Synteza (Z)-1- fenoksy-4-propoksy-2-butenu
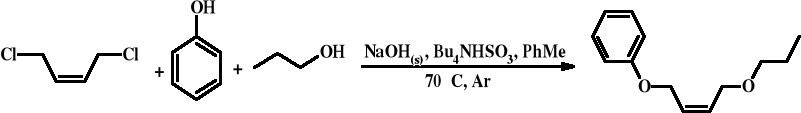
W kolbie trójszyjnej zaopatrzonej w chłodnicę zwrotną z bublerem oraz mieszadło mechaniczne, umieszczonej w łaźni wodno-lodowej, umieszczono 133 ml toluenu, 45g (38 ml) 1,4-dichloro-2-butenu (0,36 mola), 33,9 g (38,9 ml) fenolu (0,36 mola), 38 g NaOH (0,95 mola) oraz 6,1 g wodorosiarczanu(IV) tetrabutyloamoniowego (0,012 mola). Następnie wprowadzono jeszcze daną objętość toluenu, ułatwiającą mieszanie zawartości kolby. Podczas intensywnego mieszania, przez kolbę przepuszczano lekki strumień argonu. Kolbę na czas 20 minut wyjęto z łaźni obserwując czy się rozgrzewa (białe zabarwienie zawartości świadczyło o powstaniu NaCl) a następnie zaczęto powoli ogrzewać. Początkowo ogrzewano na łaźni wodnej do temperatury otoczenia a następnie na olejowej do temperatury 70- 80ႰC. W trakcie ogrzewania mieszanina zgęstniała, więc dodano ponownie toluenu. Po około 40 min grzania nastąpiła zmiana barwy na szarą a po godzinie wypadł jasnobrązowy osad i nastąpiła podział na dwie fazy (osad i ciecz). Po 90 min osad zbił się w jednolitą masę, wyłączono mieszanie, schłodzono zawartość kolby i przeniesiono ciecz do nowej kolby. Następnie dodano do niej połowę początkowej ilości NaOH i katalizatora i kontynuowano ogrzewanie oraz mieszanie mieszadłem mechanicznym przez kolejne 90 minut (Po około godzinie zawartość kolby ściemniała). Po tym czasie zawartość ochłodzono do temperatury pokojowej i dodano 43,2 g (54 ml) 1-propanolu (0,72 mola) i 6,1 g wodorosiarczanu(IV) tetrabutyloamoniowego (0,012 mola). Ponownie ogrzewano zawartość kolby jak poprzednio w atmosferze argonu oraz z intensywnym mieszaniem przez kolejne 4 godziny i rozdzielono warstwy za pomocą rozdzielacza. Warstwę organiczną przemyto sześć razy wodą i suszono bezwodnym siarczanem(VI) magnezu przez 24 godziny. Następnie mieszano przez 48 godzin z 3,8 g węgla aktywnego, odsączono na twardym sączku oraz odparowano lotne frakcje na wyparce próżniowej w temperaturze 90°C pod ciśnieniem 70 mmHg. Masa produktu wynosiła 73,4g.
Surowy żółty produkt będący mieszaniną: (Z)-1-fenoksy-4-propoksy-2-butenu, (Z)-1,4-difenoksy -2-butenu oraz (Z)-1,4-dipropoksy-2-butenu, destylowano pod zmniejszonym ciśnieniem w dwóch częściach otrzymując frakcje:
![]()
Frakcja |
Kropla/sek |
Temperatura |
Ciśnienie |
Masa |
I |
|
30 - 86°C |
|
11,55 g |
|
1/1sek |
30°C |
14 mmHg |
|
|
1/1sek |
80°C |
6 mmHg |
|
|
1/1sek |
86°C |
5 mmHg |
|
II |
|
125 - 135°C |
|
13,87 g |
|
1/1sek |
125°C |
2 mmHg |
|
|
1/1sek |
135°C |
2 mmHg |
|
Następnie destylację przerwano, roztwór ostudzono i dodano do kolby drugą część produktu. Po zmianie odbieralników produkt ponownie destylowano otrzymując kolejne frakcje:
![]()
Frakcja |
Kropla/sek |
Temperatura |
Ciśnienie |
Masa |
I' |
|
22 - 90°C |
|
5,48 g |
|
1/1sek |
22°C |
14 mmHg |
|
|
4/1sek |
89°C |
7 mmHg |
|
|
1/1sek |
90°C |
7 mmHg |
|
II' |
|
120 - 180°C |
|
7,52 g |
|
1/1sek |
120°C |
5 mmHg |
|
|
1/1sek |
134°C |
5 mmHg |
|
|
1/1sek |
147°C |
8 mmHg |
|
|
1/1sek |
157°C |
7 mmHg |
|
|
1/1sek |
162°C |
10 mmHg |
|
|
1/1sek |
166°C |
11 mmHg |
|
III |
|
180 - 215°C |
|
19,23 g |
|
1/1sek |
180°C |
12 mmHg |
|
|
2/1sek |
210°C |
14 mmHg |
|
|
3/1sek |
214°C |
14 mmHg |
|
|
4/1sek |
215°C |
14 mmHg |
|
Destylowano do temperatury 215°C gdzie nastąpiła stabilizacja, następnie ją przerwano.
Masa pozostałości (brązowy, gęsty roztwór) po destylacji wyniosła 5,94 g.
Analiza 1HNMR wykazała że frakcja II zawiera 92% (Z)-1-fenoksy-4-propoksy-2-butenu
(12,76 g), resztę stanowił (Z)-1,4-difenoksy -2-buten, zaś frakcja II' zawiera 89% (Z)-1-fenoksy-4-propoksy-2-butenu (6,69 g) oraz (Z)-1,4-difenoksy -2-buten (8%) i (Z)-1,4-dipropoksy-2-buten (3%). Frakcja III zawiera 88% (Z)-1,4-difenoksy -2-butenu i 12%
(Z)-1-fenoksy-4-propoksy-2-butenu. Pozostałe frakcje zawierały mieszaninę (Z)1,4-difenoksy -2-butenu, (Z)1-fenoksy-4-propoksy-2-butenu oraz (Z)1,4-dipropoksy-2-butenu o niewielkiej zawartości procentowej produktu właściwego.
W celu otrzymania czystszego produktu ((Z)-1-fenoksy-4-propoksy-2-butenu) zmieszano frakcję II i II' oraz poddano ponownie destylacji pod ciśnieniem 5 mmHg otrzymując frakcje:
Frakcja |
Kropla/sek |
Temperatura |
Masa |
I |
1/1sek |
130-133°C |
6,70 g |
II |
1/1sek |
133 - 136°C |
10,57 g |
III |
1/10sek |
136 - 143°C |
0,98 g |
Masa pozostałości po destylacji wyniosła 3,14 g.
(Frakcje I i II zostały połączone - jest to czysty (Z)-1-fenoksy-4-propoksy-2-buten
Frakcja III jest to (Z)-1,4-difenoksy -2-buten )
Synteza [RuCl2(PPh3)3]
[RuCl3⋅ 3H2O] + PPh3 + CH3CH2OH → [RuCl2(PPh3)3]
W kolbie trójszyjnej o pojemności 250 ml zaopatrzonej w mieszadło mechaniczne, chłodnicę zwrotna z bublerem oraz rurkę do wprowadzanie argonu, umieszczono 3,67 g trifenylofosfiny (0,014 mola) i 70 ml 96% etanolu. Przez układ przepuszczono szybki strumień argonu (około 5 minut) a następnie zmniejszono go i zaczęto ogrzewać zawartość kolby do łagodnego wrzenia za pomocą płaszcza elektrycznego. Do wrzącego, mieszającego się roztworu dodano przez chłodnicę ciepły, nasycony argonem roztwór 0,141 g trójwodnego chlorku rutenu(III) (0,002 mola) w 40 ml 96% etanolu. W celu przemycia kolby i chłodnicy dodano jeszcze 24 ml 06% etanolu. Następnie roztwór ogrzewano do wrzenia oraz intensywnie mieszano przez godzinę, a po tym czasie ochłodzono do temperatury pokojowej. Wydzielony ciemno brązowy osad kompleksu odsączono na lejku ze spiekiem i przemyto 40 ml 96% etanolem oraz 25 ml eteru dietylowego. Osad suszono na wyparce rotacyjnej. Masa produktu wyniosła 1,19 g co stanowiło 61,5% wydajności w stosunku do wydajności literaturowej, która wynosi 95%.
Synteza 2,2-dimetylo-4,5-diwinylo-1,3-dioksolanu
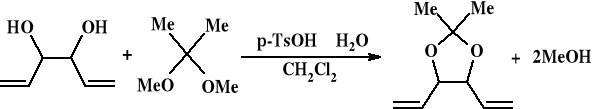
Do kolby o pojemności 10 ml z mieszadłem magnetycznym wprowadzono: 0,65g (0,64 ml) 1,5-heksadien-3,4-diolu (0,006 mola), 1,19 g (1,4 ml) 2,2-dimetoksypropanu(0,011 mola), 0,6 ml dichlorometanu oraz 0,03 g kwasu p-toluenosulfonowego. Zawartość kolby intensywnie mieszano przez 24 godziny. Roztwór przeniesiono do kolby o pojemności 50 ml i dodano 20 ml dichlorometanu oraz 0,050 g CaO (w dwóch porcjach po 0,025 g, w celu związania hydratu kwasu p-toluenosulfonowego ) mieszając intensywnie jeszcze przez dwie godziny. Następnie kwas związany przez CaO odsączono z użyciem twardego sączka i przemyto sześcioma porcjami dichlorometanu o objętości 1ml. Dichlorometan odparowano na wyparce rotacyjnej w temperaturze 25°C pod ciśnieniem 100 mmHg w czasie 1 godziny, kolbę zważono i ponownie odparowano w temperaturze 25°C pod ciśnieniem 50 mmHg w czasie 30 minut w celu sprawdzenia ubytku masy. Wydajność reakcji wyniosła 77,4%.
Synteza 4,4,5,5-tetrametylo-2-winylo-1,3-dioksolanu (EK-309)
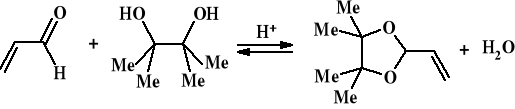
Reakcję prowadzono w kolbie kulistej o objętości 500 cm3, zaopatrzonej w nasadkę azeotropową z chłodnicą zwrotną w łaźni elektrycznej. W kolbie reakcyjnej i nasadce
azeotropowej umieszczono ok. 20 mg hydrochinonu w celu zapobieżenia polimeryzacji.
Następnie część kalibrowaną nasadki wypełniono benzenem i wprowadzono do kolby reakcyjnej kolejno: 200 cm3 benzenu, 44,5 cm3 (0,6 mola) akroleiny świeżo destylowanej, 59,1 g (0,5 mola ) pinakolu oraz 30 mg kwasu p-toluenosulfonowego. Mieszaninę ogrzewano do łagodnego wrzenia przez 2 godziny, aż do zakończenia wydzielania się wody w nasadce azeotropowej. Po ochłodzeniu do temperatury otoczenia dodano 2 g tlenku wapnia (w celu zobojętnienia kwasu) i wytrząsano kilka razy co 20 min. Osad odsączono na karbowanym sączku. Do mieszaniny reakcyjnej dodano 30 mg hydrochinonu i poddano ją destylacji prostej w celu usunięcia lotnych frakcji z użyciem krótkiej kolumny Vigreux. Następnie destylację przerwano, roztwór ostudzono i poddano destylacji próżniowej.
FRAKCJA |
KROPLA/SEK |
TEMPERATURA |
CISNIENIE |
Masa |
Ia |
1/7sek |
85-87°C do 105°C |
- |
|
II |
1/8sek |
33°C |
70 mmHg |
0,81 g |
III
|
1/4sek 1/1sek 1/2sek |
79°C 77°C 76°C |
50 mmHg 45 mmHg 45 mmHg |
5,36 g |
IV |
1/1sek |
74°C |
41 mmHg |
24,75 g |
V |
1/1sek 1/1sek |
75°C 76°C |
41 mmHg |
12,64 g |
VI |
1/1sek 1/1sek |
77°C 78°C |
41 mmHg |
9,44 g |
VII |
1/1 sek |
80°C |
41 mmHg |
2,3 g |
a - Frakcja I dotyczy destylacji prostej.
Wyszukiwarka
Podobne podstrony:
LAB1-ELEKTRO-AK KM, chemia pk rok 2
AK-REACTIVITY, CHEMIA UŁ, 3 rok, Biochemia
AK-BIOSYNTEZA, CHEMIA UŁ, 3 rok, Biochemia
Synteza a, CHEMIA
Syntezy alkenów i alkinów przykłady zadań, Chemia, Organiczna
chem org ćw 6 synteza aspiryny, chemia, organiczna, sprawozdania 2010
ROZSZCZEPIANIE I SYNTEZA JĄDROW1, Studia, chemia jądrowa
Chemia związków wanadu Synteza i analiza chemiczna otrzymanych izopoliwanadów
chemia techniczna organiczna sprawozdania synteza oranżu metylowego[1]
Chemia, Chemia - Systematyka związków nieorganicznych, OTRZYMYWANIE TLENKÓW:w drodze bezpośredniej s
Synteza do ćwiczenia z chemii metaloorganicznej, Chemia
REAKCJA SYNTEZY JĄDROWEJ, Studia, chemia jądrowa
REAKCJA SYNTEZY JĄDROWE1, Studia, chemia jądrowa
synteza i właściwości związków kobaltu z amoniakiem, chemia nieorganiczna, laboratorium, Chemia nieo
06 Synteza Indygo, Biotechnologia, chemia produktów naturalnych
SYNTEZA I BADANIE WŁAŚCIWOŚCI KATIONOWEJ ŻYWICY JONOWYMIENNEJ, CHEMIA, Synteza i badanie wł. żywicy
syntezy zwiazkow organicznych, Chemia, Organiczna
synteza wielkocząsteczkowej żywicy epoksydowej - sprawozdanie, chemia i technologia polimerów
ROZSZCZEPIANIE I SYNTEZA JĄDROWA, Studia, chemia jądrowa
więcej podobnych podstron