
Analiza biochemiczna
Dr Marcin Grąz, pok. 337
Ocena końcowa:
Egzamin (70%)
Wykłady + konwersatoria + praca semestralna (30%)
Organizmy modelowe w biochemii:
E. coli
S. cervisiae
B. subtilis
N. crassa
Ch. Pyrenoidosa
Projektowanie badań biochemicznych:
1. Identyfikacja przedmiotu badań
2. Krytyczna ocena stanu wiedzy, poznanie metodologii
3. Postawienie hipotezy badawczej
4. Wybór systemu badań (in vitro/in vivo)
5. Identyfikacja badanej zmiennej, ocean wpływu, kontrola czynników wpływających na wynik
doświadczenia (temperatura, pH, inhibitory proteaz)
6. Zaprojektowanie eksperymentu
7. Przeprowadzenie eksperymentu
8. Dokumentacja otrzymanych wyników
9. Powtórzenie eksperymentu
10. Analiza otrzymanych wyników
11. Przedstawienie wniosków na podstawie wyników
12. Przedstawienie nowych hipotez
Wszystkie doświadczenia naukowe dają wyniki, które można przedstawić statystycznie – pozwalają
one dotrzeć do zależności występujących między cząsteczkami badanych substancji.
Bazy komputerowe:
EBSCO
ELSEVIER
SPRINGER
SCOPUS
NATURE
SCIENCE AAAS
PUBMED
WE OF SCIENCE
ICM
Bazy na stronach czasopism

Wskaźniki bibliometryczne:
Bibliometria – zbiór metod służących do oceny danego zbioru czasopism, autorów,
uczelni
Indeks cytowań (Science Citation Index) znajdujący się na Web of Science na platformie
Web of Knowledge opisujący ilość cytowań danego autora
Miara oddziaływania (Impact Factor) – wskaźnik prestiżu czasopisma obliczany
na podstawie indeksów cytowań
Lista filadelfijska (ISI Master Journal List) zawiera czasopisma spełniające odpowiednie
kryteria jakości
Indeks Hirsha – wskaźnik odzwierciedla dystrybucję cytowań publikacji określonego
naukowca i liczbę jego najlepszych publikacji.
Struktura pracy naukowej:
Gatunek pracy
Zakres tematyczny
(definicja gatunku)
Szkielet
Zakres bibliografii
(redakcja może zastrzec
maksymalną
liczbę pozycji)
Naukowo-
badawcza
przebieg i wyniki
określonych badań z
zakresu medycznych nauk
podstawowych,
biomedycznych,
biotechnicznych; zwykle
tylko jeden typ badań, jeden
wynalazek albo jedno
odkrycie; każde następne
opisuje się w osobnej pracy;
wyniki badań lub odkryć
cząstkowych (etapowych)
można opisywać osobno w
kolejnych pracach wraz z
przyrostem materiału
wynikowego; wyniki
zakończonego ciągu takich
badań przedstawia się
zwykle w osobnej pracy
poglądowej jako
podsumowanie; wyniki
badania pojedynczego
podaje się wraz z opisem
badań w tym samym tekście
1. tytuł
2. nazwiska autorów (wraz z
jednostkami badawczymi)
3. słowa kluczowe
4. streszczenie
5. wstęp
a. ogólny opis przedmiotu
badań na tle
dotychczasowej wiedzy w
danej dziedzinie
b. cel badań (hipotezy,
które badacz chce
potwierdzić)
c. warunki, w jakich
wykonywano badania (w
tym miejsce, ew.
współautorzy, sponsorzy),
także trudności i
przeszkody
d. omówienie
podstawowych pojęć i
definicji i ew. objaśnienie
znaczenia tych, które w
różnych pracach są różnie
rozumiane
e. zestawienie używanych
w pracy skrótów i
skrótowców
6. metoda - szczegółowy opis
wywodu logicznego, metod
stosowanych w czasie
badań, opis modeli
eksperymentalnych,
użytych aparatów,
piśmiennictwo możliwie
najszerzej reprezentujące
tematykę

substancji, także sposobów
oceny statystycznej i
zgodności z EBM
7. materiał (opis doboru i
zrównoważenia badanych
prób, ich
reprezentatywności
względem populacji albo
innych zbiorów)
8. opis przebiegu badań,
umożliwiający innym
badaczom powtórzenie
procedur
9. wyniki badań
10. omówienie wyników
11. wnioski
Kliniczna
obserwacje statystycznie
istotnej grupy zdrowych lub
chorych w zakresie
określonego typu zjawisk
fizjologicznych lub
patologicznych, w tym
stosowane metody i
procedury diagnostyczne,
terapeutyczne,
rehabilitacyjne,
profilaktyczne,
pielęgnacyjne,
psychologiczne
1. tytuł
2. nazwiska autorów (wraz z
jednostkami badawczymi)
3. słowa kluczowe
4. streszczenie
5. wstęp
a. ogólny opis przedmiotu
badań na tle
dotychczasowej wiedzy w
danej dziedzinie
b. cel badań (hipotezy,
które badacz chce
potwierdzić)
c. warunki, w jakich
wykonywano badania (w
tym miejsce, ew.
współautorzy, sponsorzy),
także trudności i
przeszkody
6. metoda (szczegółowy opis
wywodu logicznego, metod
stosowanych w czasie
badań, opis modeli
eksperymentalnych,
użytych aparatów,
substancji, także sposobów
oceny statystycznej,
zgodności z EBM
7. materiał (opis doboru i
zrównoważenia badanych
prób, ich
reprezentatywności
względem populacji albo
innych zbiorów)
8. opis przebiegu badań,
umożliwiający innym
badaczom powtórzenie
procedur
piśmiennictwo możliwie
najszerzej reprezentujące
tematykę

9. wyniki badań
10. omówienie wyników
11. wnioski
Poglądowa
najczęściej spotykane,
powszechnie przyjęte,
oficjalne albo oparte na
ciągu własnych badań
poglądy na określony temat
albo na logicznie powiązaną
grupę zagadnień
medycznych, w tym z
zakresu nauk
podstawowych i
humanistyki lekarskiej; duże
podobieństwo do rozdziału
podręcznika, ale opis
wzbogacony rozważaniami i
komentarzami autorskimi
lub zaproszonych
specjalistów
struktura zależna od dziedziny,
najczęściej:
1. tytuł
2. autorzy (wraz z
jednostkami badawczymi)
3. słowa kluczowe
4. streszczenie
5. wprowadzenie (wstęp),
6. działy tekstu głównego
rozbudowane
piśmiennictwo dotyczące
każdego wyrażonego
poglądu i zagadnień
szczegółowych, o których
mowa w tekście, w tym
źródła cytatów lub
opisów badań, cudzych
wniosków itp.; dbałość o
uwzględnianie
pierwszeństwa w
odkryciach (o ile to
możliwe)
Skrypt
szkic podręcznika
akademickiego,
podyplomowego,
kursowego lub do
korzystania w innym
systemie edukacji;
podręcznik niekompletny,
ale zawierający materiał
wykładany (lub potrzebny
studentom do rozumienia
wykładów) autora
struktura zależna od dziedziny,
najczęściej:
1. tytuł
2. autor (autorzy), jeśli
podręcznik ma wielu
autorów
3. streszczenie (rzadko)
4. wprowadzenie (wstęp),
5. działy tekstu głównego
piśmiennictwo
zawierające tytuły
najważniejszych
podręczników z danej
dziedziny; rzadko
szczegółowe
Monografia
duża praca (zwykle w
postaci książki lub
monotematycznego serwisu
internetowego),
omawiająca jedno
zagadnienie osiowe wraz z
jego odgałęzieniami
tematycznymi i ze
wskazówkami
interdyscyplinarnymi lub
intertekstualnymi; może
mieć charakter
dokumentarny (zestawianie
materiałów, danych,
faktów), teoretyczny
(twierdzenia, hipotezy),
eseistyczny (omawianie,
rozważania, heurystyka)
albo mieszany
struktura zależna od dziedziny,
najczęściej:
1. tytuł
2. autor (autorzy), jeśli
podręcznik ma wielu
autorów
3. wprowadzenie (wstęp)
4. działy tekstu głównego
5. zakończenie z wnioskami
rozbudowane
piśmiennictwo dotyczące
każdego wyrażonego
poglądu i zagadnień
szczegółowych, o których
mowa w tekście, w tym
źródła cytatów lub
opisów badań, cudzych
wniosków, w tym
podlegających dyskusji;
dbałość o uwzględnianie
pierwszeństwa w
odkryciach (o ile to
możliwe)

Podręcznik
(rozdział
podręcznika)
najczęściej spotykane,
powszechnie przyjęte,
oficjalne albo oparte na
ciągu własnych badań
poglądy medyczne i z
zakresu nauk pokrewnych,
w tym spychologiczne i
filozoficzne, na określony
temat albo na logicznie
powiązaną grupę zagadnień
medycznych, także z
zakresu nauk
podstawowych;
najważniejsze problemy
spotykane w praktyce i
sposoby ich rozwiązywania;
nawiązanie do dziedzin
pokrewnych; także historia
zagadnienia; pewne
podobieństwo do pracy
poglądowej, ale bez
rozważań i rozwiniętych
komentarzy odautorskich
struktura zależna od dziedziny,
najczęściej:
1. układ rytmiczny, tj. każdy
typ części podręcznika
powinien mieć te same
elementy naprowadzające
czytelnika, informujące w
jakiej części dzieła się
znajduje i jakiego rodzaju
treści (wprowadzenie,
teoria, fakty, zalecenia
praktyczne itd.) może po
niej się spodziewać (stąd
także częste stosowanie
żywych pagin, sterujących
poruszanie się po książce,
lub lotnych spisów treści w
e-booku)
2. tytuł
3. autor (autorzy), jeśli
podręcznik ma wielu
autorów
4. streszczenie (rzadko)
5. wprowadzenie (wstęp),
6. działy tekstu głównego
piśmiennictwo
podstawowe i (lub)
uzupełniające albo
dokumentujące fakty
uznane za nowe (w
następnych wydaniach
takiej pozycji
piśmiennictwa może już
nie być);
bardzo rzadko
piśmiennictwo
dokumentujące
szczegółowe poglądy
(tylko w podręcznikach o
niewielkiej objętości)
Referat naukowy praca przygotowana do
wygłoszenia na konferencji
naukowej i jednocześnie do
druku w pamiętniku
konferencji; zakres
tematyczny dowolny
1. tytuł
2. autorzy (wraz z
jednostkami badawczymi)
3. krótkie wprowadzenie
(wstęp)
4. omówienie istoty
zagadnienia (bez dygresji)
5. wnioski (zwykle w formie
wykazu twierdzeń lub
postulatów)
piśmiennictwo
odpowiednie do typu
tematycznego, zwykle
ograniczone do 10-15
pozycji
Plakat naukowy
streszczenie w postaci grafu
lub listy zdań na dużym
arkuszu, przedstawiane
alternatywnie do referatu
na konferencji naukowej;
zakres tematyczny dowolny
1. tytuł
2. autorzy
3. graficzne, tabelaryczne lub
w formie wykazu
przedstawienie istoty
zagadnienia
bez piśmiennictwa,
rzadko do 5 pozycji
Podstawowe procedury i wyposażenie laboratoryjne:
umożliwiają wykonywanie doświadczenia
ustalają warunki doświadczenia
umożliwiają komputerowe opracowanie danych
oczyszczają i dezynfekują sprzęty oraz sale labolaotryjne
Normy czystości odczynników chemicznych:
odczynniki techniczne
odczynniki czyste
odczynniki chemicznie czyste
odczynniki czyste do analizy
odczynniki spektralnie czyste do specjalnych zastosowań
Stopnie jakości wody laboratoryjnej:
typ 1 (ultraczysta, MiliQ) – R > 18
typ 2 (czysta) – R > 1
typ 3 – R > 0,05
Roztwory buforowe – mieszaniny roztworów słabych kwasów lub zasad i ich soli lub mieszaniny
roztworów dwóch soli kwasu wielowodorowego o kilku stopniach dysocjacji.
Efektywny zakres buforowania wynosi pH = pK ± 1
Pojemność buforowa – zdolność przeciwstawiania się zmianom pH po wprowadzeniu kwasu
lub zasady. Wzrasta wraz ze wzrostem stężenia buforu i maleje z rozcieńczaniem. Bufory maja
największą pojemność buforową w zakresie pH zbliżonym do pK, czyli gdy stosunek stężeń
składników buforu jest zbliżony do jedności. Miarą pojemności buforowej jest stosunek liczby moli
kwasu lub zasady, która musi być dodana do 1 litra roztworu, aby spowodować zmianę pH
o jednostkę.
Bufory lotne (mrówczan amonu, octan amonu) – umożliwiają szybkie odparowanie substancji.
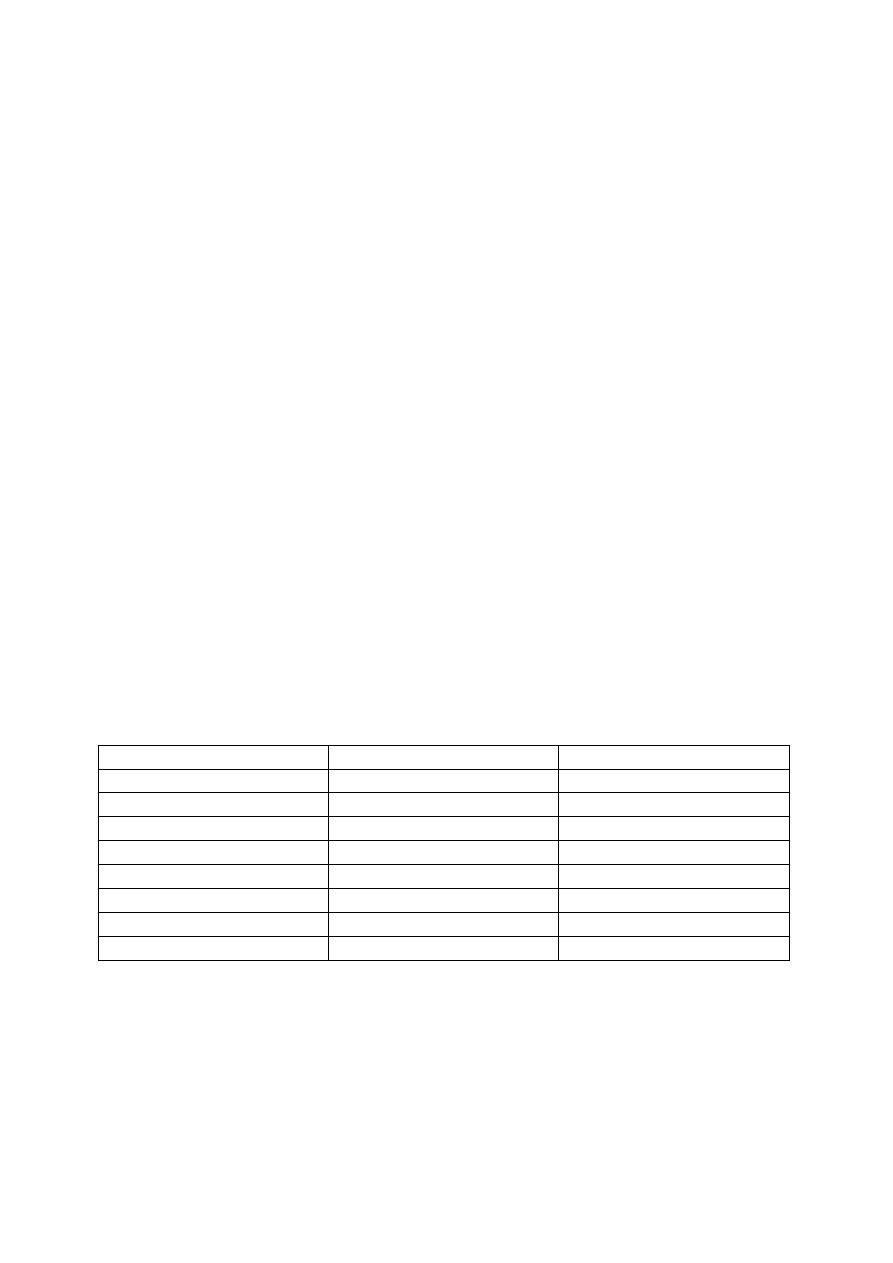
Typowe bufory stosowane w biochemii:
Efektywny zakres pH
Bufory
pK (25 C)
2,5 – 7,0
Cytrynianowe
3,13
3,0 – 4,5
Mrówczanowe
3,75
3,5 – 6,5
Bursztynianowe
4,21
4,0 – 5,5
Octanowe
4,76
6,0 – 7,0
Fosforanowe
7,20
7,5 – 9,0
Tris
8,06
8,5 – 10,0
Boranowe
9,23
9,0 – 10,5
Glicynowe
9,78
[więcej w broszurze Biological Buffer lub Stoll, V.S., Blanchard, J.S. et al. (1990) Methods Enzymol.
182, 24-38]
Dodatki do buforów:
azydek sodu
EDTA
DTT
DTE

2-merkaptoetanol
Detergenty
o Jonowe:
Kationowe (CTAB)
Anionowe (SDS)
o Niejonowe (Triton X-100)
o Obojniaczo-jonowe (CHAPS)
Usunięcie detergentu:
Wytrącanie białka
Dializa
Metody chromatograficzne
Rozcieńczenie próby
[więcej w broszurze EMD Biosciences „Guide for solubilization of membrane proteins and selecting
tools for detergent removal”]
Rodzaje elektrod:
Elektrody I rodzaju – odwracalne względem kationu (np. wodorowa)
Elektrody II rodzaju – odwracalne względem anionu (np. kalomelowa, chlorosrebrowa)
Elektrody III rodzaju – odwracalne względem kationu (np. wapniowa)
Elektrody utleniająco-redukujące (np. ninhydrynowa)
Ogniwo elektrochemiczne – układ elektroda wskaźnikowa (reagują na zmiany potencjału na obecność
jonu) – elektroda odniesienia (porównawcza, o stałym potencjale) zanurzone we wspólnym
elektrolicie.
Przygotowanie próbki:
1. Dezintegracja komórek
a. W
celu
izolacji
białek,
kwasów
nukleinowych
lub
innych
związków
niskocząsteczkowych
b. Wybór odpowiedniego materiału biologicznego i jego ochrona przed degradacją
Inhibitory proteaz
o PMSF (proteazy serynowe – 1 mM)
o Pepstein (proteazy aspartylowe – 0,1 mg/ml)
o PCMB (proteazy cysteinowe – 1 mM)
o EDTA (metaloproteazy – 5 mM)
2. Homogenizacja mechaniczna:
a. Dodatek antyspieniaczy – kontrola temperatury
b. Sonifikacja
c. Wysokie ciśnienie (prasa French’a)
d. Cykliczne zamrażanie i odmrażanie
e. Hipotoniczne bufory
f. Liza enzymatyczna komórek

3. Proces solubilizacji białek błonowych z błon komórkowych:
a. Detergenty
Wytrącanie białek i kwasów nukleinowych zależy od ich rozpuszczalności, którą warunkują m.in. pH,
temperatura, siła jonowa, czy dodatek soli. Strącanie białek może być przeprowadzane na dwa
sposoby:
Nieodwracalną agregację białek (rozfałdowanie -> denaturacja)
Odwracalna agregacja białek (strącanie białek z roztworu)
o Wysalanie białek za pomocą TCA, PEG lub siarczanu amonu
Do usunięcia soli można użycz metod:
Odsalanie SEC – chromatografia sita molekularnego, w której nanosi się na kolumnę 20-30%
jej objętości.
Dializa – metoda oczyszczania roztworów przy użyciu błony półprzepuszczalnej.
Ultrafiltracja – proces filtracji z użyciem sit molekularnych, membran i wszelkich materiałów
porowatych, o porach, których rozmiary są zbliżone do wielkości pojedynczych cząsteczek
(zwykle kilka–kilkadziesiąt nanometrów). Wielkość cząsteczek oddzielanych substancji musi
znacznie przekraczać wielkość cząsteczek rozpuszczalnika. Siłą napędową procesu jest
wysokie ciśnienie hydrauliczne roztworu rozdzielanego.
Liofilizacja – proces pozwalający na zagęszczenie próbki, ale zagęszcza także sole. Polega na suszeniu
zamrożonych substancji przez sublimację.
Chromatografia – fizykochemiczna metoda rozdzielania, w której składniki rozdzielane ulegają
podziałowi między dwie fazy: stacjonarną (nieruchomą) oraz ruchomą (poruszającą się w określonym
kierunku). Oddziaływania występują zarówno pomiędzy substancją rozdzielaną, a fazami ruchomą
i stacjonarną, jak też między oboma fazami. Równowaga podziału ma dynamiczny charakter zależny
od właściwości rozdzielanej substancji oraz rodzajów faz ruchomej i stacjonarnej, co określa
współczynnik retencji (k).
Sprawność układu chromatograficznego – zdolność kolumny do wytwarzania wąskich pików
(wyrażana liczbą półek teoretycznych). Na sprawność wpływają m.in. modyfikacje substancji
rozdzielanej, temperatura, prędkość przepływu fazy ruchomej, wielkość i kształt ziarem absorbentu
oraz jakość upakowania. Aby zwiększyć sprawność układu stosujemy ziarna absorbentu o małej
średnicy (np. 5 um) o płytkich porach oraz brak wolnych przestrzeni (tzw. dead volume) w kolumnie.
Rodzaje chromatografii [więcej w Witkiewicz Z., Klasyfikacja technik chromatograficznych]:
W zależności od rodzaju eluentu:
chromatografia cieczowa – w której eluentem jest ciekły rozpuszczalnik lub mieszanina
rozpuszczalników;
chromatografia gazowa – w której eluentem jest gaz (zwykle hel, argon lub wodór, czasem
azot);
chromatografia nadkrytyczna – w której eluentem jest substancja w stanie nadkrytycznym.

W zależności od rodzaju fazy stacjonarnej i sposobu jej przygotowania:
Chromatografia planarna
o
chromatografia cienkowarstwowa TLC (ang. thin layer chromatography) – w której
fazę rozdzielczą stanowi cienka warstwa fazy stałej naniesiona na sztywną płytkę.
Na tak spreparowaną płytkę nanosi się próbkę roztworu, po czym na skutek działania
sił kapilarnych, grawitacji lub pola elektrycznego następuje przepływ i rozdział
mieszaniny;
o
chromatografia bibułowa – w której fazę rozdzielczą stanowi pasek lub arkusz bibuły
filtracyjnej lub specjalnego typu bibuły chromatograficznej;
chromatografia kolumnowa – w której faza rozdzielcza jest umieszczona w specjalnej
kolumnie, przez którą przepuszcza się następnie roztwór badanej mieszaniny. Przepływ
roztworu przez kolumnę można wymuszać grawitacyjnie lub stosując różnicę ciśnień
na wlocie i wylocie kolumny;
chromatografia powinowactwa – w której odpowiednio spreparowana faza rozdzielcza jest
zdolna do oddziaływań chemicznych o zmiennym powinowactwie wobec rozdzielanych
substancji;
chromatografia jonowymienna – w której substancje oddziałują ze złożem za pomocą
oddziaływań jonowych.
W zależności od parametrów procesu:
HPLC
(ang.
high
performance/pressure
liquid
chromatography)
wysokosprawna/wysokociśnieniowa chromatografia cieczowa – odmiana cieczowej
chromatografii kolumnowej z użyciem eluentu pod wysokim ciśnieniem;
FPLC
(ang.
fast
protein/performance
liquid
chromatography)
szybka,
białkowa/szybkosprawna chromatografia cieczowa – odmiana HPLC działająca na niższych
ciśnieniach, stosująca prócz złóż sorpcyjnych, także zwykłe złoża typu sit molekularnych,
służąca głównie do rozdziału białek i polipeptydów. Opatentowana i wyłączna nazwa
dla firmy Pharmacia;
UPLC (ang. ultra performance liquid chromatography) ultrasprawna chromatografia cieczowa
– odmiana cieczowej chromatografii kolumnowej. Działa na wyższych ciśnieniach
i mniejszych przepływach, a kolumny mają mniejsze ziarno (1,7 – 1,8 μm). Pozwala uzyskiwać
krótsze czasy retencji i wyższe rozdzielczości. Opatentowana i wyłączna nazwa dla firmy
Waters;
GPC (ang. Gel Permeation Chromatography) chromatografia żelowa – odmiana kolumnowej
chromatografii cieczowej. Polega na rozdziale składników mieszaniny na żelu lub sitach
molekularnych w zależności od rozmiarów cząsteczek. Stosowana m.in. do określania
średnich mas cząsteczkowych polimerów.
Chromatografia adsorpcyjna:
W normalnym układzie faz – faza stacjonarna jest zawsze bardziej polarna niż faza ruchoma

W odwróconym układzie faz – faza ruchoma jest bardziej polarna niż faza stacjonarna
(pokryta „szczotką” łańcuchów alkilowych C8 lub C18)
Rodzaje chromatografii:
Sita molekularnego (ang. Size Exlusion Chromatography/ Gel Filtration)
Jonowymienna (ang. Ion Exchange Chromatography)
Chromatoogniskowanie (ang. Chromatofocusing)
Oddziaływań hydrofobowych (ang. Hydrophobic Interaction Chromatography)
Odwróconej fazy (ang. Reverse Phase Chromatography)
Powinowactwa (ang. Affinity Chromatography)
Szeregi eluotropowe – uszeregowanie rozpuszczalników według wzrastającej siły elucyjnej:
Rozpuszczalnik - Wskaźnik polarności wg Snydera (J. of Chrom. 92. 233, 1974)
n-heksan - 0,0
cykloheksan - 0,0
tetrachlorek węgla - 1,7
toluen - 2,3
dichlorometan - 3,4
butan-1-ol - 3,9
octan etylu - 4,3
chloroform - 4,4
aceton - 5,4
etanol - 5,2
metanol - 6,6
woda - 9,0
Elucja izokratyczna – niezmienny skład fazy ruchomej w trakcie całego rozdziału chromatograficznego
Elucja gradientowa – skład fazy ruchomej zmienia się systematycznie w trakcie analiz
Etapy rozdziału chromatograficznego:
1. Równoważenie kolumny (5-10 CV)
2. Aplikacja badanej próby (3-5% CV) i odpłukanie złoża
3. Elucja (wymywanie) (10-20 CV)

4. Regeneracja złoża (5-10 CV)
Budowa chromatografu HPLC:
Pompa
Dozownik
Kolumna chromatograficzna + nośnik
Detektor
Rejestrator (komputer)
Sprzęt kontrolujący warunki
Faza stacjonarna wpływa na retencje, selektywność i efektywność rozdzielania mieszanin RP HPLC.
Reaktywność żelu krzemionkowego pozwala na otrzymanie różnego rodzaju faz stacjonarnych
z różnymi grupami funkcyjnymi. Fazy stacjonarne o charakterze monomerowym są mniej trwałe,
ale zapewniają bardziej powtarzalne wyniki poprzez większą sprawność i odtwarzalność parametrów.
Fazy stacjonarne o charakterze polimerowym mają mniej zdefiniowaną strukturę, ale są bardziej
odporne i trwałe na wpływ eluentu. Typowymi rozpuszczalnikami RP HPLC są metanol, acetonitryl,
tetrahydrofuran (związki polarne!).
Sito molekularne (SEC / GF) zwykle wykorzystuje elucję izokratyczną. Pierwsze wypływają cząsteczki
większe (migrujące między ziarnami), a ostatnie najmniejsze (których droga migracji jest dłuższa, gdyż
te wchodzą do ziaren żelu). Idealna matryca powinna być:
Hydrofilowym polimerem
Obojętna chemicznie
Nienaładowana
O jednolitym rozmiarze ziaren
Matryce w chromatografii SEC:
Sephadex – usieciowany przez epichlorhydrynę dekstran
o G-10, -15, -20, -50 (odsalanie i izolacja peptydów)
o G-75, -100, -150 (dla białek i makromolekuł)
Coarse
(10 – 400 um)
Medium
(50 – 150 um)
Fine
(20 – 80 um)
Superfine
(10 – 40 um)
Rozdzielczość Szybkość
(broszura „Properties of Sephadex” – tabela „Fractionation range”)
Sepharose – polimer agarozy
o 6B, 4B (45 – 165 um), 2B (60 – 200 um) (frakcjonowanie dużych cząsteczek)
Sephacryl HR – usieciowany przez bis-akrylamid dekstran
Superose – matryca agarozowa o dużej stabilności i ujednoliconych rozmiarach ziaren
Superdex – wysoce usieciowana agaroza kowalencyjnie przyłączona do dekstranu
SEC wykorzystuje się do zastosowań preparatywnych (odsalania, wymian buforu, frakcjonowania)
oraz analitycznych (określania masy cząsteczkowej natywnego białka, kształtu białka o znanej masie

cząsteczkowej oraz badania oddziaływań międzycząsteczkowych). Technika ta jest łatwa
do zastosowania i interpretacji wyników, prosta elucja, można stosować do wszystkich rodzajów
cząsteczek, a separacja jest niezależna od składu solwentu. Ograniczeniem jest objętość nanoszonej
próbki oraz gorsza rozdzielczość w porównaniu z metodami adsorpcyjnymi.
[więcej informacji o chromatografii w broszurach GE Healthcare: „ Strategies for Protein
Purification”,
„Size
Exclusion
Chromatography”,
„Ion
Exchange
Chromatography
&
Chromatofocusing”, „Hydrophobic Interaction Chromatography”]
Elektroforeza – zjawisko elektrokinetyczne, w którym pod wpływem przyłożonego pola elektrycznego
przemieszczają się makrocząsteczki obdarzone niezrównoważonym ładunkiem elektrycznym.
Prędkość rozdziału zależy od kształtu, ładunku, rozmiaru oraz oporów ruchu środowiska.
Wysokosprawna elektroforeza kapilarna (HPCE) dzieli się na techniki:
Elektroforezę strefową – umożliwia rozdzielenie naładowanych cząstek, na podstawie różnicy
w ich ruchliwościach w wolnym roztworze. Poszczególne składniki mieszaniny ustawiają się
w oddzielnych strefach.
Elektroforezę żelową – stosowaną do rozdzielania makrocząsteczek biologicznych,
rozdzielenia mieszaniny dokonuje się na podstawie różnicy w ich wielkościach,
na odpowiednim polimerze, pełniącym rolę sita molekularnego.
Izoelektroogniskowanie – wykorzystywana jest do rozdzielania białek i peptydów
na podstawie ich punktu izoelektrycznego. Gradient pH w kapilarze powstaje dzięki
wprowadzeniu do buforu amfolitów (substancji obojnakich posiadających w swojej
cząsteczce zarówno ugrupowania o charakterze zasadowym i kwasowym).
Izotachoforeza – próbkę umieszcza się między dwoma buforami: wiodącym i zakańczającym.
Bufory dobiera się tak, aby ich efektywne ruchliwości obejmowały efektywne ruchliwości
jonów w próbce. Po przyłożeniu pola elektrycznego następuje przepływ buforu w kapilarze
i dochodzi do uszeregowania jonów zgodnie z ich ruchliwościami w przylegające do siebie
strefy, które migrują ze stałą szybkością.
Micelarna elektrokinetyczna chromatografia – umożliwia rozdzielanie cząstek obojętnych,
dzięki wprowadzeniu do roztworu buforowego związku powierzchniowo czynnego,
po przekroczeniu tzw. krytycznego stężenia micelarnego monomery surfaktanta tworzą
micele. Rozdzielanie składników mieszaniny dokonuje się na podstawie różnicy w stopniu
ich powinowactwa do miceli.
Elektroosmoza – przemieszczanie się całej masy elektrolitu wypełniającego kapilarę w kierunku
jednej z elektrod. Redukuje to czas analizy. Do detektora poruszają się więc wszystkie obecne
w roztworze jony. Podziałem tym rządzą siły wędrówki elektroforetycznej i przepływu
elektroosmotycznego.
Właściwości otoczki hydratacyjnej zależą od ładunku substancji, jej rozmiaru, a także warunków
(pH, temperatura, ciśnienie). W punkcie izoelektrycznym (pI) cząsteczka białka posiada wypadkowy
ładunek elektryczny równy zero. W związku z elektrolizą (powstanie jonów wodorowych na katodzie
i hydroksylowych na anodzie) stosuje się buforowany elektrolit. Nośniki elektryczne stabilizują

elektrolit, „zatrzymują” białko do momentu wybarwienia, warunkują lepszą separację
makrocząsteczek (sito molekularne).
Żele agarozowe używane w elektroforezie poziomej służą do separacji białek > 500 kDa
i immunoelektorforezy.
Żele poliakrylamidowe pełnią rolę sita molekularnego. Poprzez stężenie akryl amidu i proporcji
akrylamid:bisakrylamid można zmieniać wielkość porów. W detekcji można stosować światło
o długości fali >250 nm. W reakcji polimeryzacja może być inicjowana chemicznie (nadsiarczanem
amonu + TEMED) lub fotochemicznie (ryboflawina + TEMED).
Żele gradientowe charakteryzują się wraz z dystansem migracji wzrostem gęstości usieciowania,
co zwiększa dokładność żelu.
System separacji polipepytów opiera się na użyciu buforu rycynowego (a nie glicynowego).
Polimeryzacja żelu hamowana jest w pH > 6 i przez tlen cząsteczkowy.
W systemie ciągłym ten sam bufor o stałym pH znajduje się w żelu, próbie i przy elektrodach.
W systemie nieciągłym jest różnica w składzie jonów w żelu i roztworach elektrodowych, pozwala
na „zagęszczenie” próbek w początkowej fazie rozdziału. Bufor warunkuje przepływ prądu, stabilizuje
pH i odprowadza ciepło Joule’a.
Rodzaje systemów elektroforetycznych:
System Laemmeli – 5% (v/v) 2-merkaptoetanol + 2% (v/v) SDS
System Weber & Osborn’s – system ciągły z buforem fosforanowm
System Neville’a – system buforu Tris-siarczan/Tris-boran
System Ornstein-Davisa – przykład systemu nieciągłego
Bufor McLellana – do elektroforezy natywnej, pH 3,8 - 10,2 wykorzystywany do systemu ciągłego
Barwienie żelu:
Naturalne barwniki (błękit kumazyny, srebro, czerń amidowa)
Barwienie fluorochromami (bromek etydyny, barwniki fluorescencyjne)
Barwienie radioizotopowe
Barwienie enzymatyczne (substratowe)
Immunobarwienie
[więcej w broszuszach BioRad „Sub-Cell GT Agarose Gel Electrophoresis Systems” oraz „A Guide
to Polyacrylamide Gel Electrophoresis and Detection”]
Elektroforeza 2D – separacja białek w dwóch różnych kierunkach według punktu izoelektrycznego
oraz masy cząsteczkowej.
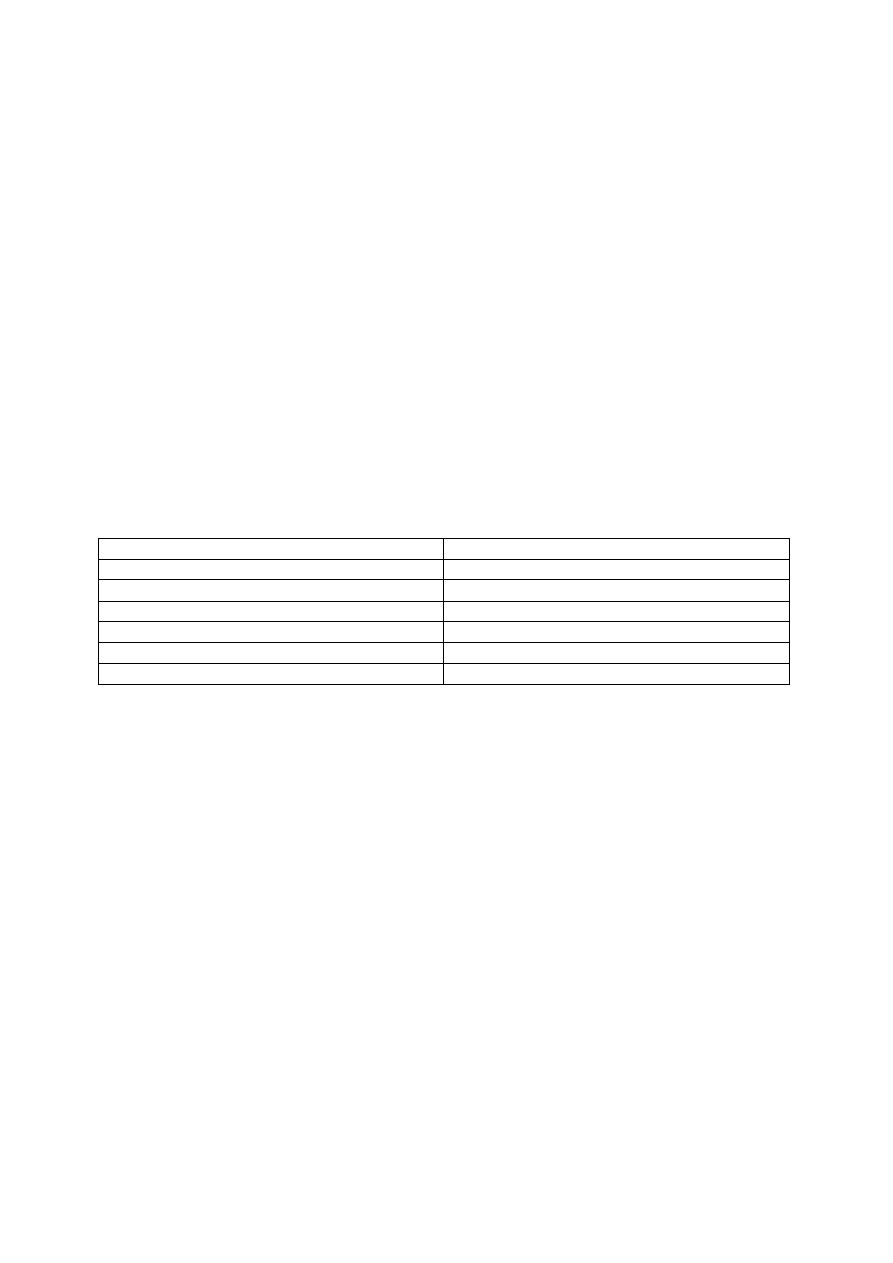
Genomika (DNA) -> Transkryptomika (mRNA) -> Proteomika (białka) –> Metabolomika
Fundamentalnym krokiem do poznania struktury białka jest enzymatyczna lub chemiczna degradacja
białkowej makromolekuły do mniejszych fragmentów, które mogą być dokładnie scharakteryzowane
w kontekście ich składu aminokwasowego i sekwencji. Obydwa sposoby fragmentacji białek muszą
spełniać kryteria stosunkowo wysokiej selektywności, powtarzalności i wydajności.
Zrealizowanie projektu dotyczącego poznania ludzkiego genomu umożliwiło naukowcom
zsekwencjonowanie wszystkich genów występujących w DNA. Zamierzeniem tych badań było
zrozumienie funkcjonowania organizmu ludzkiego. Poznanie genomu umożliwiło zdobycie wiedzy
o białkach kodowanych przez DNA, jednak nadal nie zostały wyjaśnione mechanizmy działania
komórek, organów oraz całego organizmu. Ocenia się, że w genomie ludzkim jest zapisanych około
35 tysięcy genów kodujących 200-300 tysięcy białek. Poznano strukturę oraz funkcje jedynie
nieznacznej ich części.
Przed
rozdziałem
na
podstawie
techniki
ogniskowania
izoelektrycznego
potrzebne
jest oddysocjowanie od agregatów powstałych z pasków amfolitów i denaturowanie białek.
Analiza preparatyki:
Związek niskocząsteczkowy
Sposób usunięcia
Sole, jony
Dializa, ultrafiltracja, wytrącanie, GF
Jonowe detergenty
Wytrącanie acetonem
Kwasy nukleinowe
DNAzy, RNAzy
Lipidy
Detergenty, wytrącanie acetonem
Polisacharydy
TCA, siarczan amonu
Związki fenolowe
DTT, 2-merkaptoetanol
Amfolit – związek amfoteryczny ułożony w gradientowym układzie umożliwiającym rozdział białek
na podstawie punktu izoelektrycznego. Obecnie stosuje się im mobilizowany gradient pH na paskach
(IPG strips) dostępny w różnych długościach i różnym zakresie pH.
W badaniach proteomicznych łączy się metody biochemiczne, fizykochemiczne i bioinformatyczne.
W badaniach tym możliwe jest jednoczesne analizowanie tysięcy białek z danego typu komórek
lub tkanek. Do lat 70-tych XX w. masy białek określano za pomocą takich metod jak elektroforeza,
ultrawirowanie czy chromatografia. Metody te były jednak niedokładne, błąd wynosił 10-100%.
Dopiero wprowadzenie w latach 90-tych techniki spektrometrii mas (ang. Mass Spectrometry MS)
przyczyniło się do szybkiego postępu w tej dziedzinie, dzięki wdrożeniu nowych technik jonizacji.
MS jest zdecydowanie konkurencyjna w stosunku do innych metod na ogół stosowanych
w proteomice, przede wszystkim z uwagi na możliwość analizy białek będących w bardzo małych
stężeniach, a także w skomplikowanych mieszaninach. Spektrometria masowa jest techniką
analityczną pozwalającą na precyzyjny pomiar stosunku masy do ładunku elektrycznego jonu, gdzie
przy znanym ładunku jonu daje możliwość obliczenia masy z dokładnością do pojedynczych atomów.
Typowy spektrometr masowy składa się z komory jonizacyjnej, analizatora różnicującego jony pod
względem stosunku ich masy do ładunku i detektora zliczającego liczbę jonów, które są sprzężone
z komputerowym systemem rejestracji i analizy danych.

Spektrometria masowa znalazła zastosowanie w badaniach budowy strukturalnej cząsteczek, składu
chemicznego, czystości danej substancji oraz identyfikacji zanieczyszczeń. Głównymi zaletami
spektrometrii masowej są dokładność wyznaczenia masy, rozdzielczość do kilku jednostek masy
atomowej oraz szeroki zakres stosowania, od peptydów do dużych białek (>200 kDa). Ponadto
MS umożliwia detekcję strukturalnych wariantów białek (białka zmodyfikowane posttranslacyjnie,
mutanty). Znaczenie spektrometrii mas polega na rozdzielaniu w polu elektromagnetycznym
ze względu na wartość stosunku masy m do ładunku z (m/z) zjonizowanych cząstek, jakie znajdują się
w analizatorze w wysokiej próżni. Zależnie od rodzaju analizowanych substancji, używa się różnych
technik jonizacji i rozdziału jonów. W MS do analiz białek wykorzystywane są niskorozdzielcze
analizatory (kwadrupolowe, pułapki jonowe), bądź wysokorozdzielcze (mierzące czas przelotu jonów,
cyklotronowy rezonans jądrowy z transformacją Furiera). Najbardziej rozpowszechnionymi metodami
jonizacji są desorpcja laserem w stałej matrycy MALDI (ang. matrix-assisted laser-desorption
ionization) oraz elektrorozpylanie (ESI, ang. Electrospray).
Technika ESI polega na rozpylaniu cieczy, która zawiera badaną substancję, w polu elektrycznym
o wysokim napięciu (około 1-5 kV). Na ogół metoda ta nie powoduje fragmentacji badanych
cząsteczek. Podczas wykorzystania tej techniki wzbudzania, powstające jony często mają ładunek
wielokrotny, natomiast liczba przyłączonych protonów zależna jest od liczby zasadowych
aminokwasów jakie występują w badanych cząsteczkach. Cząsteczka białka ma możliwość
przyłączenia kilkunastu, a nawet kilkudziesięciu protonów dlatego można analizować białka
o wysokich masach cząsteczkowych, które mogą wynosić kilkaset tysięcy daltonów (Da). Głównymi
zaletami tej metody są: wysoka czułość oznaczeń, możliwość analizy dużych cząsteczek do około
80 000 Da, minimalna fragmentacja próbki w czasie jonizacji, a także kompatybilność z technikami
chromatograficznymi i elektroforetycznymi. Dzięki sprzężeniu ESI, a w późniejszym czasie także
MALDI, z chromatografią cieczową uzyskano dalsze zwiększenie czułości analiz w MS.
Inną metodą jonizacji stosowaną w spektrometrach służących do analizy peptydów i białek jest
desorbcja laserowa w stałej matrycy - MALDI. W technice tej stosuje się jonizację wiązką laserową
o odpowiednio dobranej energii, by nie doprowadzać do fragmentacji cząsteczek, a tylko do ich
„wybijania” z właściwie przygotowanej matrycy. Matryca pochłania energię lasera, która z kolei
przekazywana jest do analizowanych cząsteczek. Spektrometry MALDI stosowane są do analizy białek
o masach od ~1000 Da do nawet kilkuset tysięcy Da. Technika MALDI polega na kokrystalizacji
cząstek substancji badanej z cząsteczkami matrycy absorbującej światło, zwykle kwasów
aromatycznych. Zazwyczaj jako matryce wykorzystuje się pochodne kwasu cynamonowego
(np. kwas synapinowy, kwas α-cyjano-4-hydroksycynamonowy). Cząsteczki białka w matrycy
wzbudza się za pomocą światła lasera. W wyniku tego z kryształów desorbowane są uprotonowane
cząsteczki próbki, a powstałe kationy, mające na ogół pojedynczy ładunek ([M + H]
+
), które nazywane
są jonami molekularnymi. W normalnych warunkach nie obserwuje się ich fragmentacji. W podobny
sposób działają spektrometry typu SELDI (ang. surface-enhanced laser desorption ionization).
Wykorzystują one desorbcję zjonizowanych światłem laserowym cząsteczek białek następujących
z odmiennego typu powierzchni swoiście wiążących białka. Takie powierzchnie są pokryte
substancjami będącymi odpowiednikami złóż chromatograficznych. SELDI stosowane są przede
wszystkim w proteomice klinicznej w badaniach przesiewowych. Powszechne zastosowanie
proteomiczne mają typy analizatorów m/z - czasu przelotu jonów – ToF (ang. time of flight), które
rejestrują czas przelotu zjonizowanych cząsteczek (zazwyczaj 0,01-1 ms) i są odwrotnie

proporcjonalne do wartości m/z analizowanych jonów. Z widma masowego można odczytać jaka
liczba jonów o precyzyjnie określonej masie została zliczona przez detektor.
Spektrometria mas wykorzystywana jest także do badań strukturalnych białek, a szczególnie
do ich identyfikacji. W tym celu oczyszczone białko początkowo poddaje się trawieniu na mniejsze
fragmenty przy pomocy enzymu (zastosowanie znajdują tu proteazy, jak: chymotrypsyna, trypsyna
oraz kolagenoza). Uzyskane peptydy wymywa się z żelu, a następnie poddaje analizie.
Na spektrometrze MALDI-TOF otrzymuje się widmo, które ukazuje masy poszczególnych peptydów.
Jest to mapa peptydowa, typowa dla każdego białka, co jest skutkiem specyfiki substratowej enzymu.
Jako przykład można podać trypsynę, która zrywa wiązania w białku zawsze po resztach
aminokwasowych lizyny i argininy, natomiast masy uzyskanych fragmentów zależne są od pozycji
tych dwóch aminokwasów w sekwencji białkowej. W wyniku trawienia otrzymane masy peptydów
są porównywane z masami peptydów wynikającymi z trawienia in silico sekwencji aminokwasowych,
które są obecne w białkowych bazach danych. Metoda ta umożliwia jedynie identyfikację poznanych
wcześniej białek.
[więcej o MS w Szczepaniak W., Metody instrumentalne w analizie chemicznej, PWN, Warszawa
1996]
Proteom – zbiór wszystkich białek kodowanych przez genom i produkowanych przez pojedynczą
komórkę, tkankę lub organ. Analizą proteomu zajmuje się proteomika – nauka gromadząca
informacje dotyczące identyfikacji struktur białkowych oraz zajmująca się rozpoznawaniem sekwencji
białek oraz ich funkcji.
Genom jest modelem statystycznym (niezmiennym w pewnym obszarze czasu), zaś proteom ukazuje
co dzieje się obecnie w komórce (a więc jest modelem dynamicznym). W badaniach genomiki,
transkryptomiki i proteomiki niezbędne jest korzystanie z bioinformatycznego opracowania danych.
Rozpatrywanie funkcji białka w oderwaniu od jego otoczenia może prowadzić do błędnych wniosków.
Mieszaniny białek poddawane są trawieniu proteazą (trypsyną), a następnie mogą być analizowane
w spektrometrze (metoda bottom up) lub po rozdziale MS niestrawionych proteolitycznie białek
lub peptydów o masie nieprzekraczającej 50 kDa (metoda top down).
Strategie analizy proteomu:
1. Zebranie i przechowywanie materiału
2. Rozdział białek w próbce
3. Analiza MS
4. Bioinformatyka
5. Badanie funkcji lub struktur wyższych
HPLC/MS gwarantuje wysoką czułość oraz wysoką rozdzielczość rozdziału, zapewnia ilość danych
wystarczającą do pełnej identyfikacji, jednak zastosowanie takiej techniki powoduje utratę
wprowadzonej próbki, wrażliwość na wybrane zanieczyszczenia oraz trudności w jonizacji niektórych
substancji.

Analiza mapy peptydów z udziałem systemu LC/MS:
1. Białko trawione proteazą o określonych właściwościach
2. Otrzymujemy fingerprint białka
3. Trypsyna hydrolizuje wiązanie peptydowe po C-końcowej stronie L-argininy i L-lizyny
(z wyjątkiem sytuacji, gdy kolejnym aminokwasem jest prolina)
4. Chemiczna fragmentacja (bromocyjanian, chlorowodór)
Sekwencjonowanie z udziałem tandemowej spektrometrii mas:
1. Kontrolowane rozerwanie wiązań kowalencyjnych analizowanej cząsteczki i uzyskaniu
informacji o jej strukturze na podstawie otrzymanego widma fragmentacyjnego
2. Otrzymanie etykiet peptydowych (peptide tags)
3. Uninterpreted MS serach
Bazy bioinformatyczne:
Mascot
Protein Prospector
Peptide Search
Seaquest
Protein Data Bank
KEGG
Brenda Enzyme
Działy proteomiki:
Proteomika strukturalna – poznanie struktury przestrzennej białek. Pozwala na
zlokalizowanie miejsca łączenia się leku z białkiem.
Proteomika ilościowa – charakterystyka ekspresji białek. Pozwala na weryfikowanie
markerów chorobowych (zrozumienie mechanizmów).
Proteomika funkcjonalna – ocena stanu aktywacji i wzajemnych oddziaływań między
białkami. Pozwala na nowych opracowanie technik białkowych.
Badanie funkcji białka polega na:
Identyfikacji białka
Szczegółowej charakterystyce
Próbie modyfikacji aktywności
Analizie rozmieszczenia w tkankach
Oddziaływaniu z innymi białkami
Zasady dobrej praktyki laboratoryjnej (DPL / GLP) dostępne w handbook’u Good Laboratory Practice
prezentowanym przez WHO.

Walidacja metody analitycznej – proces ustalania charakterystyki technicznej (parametrów metody)
odpowiedniego celu analitycznego. Celem walidacji jest zapewnienie, że niepewność wyniku jest
możliwa do zaakceptowania przez odbiorcę, oszacowanie wpływu różnych czynników na niepewność
wyniku. Brak jest dokładnej metodyki przeprowadzania walidacji.
Walidowane parametry:
Specyficzność – zdolność jednoznacznego określenia substancji analizowanej w obecności
składników, które mogą być zawarte w próbce
Zakres – przedział między minimalną i maksymalną zawartością/stężeniem substancji
aktywnej w badanej próbie, w którym metoda analityczna ma odpowiednią liniowość,
dokładność i precyzję [%]
Granice wykrywalności (DL) – najmniejsze stężenie (ilość) badanej substancji w próbce, które
może być wykryte, lecz niekoniecznie oznaczone z odpowiednią dokładnością
Granice oznaczalności (QL) – najmniejsze stężenie (ilość) badanej substancji w próbce, jaka
może być ilościowo oznaczona z odpowiednią dokładnością i precyzją
Dokładność – zgodność między wartością rzeczywistą (zawartością, stężeniem) a wartością
będącą wynikiem analizy [błąd systematyczny]
Zakres liniowości – zdolność do uzyskiwania wyników pomiaru analitycznego wprost
proporcjonalnych do stężenia substancji w oznaczanej próbce, w określonym zakresie
[y = ax + b]
Precyzja – stopień zgodności między pojedynczymi wynikami analizy (rozrzut wyników), gdy
dana procedura jest stosowana dla wielokrotnie powtarzanych, niezależnych oznaczeń
jednorodnej próbki [odchylenie standardowe]
Powtarzalność - wyraża precyzję oznaczeń wykonanych w krótkim odstępie czasu, przez tego
samego analityka i w tych samych warunkach [współczynnik zmienności]
Czułość
Selektywność
Odzysk
[więcej informacji w projekcie rozporządzenia Ministra Zdrowia w sprawie wymagań Dobrej Praktyki
Wytwarzania z 8 lutego 2015 r.]
Wyszukiwarka
Podobne podstrony:
analiza notatki 3 id 559208 Nieznany (2)
analiza ilosciowa 6 id 60541 Nieznany (2)
BIOCHEMIA 5 2 id 86299 Nieznany
biochemia3 id 86647 Nieznany (2)
Biochemia(1) id 86587 Nieznany
Analiza struktury id 61534 Nieznany (2)
analiza ilosciowa 2 id 60539 Nieznany
Analiza czynnikowa id 59935 Nieznany (2)
Darfur analiza kryzysu id 13186 Nieznany
Analiza Finansowa 3 id 60193 Nieznany (2)
Analiza finansowhga id 60398 Nieznany (2)
IMW W02 analiza stanow id 21233 Nieznany
biochemia id 86123 Nieznany (2)
Analiza krancowa id 60743 Nieznany (2)
analiza skupien id 61367 Nieznany
Analiza termiczna id 61671 Nieznany (2)
biochemia4 id 86651 Nieznany
analiza wzory id 61812 Nieznany (2)
biochemia 6 id 86300 Nieznany (2)
więcej podobnych podstron