
Dziedziczna neuropatia nerwu
wzrokowego -
zespół Lebera
mgr Paweł Kowalski
Warszawa, 13 stycznia 2012 r.
Zakład Genetyki Medycznej IPCZD

Historia
LHON
– zespół (neuropatia) Lebera
OMIM 535000
1871 rok
– niemiecki lekarz Teodor Leber
(1840-
1917) dokładnie opisuje klinicznie 4
rodziny, w których wzrok tracą głównie
mężczyźni.
1972 rok
– Erickson postuluje matczyny
sposób dziedziczenia LHON
1988 rok
– Wallace identyfikuje pierwszą
mutację powodującą LHON (m.11778G>A)
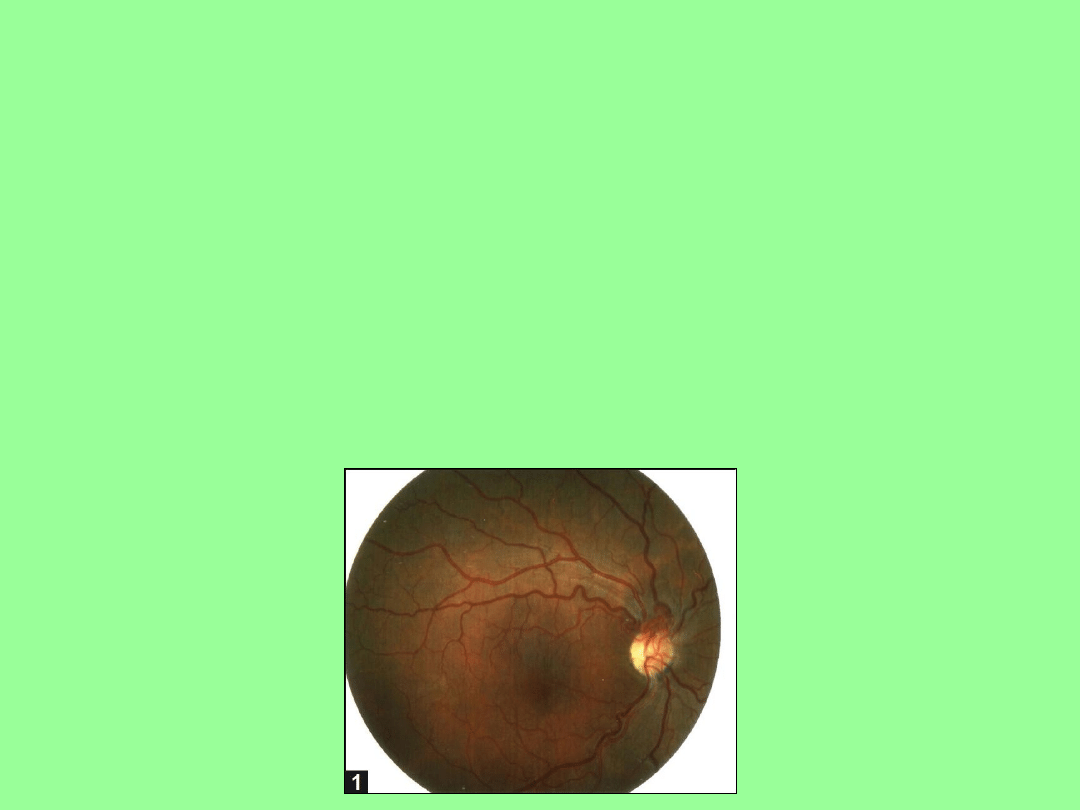
Objawy kliniczne (1)
Faza ostra choroby. Zamazany i zamglony obraz najpierw
w jednym a później w obu oczach (odstęp 2-3 miesięcy),
centralny ubytek pola widzenia, nierozróżnianie kolorów.
Faza chroniczna choroby. Nerw wzrokowy powoli degeneruje aż
całkowicie zanika. Stopień odzyskania wzroku zależy od
konkretnej mutacji, którą dany pacjent posiada. Najgorsze
prognozy dotyczą mutacji m.11778G>A. U pacjentów z mutacją
m.14484T>C obserwuje się nieznaczną poprawę wzroku.

Objawy dodatkowe. U kilku nosicielek mutacji
m.11778G>A obserwowano dodatkowo stwardnienie
rozsiane. W porównaniu ze zdrowymi kontrolami u innych
pacjentów z rozpoznaniem LHON stwierdza się m. in.
drżenie przy próbach utrzymania określonej pozycji
kończyny, neuropatię obwodową, niespecyficzną
miopatię, dystonię kurczową, ataksję, encefalopatię
dziecięcą, epilepsję, demencję, zaburzenia ruchu lub
zaburzenia rytmu serca.
Te dodatkowe objawy obserwowano m. in. pacjentów, u
których wykryto mutacje drugorzędowe (opisanych 18) w
mitochondrialnym DNA. Zwiększają one ryzyko
wystąpienia choroby lub zaostrzają jej przebieg. Mogą
występować osobno lub parami. Podobna rola 3 mutacji w
genach jądrowych: NDUFA-1, EPHX1 i TP53.
Objawy kliniczne (2)

Diagnozę LHON można postawić analizując
historię choroby pacjenta. Wykonuje się także
dodatkowe badania - regularna ocena pola
widzenia. Obrazy CT i MRI są zwykle
prawidłowe. Jeżeli postawienie
jednoznacznego rozpoznania choroby
stwarza trudności należy przeprowadzić test
genetyczny celem znalezienia jednej z trzech
powszechnych mutacji w mitochondrialnym
DNA.
Diagnostyka

Epidemiologia
LHON jest jedną z najbardziej
powszechnych genetycznych chorób
mitochondrialnych. Badania brytyjskie
– częstość 1:25000.
Pogorszenie wzroku może pojawić się
między I a VII dekadą życia, ale 95%
przypadków dotyczy osób przed 50
rokiem życia.
Mutacje de novo
są rzadkie w LHON.

Objawy biochemiczne
Zmniejszona produkcja ATP (OXPHOS) i
degeneracja komórek zwojowych siatkówki
jako wynik zaburzeń energetycznych w
komórkach. Wyniki badań potwierdzają
dysfunkcję łańcucha oddechowego u
pacjentów z LHON (m. in. obniżenie
aktywności kompleksu I o 25-65%).
Zwiększona produkcja reaktywnych form
tlenu odgrywa rolę w patofizjologii choroby.

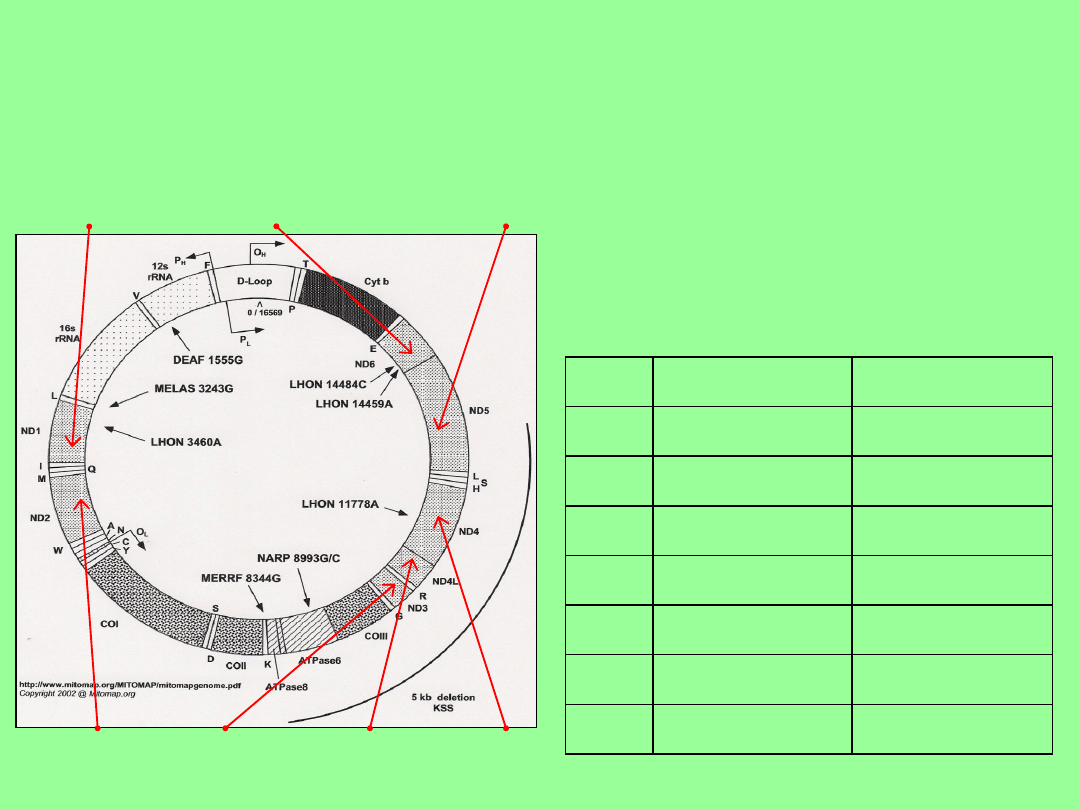
Patogenne mutacje
Nie zostały znalezione w zdrowych kontrolach.
Mutacje pierwszorzędowe w mitochondrialnym DNA:
m.3460G>A (p.Ala52Thr), m.11778G>A (p.Arg340His)
i m.14484T>C (p.Met64Val) występują w genach
kodujących podjednostki kompleksu I łańcucha
oddechowego (MTND1, MTND4, MTND6).
Wykryte u ponad 90% wszystkich przypadków LHON.
MTND1 i MTND6
– geny „hot spot” dla LHON.
Należy je badać u pacjentów, u których nie wykryto
mutacji pierwszorzędowych.

5 wieloenzymatycznych
kompleksów kodowanych
przez geny
mitochondrialne i
jądrowe + 2 nośniki
elektronów (ubichinon,
cytochrom c)
I-IV (centra red-oks)
tworzą gradient
protonowy i potencjał
błonowy, a V produkuje
ATP
Mutacje > choroby
mitochondrialne
Łańcuch oddechowy
Fosforylacja oksydacyjna
Superkompleksy I+III – poprawa katalizy
jako wynik zmniejszenia czasu dyfuzji
substratów, ochrona przed tworzeniem
patogennych superoksydantów

Charakterystyka kompleksu I
• Wielkość: ~1 MDa (największy i najbardziej złożony)
• 46 podjednostek – kontrola dwugenomowa (7 białek
mitochondrialnych, 39 białek jądrowych)
• Funkcja: transfer elektronów (flawiny, grupy Fe-S) z NADH
na ubichinon siła protonowa produkcja ATP
• Struktura L-kształtna – ramię hydrofobowe w błonie
lipidowej (translokacja protonów) i hydrofilowe w matriks
mitochondrialnej (transfer elektronów)
• 14 tzw. podjednostek rdzeniowych – kataliza transferu
elektronów z NADH na ubichinon i tworzenie potencjału
błonowego
Podjednostki mitochondrialne
kompleksu I
gen
wielkość genu
wielkość białka
ND1
955 pz
35,6 kDa
ND2
1041 pz
38,9 kDa
ND3
345 pz
13,2 kDa
ND4
1377 pz
51,4 kDa
ND4L
296 pz
10,7 kDa
ND5
1811 pz
66,6 kDa
ND6
524 pz
18,6 kDa
ND1
ND6
ND5
ND4
ND4L
ND3
ND2
Mitochondrialne DNA

U 50% mężczyzn i 10% kobiet, które
posiadają jedną z trzech
pierwszorzędowych mutacji
rozwinie się pełnoobjawowy LHON.
Zarówno to, jak i fakt, że wzrok tracą
głównie mężczyźni (80% przypadków)
sprawia, że dodatkowe czynniki
genetyczne i/lub środowiskowe muszą
regulować ekspresję fenotypową
LHON. Może to także wynikać z różnic
anatomicznych, hormonalnych (wpływ
androgenów) i/lub fizjologicznych
między kobietami a mężczyznami.

Genetyczne czynniki
mitochondrialne
W większości rodowodów z LHON mutacja
pierwszorzędowa jest homoplazmatyczna.
U 10-15% nosicieli
– heteroplazmatyczna.
Ryzyko ślepoty jest minimalne gdy poziom
mutacji jest mniejszy niż 70%. m.11778G>A
i m.14484T>C związane z haplotypem J,
m.3460G>A z K.
Haplotyp
– jednonukleotydowe substytucje
dziedziczące się jako zestaw sprzężonych
ze sobą alleli.

Czynniki środowiskowe
Palenie tytoniu, alkohol, toksyny,
głód, stres lub ostra choroba mogą
spowodować początek ślepoty w
LHON.
Podobne objawy w amblyopii
tytoniowo-alkoholowej.

Leczenie
Obecnie brak sposobów, które zapobiegałyby
lub opóźniały wystąpienie ślepoty w LHON.
Ponadto nie ma dostępnej tradycyjnej terapii,
która poprawiałaby stopień widzenia w LHON.
Podawanie witaminy B12 i C może przyspieszyć
odzyskanie wzroku.
Początki terapii genowej – syntetyczna
podjednostka MTND4, geny antyoksydantów
(modele zwierzęce).

Poradnictwo genetyczne
Jeżeli pierwszorzędowa mutacja LHON została
zidentyfikowana u probanda to innym członkom rodziny
można zaoferować testy genetyczne aby wykluczyć
możliwość powstania mutacji de novo. Nigdy nie badaliśmy
komórek płodowych. Do diagnostyki prenatalnej należy
podchodzić z rezerwą.
LHON wykazuje wyłącznie matczyny sposób dziedziczenia.
Minimum 70% poziom heteroplazmii jest wymagany do
ujawnienia się choroby.

Algorytm postępowania w
wykrywaniu mutacji
powodujących LHON
1) Izolacja DNA z krwi (pośmiertnie - nerw
wzrokowy, siatkówka oka – więcej
zmutowanego mtDNA)
2) PCR z odpowiednimi starterami
3) Sekwencjonowanie / MLPA

Diagnostyka molekularna
LHON w IPCZD
• od 2005 r. 59 pacjentów + 5 członków rodzin
• mutacje u 12/6 rodzin
- m.11778G>A
– 9 razy
- m.3460G>A
– 3 razy
- m.14484T>C
– nie wykryto, najłagodniejszy
fenotyp, mogący sprawiać trudności w
jednoznacznej identyfikacji
• Wykrywalność 20%
Wyniki (1)
a. Mutacja m.3460G>A (p.A52T) u
jednego pacjenta, gen MTND1 :
pacjent Ł.H.
zdrowa kontrola
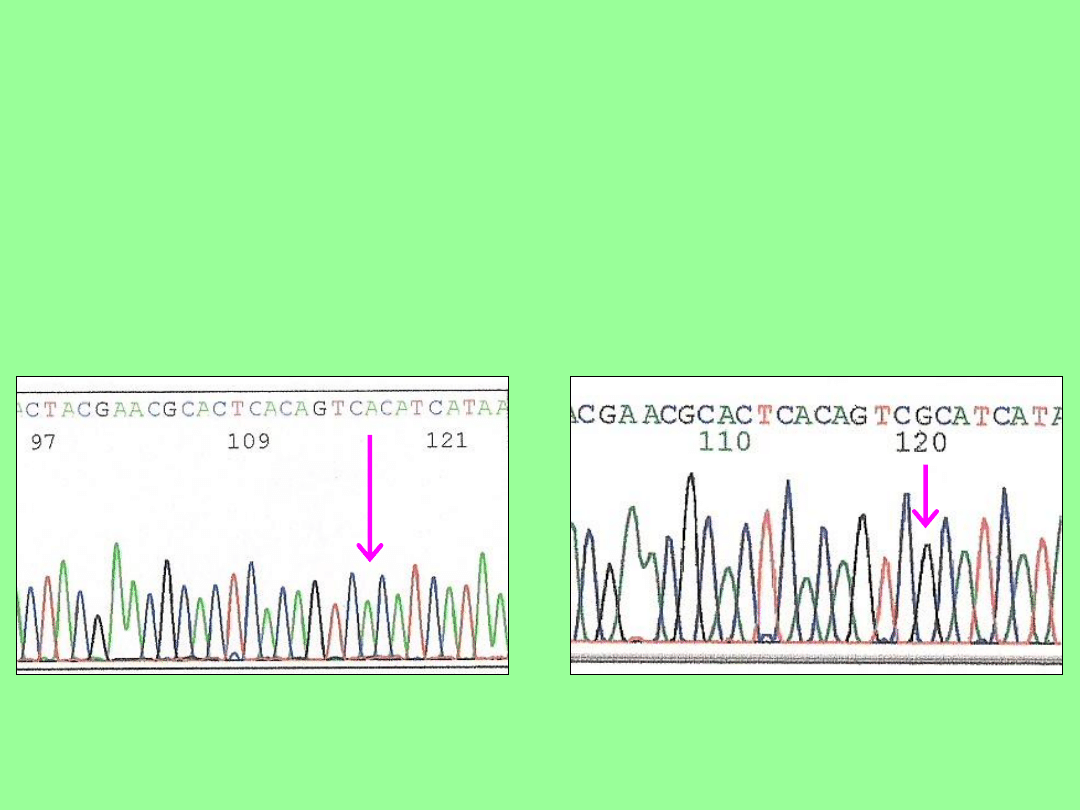
Wyniki (2)
b. Mutacja m.11778G>A (p.R340H) u
jednego pacjenta, gen MTND4 :
pacjent M.J.
zdrowa kontrola

Podsumowanie
Zastosowanie dostępnych obecnie testów
genetycznych ułatwia diagnozę kliniczną,
szczególnie nietypowych przypadków LHON.
Lepsza charakterystyka związku między
mutacjami mtDNA, biogenezą mitochondrialną i
dysfunkcją nerwu wzrokowego jest potrzebna do
wytłumaczenia niejasnej ciągle w pełni
patofizjologii LHON.
Wyszukiwarka
Podobne podstrony:
Podatek liniowy Paweł Kowalski Z6X2S1, Podatek liniowy
wyklady-Pawel
Dane i bezpieczenstwo Pawel Kowalski
Dane i bezpieczenstwo Pawel Kowalski
18 KAROLCZAK Pawel, KOWALSKI Maciej PO FORM
cz V, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
czIV, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
czII, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
77, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
ver.5 rozkad urzadzenia i systemy wytw. cnc, Polibuda MBM PWR 2012-2016, Sem. VII, CNC i roboty, Wyk
pnklimatyzacja, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
8, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
Dopuszczalne graniczne wartości, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
04.10.2011- wykl. I, Ekonomika mediów- Tadeusz Kowalski, wykład, egzamin pisemny/ test (za tydzień 1
cz V, wykłady, wentylacja, Minikowski, PAWEŁ KLIMA
S2 Historia Myśli Psychologicznej Paweł Besler wykład 4
wyklad 26 Paweł wręczył kolektę i zdał relację ze swojej działalności przed Jakubem
więcej podobnych podstron