Genom miochondrialny, choroby mitochondrialne
Anna Wawrocka
Katedra i Zakład Genetyki Medycznej, Akademii Medycznej w Poznaniu
W każdej komórce ludzkiego ciała znajdują się mitochondria (wyjątek erytrocyty), na ogół w jednej komórce jest ich kilkaset, a każde mitochondrium zawiera 4-10 cząsteczek własnego, kolistego DNA o długości 16569 par zasad (pz). Genom mitochondrialny ulega transkrypcji i translacji. Na tej kolistej cząsteczce zapisana jest informacja o syntezie 13 białek związanych z fosforylacją oksydacyjną, 22 klasach tRNA i 2 klasach rRNA.
Główna funkcja to wytwarzanie dwóch form swobodnej energii (protonowej siły motorycznej i ATP, przetworzonych z potencjału oksydo-redukcyjnego).
Fosforylacja oksydacyjna
Cząsteczki NADH i FADH2, tworzone są wskutek glikolizy, utleniania kwasów tłuszczowych, w cyklu kwasu cytrynowego. Cząsteczki NADH i FADH2 są bogate energetycznie (zawierają pary elektronów). Energia swobodna uwalniana przez przenoszenie tych elektronów na tlen cząsteczkowy O2 zostaje wykorzystana do syntezy ATP. Proces syntezy ATP zachodzący w wyniku przenoszenia elektronów z FADH2 lub NADH na O2 nazywa się fosforylacją oksydacyjną. Proces ten ma miejsce w wewnętrznej błonie mitochondrialnej.
Proces odbywa się przy udziale szeregu nośników energii (kompleksów):
reduktaza NADH-Q - I kompleks
reduktaza bursztynian-Q - II kompleks
reduktaza cytochromowa - III komleks
oksydaza cytochromowa - IV kompleks
Mutacje w mitochondrialnym DNA
W komórce występują setki kopii mtDNA (poliplazmia), które w prawidłowych komórkach są identyczne (homoplazmia). Większość mutacji dotyczy tylko części populacji mtDNA, co powoduje koegzystencję dzikiego i zmutowanego typu mtDNA (heteroplazmia)
Pierwsza opisana mutacja mtDNA - 1988r.
Defekty w genomie mitochondrialnym objawiają się najczęściej nieprawidłowym funkcjonowaniem i anomaliami struktury mitochondriów. Ujawniają się w tkankach o wysokim zapotrzebowaniu na ATP np. mięśniach szkieletowych, mózgu, sercu, siatkówce.
Mitochondrialny DNA ma znacznie wyższe tempo mutacji niż DNA jądrowy. Przyjmuje się, że wynika to z braku systemów naprawy mtDNA, z braku histonów oraz z obecności dużej ilości wolnych rodników. Mutacje pojawiające się w mitochondialnym DNA komórek linii płciowej mogą powodować choroby występujące w rodzinie, natomiast mutacje w komórkach somatycznych mogą być związane ze spadającą z wiekiem wydolnością układu fosforylacji oksydacyjnej.
Dla wielu chorób wynikających z mutacji w mt DNA objawy pojawiają się w dopiero u dorosłych, tłumaczy się to nagromadzeniem defektywnych cząsteczek DNA w pewnych tkankach w miarę rozwoju. Jednak choroby mitochondrialne występują także u dzieci. Ważnym czynnikiem jest prawdopodobnie zapotrzebowanie poszczególnych tkanek na sprawnie działającą fosforylację oksydacyjną.
Dziedziczenie mitochondrialne
Mitochondria dziedziczone są wyłącznie po matce. Mitochondria plemnika podczas zapłodnienia wnikają do oocytu, ale jest ich niewiele (około 50) w porównaniu z 200 000 mitochondriów oocytu.
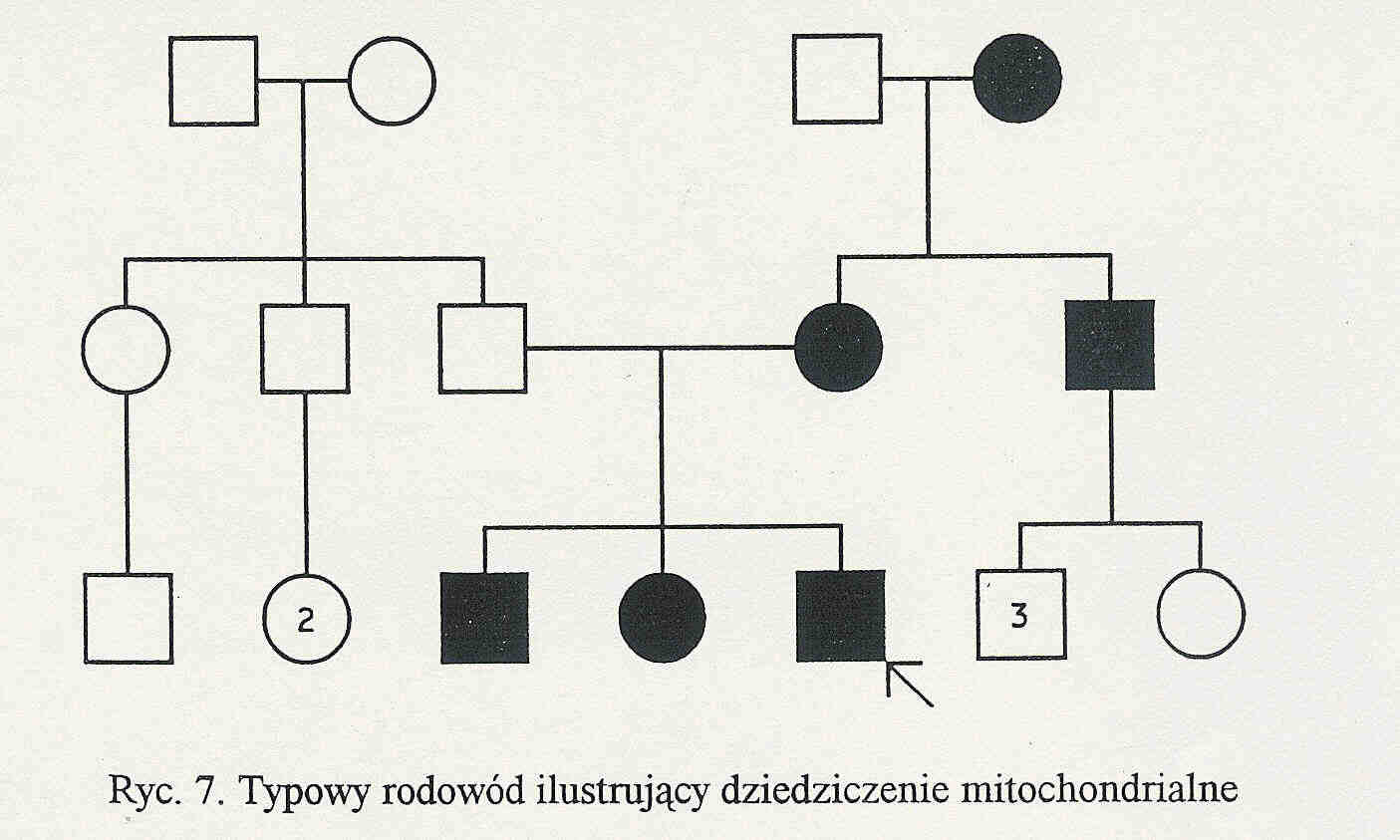
Chorują zarówno mężczyźni jak i kobiety, choroba jest przenoszona wyłącznie przez chorą kobietę. Wszystkie dzieci chorej kobiety są chore.
Choroby mitochondrialne
Choroby mitochondrialne dzielimy na:
- wywołane defektami w jądrowym DNA, kodującym białka strukturalne mitochondriów lub białka związane z regulacją ich funkcji
- wywołane defektami w mt DNA
- wywołane defektami w komunikowaniu się obu genomów
1.Pojedyncze delecje mtDNA:
Zespół Kearnsa-Sayre'a (KSS)
Objawy choroby:
-postępująca oftalmoplegia zewnętrzna (osłabienie mięśni zewnętrznych oka?)
-zwyrodnienie barwnikowe siatkówki
-początek choroby przed 20 rokiem życia
-często blok serca
-objawy móżdżkowe (ataksja)
-podwyższenie poziomu białka w płynie mózgowo-rdzeniowym
-nie stwierdza się osłabienia kończyn
-choroba występuje sporadycznie
Przewlekła postępująca oftalmoplegia zewnętrzna (CPEO) - może występować jako samodzielny zespół chorobowy, może jej towarzyszyć:
-osłabienie mięśni kończyn
-zaburzenia połykania
-zaburzenia mowy
Zespół Pearson (szpikowo-trzustkowy)
Obraz kliniczny:
-ciężka niedokrwistość makrocytowa w pierwszych tygodniach życia(% retikulocytów niski)
-podwyższony poziom hemoglobiny F
-neurotropenia (neutropenia?) i trombocytopenia (zmienne)
-obniżenie odporności-częste infekcje np. E.coli
-różny stopień dysfunkcji trzustki (stolce tłuszczowe i zespół złego wchłaniania-biegunki)
-cukrzyca
-kwasica cewkowa (wielomocz, białkomocz, cukromocz, fosfaturia, aminoaciduria) hypokaliemia i hypofosfatemia
-obniżenie napięcia mięśniowego
-KT-zanik mózgu
-dysfunkcja wątroby-podwyższony poziom transaminaz i dehydrogenazy mleczanowej, hypoprotrombinemia oporna na witaminę K
-niskorosłość
2.Mutacje punktowe
MERRF padaczka miokloniczna z włóknami „poszarpanymi” („szmatowatymi”) (myoclonic epilepsy and ragged-red fiber disease)
Mutacja mitochondrialna genów tRNA dla lizyny. Punktowa mutacja nukleotydu 8344 G-A w 80-90% MERRF
Deficyt aktywności oksydazy cytochromu. W MERRF regułą jest heteroplazmatyczność co tłumaczy olbrzymie różnice w ilości zmutowanego DNA u krewnych i różnice w ciężkości objawów (przebieg choroby zależny od % zmutowanego DNA oraz od wieku osób chorych).Opublikowana po raz pierwszy w 1973 roku. Dowody o matczynym dziedziczeniu w 1998 roku.
Pierwsze objawy w wieku późno dziecięcym lub u osób dorosłych
MERRF objawy kliniczne:
-padaczka miokloniczna współistniejąca od początku z ataksją (mogą wystąpić drgawki akinetyczne lub duże napady)
-miopatia mitochondrialna z obecnością tzw. „włókien poszarpanych” („szmatowatych”) w bioptatach mięśni
-wolno postępujące otępienie
Ze względu na znaczną zmienność fenotypową może wystąpić także:
-Niskorosłość
-Mowa skandowana
-Porażenie spastyczne
-Zanik nerwu wzrokowego
-Utrata słuchu
-Mnogie tłuszczaki na szyi i tułowiu
-Podwyższony poziom kwasu mlekowego
MELAS
-Mutacje punktowe w mitochondrialnym DNA dotyczące genów tRNA-zmiana adeniny w guaninę w 3243 pozycji (w 80% przypadków)
-mogą też być inne mutacje
Biopsja mięśni wskazuje na obecność włókien czerwonych „poszarpanych”, ze skupieniami błoniastej substancji pod błoną komórkową
Zespół MELAS (miopatia mitochondrialna, encefalopatia, kwasica mleczanowa, występowanie incydentów podobnych do udarów)
Pierwsze objawy występują w wieku dziecięcym lub wczesno młodzieńczym
Objawy kliniczne:
-zahamowanie wzrostu
-nagłe wymioty
-napady drgawkowe
-incydenty mózgowe (zaburzenia funkcji mięśni gładkich naczyń) powodujące: niedowłady połowicze, niedowidzenie połowicze a nawet objawy ślepoty korowej, otępienie, ubytki słuchu
-cukrzyca
-kardiomiopatia
Zanik nerwów wzrokowych typu Lebera
LHON (Leber's Hereditary Optic Neuropathy). U 50-74% rodzin z LHON stwierdzono mutacje punktową mtDNA dotyczącą nukleotydu 11778, zmiana G-A w efekcie zmiana argininy na histydynę w podjednostce ND4 dehydrogenazy NADH.
Znaczna przewaga chorych mężczyzn, u 50% mężczyzn z mutacją mtDNA i tylko u 20% kobiet z mutacją, występują objawy chorobowe (obniżona penetracja i opóźniona manifestacja objawów u kobiet).
Podatność na rozwój zaniku nerwów wzrokowych uwarunkowana interakcją mitochondrialno-jądrową.
NARP
(neurodegeneration, ataxia and retinitis pigmentosa) najczęstsza mutacja w nukleotydzie 8993 genomu mitochondrialnego lub zmiana T-G w 8993, co prowadzi do zmiany leucyny na argininę w podjednostce 6 mitochondrialnej ATP-azy a w konsekwencji do zaburzeń syntezy ATP (tzw. mutacja NARP).
Cechy kliniczne NARP:
-różnorodna kombinacja objawów
-retinitis pigmentosa
-wiotkość mięśni proksymalnych neurogenna
-ataksja
-opóźnienie rozwoju
-drgawki
-otępienie
-ostre zagrażające życiu epizody związane z kwasicą mleczanową
Diagnostyka chorób mitochondrialnych
-Analiza rodowodu
-Biopsja mięśnia i analiza bioptatu pod kątem zawartości enzymów mitochondrialnych (odpowiednie techniki barwienia) i obecności czerwonych poszarpanych włókien (metoda histochemiczna)
-Badania molekularne - prowadzone w Polsce od 1996 roku w Zakładzie Genetyki Uniwersytetu Warszawskiego
Wyszukiwarka
Podobne podstrony:
2 Genom mitochondrialny, choroby mitochondrialne
Genom mitochondrialny
genom mitochondrialny
Genom mitochondrialny ćw 4, IV rok
LUDZKI GENOM MITOCHONDRIALNY(1)
Choroby układu immunologicznego, Szkoła, przydatne w szkole
Choroby z autoagresji, immunologia kliniczna
Choroby układu immunologicznego
16 Choroby układu immunologicznego AIDS
choroba Alzheimera immuno
choroby mitochondrium peroksysomy lizosomy
MITOCHONDRIALNE CHOROBY
Biochemia.Choroby mitochondrialne, Dietetyka CM UMK, Biochemia
choroby mitochondrium peroksysomy lizosomy
CHOROBY DZIEDZICZONE MITOCHONDRIALNIE
Choroby mitoch2011
więcej podobnych podstron