Spektrofotometria.
Podstawy spektrofotometrii absorpcyjnej UV/VIS
Spektroskopia jest interdyscyplinarną dziedziną nauki zajmującą się analizą widmową - jej podstawami teoretycznymi, sposobami otrzymywania widm oraz korelacją widm z budową badanego układu. W jej ramach opracowane zostały metody badawcze nazywane metodami analizy spektroskopowej. Zalicza się do nich: spektrometrię optyczną, spektrometrię magnetycznego rezonansu jądrowego NMR, spektrometrie elektronów i jonów.
Najwcześniej rozwinęła się spektrometria optyczna; w jej skład wchodzą: spektrofotometria absorpcyjna w zakresie widzialnym (VIS), nadfioletu (UV) i podczerwieni, spektrometria Ramana oraz spektrofłuorymetria.
Spektrofotometria jest zespołem optycznych metod analitycznych w których podstawowym układem badanym jest cząsteczka. Powiązanie widma z budową cząsteczki, ze zmianami zachodzącymi w układzie elektronów na jej orbitalach wymaga znajomości przyjętych modeli, praw i reguł mechaniki kwantowej. Zostaną one w dużym skrócie i uproszczeniu przedstawione w dalszej części wykładu..
Budowa atomu
Pojęcie atomu liczy sobie ponad 2400 lat. Starożytni filozofowie greccy wysuwali tezę, że materia ma strukturę ziarnistą. Wszystkie ciała zbudowane są z atomów które, według nich, stanowią najmniejsze, niepodzielne cząstki materii. Odkrycia liniowego widma pierwiastków, promieni katodowych {elektronów), promieniotwórczości naturalnej oraz jądra atomowego w drugiej połowie XIX i na początku XX wieku spowodowały powrót koncepcji atomowej budowy materii; by wytłumaczyć obserwowane zjawiska należało rozszerzyć teorię o wewnętrzną strukturę atomów.
Pierwszym modelem budowy atomu była teoria budowy atomu wodoru duńskiego fizyka Nielsa Bohra opublikowana w 1913 r. Opiera się ona o wcześniejszy, tak zwany planetarny model atomu zaproponowany w !911 r. przez Ernesta Rutherforda.
Model Bohra atomu wodoru
W modelu tym atom zbudowany jest z ciężkiego, dodatnio naładowanego jądra i krążącej wokół niego po orbicie kołowej lekkiej naładowanej ujemnie cząstki — elektronu. Aby wyjaśnić trwałość takiego układu Bohr, jako pierwszy, nałożył dodatkowe warunki kwantowe tzw. postulaty, będące uogólnieniem teorii Plancka kwantowego oscylatora liniowego.
I postulat
W atomie wodoru elektron porusza się wokół jądra wyłącznie po. orbitach kołowych, dla których jego moment pędu (kręt) jest całkowitą wielokrotnością pewnej stałej:
We wzorze: m oznacza masę elektronu. v -jego prędkość na orbicie o promieniu r, n jest liczbą całkowitą, zaś h - stałą Plancka równą 6.625x10-34 Js). Tak zdefiniowane orbity nazywamy dozwolonymi.
Powyższy warunek umożliwia obliczenie promieni kolejnych orbit; są one proporcjonalne do kwadratu numeru orbity. Poruszający po n-tej orbicie dozwolonej elektron znajduje się w stanie stacjonarnym, czyli ma stałą energię. Jest ona odwrótnie proporcjonalna do n2. Jeśli elektron krąży na pierwszej orbicie, atom ma najniższą z możliwych energię. Taki stan nazywamy stanem podstawowym atomu.
Inne stany dozwolone nazywamy stanami wzbudzonymi. Wprowadzenie atomu w stan wzbudzony wymaga dostarczenia mu energii z zewnątrz.
Elektronowi poruszającemu się po orbicie z prędkością v można przypisać elementarny prąd o wartości
I = e/T=ev/2ၰ r = (e/2m) (mvr/ ၰ r2)
gdzie: T— okres obiegu elektronu, e - ładunek elektronu, m -jego masa, r - promień orbity kołowej.
Jeżeli wprowadzimy oznaczenia: mvr = K (orbitalny kret elektronu), ၰr2= S (pole powierzchni ograniczone orbitą) to wzór można będzie zapisać w postaci:
I = (e/2m)(K/S lub I x S = (e/2m)K
Wyrażenie I x S = pm oznacza moment magnetyczny obwodu z prądem; stosując ten wzór do atomu jako do orbitalnego momentu magnetycznego mikroobwodu z prądem, otrzymamy:
pm = (e/2m)K
Wzór ten łączy orbitalny moment pędu K elektronu ze związanym z nim orbitalnym momentem magnetycznym Uwzględniając ujemny znak ładunku elektronu wyrażenie to można zapisać w postaci wektorowej.
Wektory pm i K mają zwroty przeciwne a wielkość
ၭB = e/2m = 9,222741 x 10-24 Am2
nazywana jest magnetonem Bohra i przyjęta jako atomowa jednostka momentu magnetycznego.
Orbitalne momenty: pędu i związany z nim magnetyczny są skwantowane. Oznacza to. że ich rzuty na wyróżniony w przestrzeni kierunek przyjmują ściśle określone wartości będące wielokrotnością wielkości podstawowej (magnetonu Bohra). Wektory momentów tworzą więc z wyróżnionym kierunkiem ściśle określone kąty.
II postulat
Przejściu elektronu z n-tej orbity stacjonarnej na m-tą towarzyszy emisja lub pochłonięcie kwantu energii równe różnicy energii elektronu na tych orbitach:
hၮ = En - Em
ၮ jest częstotliwością promieniowania elektromagnetycznego absorbowanego lub emitowanego przez atom. Z powyższego warunku można wyliczyć z dużą dokładnością częstotliwości wszystkich linii widmowych wodoru.
Teoria Bohra dobrze opisuje atom wodoru i układy wodoropodobne (jednoelektronowe), natomiast nie daje poprawnych wyników ilościowych przy zastosowaniu do atomów wieloelektronowych. Udoskonalana w latach następnych np. przez Sommerfelda, została współcześnie zastąpiona przez modele całkowicie oparte o mechanikę kwantową.
Ujęcie kwantowe
W ujęciu kwantowej mechaniki falowej elektronowi przypisuje się właściwości falowe, co prowadzi do zastąpienia jego ruchu po torze przez określenie prawdopodobieństwa (ściślej gęstości prawdopodobieństwa) znalezienia się elektronu w danym punkcie przestrzeni wokół jądra.
Można powiedzieć, że porusza się on wokół jądra w określonych obszarach nazywanych orbitalami - obrazowo określa się je jako chmury elektronów.
Orbitale opisane są liczbami kwantowymi, które wyznaczają energię elektronu na orbitalu nazywanym też poziomem energetycznym, stanie energetycznym lub stanie kwantowym.
Własny moment pędu (spin) elektronu i związany z nim własny moment magnetyczny (spin magnetyczny) elektronu pojawiają się w opisie kwantowym jako kwantowo-mechaniczna właściwość elektronu. Zwroty wektorów momentów są przeciwne, a stosunek spinu magnetycznego do spinu jest dwukrotnie większy niż w przypadku momentów orbitalnych.
Emisja i absorpcja energii związane są z przejściami elektronu między orbitalami podobnie jak w modelu Bohra.
Stan elektronu w atomie określają następujące liczby kwantowe:
n - główna liczba kwantowa. Decyduje ona o energii elektronu na orbitalu; zespól możliwych do obsadzenia przez elektron stanów dla danej liczby n nazywa się powloką elektronową i oznacza dużymi literami: K dla n = 1; L dla n = 2; M dla n = 3; N dla n = 4; O dla n = 5;
l - poboczna liczba kwantowa. Określa ona orbitalny moment pędu elektronu:
z którym związany jest kształt orbitali (kształt chmury elektronowej). Poboczna liczba kwantowa l przyjmuje wartości l = 0, 1, 2,....n-1.
Stany energetyczne odpowiadające danej liczbie l nazywa się podpowłokami (podpoziomami) i oznacza:
dla l = 0 podpowłoka s,
dla l = 1 podpowłoka p,
dla l = 2 podpowłoka d,
dla l = 3 podpowłoka f;
m - magnetyczna liczba kwantowa określa ilość orbitali w podpowłoce dla danej wartości l i ich wzajemne położenie. Odpowiada za ustawienie względem wyróżnionego w przestrzeni kierunku (na przykład zewnętrznego pola elektrycznego lub magnetycznego) wektora orbitalnego momentu magnetycznego, a więc i wektora orbitalnego momentu pędu - czyli za tzw. kwantowanie przestrzenne.
Magnetyczna liczba kwantowa może przyjmować dowolne wartości całkowite z przedziału (+1, 0 -1);
s - spinowa liczba kwantowa. Określa ona rzut spinu (a więc i spinu magnetycznego) na wyróżniony kierunek w przestrzeni. Na orbitalu mogą znajdować się najwyżej dwa elektrony o spinach przeciwnych, tzw. elektrony sparowane. Spinowa liczba kwantowa przyjmuje zatem dwie wartości -1/2 i +1/2, a wartość spinu elektronu wynosi
Energia na orbitalu (energetyczny stan elektronu) jest zależna przede wszystkim od głównej liczby kwantowej n. Całkowitą energię określają cztery liczby kwantowe: n, l, m, s.
W atomie nie może być dwóch elektronów w tym samym stanie (o takich samych liczbach kwantowych) tę prawidłowość odkrył Wolfgang Pauli i przedstawił w postaci prawa nazywanego zakazem Pauliego.
Wiązania chemiczne
Podstawą wszystkich wiązań chemicznych jest posiadanie wspólnych elektronów wiążących połączone atomy.
Atomy musza zbliżyć się do siebie na tyle, aby ich orbitale walencyjne nałożyły się na siebie.
Elektrony na orbitalach atomów muszą być tak rozmieszczone, aby mogły z nich powstać wiążące pary elektronów sparowanych.
Powstanie wiązania chemicznego np. w cząsteczce wodoru przedstawić można obrazowo za pomocą rysunku orbitali lub przy pomocy symboli kwadratów.
Jeżeli miejscem maksymalnego występowania elektronów w cząsteczce np. dwuatomowej są jej krańce, to odpowiadający orbilal cząsteczkowy nosi nazwę orbitalu antywiążącego ၳ. Orbitale wiążące decydują o trwałości cząsteczki, orbitale antywiążące osłabiają wiązanie znosząc przyciągające działanie chmury elektronowej między jądrami atomów, obiekty wiążące i antywiążące są tym większe im bardziej przenikają sie orbitale.
Jeżeli łączą się atomy których nakładające się orbitale zawierają po jednym elektronie, utworzone wiązanie chemiczne nosi nazwę wiązania kowalencyjnego. Jest to wiązanie pojedyncze. Jego długość (odległość miedzy jądrami) ustala się po zrównoważeniu sił przyciągania i odpychania między ładunkami atomów.
Istnieją także wiązania chemiczne utworzone tylko przez jeden elektron, Przykładem może być wiązanie w kationie H2+. który powstaje w wyniku nałożenia orbitalu wodoru zawierającego jeden elektron z pustym orbitalem kationu wodoru-(protonu), (pozyton).
Przyjmuje się. że w tworzeniu wiązania chemicznego uczestniczą tylko orbitale powłok walencyjnyeh; przenikanie orbitali powłok wewnętrznych zachodzi w minimalnym stopniu
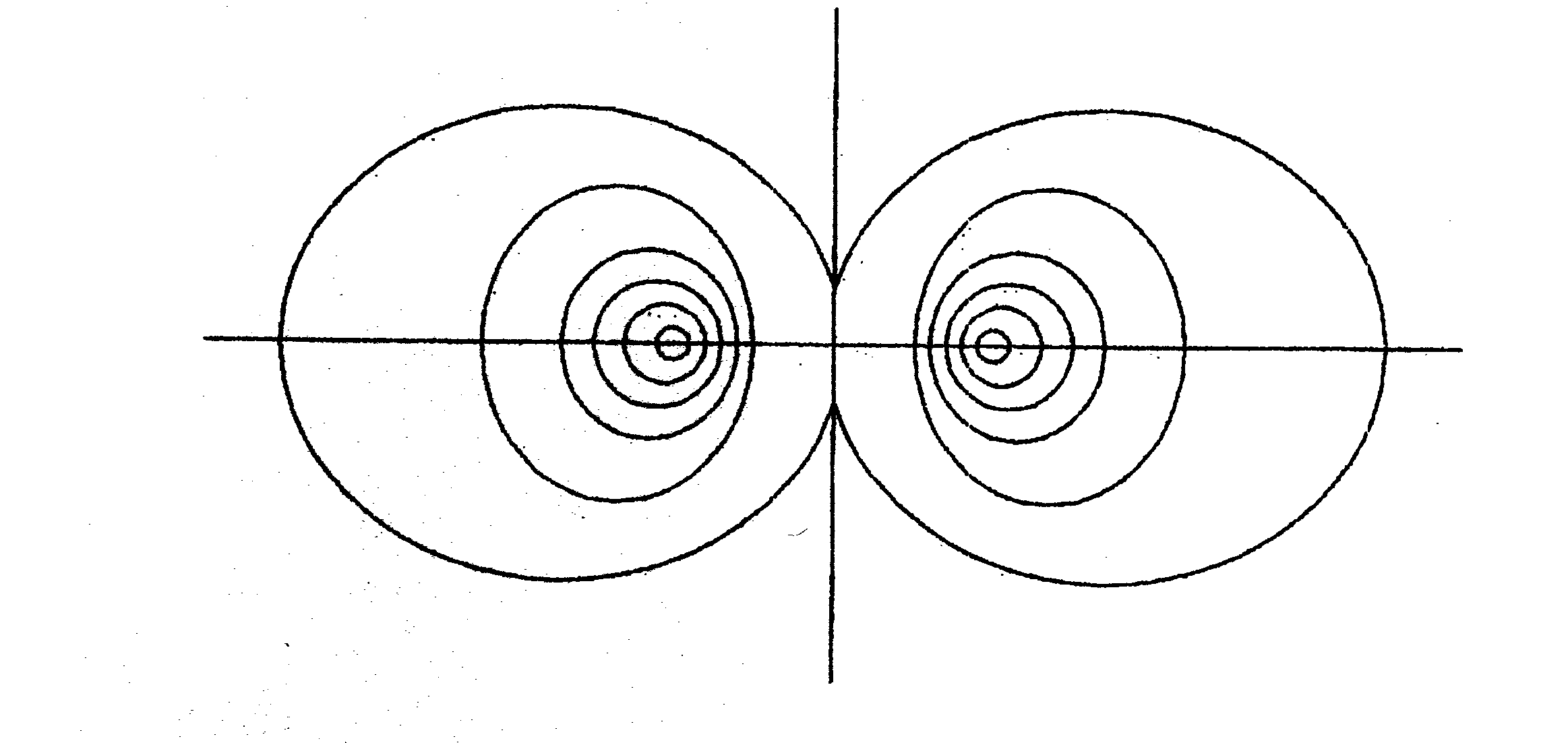
Rozmieszczenie gęstości chmury elektronowej w stanie przyciągania atomów wodoru w cząsteczce H2.
Poza wiązaniem pojedynczym istnieją także wiązania podwójne i potrójne: powstają wtedy gdy miedzy atomami dochodzi do nałożenia się dwóch (trzech) par orbitali i powstają dwie (trzy) wspólne pary elektronów. Dalej zamieszczone zostaną przykłady wiązań pojedynczych, podwójnych i potrójnych.
W cząsteczce wodoru istnieje proste wiązanie chemiczne, ponieważ każdy alom wodoru posiada konfigurację elektronów 1s1. Oznacza to, że między atomami wodoru może dojść do nałożenia się jedynie dwóch orbitali przy powstaniu jednej pary elektronów uwspólnionych. Miejscem maksymalnego prawdopodobieństwa występowania elektronów uwspólnionych jest linia łącząca oba jądra atomowe
Wiązanie o takim rozkładzie przestrzennym uwspólnionej pary elektronów nosi nazwę wiązania sigma ၳ, a odpowiadający temu wiązaniu orbital cząsteczkowy ma charakter orbitalu wiążącego er.
Oprócz orbitalu (wiązania) sigma występują jeszcze orbitale (wiązania) pi ၰ, tau ၴ, delta ၤ również jako crbitale wiążące i anty wiążące.
Wiązanie ၰ charakteryzuje się występowaniem maksymal-nego zagęszczenia uwspólnio-nych par elektronów nad linią łączącą jądra atomów, na płaszczyźnie przechodzącej przez linię łączącą oba jądra.
Obok schemat powstania takiego wiązania w wyniku nałożenia się dwóch orbitali p.
![]()
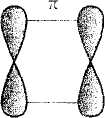
a) b)
a) powstawanie wiązania ၰ;
b) schemat tego wiązania
Wiązanie ၰ powstaje na ogół dopiero po powstaniu wiązania sigma.
Na schematach obok przedstawione są wiązania podwójne i potrójne na przykładzie cząsteczki etenu i cząsteczki azotu.
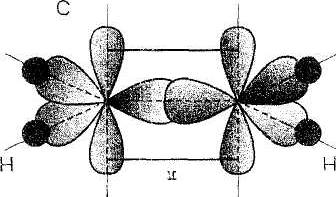
Powstawanie wiązań w cząsteczce etenu.
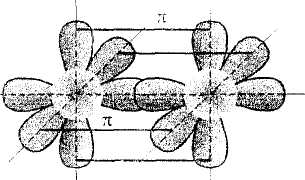
Powstawanie wiązań w cząsteczce azotu.
Cechą charakterystyczną wiązania chemicznego jest także odległość jąder połączonych atomów, tzw. długość wiązania.
Długość wiązania chemicznego zależy nie tylko od wielkości atomu, ale także od typu wiązania między atomami. Wraz z rosnącą wielokrotnością wiązania chemicznego między jednakowymi atomami dochodzi do skracania wiązania chemicznego.
Poza wiązaniami pojedynczymi, podwójnymi i potrójnymi istnieje szereg wiązań o charakterze przejściowym. Liczba wyrażająca ilość par elektronów tworzących w rzeczywistości wiązanie chemiczne nazywa się rzędem wiązania.
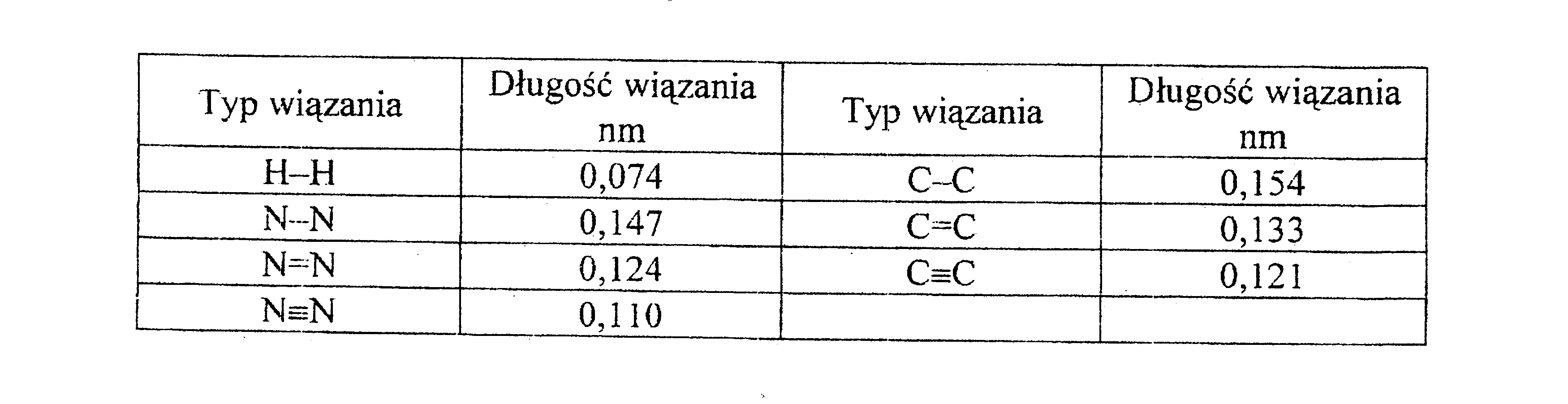
Długość wiązania podwójnego jest mniejsza niż wiązania pojedynczego, ale większa niż wiązania potrójnego.
Widma absorpcyjne w zakresie UV
Natężenie promieniowania monochromatycznego I0 ulega osłabieniu do wartości It, przy przejściu przez ośrodek absorbujący.
Następuje to przede wszystkim w wyniku absorpcji promieniowania przez układ elektronów, również częściowo na skutek rozproszenia promieni na pojedynczych cząsteczkach i ich skupiskach oraz na skutek odbicia promieniowania od ścianek naczynia pomiarowego (pomiary w zakresie widzialnym i nadfioletu prowadzi się najczęściej dla substancji ciekłych lub rozpuszczonych).
Stosunek I0 / It nazywany był przepuszczalnością a obecnie transmitancją T; transmitancja wskazuje jaka część promieniowania padającego została przepuszczona przez roztwór. Logarytm dziesiętny odwrotności transmitancji określony został mianem absorbancji A.
A = log I0 / It lub A = log l/T.
W sensie fizycznym absorbancja jest miarą strat energii w ośrodku absorbującym w porównaniu do strat w ośrodku stanowiącym odnośnik.
Jeżeli transmitancja wyrażona jest w procentach, to absorbancję można wyrazić wzorem:
A=log100/T.
zależność odwrotną podaje wzór: T= 1/ 10A
a w procentach: T=100/10A
Oprócz terminu absorbancja używane są także inne nazwy: wartość absorpcji, ekstynkcja lub gęstość optyczna.
W podręcznikach podaje się czasami pięć szczegółowych praw odnoszących się do absorpcji. W tym wykładzie zostaną podane dwa prawa najbardziej przydatne,
Prawo Bouquera-Lamberta-Beera-Waitera
Prawo addytywności absorbancji
Prawo Bouquera-Lamberta-Beera-Waitera nazywane prawem absorpcji podaje zależność między absorbancja, stężeniem roztworu i grubością warstwy absorbującej :
A = ၡcl
gdzie: ၡ jest współczynnikiem absorpcji, c stężeniem roztworu, l grubością warstwy absorbującej.
Współczynnik absorpcji a zależnie od zastosowanych jednostek stężenia roztworu nosi nazwę współczynnika absorpcji właściwej lub molowego współczynnika absorpcji :
jeżeli stężenie wyrażone jest jako stężenie masowe ၲ (stosunek masy składnika do objętości roztworu zawierającego ten składnik) to:
[a]= [g-1cm-1];
współczynnik ၡ nosi nazwę współczynnika absorpcji właściwej.
jeżeli stężenie substancji wyrażone jest w molach/dm3 wtedy współczynnik absorpcji oznaczany jest przez ၥ i nosi nazwę molowego współczynnika absorpcji: [ၥ]=[M-1cm-1]. Jest on liczbowo równy absorbancji, jeżeli roztwór ma stężenie mol/dm3 a światło monochromatyczne przechodzi przez kuwetę o grubości 1 cm.
Wartość molowego współczynnika absorpcji zależna jest od długości fali padającego promieniowania: doświadczalnie wyznacza się go przy długości fali odpowiadającej maksimum absorpcji promieniowania ၬm. Dla tej samej substancji współczynnik ten może mieć kilka wartości, jeżeli widmo zawiera kilka maksimów.
Mając do wyboru kilka wartości współczynnika ၥ należy do pomiarów wybrać wartość największą i związaną z nią analityczną długość fali), ponieważ zwiększa to czułość metody.
Miarą czułości metody jest stosunek ၄A/၄c, to znaczy stosunek przyrostu absorbancji do przyrostu stężenia wywołującego przyrost absorbancji.
Im wyższy jest współczynnik absorpcji, tym stężenia odpowiadające dopuszczalnej dolnej granicy absorbancji tj. najmniejszej mierzonej wartości są niższe.
Dolna dopuszczalna granica absorbancji zależy od dopuszczalnego błędu oznaczenia.
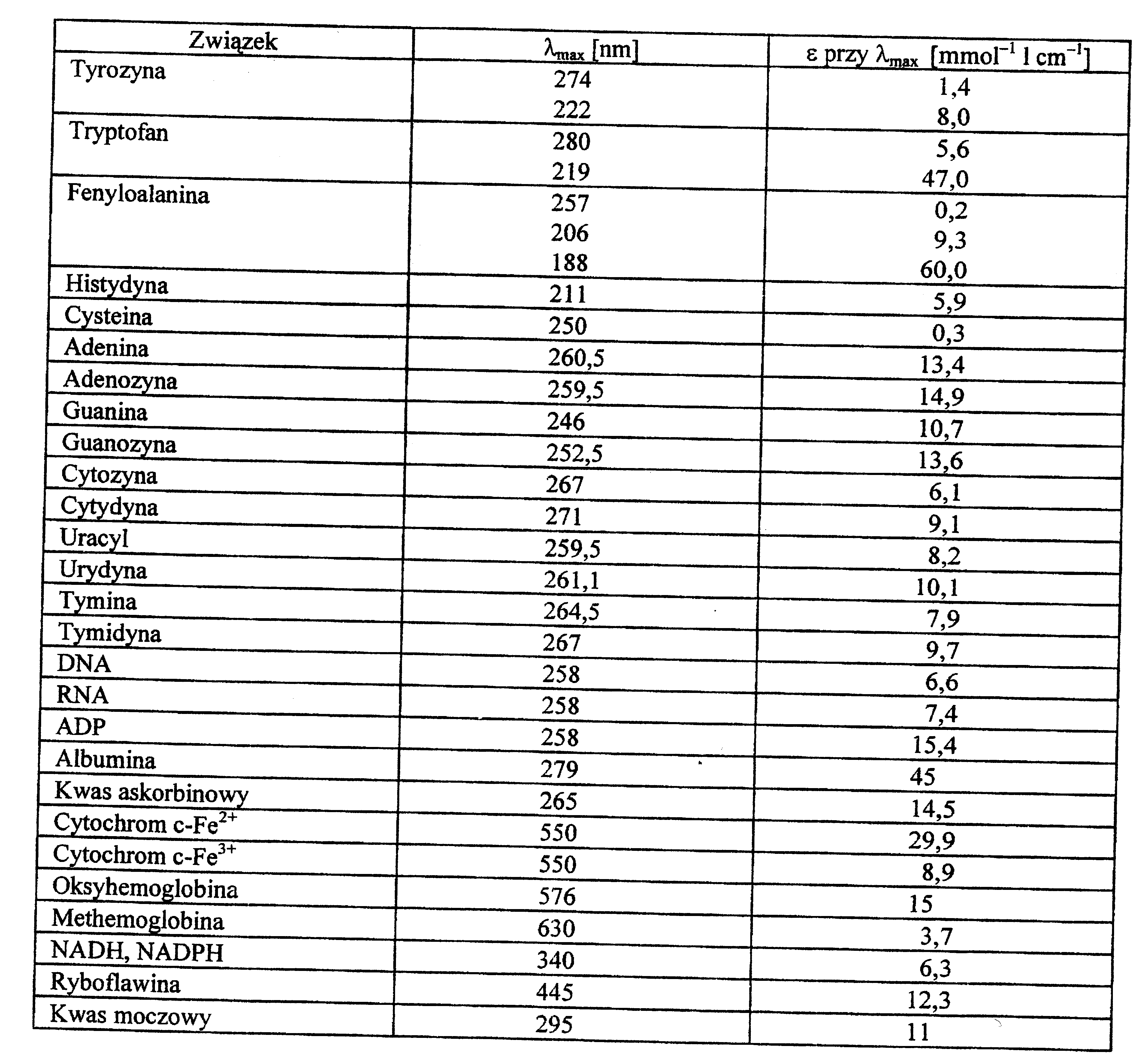
Maksima pochłaniania i molowe współczynniki absorpcji niektórych biologicznie ważnych chromoforów podane są w tabeli poniżej
Prawo addytywności absorbancji: dotyczy roztworów i mieszanin wieloskładnikowych - absorbancja całkowita roztworu jest równa sumie absorbancji poszczególnych składników.
Musi być przy tym spełniony warunek brak oddziaływań między składnikami środowiska absorbującego.
Najprostszym układem wieloskładnikowym jest układ dwuskładnikowy. Aby oznaczyć dwa składniki jednocześnie układ musi spełniać poniższe warunki:
prawo absorpcji dla badanego zakresu długości fal i stężeń,
prawo addytywności absorpcji
maksima absorpcji składników nie mogą pokrywać się co pozwoli wybrać dwie analityczne długości fal,
nie ma innych składników absorbujących.
Maksima pochłaniania i milimolowe współczynniki absorpcji niektórych biologicznie ważnych chromoforów.
Odstępstwa od praw absorpcji:
Liniowa zależność absorbancji w funkcji stężenia badanej substancji jest zachowana jedynie dla niskich stężeń roztworów c < 10-2 mol/dm3.
Przy wyższych stężeniach ujawnia się zależność molowego współczynnika absorpcji od współczynnika załamania światła; ten ostatni ma inną wartość w próbce niż współczynnik załamania rozpuszczalnika.
Czynnikiem wprowadzającym zakłócenia są również wzajemne oddziaływania między cząsteczkami prowadzące do deformacji iub tworzenia agregatów.
W roztworach kwasów i zasad, zależnie od pH mogą ustalać się różne stany równowagi kwasowo-zasadowej (forma zdysocjowana i niezdysocjowana). Dla takiego układu prawo absorpcji jest spełnione tylko w ściśle określonym pH roztworu (i przy stałej sile jonowej).
Niedoskonałości w budowie aparatury są kolejną przyczyną nieliniowości mierzonej absorbancji próbki.
Zbyt duża szerokość spektralna wiązki i zbyt duży poziom światła rozproszonego, niska czułość detektora i nieliniowa zależność między wielkością sygnału detektora i natężeniem padającego promieniowania
Te przyczyny instrumentalne ograniczają znacznie zakres stężeń, które możemy stosować.
Ograniczenia możemy ominąć stosując przyrząd wyższej klasy, co nie zawsze jest wykonalne.
W każdym przypadku należy upewnić się czy możemy pracować w danym zakresie stężeń sprawdzając zakres stosowalności naszego układu do praw absorpcji.
Parametry widm absorpcyjnych:
Krzywą absorpcji zwaną inaczej widmem absorpcyjnym otrzymuje się w postaci wykresu zależności natężenia wiązki promieniowania elektromagnetycznego po przejściu przez absorbent w funkcji długości fali padającego na absorbent promieniowania.
Zależnie od potrzeb osie układu współrzędnych można opisać przy pomocy innych wielkości, przy czym wykres tak opisany również nazywany jest widmem.
Najczęściej na osi odciętych odkłada się długość fali ၬ, częstotliwość ၮ lub liczbę falową, natomiast na osi rzędnych - przepuszczalność (ang. transmittance) T, absorbancje A lub molowy współczynnik absorpcji ၥ; czasami spotkać można logarytmy dwóch ostatnich wielkości.
Widmo cząsteczkowe (w stanie roztworu) ma skomplikowany kształt, zawiera kilka lub wiele maksimów i minimów zawierających informację o zmianach zachodzących w układzie wewnętrznym cząsteczki.
Cząsteczka jest układem atomów w których zachodzi jednocześnie wiele ruchów: jądra atomów wchodzących w skład cząsteczki wykonują ruchy oscylacyjne wokół wiązań chemicznych, elektrony na orbitalach przemieszczają się zależnie zaabsorbowanej lub wyemitowanej energii, cząsteczka zaś jako całość podlega ruchom rotacyjnym polegającym obrotach wokół osi przechodzącej przez jej środek masy.
Powstanie widma absorpcyjnego wiąże się z zaabsorbowaniem energii: jej wielkość decyduje, jakiego rodzaju przejście (ruch) nastąpi i w jakim zakresie fal (obszarze widmowym) będzie rejestrowany sygnał spektralny.
Energia przejść elektronowych jest wielokrotnie większa od energii oscylacyjnej która, z kolei, jest większa od energii rotacyjnej.
Różnica energii między poziomami elektronowymi wynosi kilka eV, między poziomami oscylacyjnymi dziesiąte części eV, a między poziomami rotacyjny tysięczne części eV.
Przejścia elektronowe może wywołać jedynie absorpcja promieniowania z zakresu widzialnym i nadfioletu.
Zmiana rozkładu ładunków w cząsteczce spowodowana absorpcją promieniowania powoduje również zmiany w jej ruchach oscylacyjnych i rotacyjnych.
W efekcie otrzymuje się złożone widmo elektronowo-oscylacyjno-rotacyjne.
Na ogół widma te można wyodrębnić stosując wąskie zakresy długości fal i aparaturę o dużej zdolności rozdzielczej, a także badając widma w różnych stanach skupienia cząsteczek.
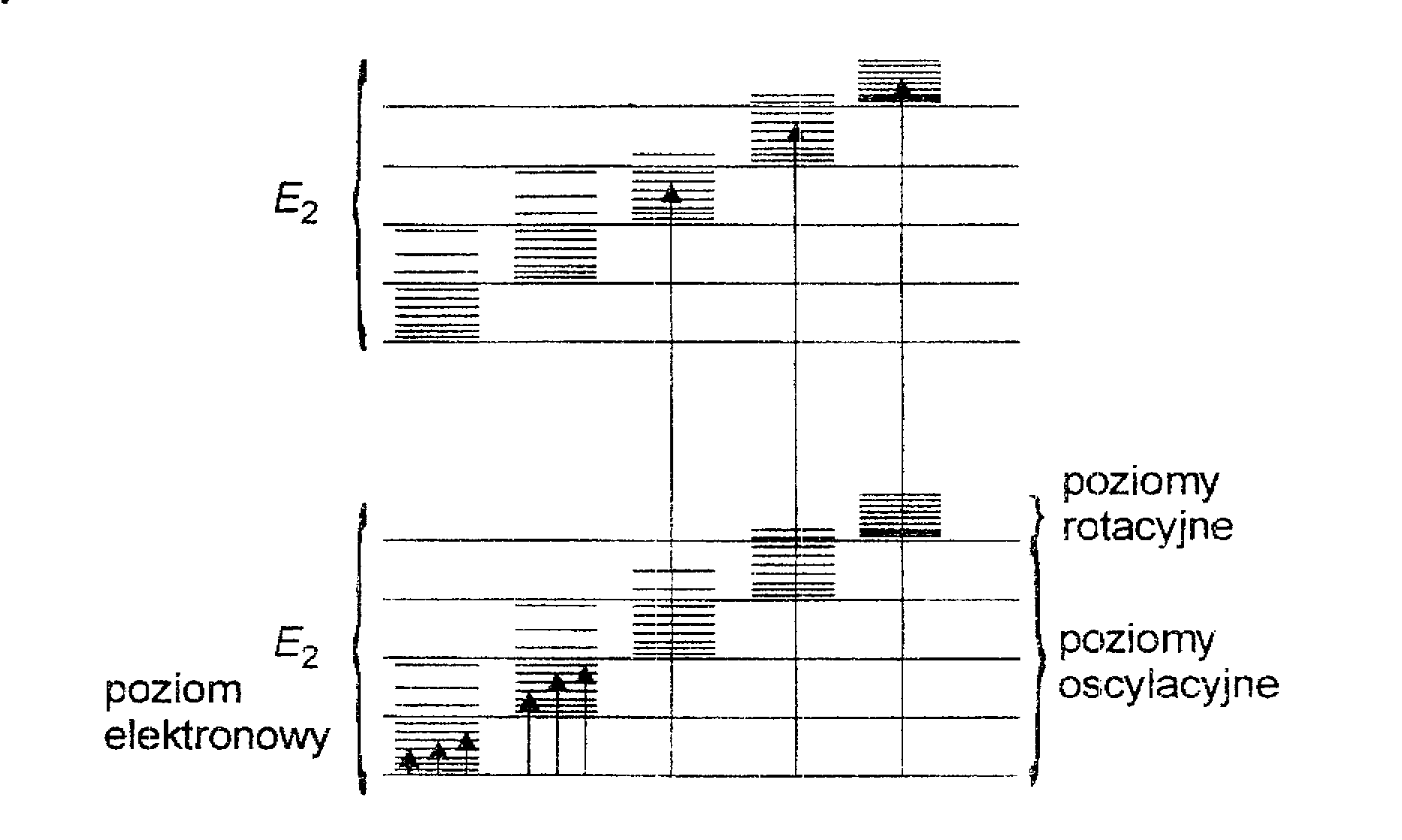
Schemat poziomów energetycznych dwuatomowej cząsteczki z zaznaczeniem poziomów elektronowych, oscylacyjnych i rotacyjnych.
Każdemu stanowi elektronowemu odpowiada układ poziomów oscylacyjnych, a każdy poziom oscylacyjny obejmuje układ poziomów rotacyjnych.
Na skutek nakładania się tych przejść energetycznych składa się z szeregu pasm oscylacyjno-elektronowych, które obejmują blisko siebie położone ale różnie odległe pasma rotacyjne.
Otrzymujemy rozmyte widmo elektronowe które, jak wspomniano wyżej, można rozdzielić.
Pobudzenie poziomów rotacyjnych zachodzi pod wpływem promieniowania z zakresu dalekiej podczerwieni i mikrofal.
Widma rotacyjne mają zastosowanie np. przy oznaczaniu odległości międzyjądrowych.
Elektronowe widma absorpcyjne umożliwiają m.in. określenie wiązań wielokrotnych, liczbę i położenia podstawników a także ich wzajemne oddziaływania.
Przejścia elektronowe w widmie absorpcyjnym
Absorpcja promieniowania w zakresie UV/V1S związana jest z przejściami elektronów walencyjnych wiązań pojedynczych sigma, wiązań wielokrotnych pi oraz elektronów wolnych par elektronowych n — nie należących do żadnego wiązania.
Wyróżnia się trzy typy przejść elektronowych:
przejścia z wiążących orbitali w stanie podstawowym na odpowiadające im orbitale antywiążące zapisywane jako a—> ၳ* i ၰ —> ၰ*; .
przejścia z niewiążącego orbitalu atomowego na molekularny orbital o wyższej energii zapisywane jako przejścia n -> ၳ* i n -> ၰ*;
przejścia z orbitalu w stanie podstawowym na orbital o bardzo wysokiej energii prowadzące do jonizacji cząsteczki; pasma tego typu leżą w zakresie nadfioletu.
W cząsteczkach odległości między wymienionymi poziomami mogą być różne, różna też może być liczba iść elektronowych.
Diagram zamieszczony niżej ilustruje, w sposób ogólny, możliwe przejścia elektronowe w cząsteczce z uwzględnieniem różnicy energii.
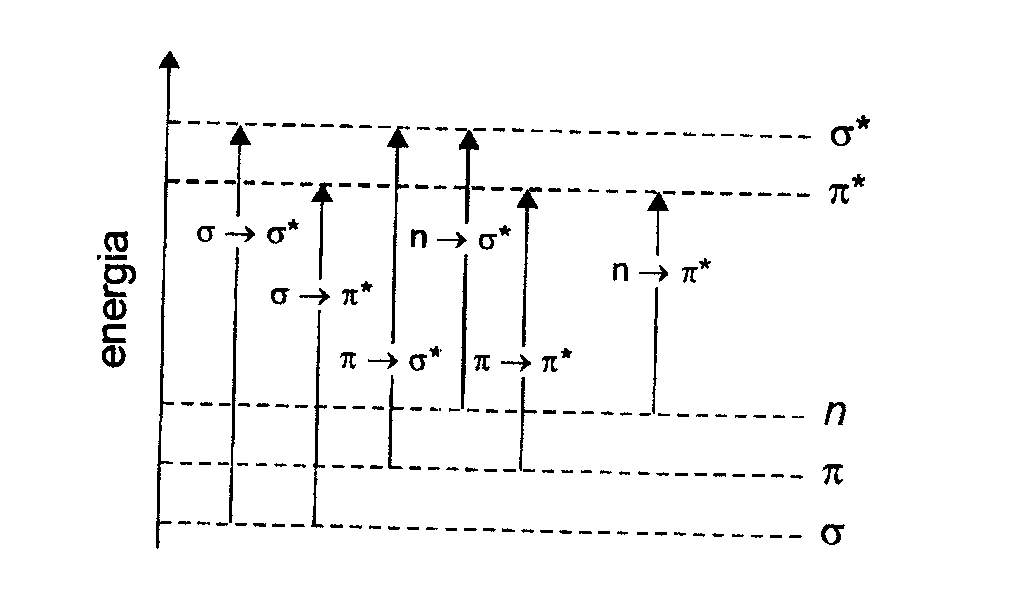
Największych energii wymagają przejścia między orbitalami ၳ Ⴎ ၳ; odpowiadające tym przejściom pasma leżą w zakresie nadfioletu poniżej 200 nm.
Przejściami o niższej energii są przejścia z orbitalu niewiążącego n na poziom antywiążący ၳ*; odpowiednie pasma pojawiają się w zakresie około 180 nm.
Przejściom między orbitalami ၰ Ⴎ ၰ* odpowiada absorpcja na pograniczu UV i pasma leżące w tym zakresie.
Ulegają one przesunięciu w kierunku długofalowym, jeżeli w cząsteczce są podstawniki.
Najmniejsza energia odpowiada przejściom n —> ၰ* i w dużym stopniu zależy od rodzaju związku.
Pasma absorbcyjne tego przejścia leżą przeważnie w obszarze widzialnym, a ich natężenie jest znacznie niniejsze niż pasm stałych przejść.
Szczególnym rodzajem przejść są przejścia przeniesienia ładunku (ang. charge transfer):
W wyniku absorpcji promieniowania następuje przeniesienie elektronu z orbitalu cząsteczki - „donora" na nie ząjęty orbital cząsteczki „akceptora" przy czym nie jest to proces utleniania-redukcji.
Międzycząsteczkowe przeniesienie ładunku towarzyszy powstawaniu związków kompleksowych w wyniku najrozmaitszych oddziaływań: kationów i anionów, między jonami i obojętnymi cząsteczkami oraz jako wynik oddziaływań międzycząsteczkowych.
Pasma przeniesienia ładunku są szerokie i rozmyte, mają duże natężenie i pojawiają się w widmie najczęściej jako nowe, poprzednio nie istniejące pasmo.
Krzywą absorpcji zwaną inaczej widmem absorpcyjnym otrzymuje się w postaci wykresu zależności natężenia wiązki promieniowania elektromagnetycznego po przejściu przez absorbent w funkcji długości fali padającego na absorbent promieniowania.
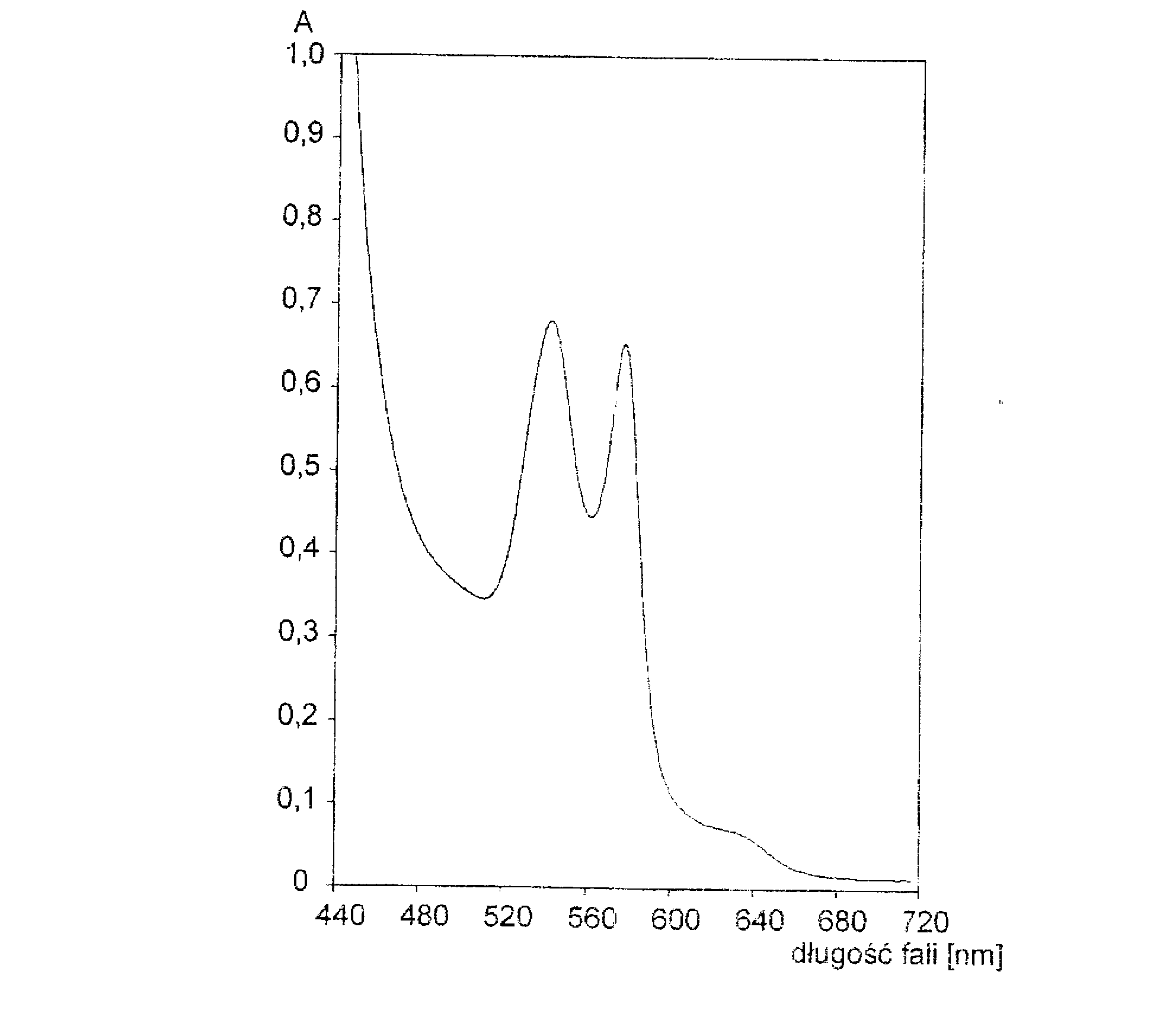
Zależnie od potrzeb osie układu współrzędnych można opisać przy pomocy innych wielkości, przy czym wykres tak opisany również nazywany jest widmem. Najczęściej na osi odciętych odkłada się długość fali ၬ, częstotliwość ၮ lub liczbę falową , natomiast na osi rzędnych - przepuszczalność (ang. transmittance) T, absorbancje A lub molowy współczynnik absorpcji ၥ; czasami spotkać można logarytmy dwóch ostatnich wielkości.
Widmo cząsteczkowe (w stanie roztworu) ma skomplikowany kształt, zawiera kilka lub wiele maksimów i minimów zawierających informację o zmianach zachodzących w układzie wewnętrznym cząsteczki.
Cząsteczka jest układem atomów w których zachodzi jednocześnie wiele ruchów: jądra atomów wchodzących w skład cząsteczki wykonują ruchy oscylacyjne wokół wiązań chemicznych, elektrony na orbitalach wiąże się to z zaabsorbowaniem energii: jej wielkość decyduje, jakiego rodzaju przejście (ruch) nastąpi i w jakim zakresie fal (obszarze widmowym) będzie rejestrowany sygnał spektralny.
Otrzymujemy rozmyte widmo elektronowe które, jak wspomniano wyżej, można rozdzielić. Pobudzenie poziomów rotacyjnych zachodzi pod wpływem promieniowania z zakresu dalekiej podczerwieni i mikrofal. Widma rotacyjne mają zastosowanie np. przy oznaczaniu odległości międzyjądrowych.
Elektronowe widma absorpcyjne umożliwiają m.in. określenie wiązań wielokrotnych, liczbę i położenia podstawników a także ich wzajemne oddziaływania.
Chromofory i auksochromy
W początkowym okresie rozwoju spektroskopii relacje między kształtem widma a budową cząsteczki ustalano doświadczalnie. Zaobserwowano wówczas, że barwne związki mają wiązania nienasycone, a barwa związku zależna jest od grupy atomów powiązanych tymi wiązaniami; taką grupę atomów nazwano chromoforem.
Nazwa ta utrzymana została do dzisiaj i oznacza grupę funkcyjną nienasyconą, która łatwo polaryzuje się i absorbuje selektywnie w zakresie 200-800 nm. Z punktu widzenia budowy elektronowej grupa ta zawiera elektrony ၰ o ich szczególnym układzie zarówno w stanie podstawowym jak i wzbudzonym.
Każdy chromofor ma charakterystyczne dla siebie pasmo absorpcyjne niezależnie od tego w jakim związku występuje. Rozróżnia się chromofory proste i złożone; złożone są układem chromoforów prostych w cząsteczkach o skomplikowanej budowie.
Układy chromoforów mają charakterystyczne dla siebie maksima absorpcji. Przykłady prostych chromoforów pokazane są poniżej:
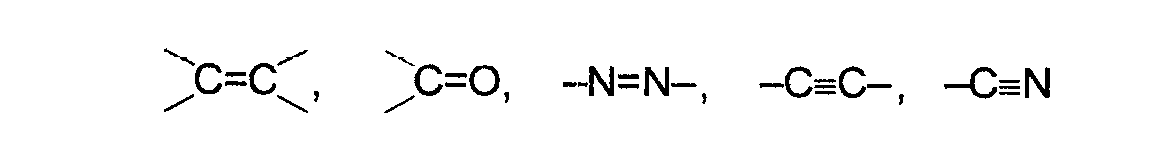
Grupy atomów powodujące zmiany widma chromoforu noszą nazwę auksochromów. Auksochromy nie absorbują promieniowania ale ich obecność wywiera silny wpływ na kształt widma chromoforu. Auksochromanf między innymi grupy:
-NH2, -NR2, -SH, -OH, -OR
Kształt widma dla danego rodzaju cząsteczek zależy od wielu czynników: stanu skupienia, temperatury, rozpuszczalnika, pH, zanieczyszczeń używanych odczynników i szkła a także czynników aparaturowych.
Przy przejściu od widma cząsteczek w fazie gazowej do widma cząsteczek w roztworze zawsze występują chromowe przesunięcia maksimów absorpcji. Powoduje to wprowadzenie rozpuszczalnika.
Wpływ temperatury uwidacznia się w jeden sposób: zmienia wartość absorbancji maksimów i wprowadza błędy przy obliczaniu wartości współczynników absorpcji badanych cząsteczek. Wahania temperatury pomieszczenia w których przeprowadza się pomiary a także nagrzewanie się komory pomiarowej
Wpływ temperatury uwidacznia się w jeden sposób: zmienia wartość absorbancji maksimów i wprowadza błędy przy obliczaniu wartości współczynników absorpcji badanych cząsteczek.
W przypadku roztworów bardzo rozcieńczonych, używanych przy pomiarach UV/VIS błąd współczynników absorpcji przy różnicy temperatur 5°C może dochodzić do 25%.
Wpływ pH na kształt widma - ze względu na duże zmiany, jakie występują pod wpływem wahań pH w widmie wielu cząsteczek należy upewnić się doświadczalnie, czy wybrana do pomiarów wartość pH znajduje się w zakresie stabilnym (niezależność absorpcji od pH).
Charakterystykę widma absorpcyjnego stanowią:
- liczba pasm absorpcji (maksimów),
- długość fali (lub częstotliwość) odpowiadająca maksimum,
- natężenie i kształt pasm absorpcji.
Pasmo absorpcyjne ma kształt krzywej dzwonowej opisanej funkcją Gaussa. Jako wielkość charakteryzującą podaje się szerokość połówkową q krzywej.
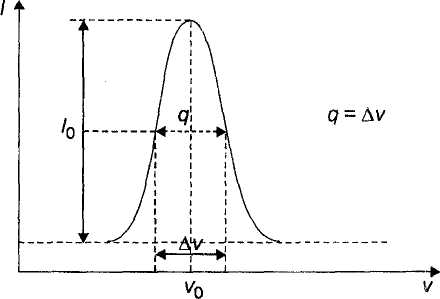
Kontur pasma spektralnego i jego parametry
Najczęściej dość wąskie pasmo absorpcji ulega poszerzeniu, a powodów jest kilka :
sprzężenie elektronowo-oscylacyjno-rotacyjne ,
stan skupienia badanej próbki; wkład do poszerzenia pasma wnosi rozpuszczalnik oraz oddziaływanie cząsteczka-rozpuszczalnik,
czas życia cząsteczki na danym poziomie energetycznym; im krótszy jest ten czas tym większe rozmycie poziomu,
wady aparatury.
Częstotliwość ၮ0 odpowiadająca maksimum absorpcji jest parametrem określającym położenie danego pasma względem innych pasm w widmie. Natężenie pasma odzwierciedla przejścia elektronowe:
- dozwolone (spełniające reguły wyboru) o dużym natężeniu,
- wzbronione (nie spełniające reguł wyboru) o małym natężeniu pojawiające się w wyniku deformacji występujących wewnątrz cząsteczki, które częściowo znoszą zakazy wynikające z reguł wyboru.
Oznaczenia spektrofotometryczne — sposób wykonania
Czynniki wpływające na dokładność wyników
Planując oznaczenia spektrofotometryczne należy wziąć pod uwagę niżej wymienione czynniki, które mogą ułatwić interpretację wyników :
wartość współczynnika absorpcji badanej substancji - im większa jego wartość, tym większa czułość i wykrywalność metody,
„trwałość" absorpcji mierzonego roztworu; przy dużej szybkości zmian konieczne jest mierzenie absorbancji po określonym czasie od zmieszania składników,
zależność absorpcji od pH środowiska - im mniejsza zależność tym bardziej dokładne wyniki: należy zatem sprawdzać zależność badanego układu od pH,
zależność absorpcji od temperatury - najczęściej wykonuje się pomiary w tzw. temperaturze pokojowej której wahania mogą dochodzić do 10°C.
szybkość reakcji - należy wybierać reakcje na tyle szybkie, aby nie czekać na ustalenie się stanu równowagi.
Przygotowanie próbek
Oznaczenie spektrofotometryczne wymaga przeprowadzenia procedury składającej się z kilku następujących po sobie etapów :
pobrania próbki,
przygotowania próbki do pomiaru,
pomiaru,
przeprowadzenia obliczeń i uzyskania wyniku końcowego.
Kolejnym bardzo ważnym krokiem przygotowawczym jest właściwe dobranie odnośnika.
Odnośnikiem może być :
powietrze /lub kuweta pusta/
rozpuszczalnik
ślepa próba
roztwór oznaczanej substancji o znanym stężeniu.
Ślepa próba to roztwór zawierający wszystkie składniki próbki oprócz substancji oznaczanej. Ślepa próba kompensuje absorbancję pochodzącą od składników nie oznaczanych.
Zanieczyszczenia szkła laboratoryjnego są plaga wszystkich laboratoriów!
Mycie detergentami jest niedozwolone przy pracy w zakresie nadfioletu. Detergenty bardzo trudno usunąć; można tego dokonać myjąc naczynie mieszaniną kwasu azotowego i siarkowego a następnie wielokrotnie płucząc wodą destylowaną.
Można też naczynia! moczyć w mieszaninie chromowej pamiętając jednak, że chrom przechodzi ze ścianek do próbki i jest środkiem kancerogennym. Ostatnio stosuje się zastępczo stężony kwas siarkowy cz.d.a.
Czynnością poprzedzającą pomiar jest wybranie analitycznej długości fali - wartości długości fali odpowiadającej maksimum (najwyższemu lub niższemu) w widmie absorpcyjnym.
Dokonuje się tego po sporządzeniu widma, a sprawdza się dodatkowo korzystając z tablic spektrofotometrycznych. Porównanie widma i danych tablicowych jest konieczne ze względu na możliwość wystąpienia przesunięć w widmie uzyskanym doświadczalnie.
Absorpcja rozpuszczalnika ma tu duże znaczenie; należy wybrać taką długość fali przy której rozpuszczalnik ma mniejszą absorpcję, nawet wtedy, gdy jest to niższe maksimum widma. Niekiedy wybiera się kilka analitycznych długości fal - postępuje się tak przy pracy z substancją mającą szereg maksimów o różnych współczynnikach absorpcji, którą bada się w szerokim zakresie stężeń.
Ustawienie zakresu mierzonych absorbancji w porównaniu z poprzednio wymienionymi czynnościami jest sprawą prostą: należy dobrać takie stężenie próbki aby absorbancja zawierała się w przedziale alecanym przez instrukcję aparatury.
Zarówno mniejsze jak i większe wartości absorbancji mogą wiązać się z wprowadzeniem błędów pomiarowych zwłaszcza w przypadku korzystania z aparatury cechującej się „wysługą lat".
Dlatego konieczna jest legalizacja aparatury minimum co dwa lata.
Sprawdzenie, czy przy wybranym stężeniu próbki spełnione jest prawo absorpcji w zalecanym zakresie absorbancji przeprowadzić można w dwojaki sposób. Dla trzech znacznie różniących się stężeń (w pięciu powtórzeniach) należy wyznaczyć wartości współczynników absorpcji obliczając błędy pomiarowe.
Jeżeli wartość współczynników absorpcji w granicach błędu jest stała — układ spełnia prawo absorpcji. Znacznie częściej wybiera się prostszy sposób - wykreśla się krzywą wzorcową. Otrzymany wykres pozwala sprawdzić liniowość a dodatkowo pokazuje w jakim zakresie stężeń układ spełnia prawo absorpcji.
Wygrzanie aparatu jest czynnością o której nie należy zapominać pomimo presji czasu i kolejki użytkowników aparatu czyhających za plecami na każde nasze potknięcie!
Stabilne warunki pracy lampy, ustabilizowanie się temperatury wewnątrz komory pomiarowej to czynniki wpływająca na dokładność przeprowadzanych pomiarów.
W aparatach pracujących wiele lat zdarza się, że widma tych samych próbek uzyskane w różnych porach dnia są zmienione z powodu zmian widma lampy w czasie długotrwałej pracy oraz zmian temperatury wewnątrz komory pomiarowej.
Ostatnią czynnością przed wykonaniem pomiaru jest zapisanie ustawionych parametrów aparatu - nie należy polegać na pamięci !!! Przy wykonywaniu oznaczeń różnych próbek przez wiele miesięcy przy zmieniających się zależnie od potrzeb parametrach, zapisanie warunków pomiaru ułatwia kontynuowanie pracy a często wyjaśnia wstające wątpliwości.
Źródła błędów w oznaczeniach spektrofotometrycznych
Do najważniejszych źródeł błędów systematycznych w pomiarach w zakresie UV/VIS zalicza się:
nieliniowość układu fotometrycznego - tj. odchylenia od liniowego charakteru zależności między stężeniem wiązki promieniowania padającego na detektor a wartością sygnału elektrycznego rejestrowanego przez układ.
Uwidacznia się ona jako odstępstwo od prawa absorpcji w pewnych zakresach stężeń.
jakość i stan techniczny układu mechanicznego - w miarę upływu czasu, na skutek zużycia, metalowi uchwyty kuwet i mechanizm przesuwu tracą pierwotną precyzję co powoduje m. in. zmiany nachylenia ścian kuwet w stosunku do padającej wiązki światła i zwiększa efektywną grubość roztworu, a także powoduje odchylenie wiązki wychodzącej z próbki.
promieniowanie rozproszone - może to być promieniowanie spowodowane odbijaniem i rozpraszaniem promieni na powierzchniach części składowych monochromatora, defektami wykonania pryzmatów lub siatek dyfrakcyjnych itd.
Może to być również promieniowanie powstające w wyniku odbijania i rozpraszania promieni na elementach optycznych komory pomiarowej w tym na powierzchniach kuwet i badanych próbek.
Wyeliminowanie tego promieniowania jest praktycznie niemożliwe; dbając o właściwą eksploatację przyrządu można jedynie przedłużyć istniejący stan.
Ilość promieniowania rozproszonego wzrasta wraz ze starzeniem się aparatu tj. wraz z przenikaniem kurzu i par substancji chemicznych, starzeniem się detektorów i lamp itd.
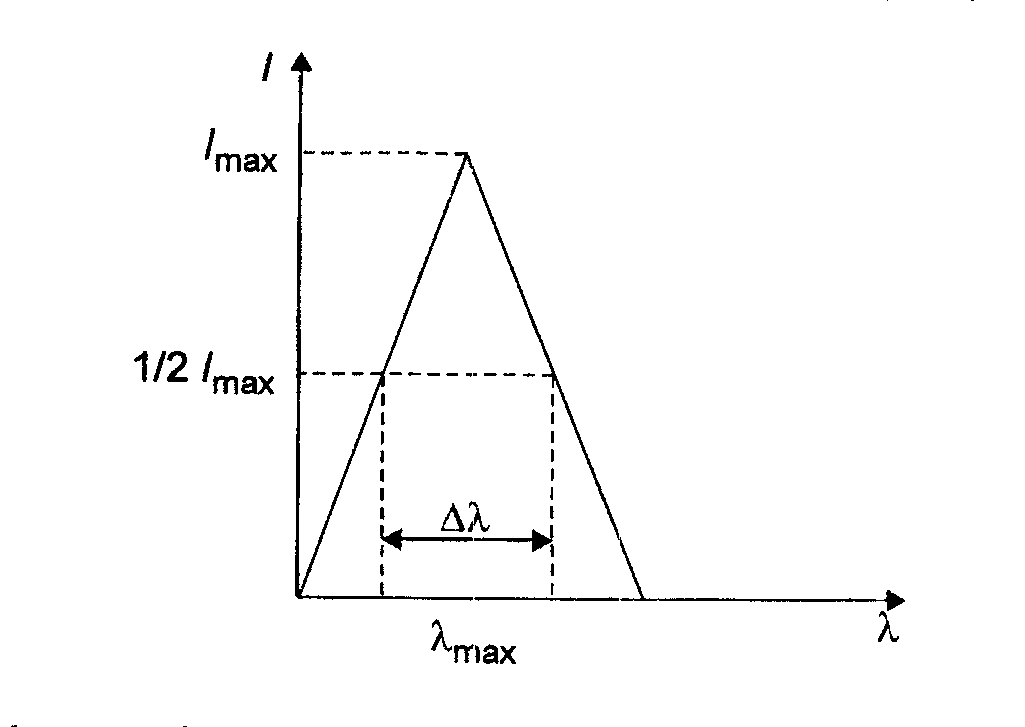
niewłaściwie dobrana szerokość spektralna wiązki promieniowania - szerokość spektralna wiązki przy danej długości fali pełni rolę zdolności rozdzielczej przyrządu.
Definicja szerokości spektralnej wiązki wyprowadzona jest przy założeniu, że monochromator ma równe szczeliny wejściową i wyjściową, a zależność natężenia promieniowania przy danej długości fali w funkcji ၬ ma kształt trójkątny
Aparatura pomiarowa
Spektrofotometry najnowszej generacji, w pełni skomputeryzowane z możliwością analizowania kilkunastu próbek w szerokim zakresie temperatur, pomiaru kilku parametrów, rejestracją widm i ich pochodnych należą do przyrządów bardzo drogich.
Konstruowaniem tego typu aparatów zajmuje się kilka firm wyspecjalizowanych od dziesięcioleci w produkcji przyrządów naukowych: Beckman, Cary Instruments, Pay Unicam, Perkin-Elmer itd.
Znacznie tańsze, wystarczająco dokładne spektrofotometry rejestrujące widma w zakresie widzialnym i ultrafioletu kupić można np. w firmie Amersham Pharmacia Biotech lub polskiej firmie Marcel (Warszawa).
Pierwsze przyrządy pozwalające mierzyć stężenie roztworów barwnych nosiły nazwę kolorymetrów, a pomiar przeprowadzany był metodą zrównania barw między roztworem badanym i roztworem wzorcowym.
Szerokość trójkąta w połowie jego wysokości określa spektralną szerokość wiązki ၬ၄. Trójkątny kształt funkcji szczelinowej może być zachowany przy dużych szerokościach szczelin; najmniejszą szerokość geometryczną, szczeliny monochromatora, poniżej której szerokość spektralna wiązki pozostaje stała można obliczyć ze wzoru:
D =f ၬ/S
w którym: D — szerokość geometryczna szczeliny, f - ogniskowa kolimatora w układzie monochromatora, S - przekrój wiązki padającej na układ rozszczepiający.
Spektralną szerokość wiązki ၬ၄ można obliczyć ze wzoru:
ၬ၄ = (dၬ /dl) x D
w którym wartości (dၬ /dl) w funkcji długości fali podawane są w instrukcji obsługi jako krzywe dyspersji.
Detektorem było oko badacza czułe na zakres promieniowania 400-750 nm. z maksimum około 550 nm.
Z czasem oko zostało zastąpione przez fotoogniwo, wprowadzono też filtry świetlne umożliwiające częściową monochromatyzację promieniowania.
W ten sposób powstały fotokolorymetry.
Po zastosowaniu monochromatora fotokolorymetry przekształciły się w spektrokolorymetry.
Dalsze udoskonalenia spektrokolorymetrów dokonywane równolegle z rozwojem mikroelektroniki, optyki, mechaniki i automatyki doprowadziły do zbudowania spektrofotometrów którymi posługujemy się dzisiaj.
Pierwszymi aparatami pracującymi w zakresie widzialnym, produkowanymi na skalę przemysłową były m.in. spektrokolorymetry Spekol firmy C. Zeiss (Jena) produkowane w latach 1945-1970.
Następnym modelem spektrofotometru firmy C. Zeiss był spektrokolorymetr Spekol 20 służący do pomiaru absorbancji i transmitancji w zakresie widzialnym z odczytem wartości na wyświetlaczu 4-cyfrowym.
Kolejny model spektrokolorymetru firmy C. Zeiss oznaczony jako Spekord UV/VIS rejestrował widma w postaci wykresów na odpowiednio przygotowanych arkuszach.
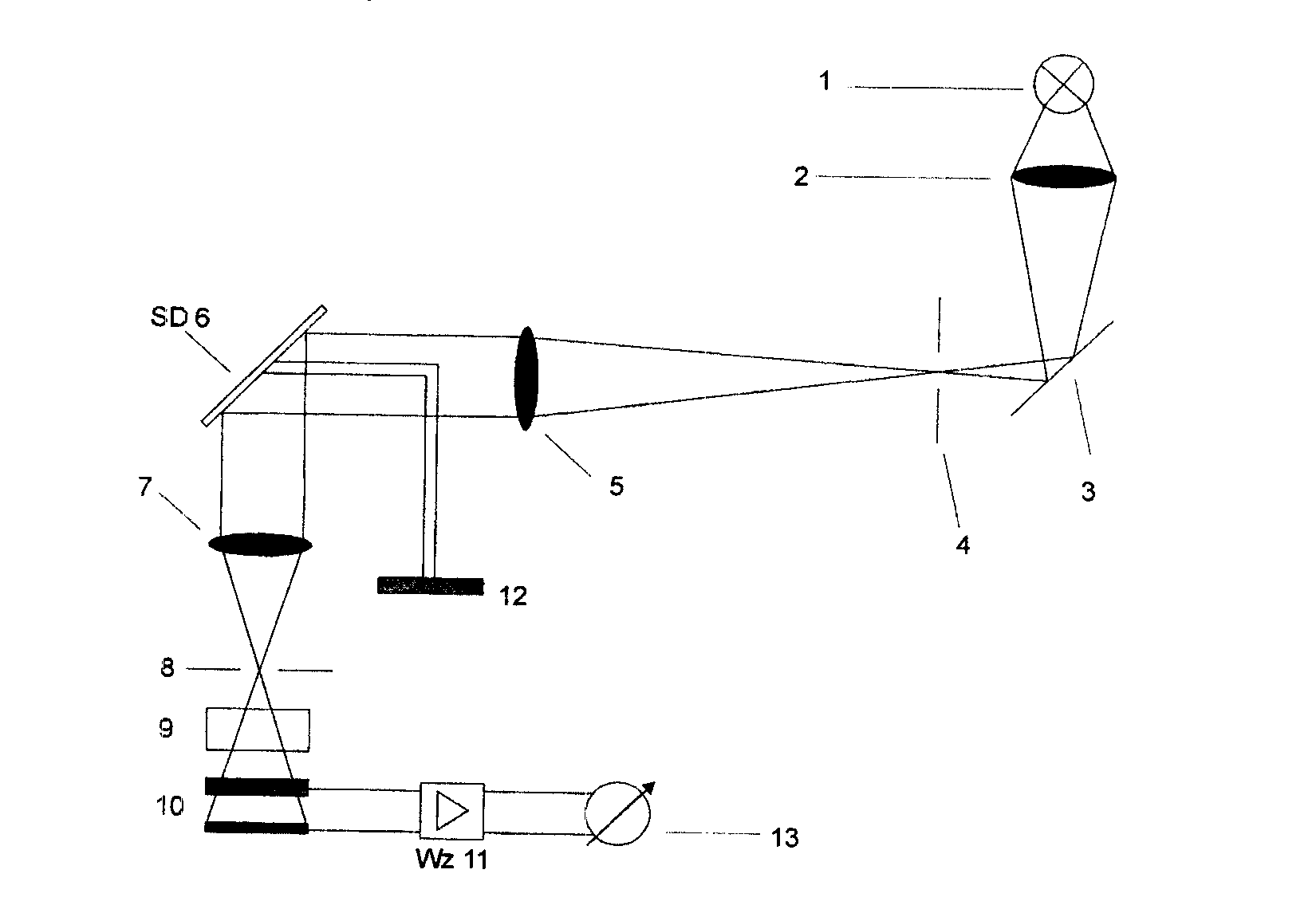
Światło emitowane przez żarówkę (1) przechodzi przez kondensor (2) i po odbiciu od zwierciadła (3) wchodzi do monochromatora przez szczelinę wejściową (4). Po przejściu przez układ achroma-tyczny (5) wiązka promieni równoległych pada na siatkę dyfrakcyjną (SD 6), skąd odbita przechodzi przez obiektyw (7) i trafia na szczelinę wyjściową monochromatora (8). Za pomocą odpowiedniego układu dźwigni poruszających bębnem (12) można obracać siatką dyfrakcyjną i dzięki temu przepuszczać przez szczelinę wyjściową wiązki światła o różnych długościach fal. Szerokość spektralna przepuszczanych przez szczelinę wiązek jest jednakowa w całym zakresie widma. Wiązka świetlna po przejściu przez kuwetę z roztworem (9) pada na ogniwo selenowe (10). Powstały tutaj fotoprąd dostaje się poprzez wzmacniacz (Wz 11) do urządzenia pomiarowego (13)
Klasę spektrofotometru określa jakość monochromatora i układ detekcji. W pierwszych przyrządach monochromatorami były pryzmaty wykonane ze szkła lub kwarcu.
Pryzmaty zastąpione zostały przez siatki dyfrakcyjne: początkowo płaską, następnie odbiciową i ostatnio holograficzną.
Siatki holograficzne dają wiązkę promieniowania o szerokości spektralnej 0,2—1,0 nm. w zakresie widzialnym.
Monochromatyczna wiązka światła może być rozdzielona na dwie wiązki równoległe, z których jedna przechodzi przez odnośnik a druga przez roztwór badany.
Pomiaru natężeń przechodzących dokonują dwa niezależne detektory a rejestrator wykazuje różnicę natężeń prądu wytworzonego w obu detektorach (przeliczoną na absorbancję).
Ten typ spektrofotometru nosi nazwę dwuwiązkowego i cechuje go duża szybkość rejestracji.
Ze względu na sposób rejestracji wyników pomiarów spektrofotometry dzieli się na punktowe i rejestrujące.
W spektrofotometrze punktowym pomiar dokonywany jest „punkt po punkcie" w wybranym zakresie długości fal.
Są to zazwyczaj przyrządy jednowiązkowe; mierzony jest kolejno odnośnik i próbka badana.
Spektrofotometry punktowe cechuje duża dokładność pomiaru absorbancji.
Spektrofotometry rejestrujące, przeważnie dwuwiązkowe cechuje duża szybkość rejestracji; nie ma przesłanek, które wskazywałyby na „gorszą jakość" uzyskiwanych na tych przyrządach wyników w stosunku do pomiarów wykonywanych na spektrofotometrach punktowych.
Nie można jednoznacznie określi typu spektrofotometru, który przy założonych przez nabywcę parametrach byłby przyrządem kupionym z najlepszego wyboru.
Przykłady zastosowań spektrofotometrii
I. Oznaczanie stężenia substancji
Jeśli roztwory substancji stosują się do prawa Lamberta-Beera, pomiar absorbancji substancji pochłaniającej światło w zakresie widzialnym lub w zakresie nadfioletu pozwala na szybkie wyznaczenie jej stężenia, o ile w roztworze nie ma innych substancji pochłaniających w zakresie długości fali, w jakim prowadzimy pomiar i jeśli znamy współczynnik pochłaniania światła badanej substancji ၥ.
Stężenie oksyhemoglobiny w roztworze możemy określić oznaczając absorbancję jej roztworu przy długości fali 576 nm. Jeśli roztwór hemoglobiny (mierzony w kuwetach grubości 1 cm) ma absorbancję A576, to stężenie hemoglobiny w roztworze równe jest:
c = (A576 /ၥ576 x 1 cm) = A 576 /15 mmol l-1
Często musimy określić stężenie roztworu zbyt stężonego, by można było bezpośrednio dokonać pomiaru jego absorbancji.
Wtedy albo mierzymy absorbancję w kuwetce o grubości mniejszej niż standardowa (np. 0,1 cm lub 0,2 cm) i wtedy oczywiście:
c = A/(ၥ x l)
gdzie l grubość kuwetki, w której dokonujemy pomiaru (w cm), ၥ - molarny współczynnik absorbcji związku. Bądź też rozcieńczamy roztwór R razy i mierzymy absorbancję rozcieńczonego roztworu w standardowej (1 cm) kuwetce.Wtedy:
c = R x (A / (ၥ x 1) [cm]
Jeśli do 0,1 ml roztworu hemoglobiny dodaliśmy 0,9 ml wody (R = 10) i absorbancja rozcieńczonego roztworu wynosi A 576 = 0,3 to :
c = 10 x (0,3 / 15 mmol-1 l cm-1 x 1 cm) = 0,2 mmol l-1
II. Identyfikacja substancji
Widmo pochłaniania światła czystej substancji lub jej roztworu może być jedną z metod identyfikacji substancji. Częściej pozwala ono na określenie formy, w jakiej znajduje się substancja.
Widma form: utlenionej i zredukowanej dinukleotydu nikotynoamidoadeninowego (NAD+ i NADH) różnią się znacznie.
Pomiar widma roztworu pozwala na stwierdzenie, w jakiej formie znajduje się nukleotyd, a spektrofotometryczny pomiar stopnia czy szybkości jego redukcji (utlenienia) jest podstawą wielu testów enzymatycznych.
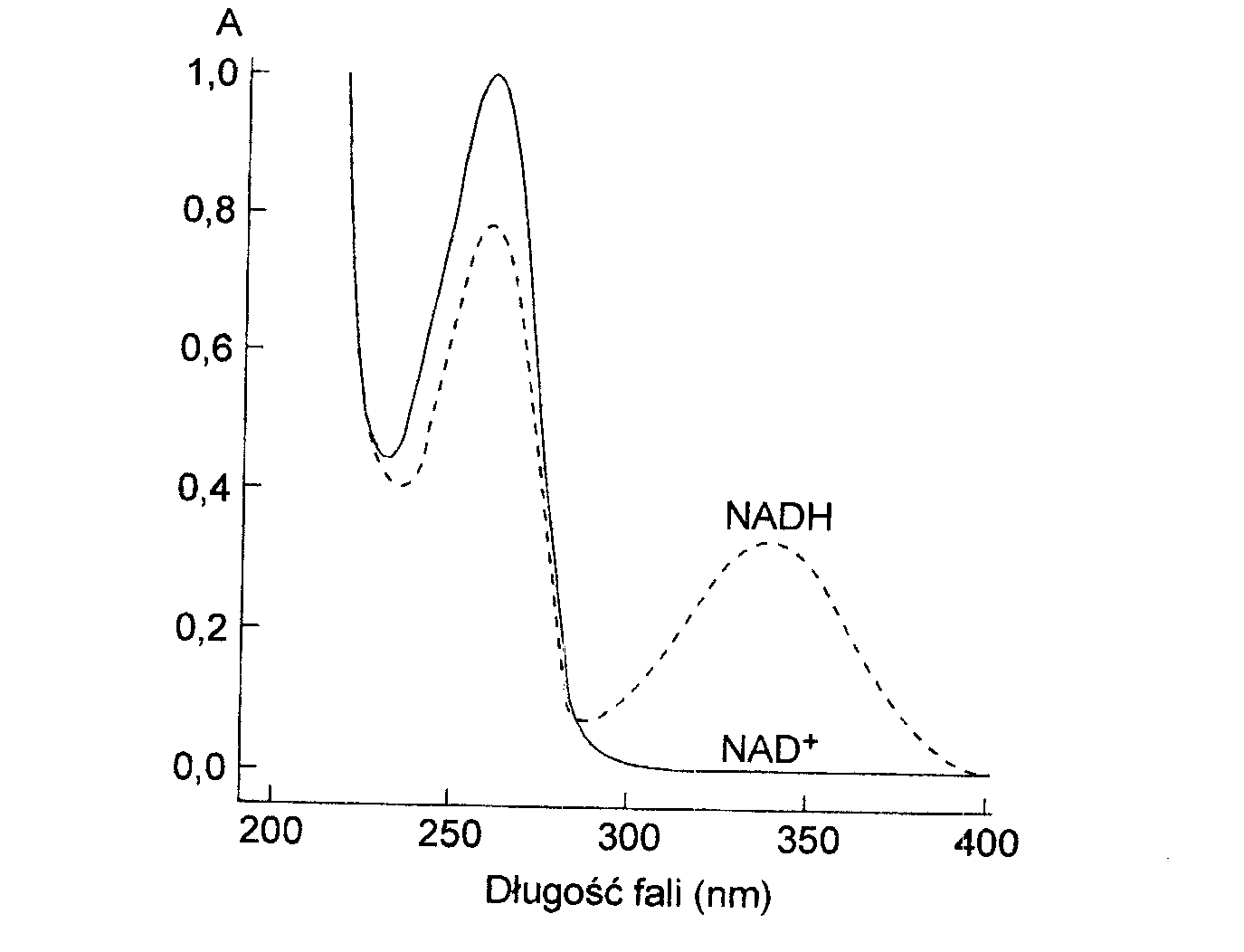
Widma pochłaniania światła przez utlenioną (NAD1) i zredukowaną (NADH) formę dinukleotydu nikotynoamidoadeninowego.
III. Określenie stopnia czystości substancji
Pomiary spektrofotometryczne są często stosowane dla określenia stopnia oczyszczenia substancji biologicznych w toku preparatyki lub stopnia czystości uzyskanego preparatu.
W metodzie otrzymywania DNA drogą odsalania białek stopień czystości uzyskanych preparatów DNA charakteryzuje się poprzez pomiar stosunku absorbancji preparatów przy długości fali 260 nm bliskiej maksimum pochłaniania dla DNA do absorbancji przy długości fali 280 nm (maksimum pochłaniania dla większości białek).
Preparat DNA uważa się za dobrze oczyszczony od białek, jeśli stosunek A260/A280 zawarty jest w granicach 1,8-2,0.
IV. Określenie składu mieszaniny
Jeśli mamy do czynienia z mieszaniną dwu substancji o różnych właściwościach spektralnych, możemy ilościowo określić zawartość obu składników na podstawie pomiaru pochłaniania światła przy dwu długościach fal (jeśli mamy współczynniki pochłaniania obu substancji dla tych długości fal).
Analogiczna zasada obowiązuje dla mieszaniny złożonej z 3 składników.
Przykładem może być metoda wyznaczania zawartości trzech form mioglobiny (czerwonego barwnika mięśni) - oksymioglobiny (oxyMb), metmioglobiny (metMb) i ferrylomioglobiny (ferry-lb) w oparciu o pomiary absorbancji mieszaniny przy długościach fal: 490 nm, 560 nm i 580 nm:
oxyMb] = 2,8 A490 - 127 A560 + 153 A58O
metMb] = 146 A490 - 108 A560 + 2,1 A580
ferryloMb] - 62 A490 + 242 A560 - 123 A580
V. Pomiar kinetyki reakcji (bio)chemicznej
Jeśli w reakcji chemicznej powstaje produkt o innych właściwościach spektralnych niż substrat, to zmiany pochłaniania światła przez substrat czy produkt mogą stanowić wygodną metodę badania kinetyki reakcji.
Przykład oznaczenia tego typu może być pomiar szybkości rozkładu nadtlenku wodoru przez katalazę, oparty na określę kinetyki spadku absorbancji substratu enzymu w zakresie nadfioletu .
VI. Pomiar temperatury topnienia DNA
Absorbancja roztworów DNA jest mniejsza (o ok. 40%) niż suma absorbancji nukleotydów wchodzących w jej skład (co jest określane jako efekt hipochromowy).
Podczas ogrzewania roztworów DNA zachodzi denaturacja termiczna polimeru, naruszone zostają oddziaływania pomiędzy zasadami wchodzącymi w skład DNA, w wyniku czego pochłanianie promieniowania nadfioletowego przez roztwór DNA, zwiększa się.
Zjawisko to nosi nazwę efektu hiperchromowego. Pomiar zależności absorbancji roztworu DNA od temperatury pozwala na wyznaczenie temperatury, w której przyrost absorbancji osiąga wartość połowy wartości maksymalnej.
Temperatura ta określana jest jako temperatura topienia DNA - `(Tm Rys. poniżej).
W stałych warunkach środowiska Tm jest funkcją składu nukleotydowego DNA.
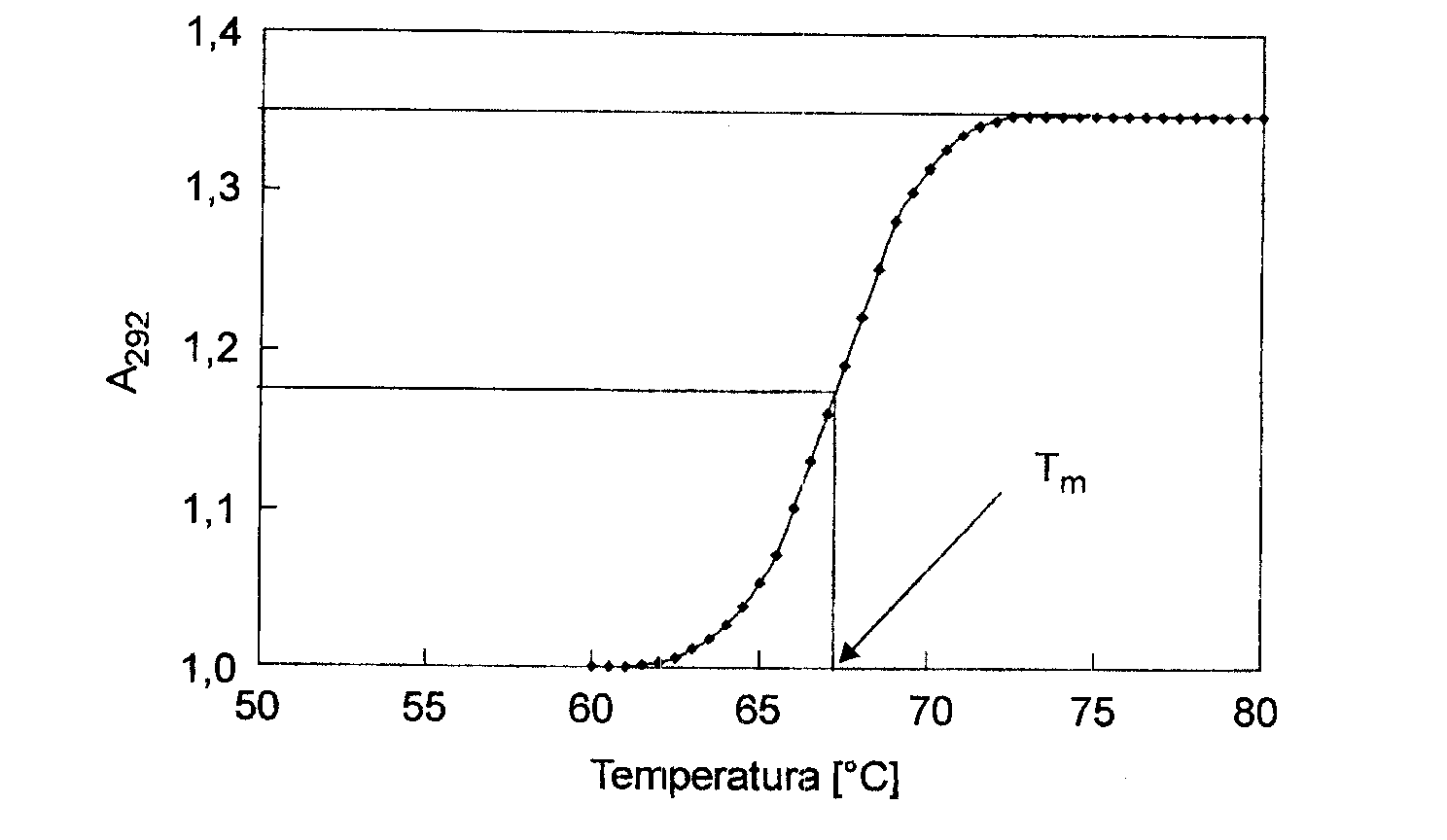
Przebieg „ krzywej topnienia” DNA
VII. Badanie zmian konformacji białka
Widma pochłaniania chromoforów wchodzących w skład cząsteczek białek zależą od ich mikrootoczenia. Długości fal odpowiadających maksimum pochłaniania światła ၬmaks oraz wartości molarnych współczynników absorbcji ၥ reszt fenyloalaniny, tyrozyny, tryptofanu i histydyny wzrastają, jeśli reszty te znajdą w otoczeniu mniej polarnym.
Zatem jeśli wartości ၬmaks i ၥ reszty któregoś z tych aminokwasów wchodzących w skład białka okażą się mniejsze niż odpowiednie wartości dla wolnego aminokwasu znajdującego się w tym samym rozpuszczalniku, oznacza to, że reszta aminokwasowa znajduje się we wnętrzu cząsteczki białkowej w otoczeniu niepolarnych reszt innych aminokwasów.
Natomiast jeżeli widmo pochłaniania białka jest wrażliwe na zmiany polarności roztworu, oznacza to, że reszta aminokwasu, dla której obserwuje się zmiany ၬmaks i ၥ znajduje się na powierzchni cząsteczki białka, w kontakcie z rozpuszczalnikiem.
Często określa się położenie chromoforu w cząsteczce białka poprzez porównanie widma roztworu białka w roztworze wodnym i w roztworze mniej polarnym. Można przy tym stosować roztwory, w których białko pozostaje rozpuszczalne i które nie powodują jego denaturacji.
Stosowane są w tym celu np. dimetylosulfotlenek, dioksan, etanol, glikol etylenowy i gliceryna (zwykle w proporcji 20 objętości/100 objętości wodnego roztworu białka).
Aby łatwiej uchwycić drobne zmiany widma pochłaniania białka, zazwyczaj dokonuje się pomiaru widma różnicowego, mierząc widmo pochłaniania światłą przez roztwór białka w rozpuszczalniku mniej polarnym stosując wodny roztwór białka o dokładnie tym samym stężeniu jako odnośnik.
VIII. Badanie denaturacji białka
Często podczas denaturacji białka chromofor niedostępny dla rozpuszczalnika wchodzi w kontakt z rozpuszczalnikiem i absorbancja roztworu ulega zmianie.
Jeśli białko zawiera reszty tryptofanowe, część których znajduje się wewnątrz cząsteczki, w procesie przejścia (denaturacji białka) wzrasta absorbancja roztworu przy długości fali 292 nm.
Bardziej czułą metodą jest wyznaczanie różnicy absorbancji pomiędzy roztworem białka w rozpuszczalniku mniej polarnym i w roztworze wodnym (poprzez pomiar widm różnicowych).
Pomiary zależności absorbancji od temperatury umożliwiają badanie wpływu czynników modyfikujących na przebieg denaturacji termicznej białka
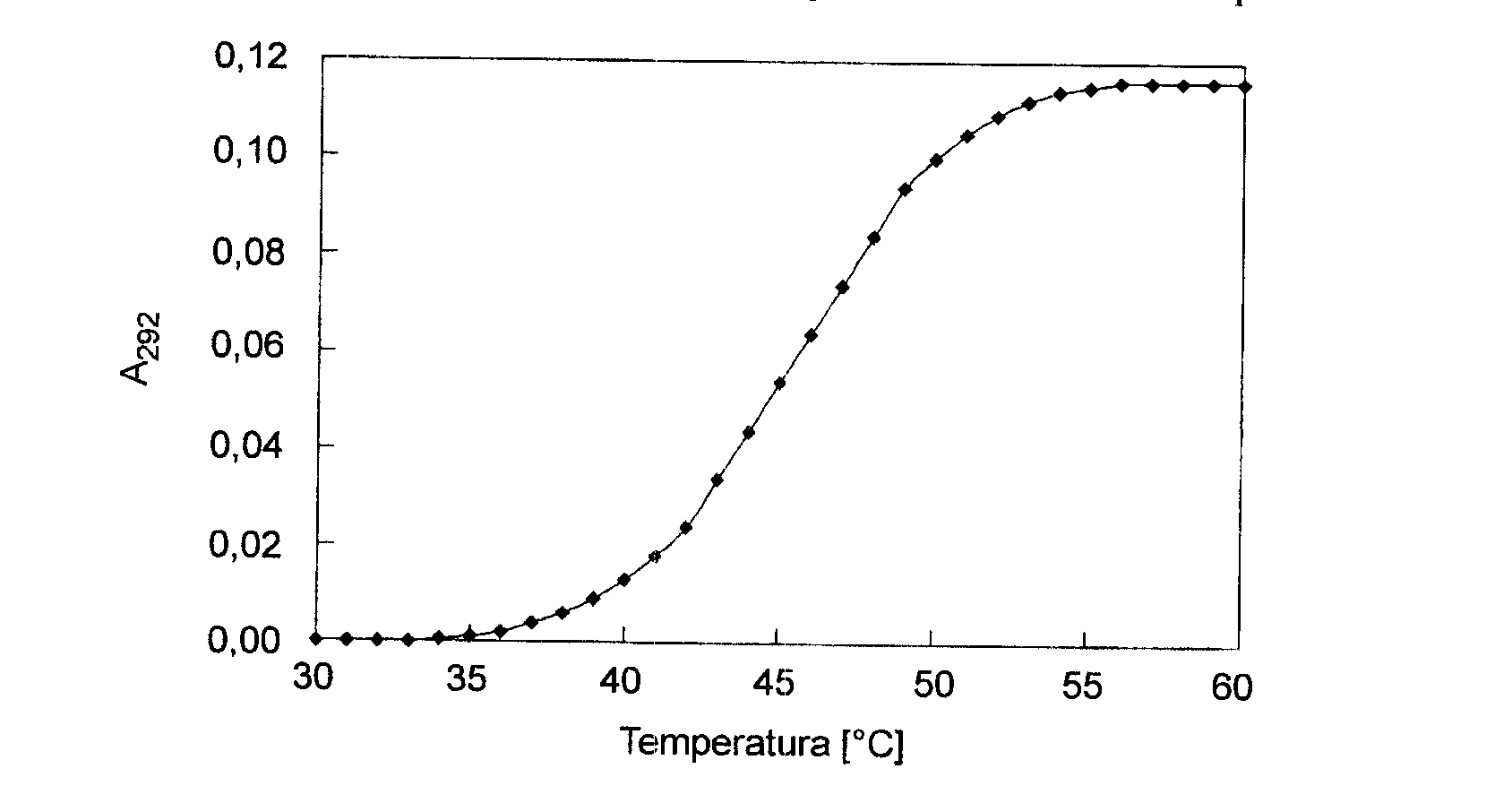
Przebieg denaturacji termicznej białka. Na osi rzędnych różnica absorbancji roztworu białka w roztworze zawierającym 20% glikol etylenowy i w roztworze wodnym
Na tej samej zasadzie można spektrofotometrycznie śledzić inne zmiany konformacyjne białek, z którymi związana jest zmiana dostępności chromoforów dla rozpuszczalnika.
IX. Spektrofotometryczne wyznaczanie pK
Właściwości spektralne szeregu chromoforów zmieniają się w wyniku jonizacji.
W takim przypadku możliwe jest wyznaczenie wartości pK (ujemnego logarytmu stałej dysocjacji chromoforu) na podstawie przebiegu zależności absorbancji odpowiadającej jednej z form (zjonizowanej lub niezjonizowanej) w funkcji pH.
Wartość pK odpowiada tej wartości pH, dla której absorbancja formy barwnej (lub różnica absorbancji pomiędzy obiema formami) osiąga połowę wartości maksymalnej.
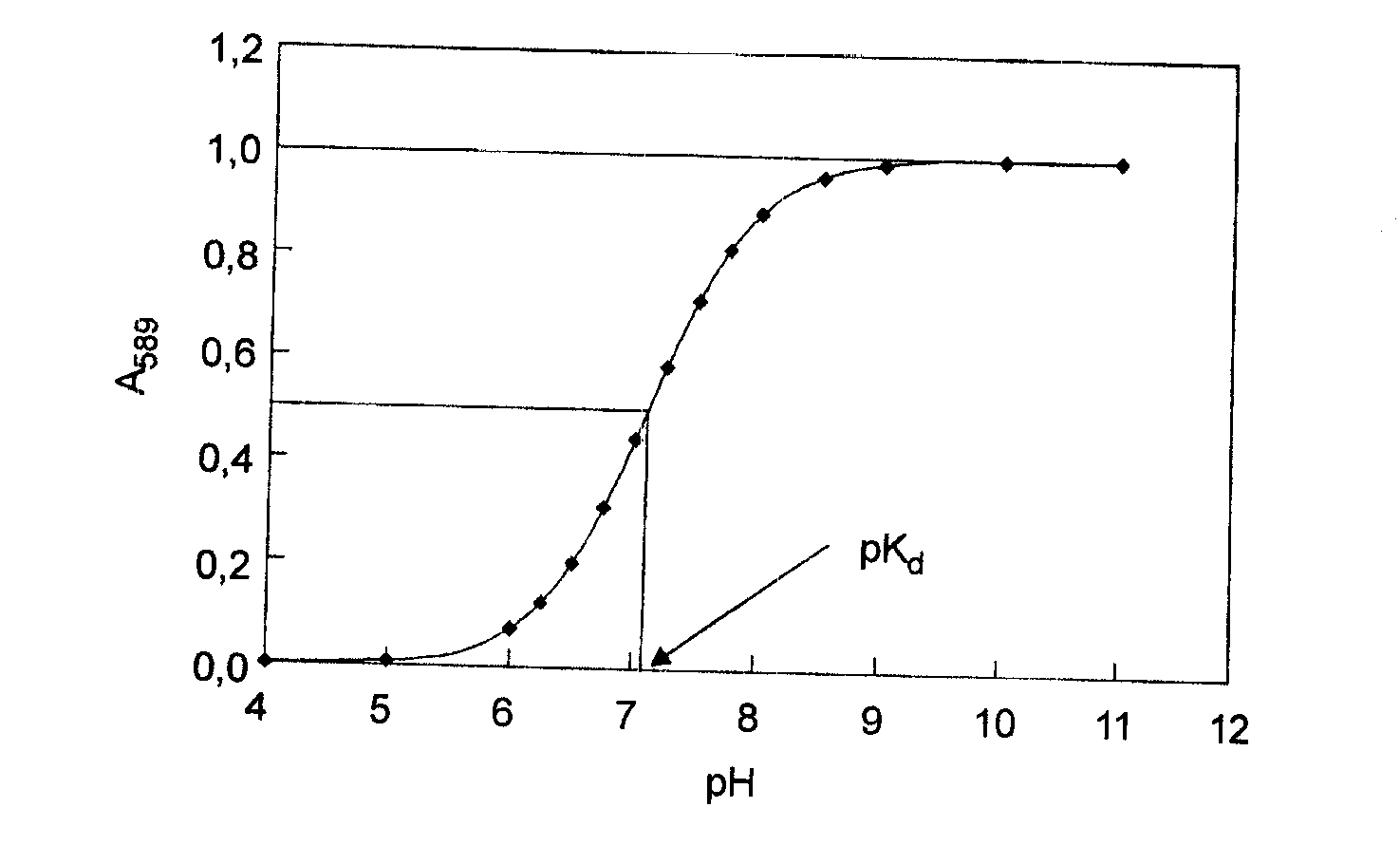
Wyznaczanie pK dysocjacji błękitu bromotymolowego na podstawie pomiaru zależności absorbancji roztworu barwnika od pH.
23
Wyszukiwarka
Podobne podstrony:
Spektroskopia NMR
SPEKTROSKOPIA ROTACYJNA
Spektrometria mas NMAZ
instr 2011 pdf, Roztw Spektrofoto
analityka podstawy spektroskopii 2012 2013
CHEMIA FIZYCZNA- spektrografia sc, Ochrona Środowiska pliki uczelniane, Chemia
spektro6, Technologia chemiczna pw, 2rok, spektra
Spektrometr-76, Studia, Fizyka, Sprawozdania, 76a
Analiza spektralna widm (2), Matematyka - Fizyka, Pracownia fizyczna, Analiza spektralna widm
SPEKTROFOTOMETRYCZNE OZNACZENIE ŻELAZA W POSTACI TIOCYJANIANU ŻELAZA, NAUKA, WIEDZA
Spektroskopia Jądrowego Rezonansu Magnetycznego
pwsz ioś kalisz Ćw 4 Spektrofotometria, inżynieria ochrony środowiska kalisz, a pwsz kalisz ioś, ana
Analiza Instrumentalna Miareczkowanie spektrofotometryczne Sprawozdanie 3 x
Spektro 4
Badanie aktywności dehydrogenaz mikroorganizmów osadu czynnego metodą spektrofotometryczną z TTC
materiały spektroskopia UV VIS
Algorytmy sumowania w metodzie spektrum odpowiedzi i ich wpływ na obliczaną odpowiedź budynku wysoki
Biopsja pod kontrolą USG ROZSZERZENIE SPEKTRUM DIAGNOSTYCZNEGO ULTRASONOGRAFII
Absorpcyjna Spektrofotometria czasteczkowa
więcej podobnych podstron