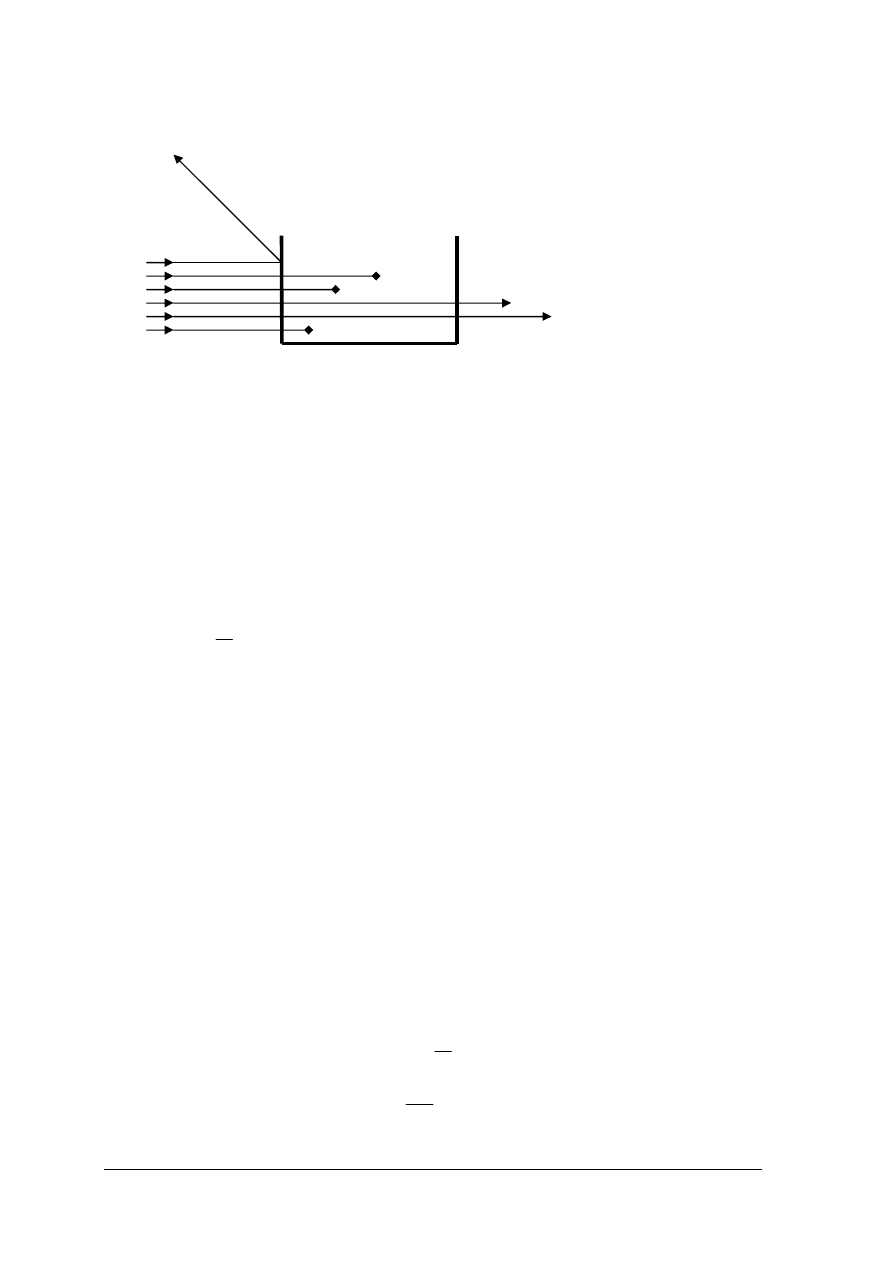
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
20
Rys. 12. Podział strumienia światła na frakcje przy przechodzeniu przez badany roztwór [2, s. 227]
I
0
– natężenie światła padającego, (I
odb
) – natężenie światła odbitego, I
a
– natężenie światła podlegającego
absorpcji, I
r
– natężenia światła polaryzującego rozproszenie, I
t
– natężenie światła przechodzącego przez
roztwór
Zapisać to można wzorem:
I
0
= I
odb
+ I
r
+ I
a
+I
t
Oznaczenia kolorymetryczne prowadzone są na klarownych roztworach substancji barwnych
oraz przy użyciu tych samych kuwet. wartości natężeń promieniowania odbitego
i rozproszonego można pominąć i wówczas:
I
0
= I
a
+ I
t
Stosunek
T
I
I
t
=
0
nosi nazwę przepuszczalności lub transmisji. Informuje on jaka część
promieniowania świetlnego zostaje przepuszczona przez roztwór. Przepuszczalność wyrażana
jest w procentach. Innym wskaźnikiem jest absorpcja, której wyrażenie liczbowe stanowi
absorbancja.
W praktyce do przeliczania wartości liczbowych służą tabele przeliczeniowe określające
zależności liczbowe pomiędzy przepuszczalnością (transmisją) roztworu (T) i absorbancją (A).
Do oznaczania ilościowego składników barwnych zawartych w badanych roztworach można
stosować metodę algebraiczną i graficzną, ale tylko w przypadku, gdy spełnione jest prawo
Lamberta - Beera, mówiące, że absorpcja światła przez roztwór jest wprost proporcjonalna do
grubości warstwy roztworu i jego stężenia, co można wyrazić równaniem:
A = k · l · c
Gdzie: k- współczynnik absorpcji, którego wartość jest stała dla danej substancji
i rozpuszczalnika, przy określonej długości fali światła,
l- grubość warstwy roztworu,
c- stężenie roztworu.
W metodzie algebraicznej stężenie roztworu oblicza się ze wzoru będącego
przekształceniem równania obrazującego prawo Lamberta-Beera:
l
k
A
c
c
l
k
I
I
A
t
⋅
=
⋅
⋅
=
=
0
log
I
0
I
odb.
I
t
I
a
I
r
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
21
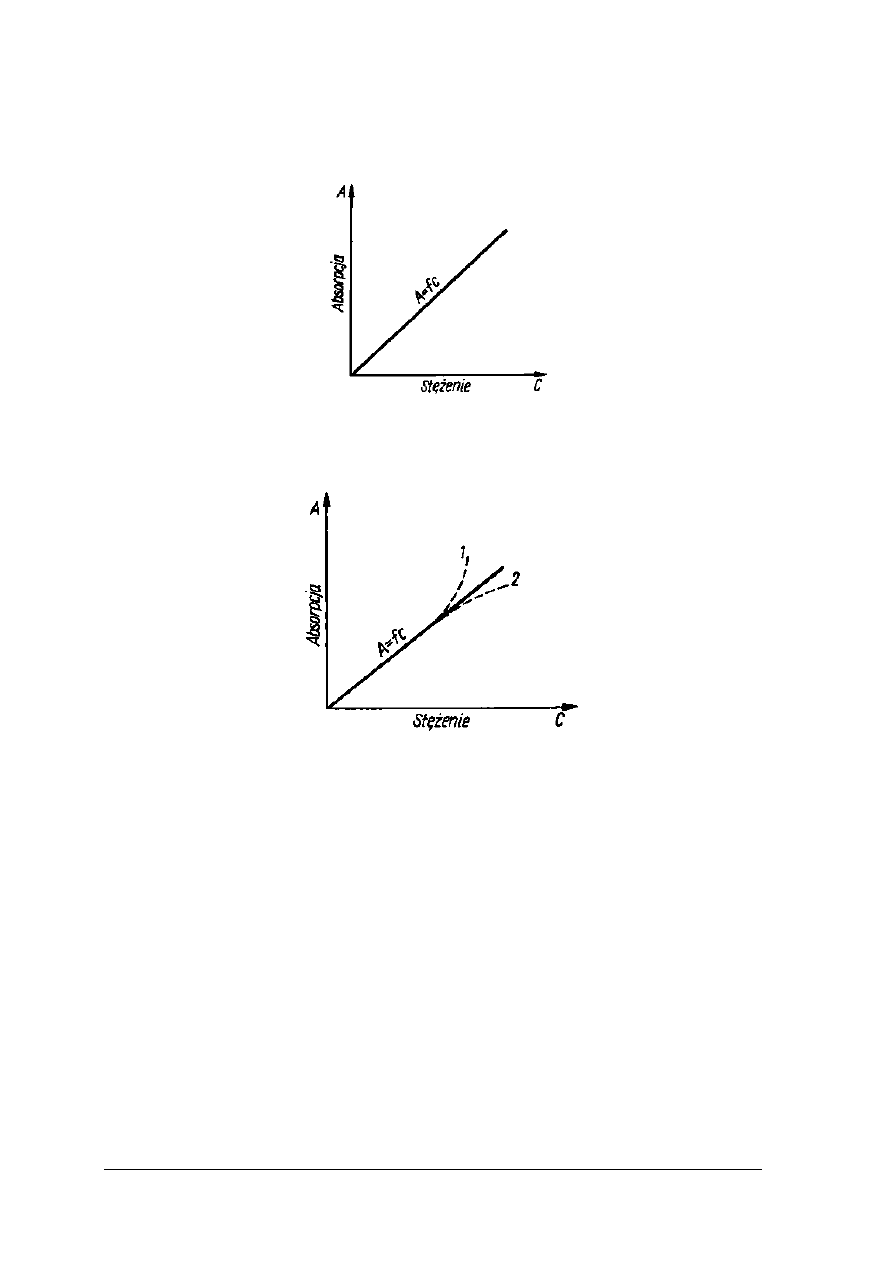
W metodzie graficznej należy wykreślić krzywą wzorcową w układzie współrzędnych,
nanosząc na oś odciętych (x) wartości liczbowe stężenia, a na oś rzędnych (y) wartości
liczbowe absorbancji. Zależności przedstawia rys. 13.
Rys. 13. Wykres zależności absorpcji roztworu od jego stężenia [2, s. 231]
Rys. 14. Wykres zależności absorpcji roztworu od jego stężenia 1,2 – odchylenia od prawa Lamberta-
Beera [2,s.231]
Odchylenia od prawa Lamberta-Beera są wynikiem zmian zachodzących w roztworach np.
asocjacja cząsteczek, polimeryzacja.
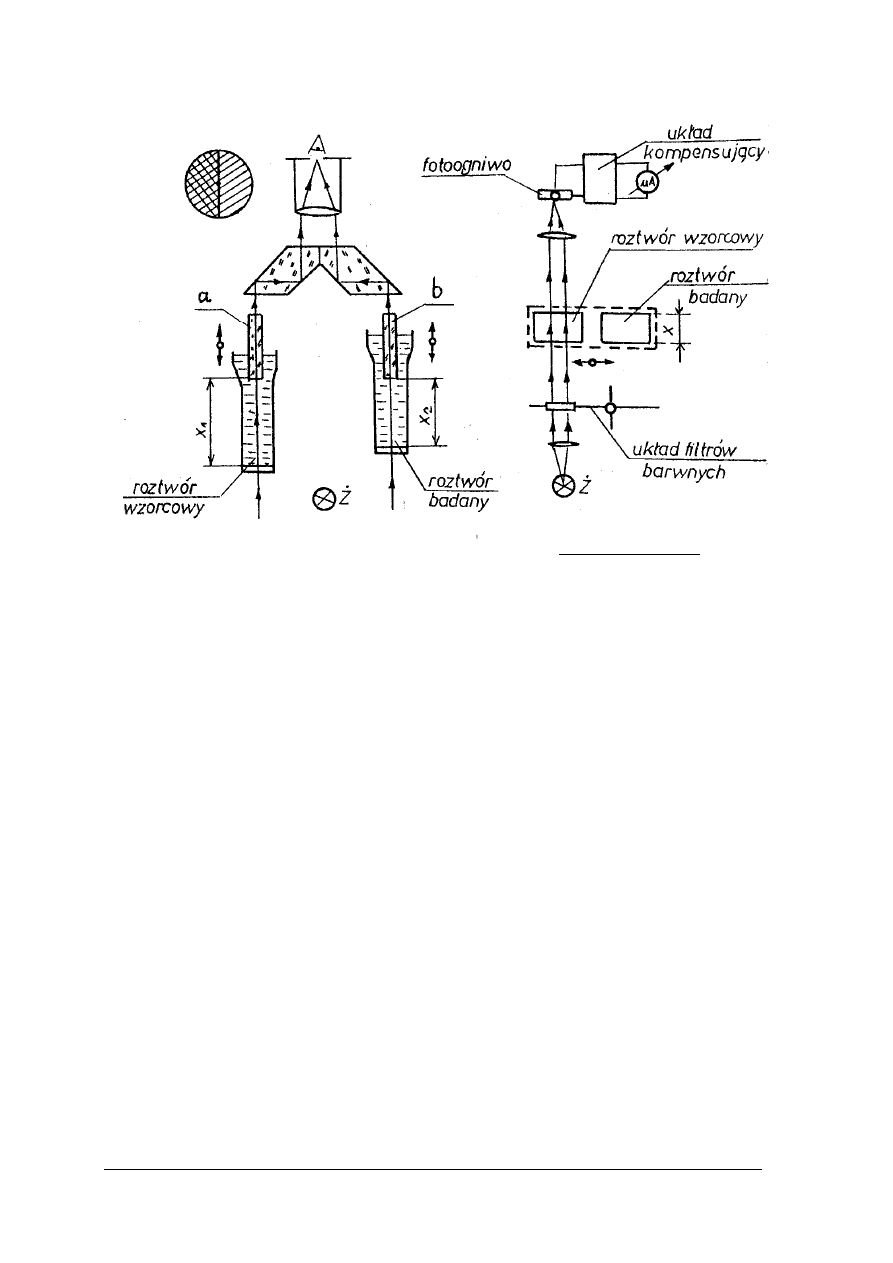
Stężenia roztworów związków barwnych można określać metodą porównania
z roztworami barwnymi o znanych stężeniach. Porównywania barwy roztworów badanych
z wzorcowymi można dokonywać w cylindrach Hehnera rys. 14 lub w kolorymetrze Dubosqa
rys. 15.
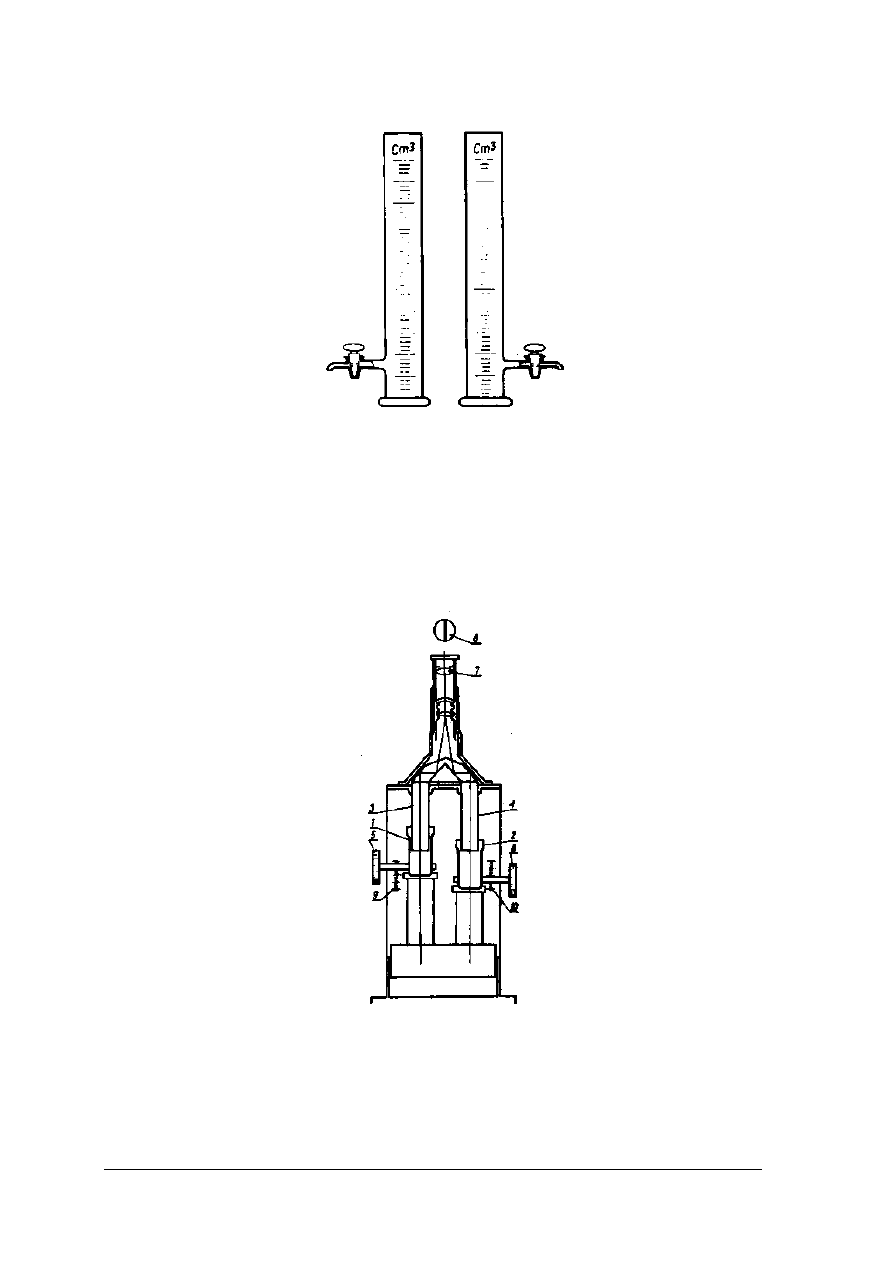
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
22
Rys. 15. Cylindry Hehnera [2, s. 239]
Cylindry Hehnera posiadają kurki spustowe oraz wyskalowane podziałki. Dokonując
pomiaru należy wlać do cylindrów jednakowe ilości roztworów: badanego i wzorcowego.
Z roztworu o silniejszym zabarwieniu należy odlewać go przez kran spustowy do momentu
zrównania intensywności zabarwień w obu cylindrach. Po tej czynności należy odczytać na
wyskalowanych podziałkach równoważne grubości warstw obydwu roztworów.
Rys. 16. Kolorymetr Dubosqa [2, s. 239]
1,2 - naczynia szklane, 3,4 – pręty zanurzeniowe, 5, 6 – śruby, 7 – okular, 8 – pole widzenia, 9,10, - skale
Kolorymetr Dubosqa składa się z dwóch cylindrów o płaskich dnach, które wykonane są
z przezroczystego szkła. Cylindry są umieszczone w ruchomych metalowych uchwytach.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
23
Przekładnia zębata pozwala na przesuwanie cylindrów w kierunku pionowym. W tym czasie
pręty szklane zanurzają się w cylindrach na odpowiednią głębokość. Po osiągnięciu położenia,
przy którym zabarwienie roztworów jest jednakowe należy odczytać na skalach równoważne
grubości warstw roztworów badanego i wzorcowego.
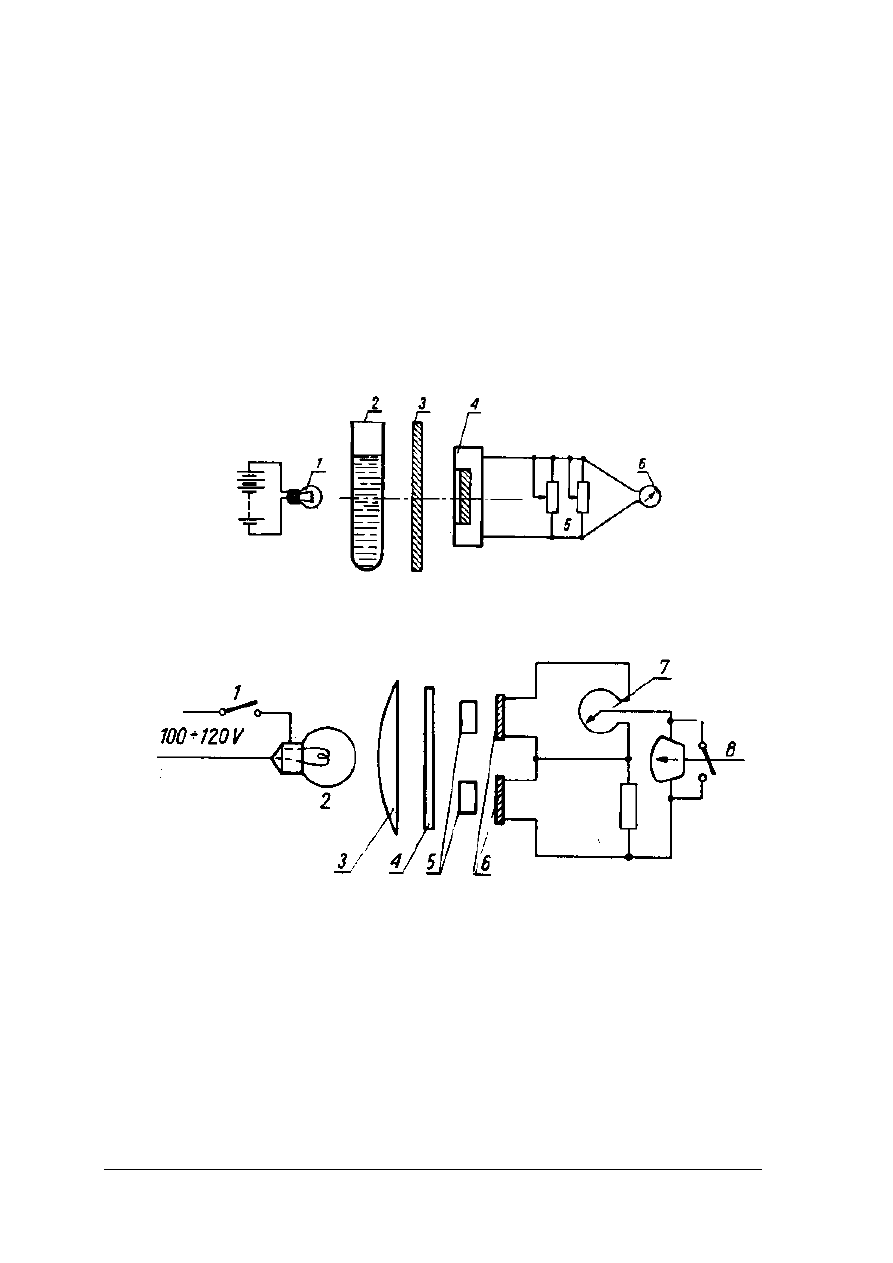
Urządzeniami, w których absorpcję światła mierzy się za pomocą fotokomórek,
a natężenie fotoprądu za pomocą wyskalowanego w wartościach liczbowych absorbancji
galwanometru jest elektrofotokolorymetr. Elektrofotokolorymetr jednokomórkowy posiada
jedną kuwetę, którą należy napełnić tzw. ślepą próbą lub rozpuszczalnikiem, a drugą
identyczną roztworem badanym. Po nastawieniu odpowiedniego filtru kuwetę z próbą ślepą
należy wstawić do gniazda elektrofotokolorymetru. Wskazania galwanometru należy ustawić
na 100% transmisji przez pokrętła sprzężone z przesłoną lub opornikiem. Następnie po wyjęciu
ślepej próby wstawić należy w to samo miejsce kuwetę z badanym roztworem i odczytać na
skali galwanometru wskazania absorbancji lub transmisji.
Rys. 17. Schemat elektrofotokolorymetru jednokomórkowego [2, s. 242]
1 – żarówka, 2 – probówka z badanym roztworem, 3 – filtr, 4 – fotokomórka, 5 – oporniki, 6 –
galwanometr
Rys. 18. Schemat elektrofotokolorymetru dwukomórkowego [2, s. 243]
1 – źródło prądu, 2 – żarówka, 3 – soczewka, 4 – filtr, 5 – kuwety, 6 – fotokomórki, 7 – opornik, 8 –
galwanometr
W elektrokolorymetrze dwukomorowym kuwety wstawia się jednocześnie i sprowadza
ruchomą skalę absorbancji do takiego położenia, przy którym galwanometr nie wskazuje
przepływu prądu.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
24
Rys. 19. Kolorymetr fotoelektryczny SPEKOL 1300 [http://pl.wikipedia.org/]
Kolorymetr fotoelektryczny wykorzystywany w laboratoriach chemicznych działa
następująco: Wiązka światła białego z żarówki, pada na odbiciową siatkę dyfrakcyjną, która
rozszczepia światło na różne długości fal (barwy). Siatka jest sprzężona z bardzo precyzyjnym
mechanizmem
pozwalającym obracać ją tak, aby dokładnie skierować promień określonej
barwy do układu optycznego, a następnie na badane ciało (najczęściej jest to, umieszczona
w specjalnej kuwecie szklanej lub kwarcowej, ciecz). Po przejściu przez badaną substancję
światło pada na fotoogniwo, w którym powstaje napięcie, zależne od natężenia padającego
światła. Do pomiaru ekstynkcji nie wystarcza jeden pomiar, gdyż nie jest znane natężenie
wiązki padającej na substancję (które jest zresztą różne dla różnych długości fal i zależy od
charakterystyki oświetleniowej danej żarówki). Pomiarów przy pomocy SPEKOLA należy
dokonywać przy wykorzystaniu substancji wzorcowej, którą najczęściej jest woda
destylowana. Najpierw dla wody destylowanej ustawiane jest wskazanie przyrządu na zero
ekstynkcji (czyli 100% transmisji), a następnie dla tych samych ustawień umieszcza się na
drodze wiązki świetlnej zamiast wody badaną substancję. Teraz już można bezpośrednio ze
skali odczytać ekstynkcję bądź transmisję. Stosując powyższą metodę eliminujemy
pochłanianie światła przez sam rozpuszczalnik (wodę).
Aby z kolei określić stężenie danego roztworu, należy najpierw sporządzić zależność
ekstynkcji roztworu od jego stężenia. W tym celu sporządza się roztwory wzorcowe o znanych
stężeniachc , mierzy ich ekstynkcje E dla wybranej długości fali (przeważnie dla tej, przy której
absorpcja jest maksymalna) i sporządza wykres zależności E(c). Teraz mierząc ekstynkcję
roztworu o nieznanym stężeniu można w prosty sposób odczytać z wykresu jej stężenie.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
25
Rys. 20. Schemat działania fotokolorymetru SPECOL 1300 [http://pl.wikipedia.org/]
4.2.2.Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń
1. Jakie zjawiska fizyczne wykorzystywane są w pomiarach refraktometrycznych?
2. Jak działa refraktometr Abbego?
3. Kiedy odczyty refraktometryczne są prawidłowe?
4. Jak działa refraktometr zanurzeniowy Zeissa?
5. Jakie roztwory sacharozy mogą być badane za pomocą refraktometrów?
6. Jaka jest zasada działania polarymetrów?
7. Na czym polega zasada półcienia wykorzystywana w nowszych polarymetrach?
8. Jakie roztwory mogą być oznaczane polarymetrycznie?
9. Jakich przyrządów należy użyć do oznaczeń kolorymetrycznych i jak z nich korzystać?
4.2.3. Ćwiczenia
Ćwiczenie 1
Oznacz zawartość wody w galaretkach.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować refraktometr: nastawić na temperaturę 20°C, sprawdzić wartość
współczynnika załamania światła dla wody destylowanej, w przypadku odchyleń skalę
odpowiednio poprawić,
2) wytarować zlewkę,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
26
3) odważyć 25 g galaretki,
4) dodać 20 cm
3
wody destylowanej i podgrzewać stale mieszając do całkowitego
rozpuszczenia produktu,
5) wystudzić do temperatury pokojowej, zważyć i dodać wody destylowanej tyle, aby ogólna
masa zawartości zlewki wynosiła 50 g (dokładność ważenia 0,01g),
6) zawartość zlewki dokładnie wymieszać,
7) przenieść kroplę roztworu na pryzmat, odczekać 30s, docisnąć obydwa pryzmaty,
8) wyregulować okulary na ostrość pola widzenia,
9) pryzmat refraktometru nastawić tak, aby trzy przerywane kreski (w refraktometrze Zeissa)
stanowiły linię graniczną oddzielającą jasną część pola widzenia od ciemnej
(w refraktometrze Abbego linia graniczna powinna przechodzić dokładnie przez punkt
skrzyżowania)
10) odczytać na skali procentowej zawartość ekstraktu badanego roztworu,
11) wykonać obliczenie wg wzoru: x = 100 – 2E, w którym x – procentowa zawartość wody,
E – odczytana zawartość ekstraktu w %, odejmujemy podwójną odczytaną zawartość
ekstraktu 2E, ponieważ galaretka została rozcieńczona dwukrotnie wodą.
Wyposażenie stanowiska pracy
−
waga techniczna,
−
refraktometr z instrukcją obsługi,
−
ultratermostat Höpplera (warunkowo),
−
zlewka o pojemności 100cm
3
,
−
bagietka,
−
trójnóg,
−
palnik,
−
próbka galaretki
Ćwiczenie 2
Oznacz skręcalność miodu za pomocą polarymetru.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) użyć do oznaczenia 20-procentowego roztworu podstawowego miodu,
2) rozpuścić 20g miodu w ciepłej wodzie destylowanej,
3) przenieść ilościowo po oziębieniu do kolby miarowej o pojemności 100cm
3
i uzupełnić
wodą destylowaną do kreski,
4) wlać 25cm
3
roztworu podstawowego do kolby o pojemności 50cm
3
dodać 2cm
3
roztworu
zasadowego octanu ołowiu, 4cm
3
nasyconego roztworu fosforanu dwusodowego i 1-2
krople amoniaku,
5) wymieszać zawartość kolbki i dopełnić do kreski wodą destylowaną,
6) wymieszać zawartość kolby i po 30 minutach przesączyć,
7) określić w przesączu polaryzację, w rurce 200mm,
8) odczytać wynik,
9) pomnożyć wynik przez 2, ponieważ otrzymasz w ten sposób skręcalność roztworu
20-procentowego.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
27
Wyposażenie stanowiska pracy:
–
kolba miarowa o pojemności 100cm
3
,
–
kolba miarowa o pojemności 50cm
3
,
–
20-procentowy roztwór podstawowy miodu ,
–
zasadowy octan ołowiu,
–
fosforan dwusodowy roztwór nasycony,
–
amoniak,
–
polarymetr z instrukcją obsługi.
Ćwiczenie 3
Oznacz zawartość żelaza metodą kolorymetryczną.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
przygotować wzorcowy roztwór żelaza zawierający 0,1mg Fe w 1cm
3
roztworu:
1) odważyć na wadze analitycznej 0,8634g ałunu żelazowo-amonowego i zadać go 10cm
3
roztworu kwasu siarkowego,
2) przenieść zawartość naczyńka do kolby miarowej o pojemności 200cm
3
, popłukując
naczyńko, lejek i bagietkę wodą destylowaną,
3) uzupełnić zawartość kolby wodą destylowaną i dokładnie wymieszać,
4) pobrać pipetą 20cm
3
przygotowanego roztworu, przenieść do kolby miarowej
o pojemności 100 cm
3
, uzupełnić do kreski wodą destylowaną i dokładnie wymieszać
(roztwór zawiera 0,1g Fe w 1cm
3
),
sporządzić krzywą wzorcową:
5) odmierzyć do sześciu kolb miarowych o pojemności 100 cm
3
kolejno 1, 2, 3, 4, 5 i 6cm
3
wzorcowego roztworu żelaza,
6) dodać po 2 cm
3
roztworu kwasu solnego o stężeniu 2 mol/dm
3
, po 2cm
3
wody utlenionej
i po 5cm
3
5 % tiocyjanianu potasu,
7) uzupełnić zawartość kolb wodą destylowaną do 100 cm
3
i dokładnie wymieszać,
8) oznaczyć absorbancję każdego z roztworów w fotokolorymetrze przy filtrze
niebieskozielonym, nastawiając 100% przepuszczalności na wodę destylowaną,
9) sporządzić wykres krzywej wzorcowej w układzie współrzędnych nanosząc wartości
liczbowe absorbancji na oś rzędnych (y), a na oś odciętych (x) stężenie żelaza
w miligramach na 100cm
3
kolorymetrowanych płynów,
wykonać oznaczenia:
10) odmierzyć pipetą 10cm
3
badanego roztworu żelaza do kolby miarowej o pojemności
100cm
3
,
11) dodać 2cm
3
roztworu kwasu solnego o stężeniu 2mol/dm
3
, 2cm
3
wody utlenionej i 5cm
3
5-procentowego roztworu rodanku potasu (tiocyjanian potasu),
12) uzupełnić do kreski zawartość kolby wodą destylowaną i dokładnie wymieszać,
13) użyć płyn do oznaczenia absorbancji,
14) nastawić elektrofotokolorymetr na 100% przepuszczalności przy filtrze niebieskozielonym
na wodę destylowaną,
15) oznaczyć absorbancję zabarwionego płynu, przygotowanego z badanego roztworu żelaza
16) obliczyć wyniki posługując się krzywą wzorcową.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
28
Wyposażenie stanowiska pracy:
−
elektrofotokolorymetr z instrukcją obsługi,
−
kolby miarowe o pojemności 100 i 200cm
3
−
pipety jednomiarowe o pojemności 2, 5, 10 i 20cm
3
,
−
pipeta wielomiarowa o pojemności 10cm
3
,
−
naczyńko wagowe,
−
bagietka szklana,
−
waga analityczna,
−
badany roztwór żelaza o zawartości 5÷20mg w 1000cm
3
,
−
wzorcowy roztwór żelaza o zawartości 0,1mg Fe w 1cm
3
,
−
kwas solny, roztwór o stężeniu 2mol/ dm
3
−
kwas siarkowy, roztwór rozcieńczony w stosunku objętościowym 1:1
−
rodanek potasu, roztwór 5-procentowy,
−
nadtlenek wodoru, roztwór 3-procentowy,
−
ałun żelazowo-amonowy, krystaliczny.
4.2.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) zdefiniować współczynnik załamania światła, i kąt graniczny?
2) zdefiniować prawo Lamberta-Beera,?
3) wykonać oznaczenia używając refraktometru Abbego?
4) wyjaśnić budowę i zasadę działania refraktometrów: Abbego,
zanurzeniowego i ręcznego?
5) wyjaśnić zasadę działania polarymetru?
6) określić wielkość charakterystyczną dla związków optycznie czynnych?
7) posłużyć się kolorymetrem Dubosqa?
8) wykonać oznaczenia w cylindrach Hehnera?
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
29
4.3. Oznaczenia potencjometryczne i chromatograficzne
4.3.1. Materiał nauczania
Potencjometria
Analiza potencjometryczna polega na określeniu stężenia jonów w roztworze na
podstawie pomiaru potencjału elektrody zanurzonej do roztworu.
W laboratoriach analitycznych bardzo często oznaczana jest kwasowość artykułów
spożywczych. Z chemicznego punktu widzenia kwasowość można podzielić na potencjalną
czyli bierną i na aktywną zależną od stężenia lub aktywności jonów wodorowych H
+
lub
hydroniowych H
3
O
+
. Kwasowość potencjalna informuje o zawartości kwasu lub kwasów
w środowisku. Przy oznaczaniu kwasowości potencjalnej nie można oznaczyć absolutnej
zawartości poszczególnych kwasów występujących w surowcach i produktach roślinnych lub
zwierzęcych. Oznaczenie sprowadza się więc do umownego wyrażania kwasowości
środowiska w przeliczeniu na jeden z kwasów tam występujących, reguły w największych
ilościach. Z tego względu podając wynik oznaczenia kwasowości podajemy, że jest to
kwasowość w przeliczeniu na kwas mlekowy, jabłkowy, cytrynowy.
Kwasowość aktywna podaje faktyczne stężenie jonów wodorowych lub hydroniowych.
Uzależniona jest ona od rodzaju kwasu zawartego w roztworze. Kwasowość aktywną wyraża
się jako wykładnik wodorowy (pH), którego wartość liczbowa jest równa ujemnemu
logarytmowi dziesiętnemu ze stężenia jonów wodorowych. W analizie spożywczej wyróżnia
się także kwasowość lotną, kwasowość związaną oraz kwasowość całkowitą, która jest sumą
kwasowości potencjalnej i związanej.
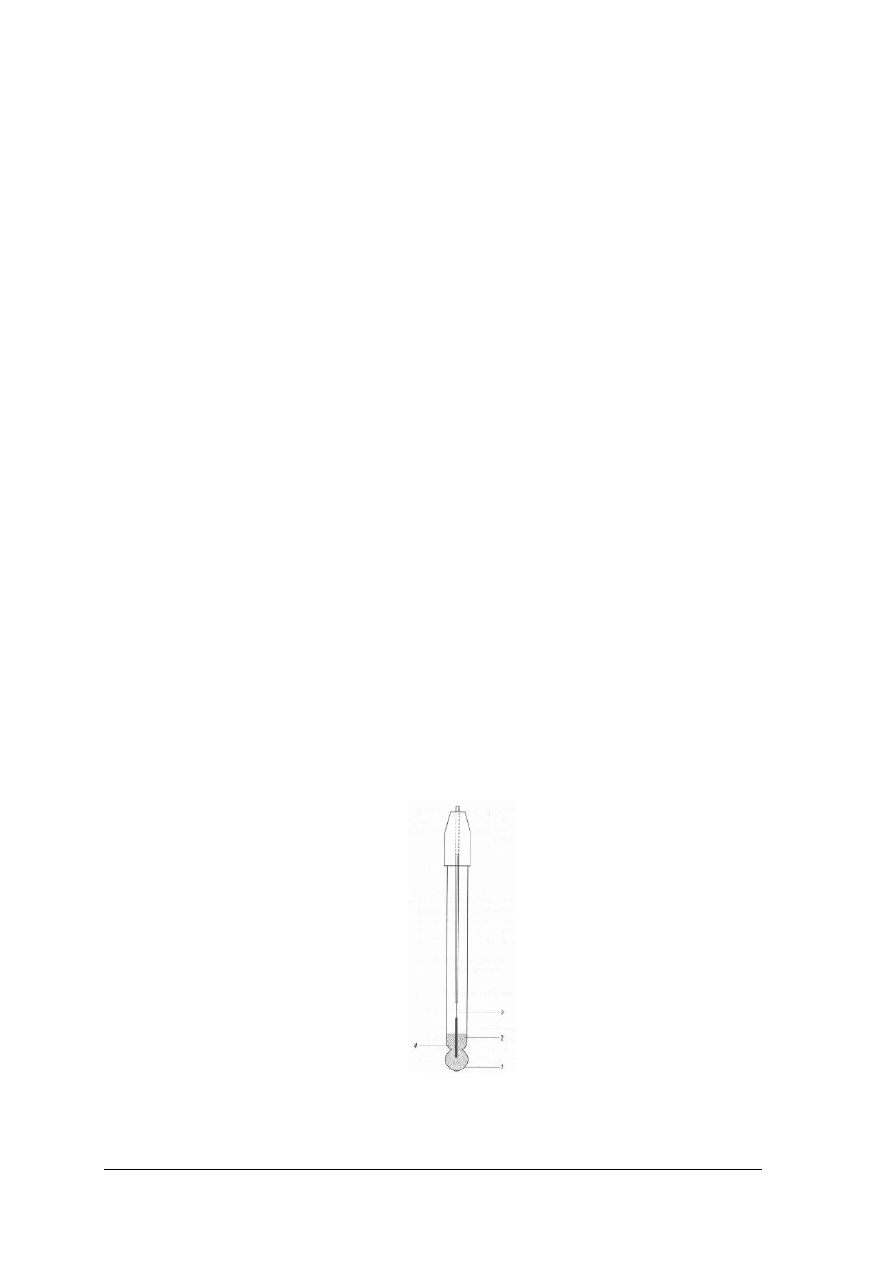
Potencjometryczny pomiar wykładnika wodorowego (pH)jest dokładniejszy niż
oznaczenie kolorymetryczne. Do przeprowadzenia tego oznaczenia potrzebna jest elektroda,
której potencjał zależy od stężenia jonów wodorowych w roztworze, np. elektroda wodorowa,
określana jako wskaźnikowa. W celu wykonania potencjometrycznego pomiaru pH elektrodę
wskaźnikową wraz z elektrodą porównawczą, która ma stały potencjał zanurza się w badanym
roztworze. W ten sposób otrzymuje się ogniwo, które można zapisać w postaci:elektroda
wskaźnikowa roztwór badany elektroda kalomelowa.
Siła elektromotoryczna utworzonego w ten sposób ogniwa jest równa różnicy
potencjałów poszczególnych elektrod: kalomelowej i wskaźnikowej.
Rys. 21. Konstrukcja elektrody szklanej; 1 – membrana szklana, 2 - roztwór wewnętrzny np. 0,1 molowy HCl,
3 - przewód wyprowadzajacy, 4 – Ag/AgCl [2, s. 333]
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
30
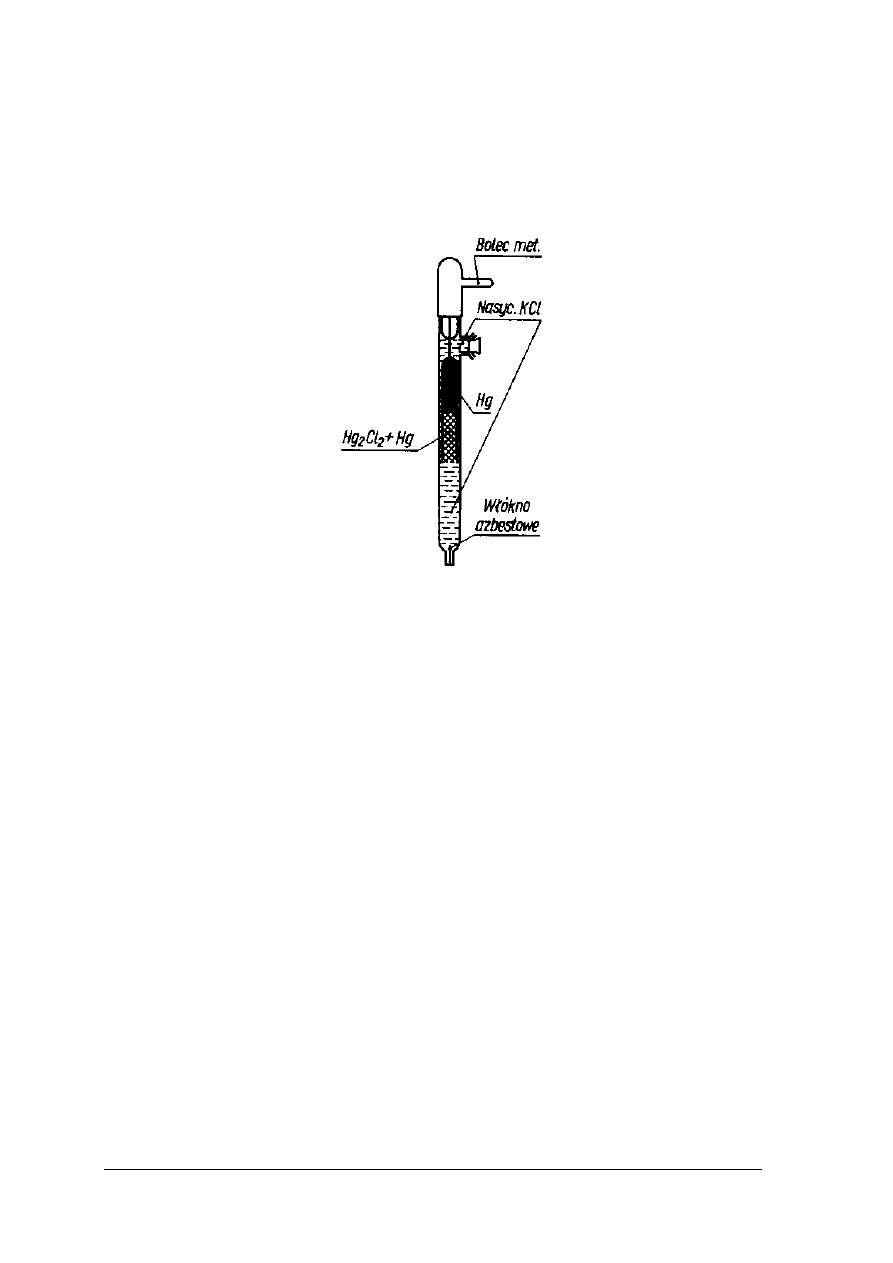
Elektroda kalomelowa
Elektroda kalomelowa należy do elektrod, których potencjał zależy od stężenia anionu.
Jest elektrodą drugiego rodzaju. Zbudowana jest z metalu pokrytego warstwą jego trudno
rozpuszczalnej soli oraz roztworu zawierającego anion tej soli. Elektroda złożona jest z rtęci,
warstwy kalomelu (Hg
2
Cl
2
) i wodnego roztworu chlorku potasu (KCl).
Rys.22. Elektroda kalomelowa [2, s. 330]
Reakcja przedstawiająca działanie elektrody kalomelowej jest następująca:
Hg
2
Cl
2(s)
+ 2 e
-
→
2 Hg
(c)
+ 2 Cl
-
Zaletą elektrody kalomelowej jest stałość jej potencjału wynikająca z faktu, że gdy
odbijemy z niej elektrony odpowiednia ilość rtęci przechodzi do roztworu.
2 Hg
→
Hg
2
2+
+ 2e
-
Wobec stałości iloczynu rozpuszczalności kalomelu i przy nadmiarze jonów chlorkowych
wytrąca się osad kolonelu, zgodnie z reakcja:
Hg
2
2+
+ 2Cl
-
→
Hg
2
Cl
2(s)
Jeżeli elektrony zostaną doprowadzone do układu, jony rtęci przechodzą w rtęć
metaliczną, a w to miejsce odpowiednia ilość kalomelu ulega rozpuszczeniu:
Hg
2
2+
+ 2 e
_
→
2 Hg
Hg
2
Cl
2
→
Hg
2
2+
+ 2 Cl
-
W obydwu przypadkach stężenie jonów rtęciawych i chlorkowych pozostają stałe, a więc
potencjał nie ulega zmianie. Potencjał elektrody kalomelowej z 1-molowym roztworem KCl w
temperaturze 20°C wynosi +0,2812 wolta, a potencjał elektrody kalomelowej z nasyconym
roztworem KCl w tej samej temperaturze wynosi + 0,2476 wolta. Elektroda kalomelowa jest
powszechnie stosowana jako elektroda porównawcza.
Elektroda szklana składa się z banieczki wypełnionej roztworem HCl o stężeniu 0,1
mol/dm
3
, w którym zanurzony jest drucik pokryty chlorkiem srebra. Potencjał elektrody zależy
od stężenia jonów wodorowych w roztworze oraz od składu jakościowego i ilościowego
szkła, a także grubości banieczki szklanej. Ponieważ nie udaje się produkować banieczek o tej
samej grubości oraz ze szkła o tym samym składzie ilościowym ilościowego jakościowym,
dlatego wartość potencjału zależna od rodzaju szkła i grubości banieczki jest zmienna dla
różnych elektrod. Należy ją wyznaczyć doświadczalnie dla każdej elektrody, poddając ją

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
31
cechowaniu
na
roztworach
o
znanych
roztworach
buforowych
o znanych wartościach pH. Bufory to mieszaniny słabych kwasów i ich soli z mocnymi
kwasami. Zachowują one w pewnym zakresie stałe wartości pH mimo rozcieńczania wodą
oraz podczas dodawania do nich pewnych ilości mocnych kwasów lub mocnych zasad.
Przykładem roztworu buforowego jest mieszanina kwasu octowego i octanu sodu.
Elektroda szklana znajduje powszechne zastosowanie jako elektroda wskaźnikowa. Może
być stosowana do pomiarów wartości wykładnika wodorowego mieszczących się w zakresie
od 1÷ 9.
Przed użyciem elektroda wymaga 10-godzinnego moczenia w wodzie destylowanej.
Do wyznaczania pH roztworu (odczynu roztworu wodnego) służy pehametr. Jest to
woltomierz elektroniczny służący w codziennej praktyce laboratoryjnej, badaniach naukowych
lub zastosowaniach polowych. Wartość pH jest określana na podstawie pomiaru siły
elektromotorycznej ogniwa (SEM). Pehametr wyposażony jest najczęściej w elektrodę szklaną,
lub kombinowaną.
Rys.23. Pehametr przenośny IQ 140
Metody chromatograficzne
Chromatografia to metoda analizy stosowana do wydzielania, rozdzielania i identyfikacji
substancji o podobnych właściwościach chemicznych. Proces chromatograficzny może być
prowadzony z dowolnego układu wieloskładnikowego, w którym faza rozpraszająca jest
gazem lub cieczą o nieograniczenie małych stężeniach. Analiza chromatograficzna pozwala
rozdzielić składniki mieszaniny o bardzo zbliżonych właściwościach chemicznych. Oparta jest
na zjawisku sorpcji. Sorpcja to zjawisko samorzutnej zmiany stężenia składnika w układach
złożonych heterofazowych wskutek oddziaływania drugiego składnika znajdującego się w
odmiennej fazie.
Składnik, który zmienia swoje stężenie wskutek oddziaływania drugiego składnika nazywa
się sorptywem. Składnik, który wywołuje zmianę stężenia sorptywu nosi nazwę sorbenta.
Urządzenie, w którym zachodzi sorpcja nazywa się sorberem.
Szczególne zastosowanie sorpcja znajduje w procesach chromatograficznych.
Sprowadzają się one do wykorzystania zjawisk sorpcyjnych przy rozdzielaniu układów
złożonych.
Układ chromatograficzny składa się z trzech elementów: fazy nieruchomej, fazy ruchomej
i substancji rozpuszczonej w tej ostatniej. Proces chromatograficzny sprowadza się do podziału
substancji rozpuszczonej między fazę ruchomą i nieruchomą. Biorąc pod uwagę mechanizm
podziału rozdzielanych substancji pomiędzy fazę ruchomą i nieruchomą rozróżnia się cztery
rodzaje chromatografii:
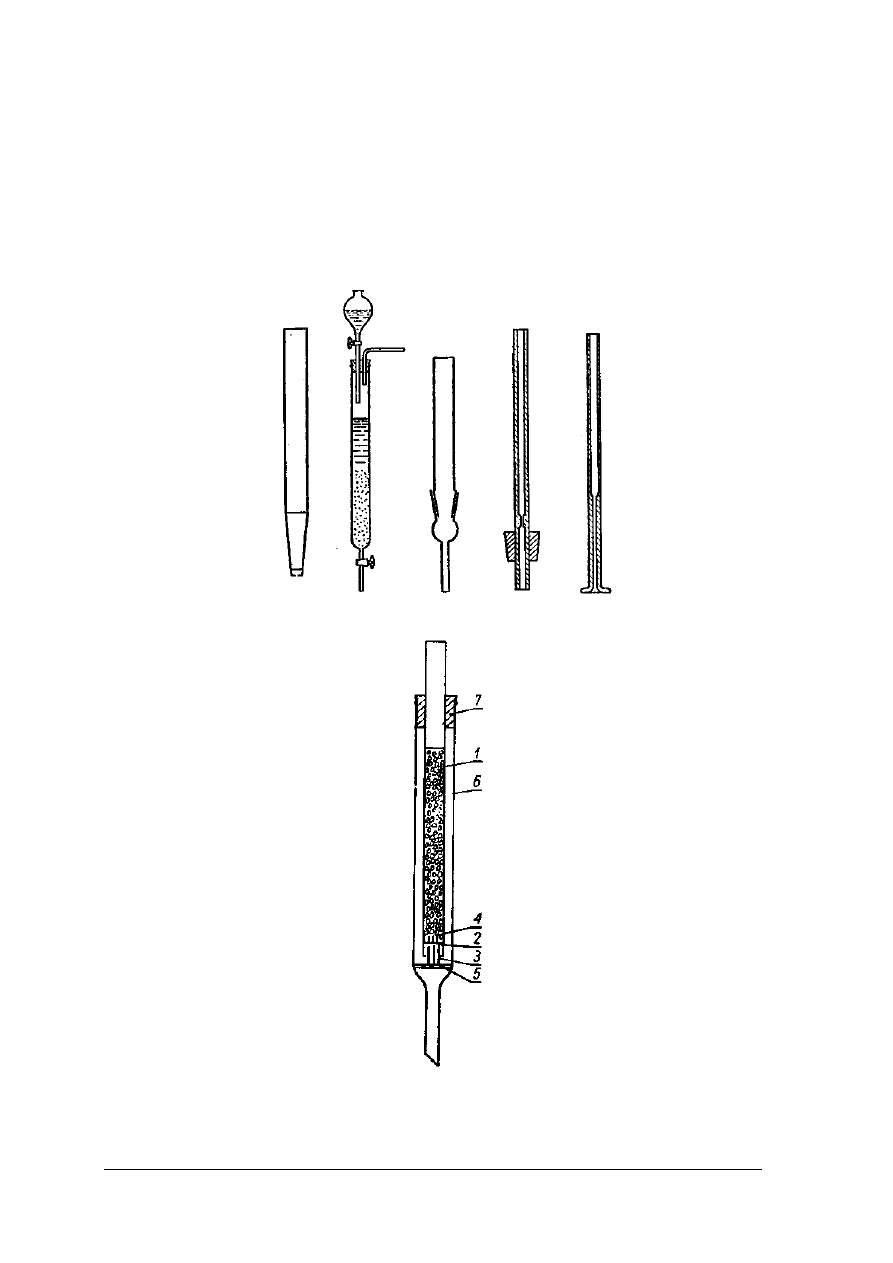
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
32
−
chromatografię adsorpcyjną,
−
chromatografię jonowymienną
−
chromatografię podziałową,
−
chemichromatografię.
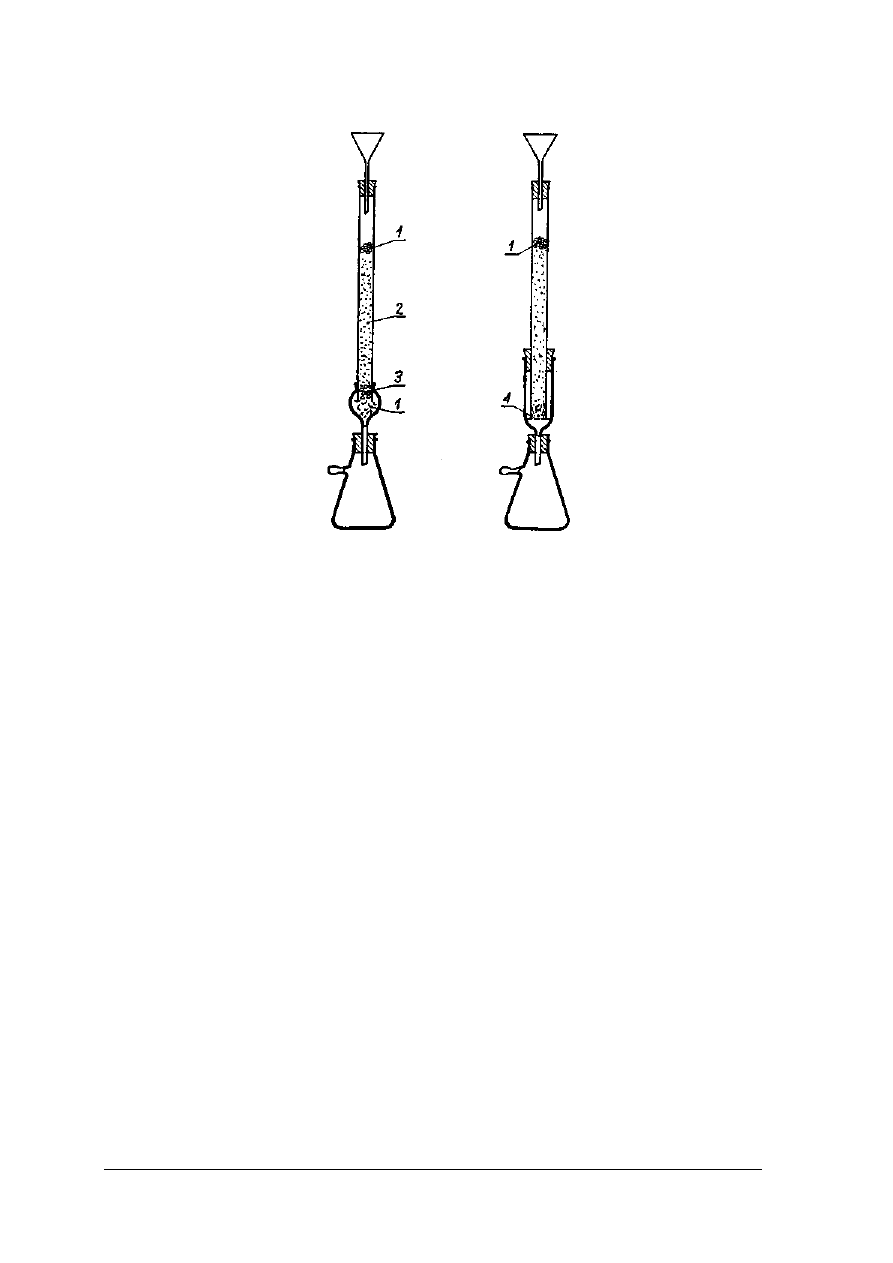
Chromatografia kolumnowa
Chromatografia kolumnowa prowadzona jest w naczyniach cylindrycznych o różnych
wymiarach od kilku milimetrów do kilku metrów, najczęściej o jednostronnym przewężeniu,
w których wmontowane są różnego rodzaju zawory regulujące.
Rys. 24. Różne typy kolumn chromatograficznych [1. s. 99]
Rys. 25. Wypełniona kolumna chromatograficzna [1, s. 100]
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
33
Rys.26. Zestawy do chromatografii kolumnowej [ 1, s.100]
1 – wata, 2 – Al
2
O
3
, 3 – szlif szklany, 4 – szkło piankowe
Wypełnienie kolumny chromatograficznej musi być równomierne i bardzo dokładne, aby
eliminowało powstawanie rys i pęknięć w sorbencie, które mogą doprowadzić do
niewłaściwego rozdziału substancji. Sorbent należy wprowadzać wraz z rozpuszczalnikiem
małymi porcjami tak, aby zapewnić usunięcie wszystkich ewentualnych pęcherzyków
powietrza.
W opisywanej chromatografii kolumnowej fazą ruchomą są mieszaniny ciekłe roztwory.
W przypadku gdy fazą ruchomą są gazy, mimo że proces zachodzi w kolumnach metodę
wyodrębnia się i nazywa chromatografią gazową.
Roztwór przeznaczony do rozdziału chromatograficznego wprowadza się na wierzchołek
kolumny wypełnionej odpowiednim sorbentem i rozpuszczalnikiem tak, aby cała górna
powierzchnia sorbentu została nakryta nim równocześnie i równomiernie. Podczas
przemieszczania się roztworu rozdzielanego przez kolumnę, pierwsze porcje składnika
o większym powinowactwie sorpcyjnym ulegają sorpcji tuż na jej powierzchni. Roztwór
przechodzący do dalszych warstw kolumny jest uboższy w składnik o większym
powinowactwie sorpcyjnym do sorbentu. Nadchodzące nowe warstwy sorbentu o stężeniu
początkowym, dosycają powierzchnie sorbentu w składnik o większym powinowactwie
sorpcyjnym do stanu równowagi według stosunku podziału dla danego składnika
rozdzielanego. Czołowe porcje roztworu zawierające „nadmiar” składnika rozdzielanego
w warstewce powierzchniowej przemieszczają się w głąb kolumny. Napotykają nowe
powierzchnie sorbentu, które wchłaniają rozpatrywany składnik. Jeżeli nowo napotkane
warstwy sorbentu są wypełnione innym sorptywem o mniejszym powinowactwie sorpcyjnym
to przemieszczający się składnik wypiera z poszczególnych powierzchni napotkany sorptyw
i zajmuje jego miejsce. W ten sposób z roztworu przechodzi całkowicie na sorbent składnik
o większym powinowactwie sorpcyjnym do sorbentu już w pierwszych warstwach. Tuż za nim
przechodzi na tych samych zasadach składnik o mniejszym powinowactwie sorpcyjnym itd.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
34
Trwa to do momentu, aż wychodzące porcje o stężeniu początkowym nie wysycą całego
wypełnienia kolumny.
Chromatografia bibułowa to rodzaj chromatografii cieczowej. Metoda głównie
analityczna, lecz służąca do oczyszczania i identyfikacji mieszanin substancji chemicznych.
Fazę rozdzielczą, czyli fazę stacjonarną stanowi bibuła. Zwykła filtracyjna lub
chromatograficzna, o wysokiej czystości i określonych parametrach. Prędkość ruchu
poszczególnych składników rozdzielanej mieszaniny jest uzależniona od oddziaływań
międzycząsteczkowych między związkami chemicznymi tworzącymi analizowaną próbkę,
a fazą rozdzielczą i eluentem. Substancje rozdzielane nakrapia się punktowo przy dolnej
krawędzi bibuły, po czym umieszcza się ją w komorze chromatograficznej zanurzoną na kilka
milimetrów w eluencie (zwanym często tu, i w chromatografii cienkowarstwowej (TLC),
układem rozwijającym), tak by nakropione substancje nie zostały zanurzone. Eluent stopniowo
wznosi się po bibule (przy udziale sił kapilarnych) powodując ruch substancji rozdzielanych w
górę („rozwijanie się” chromatogramu). Gdy czoło eluenta dotrze do górnej krawędzi bibuły
rozdział jest zakończony. Jest to tak zwana chromatografia bibułowa wstępująca. Istnieje
jeszcze chromatografia bibułowa zstępująca, gdzie w komorze chromatograficznej o nieco
innej budowie, ruch eluentu następuje w kierunku dolnej krawędzi bibuły (mieszanina
rozdzielana umieszczona jest wtedy przy górnej krawędzi). Zależnie od swojej natury
fizykochemicznej - składniki rozdzielanej mieszaniny dotarły na różną wysokość (współczynnik
Rf). W przypadku, gdy nie są naturalnie barwne, ich obecność można wizualizować używając
zależnie od ich właściwości światła UV, roztworów wywołujących (w których bibuła jest
zanurzana lub nimi spryskiwana reagują one specyficznie i barwnie z rozdzielanymi
substancjami) lub innych metod (np. wywoływanie w parach jodu). W celu odzyskania
rozdzielonych substancji można je wymyć z określonego fragmentu bibuły. Obecnie rzadko
stosowana metoda. Zwykle stosowana do substancji naturalnie barwnych. Jej nowocześniejszą
wersją jest TLC.
Metody technik chromatograficznych stosowane są w analizie żywności między innymi do
wykrywania oznaczania mieszaniny związków konserwujących w produktach spożywczych,
wykrywania zafałszowań wina np. czerwienią Bordeaux, wykrywania i oznaczania kwasu
propionowego, który bywa używany jako środek przeciwpleśniowy w produktach mącznych,
pieczywie, ciastach itp.
4.3.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń
1. W jaki sposób przygotowywane są roztwory buforowe?
2. Jaką rolę pełni elektroda kalomelowa w oznaczeniach i jak jest zbudowana?
3. Jakie jest znaczenie elektrody szklanej?
4. Jak działa potencjometr?
5. Jakie oznaczenia można wykonywać metodami chromatograficznymi?
6. Jak obsłużyć kolumnę chromatograficzną?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
35
4.3.3. Ćwiczenia
Ćwiczenie 1
Oznacz wykładnik wodorowy (pH) soku owocowego.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować próbkę do soku owocowego do badania,
2) sprawdzić uziemienie pehametru,
3) podłączyć pehametr do sieci elektrycznej i odczekać 15 minut (nie dotyczy pehametrów
tranzystorowych),
4) zanurzyć elektrody do zlewki z roztworem buforowym o pH ok. 3,
5) sprowadzić wskazania miernika skali dokładnie do wartości pH buforu,
6) odstawić zlewkę z roztworem buforowym,
7) popłukać elektrody wodą destylowaną z tryskawki,
8) osuszyć paskiem bibuły,
9) zanurzyć elektrody w zlewce z próbą badanego soku owocowego,
10) dokonać odczytu, zlewkę odstawić, popłukać elektrody i osuszyć je bibułą,
11) wyskalować pehametr na bufor o pH ok.6.
Wyposażenie stanowiska pracy:
–
pehametr wraz z elektrodami (szklaną i kalomelową),
–
kolby miarowe o pojemności 100cm
3
– 2 sztuki,
–
zlewki o pojemności 25cm
3
– 4 sztuki,
–
kolba stożkowa o pojemności 100cm
3
z doszlifowanym korkiem,
–
zlewki o pojemności 30cm
3
– 3 sztuki,
–
bagietki szklane,
–
lejki szklane,
–
cylinder miarowy o pojemności 100cm
3
,
–
tryskawka z wodą destylowaną,
–
roztwory buforowe o wartościach pH 3 i 6,
–
sok owocowy.
Ćwiczenie 2.
Oznacz potencjometryczną wartość pH wina czerwonego.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować próbkę wina do badania,
2) wyzerować aparat przed pomiarem,
3) wyskalować pehametr w temperaturze 20ºC stosując roztwory buforowe o pH = 6,88
i pH=3,57. Do sprawdzenia kalibracji skali użyć roztworu buforowego opH = 4,00
o temperaturze 20º,
4) zanurzyć elektrodę w badanej próbce, której temperatura wynosi od 20º÷25ºC,
5) odczytać wartość pH na skali pehametru,
6) wykonać co najmniej dwa równolegle oznaczenia badanej próbki,
7) obliczyć wynik końcowy jako średnią arytmetyczną z dwóch oznaczeń,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
36
8) wartość pH wina podać z dokładnością do dwóch miejsc po przecinku.
Wyposażenie stanowiska pracy:
−
pehametr wyskalowany w jednostkach pH,
−
elektrody; szklana przechowywana w wodzie destylowanej,
−
kalomelowa nasycona elektroda odniesienia przechowywana w nasyconym roztworze
chlorku potasowego,
−
ewentualnie elektroda kombinowana, przechowywana w wodzie destylowanej,
−
roztwory buforowe: nasycony roztwór wodorowinianu potasu, 0,05-molowy roztwór
wodoroftalanu potasu,
−
wzorcowy roztwór buforowy.
Ćwiczenie 3
Oznacz wykładnik wodorowy (pH) mięsa.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować próbkę mięsa do badania: odważyć w zlewce 50cm
3
25g drobno
rozdrobnionej (w maszynce do mięsa) próbki mięsa. Naważkę przenieść ilościowo do
kolby stożkowej o pojemności 100cm
3
popłukując zlewkę 25cm
3
świeżo przegotowanej
i ostudzonej wody destylowanej. Kolbę zamknąć doszlifowanym korkiem i wytrząsać 15
minut. Zawiesinę przesączyć przez sączek z bibuły filtracyjnej. Przesącz użyć do
oznaczenia pH,
2) odważyć w suchej zlewce o pojemności 50cm
3
na wadze technicznej 25g rozdrobnionej
próbki mięsa,
3) przenieść naważkę do kolby stożkowej o pojemności 100cm
3
,
4) popłukać zlewkę 25cm
3
świeżo przegotowanej i ochłodzonej wody destylowanej,
popłuczyny dodać do kolby z oznaczoną próbką mięsa,
5) wytrząsać zamkniętą kolbę przez 15 minut,
6) sączyć zawiesinę przez sączek z bibuły filtracyjnej,
7) wykonać oznaczenie na pehametrze jak w poprzednim ćwiczeniu.
Wyposażenie stanowiska pracy:
−
pehametr wraz z elektrodami (szklaną i kalomelową),
–
kolby miarowe o pojemności 100cm
3
– 2 sztuki,
–
zlewki o pojemności 25cm
3
– 4 sztuki,
–
kolba stożkowa o pojemności 100 cm
3
z doszlifowanym korkiem,
–
zlewki o pojemności 30cm
3
– 3 sztuki,
–
bagietki szklane,
–
lejki szklane,
–
cylinder miarowy o pojemności 100cm
3
,
–
waga techniczna, komplet odważników,
–
tryskawka z wodą destylowaną,
–
roztwory buforowe o wartościach pH 3 i 6.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
37
Ćwiczenie 4
Wprowadź roztwór z liści na kolumnę chromatograficzną i zaobserwuj przebieg procesu.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować roztwór z liści przeznaczony do rozdziału chromatograficznego,
2) rozetrzeć w moździerzu kilka młodych liści z odrobiną piasku,
3) mieszaninę przenieść do kolby i dodać trochę acetonu i odstawić na kilka minut,
4) przygotować kolumnę, koniec szklanej rurki o długości ok. 20cm zatkać z jednej strony
korkiem z rurką, na dno włożyć mały zwitek waty,
5) wypełnić rurkę cukrem pudrem, do wysokości 15cm
6) przesączyć mieszaninę przez bibułę,
7) wprowadzić roztwór na wierzchołek kolumny,
8) wlewać kolejne niewielkie porcje acetonu kiedy roztwór z liści spłynie w dół kolumny,
9) prowadzić obserwację do momentu kiedy wychodzące porcje roztworu o stężeniu
początkowym nie wysycą całego wypełnienia kolumny,
10) wyjaśnić zjawiska zachodzące w kolumnie chromatograficznej.
Wyposażenie stanowiska pracy
−
kolumna chromatograficzna,
−
zwitek waty, cukier puder do wypełnienia kolumny,
−
liście dowolnego warzywa,
−
aceton,
−
lejek, bibuła.
Ćwiczenie 5
Wykryj pary jonów Cu
2+
i Cd
2+
lub inne pary jonów.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) wyciąć paski bibuły rozmiaru 1x13cm,
2) nanieść na bibułę (w odległości 1cm od brzegu) kroplę badanego roztworu,
3) wysuszyć otrzymaną plamę,
4) nalać na dno probówki (długości ok. 12cm) 0,5÷1cm
3
mieszaniny eluenta: alkoholu
n-butylowego i kwasu solnego w stosunku 4:1,
5) zanurzyć pasek bibuły w eluencie na głębokość około 5mm i zamknąć probówkę (plama
badanego roztworu znajduje się tuż nad eluentem) korkiem w ten sposób, aby korek
przytrzymywał pasek bibuły, wymywać około dwóch godzin,
6) wysuszyć bibułę i wywołać chromatogram,
7) spryskiwać bibułę roztworem siarczku amonowego (z rozpylacza), miedź tworzy czarne
pasmo u dołu, a kadm żółte w górze chromatografu
8) podać skład jakościowy badanego roztworu na podstawie wywołanego chromatogramu.
Uwaga: Chromatograf można wywołać inaczej. W odległości około 4,5 cm od dolnego
brzegu nanieść kroplę roztworu żelazicyjanku potasowego – w obecności miedzi powstanie
czerwono-brunatna plama . W odległości około 8,5cm od dolnego brzegu nanieść kroplę
roztworu siarczku amonowego – w obecności kadmu powstanie żółta plama.
Możliwości dalszego rozwinięcia ćwiczenia:

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
38
Opracować rozdzielenie w tych samych warunkach mieszanin: Fe
3+
- Co
2+
, Hg
2+
- Pb
2+
,
Hg
2+
- Ni
2+
, Hg
2+
- Cu
2+
, Ni
2+
- Bi
3+
, i sposób identyfikacji plam.
Wyposażenie stanowiska pracy:
−
alkohol n-butylowy,
−
kwas solny, stężony,
−
siarczek amonowy: nasycić roztwór amoniaku o stężeniu 2mol/dm
3
gazowym
siarkowodorem (do chwili, gdy dodanie tego roztworu do roztworu MgCl
2
nie wywoła
powstania osadu) i rozcieńczyć go równą objętością amoniaku o stężeniu 2mol/dm
3
,
−
żelazicyjanek potasowy, roztwór nasycony.
4.3.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) zdefiniować pojęcie buforu i pojemności buforowej ?
2) zdefiniować pojęcie metod elektrometrycznych?
3) określić działanie elektrod?
4) oznaczyć wartość wykładnika wodorowego (pH)?
5) określić zastosowanie metod chromatograficznych?
6) wyjaśnić zasady procesu chromatograficznego?
7) wyjaśnić budowę i zasadę działania kolumny chromatograficznej?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
39
5. SPRAWDZIAN OSIĄGNIĘĆ
INSTRUKCJA DLA UCZNIA
1. Przeczytaj uważnie instrukcję.
2. Podpisz imieniem i nazwiskiem kartę odpowiedzi.
3. Zapoznaj się z zestawem pytań testowych.
4. Udzielaj odpowiedzi tylko na załączonej karcie odpowiedzi.
5. Pracuj samodzielnie, pamiętaj, że w przyszłej pracy nie możesz liczyć na podpowiedzi
kolegów.
6. Na udzielenie odpowiedzi masz 45 minut.
Powodzenia!

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
40
ZESTAW ZADAŃ TESTOWYCH
1. W analizie żywności do mierzenia gęstości cieczy używa się:
a) polarymetru.
b) refraktometru.
c) areometru.
d) kolorymetru.
2. Badania kolorymetryczne wykorzystuje się do oznaczania:
a) masy ciał.
b) stężeń roztworów.
c) pH roztworów.
d) gęstości ciał.
3. Metody refraktometryczne stosowane są do:
a) oznaczania zawartości cukrów i wody.
b) wykrywania zafałszowań.
c) identyfikacji tłuszczów.
d) do wszystkich trzech oznaczeń.
4. Do kolorymetrycznych pomiarów stężeń służą urządzenia:
a) cylindry Hehnera.
b) spektrofotometr.
c) absorpcjometr.
d) wszystkie wyżej wymienione.
5. W oznaczeniach piknometrycznych wykorzystywany jest piknometr:
a) próżniowy.
b) Geisslera.
c) Reischauera.
d) wszystkie odpowiedzi są prawdziwe.
6. Dobra waga hydrostatyczna pozwala określić gęstość z dokładnością do:
a) 0,0001.
b) 0,001.
c) 0,01.
d) 0,1.
7. Na jakość oznaczeń polarymetrycznych ma wpływ:
a) ciśnienie.
b) gęstość.
c) objętość.
d) temperatura.
8. Do oznaczania gęstości mleka używa się:
a) laktodensymetru.
b) piknometru.
c) wagi hydrostatycznej.
d) sacharymetru.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
41
9. Waga Mohra-Westphala służąca do hydrostatycznego pomiaru gęstości wyskalowana jest
w temperaturze:
a) 5°C.
b) 10°C.
c) 15°C.
d) 25°C.
10. Gęstość ciała określa się jako:
a) iloczyn masy ciała i objętości.
b) stosunek masy ciała do objętości.
c) stosunek objętości do ciężaru ciała.
d) iloczyn ciężaru i gęstości.
11. Zasada działania areometru oparta jest na prawie:
a) Ballinga.
b) Trallesa.
c) Gay-Lussaca.
d) Archimedesa.
12. W celu uniknięcia spieniania płynów należy przelewać je:
a) szybko.
b) ostrożnie po ściance do zlewki.
c) bardzo szybko.
d) jakkolwiek.
13. Elektrodę szklaną można stosować w roztworach o pH:
a) 0÷12.
b) 0÷12,5.
c) 0÷13.
d) 0÷14.
14. Refraktometr zanurzalny posiada:
a) jeden pryzmat.
b) dwa pryzmaty.
c) trzy pryzmaty.
d) cztery pryzmaty.
15. Dokładność pomiaru piknometrem zależy od:
a) temperatury.
b) wielkości piknometru.
c) czystości piknometru.
d) wszystkich wymienionych czynników.
16. Miody pitne mają skręcalność:
a) 3,0÷5,0°.
b) 1,40÷ 1,43°.
c) 2,0÷2,5°.
d) 3,0÷4,0º.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
42
17. Oznaczenia spektrofotometryczne pozwalają:
a) charakteryzować substancje jakościowo.
b) oznaczać ilościowo stężenie substancji.
c) wnioskować o charakterze substancji.
d) spełniać wymienione wyżej funkcje.
18. Elektroda szklana może być stosowana do pomiaru pH w zakresie:
a) 0÷5.
b) 0÷7.
c) 0÷10.
d) 0÷12.
19. Punktem zerowym polarymetru jest pozycja analizatora dla której:
a) obydwa pola widzenia są jasne.
b) obydwa pola widzenia są najbardziej ciemne.
c) pola widzenia są podzielone na trzy części.
d) pola widzenia są lekko przyciemnione.
20. Chromatografia kolumnowa obejmuje metody, w których fazą ruchomą są:
a) substancje stałe.
b) substancje jednorodne.
c) mieszaniny ciekłe, roztwory.
d) substancje gazowe.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
43
KARTA ODPOWIEDZI
Imię i nazwisko........................................................................................
Wykonywanie instrumentalnej analizy żywności
Zakreśl poprawną odpowiedź, wpisz brakujące części zdania lub wykonaj rysunek.
Nr
zadania
Odpowiedź
Punkty
1.
a
b
c
d
2.
a
b
c
d
3.
a
b
c
d
4.
a
b
c
d
5.
a
b
c
d
6.
a
b
c
d
7.
a
b
c
d
8.
a
b
c
d
9.
a
b
c
d
10.
a
b
c
d
11.
a
b
c
d
12.
a
b
c
d
13.
a
b
c
d
14.
a
b
c
d
15.
a
b
c
d
16.
a
b
c
d
17.
a
b
c
d
18.
a
b
c
d
19.
a
b
c
d
20.
a
b
c
d
Razem:

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
44
6. LITERATURA
1. Budsławski J., Drabant Z.: Metody analizy żywności. WNT, Warszawa 1999
2. Drzazga B.: Analiza techniczna w przemyśle spożywczym. WSiP, Warszawa 1992
3. Jarosz M., Malinowska E.: Pracownia chemiczna. Analiza instrumentalna. WSiP,
Warszawa 200
4. Łada Z. Różyczki C.: Pracownia chemii analitycznej analiza techniczna i insrymentalna
WSiP 1984
5. Witkiewicz Z.: Podstawy chromatografii. WNT, Warszawa 2000
6. http://pl.wikipedia.org/
Wyszukiwarka
Podobne podstrony:
34 Wykonywanie instrumentalnej analizy żywności
32 Wykonywanie wagowej analizy żywności
33 Wykonywanie objętościowej analizy żywności
32 Wykonywanie wagowej analizy żywności
32 Wykonywanie wagowej analizy żywności
Metody reologiczne w analizie żywności
Zagadnienia na kolokwium OEBHP, (Sylwia) studia semestr 3, Analiza żywności, Bhp i ergonomia
Oznaczenie zawartości sacharydów, Technologia żywnosci i Żywienie człowieka, 4 SEMESTR, Analiza żywn
Analiza żywności ćw 4 kwasowość, Tż, Analiza żywności II, Sprawozdania
Analiza żywności egzamin 13
dr Szczapa, Analiza żywności, Karotenoidy
Polarymetryczne oznaczanie zawartości skrobi, Tż, Analiza żywności II, Sprawozdania
TiAZ- produkcje, studia, bio, 3rok, 5sem, technologia i analiza żywności, wykład
Kalend.-Ćwiczeń-z-Now.-Met.-Anal.-Żywn.-13-14, Nowoczesne metody analizy żywności
sprawko tran, Nowoczesne metody analizy żywności
Oznaczanie cukrow prostych metoda Antronowa, Tż, Analiza żywności II, Sprawozdania
OZNACZANIE ZAWARTOCI POLISACHARYDW1, 2 rok, analiza, Analiza żywności, analiza cd, sprawka
więcej podobnych podstron