
Zasady diagnostyki
chorób mitochondrialnych
u dzieci
Dr Dorota Piekutowska-Abramczuk
Zakład Genetyki Medycznej IPCZD
Warszawa, 02.12.2011

Chorobą mitochondrialną
nazywamy stan chorobowy wywołany
uwarunkowaną genetycznie zmianą budowy
białka, pierwotnie zaburzającą przebieg
procesu fosforylacji oksydacyjnej w komórce.
45
7
38
4
4
11
1
10
13
3
10
16
2
14
Podjednostki
mtDNA
nDNA
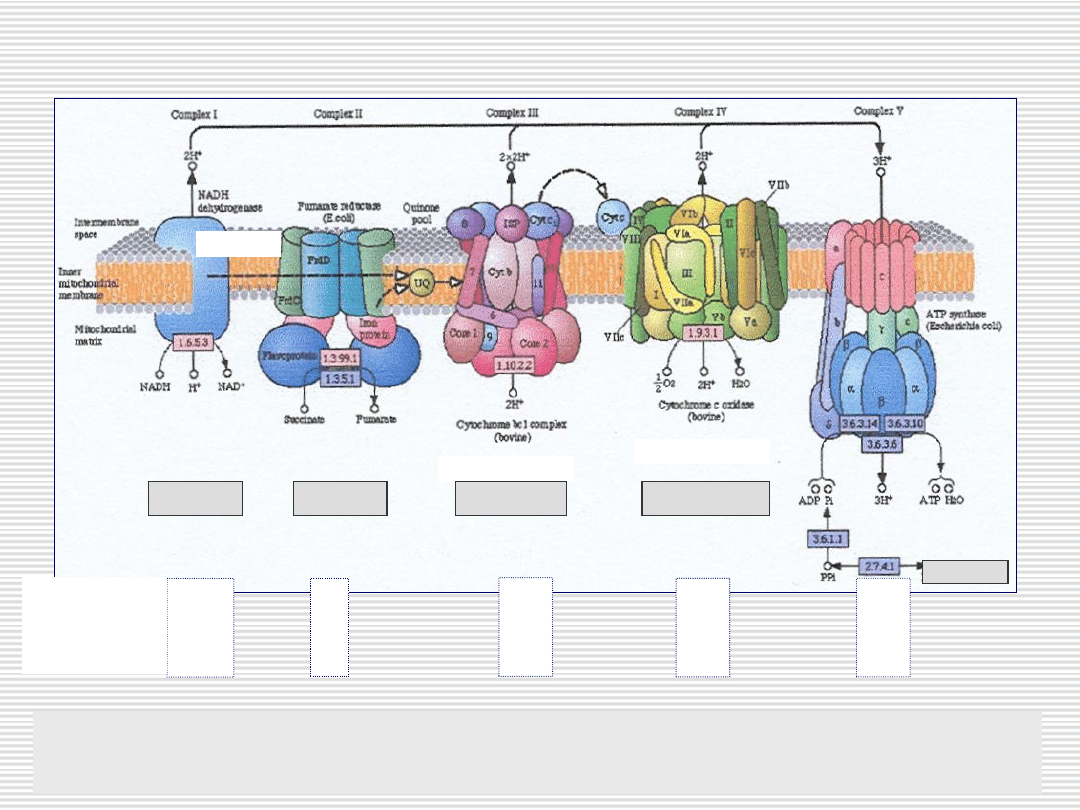
System fosforylacji oksydacyjnej (OXPHOS)
Dehydrogenaza
bursztynianowa
Dehydrogenaza
NADH
Kompleks
cytochromów bc1
Oksydaza
cytochromu c
Syntaza ATP
Zaburzenia aktywności kompleksów – izolowane lub złożone
najczęściej – KI i KIV

Choroby mitochondrialne
Częstość występowania 1:8500 - 1:5000
Prawdopodobnie wiele przypadków pozostaje nierozpoznanych
Heterogenność kliniczna, biochemiczna i genetyczna
Patomechanizm
obniżenie efektywności syntezy ATP,
wzrost produkcji reaktywnych form tlenu,
zaburzenia wewnątrzkomórkowej homeostazy wapnia.

Choroby mitochondrialne
Uwarunkowane mutacjami w genach:
› mitochondrialnych
» strukturalne podjednostki OXPHOS
» tRNA
» rRNA
› jądrowych
» strukturalne podjednostki OXPHOS
» niestrukturalne
»› czynniki towarzyszące (
assembly factors
) (I-V)
»› czynniki zaburzające replikację mtDNA
»› zaburzające transkrypcję i translację genów OXPHOS
»› zaburzające import białek mitochondrialnych i in.
>50% wszystkich mutacji w mtDNA (gł.
MTTK, MTTL1, MTTI)

Dziedziczenie
matczyne
autosomalne recesywne
autosomalne dominujące
sprzężone z chromosomem X
Przypadki sporadyczne
Występuje w 10
3
– 10
5
liczby kopii
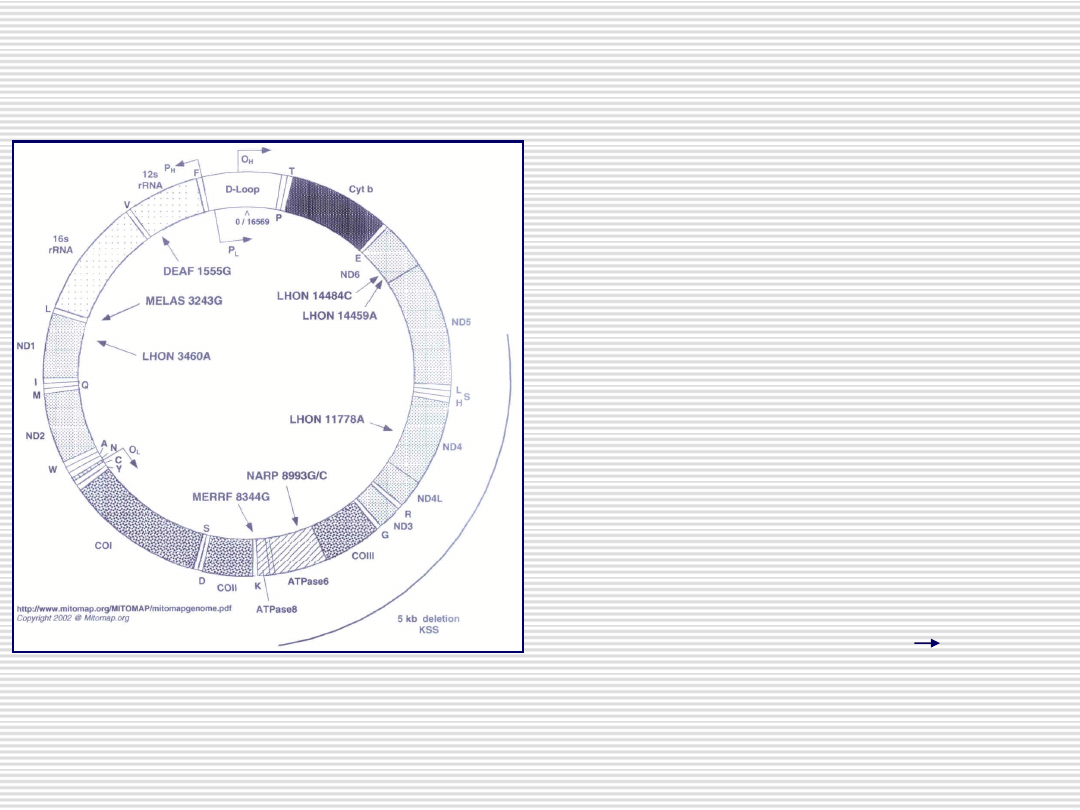
Kolista cząsteczka o wielkości 16 569
kpz (nić H i L)
Zawiera 37 genów (13 strukturalnych
podjednostek systemu OXPHOS, 2 rRNA,
22 tRNA)
Małe lub brak przestrzeni
międzygenowych
Brak intronów
2 małe obszary niekodujące: pętla D –
zawiera miejsca inicjacji transkrypcji i
replikacji nici H i miejsce startu replikacji
nici L
Odstępstwa od uniwersalnego kodu
genetycznego (np. TGA (Stop) Trp)
Mitochondrialne DNA

Genetyka mitochondrialna
• Dziedziczenie matczyne
Tylko kobieta przekazuje zmutowany gen swemu potomstwu, zarówno córce
jak i synowi (nieliczne mtDNA plemnika są aktywnie usuwane we wczesnych
podziałach zygoty).
• Poliploidalność mitochondriów (setki mitochondriów, a w każdym do 10
cząsteczek mtDNA)
• Szybkie tempo ewolucji (mała wydajność systemów naprawy, brak
histonów, brak zjawiska rekombinacji, nadprodukcja ROS)
• Heteroplazmia i wartość progowa
Prezentacja objawów zależy od poziomu heteroplazmii i wartości progowej.
Segregacja mitotyczna (efekt
bottleneck
) w oogenezie tłumaczy częste
zmiany poziomu mutacji w kolejnych pokoleniach i znaczne zróżnicowanie
fenotypowe.

Cechy mutacji mtDNA
Heteroplazmatyczne
Homoplazmatyczne
Stabilne
• silna korelacja genotyp-fenotyp
• mała/brak zmienność poziomu mutacji w różnych tkankach
• mała/brak zmienność poziomu w czasie (podczas rozwoju płodowego i
pourodzeniowego)
m.8993T>G, m.8993T>C
Niestabilne
• bardzo zróżnicowany obraz kliniczny
• niejednorodne tkankowe rozmieszczenie mutacji
• duża zmienność poziomu mutacji w czasie
m.3243A>G

Choroby mitochondrialne u dzieci
Częściej wynikają z mutacji w jądrowym DNA
Cięższe od objawiających się w wieku późniejszym i częściej
wielonarządowe;
Dotyczą dysfunkcji wątroby (MDS), choroby nerek (MDS, deficyt
KIII) i zaburzeń hemopoezy (zespół Pearsona)
Opóźnienie rozwoju w połączeniu z kwasicą mleczanową
Postępujący regres rozwojowy, utrata posiadanych umiejętności
(zespół Leigha)
Niespecyficzne objawy zaburzenia w odżywianiu, wzrastania,
drgawki, częste infekcje
Rzadko obecne RRF

Rozpoznanie choroby mitochondrialnej
Zdefiniowane, wysoce prawdopodobne
analiza mutacji
Prawdopodobne
biopsja mięśnia
Możliwe
obserwacja, biopsja mięśnia, zabezpieczenie
materiału w wypadku zgonu

Materiał biologiczny
mięsień
krew obwodowa
wymaz z nabłonka jamy ustnej
mocz
wątroba
fibroblasty
włosy
płyn owodniowy
kosmki kosmówki
Oragene DNA
www.dnagenotek.com

Biopsja mięśnia
kompleksowe potwierdzenie rozpoznania
Ocena morfologiczna (włókna RRF)
Badanie histochemiczne (oksydaza cytochromu c mozaikowość,
dehydrogenaza bursztynianowa)
Badanie proteomiczne (obecność i aktywność kompleksów łańcucha
oddechowego i ich poszczególnych podjednostek)
Analiza enzymatyczna (aktywność kompleksów łańcucha oddechowego i
syntazy cytrynianowej)
Analiza molekularna

Diagnostyka molekularna chorób
mitochondrialnych w ZGM IPCZD
1. Identyfikacja powszechnych mutacji
• m.8993T>C/G (
MTATP6
) - NARP/MILS
• m.3243A>G (
MTTL1
) - MELAS
• m.8344A>G (
MTTK
) - MERRF
• delecja 4977pz (m.8470_13446del) KSS i in. zespoły delecyjne
• m.11778G>A (
MTND4
), m.3460G>A (
MTND1
), m.14484T>C (
MTND6
) - LHON
• g.1541G>A (
SCO2
) - deficyt białka SCO2
• c.845_846delCT, c.311_312insAT312_321del10(
SURF1
) – LS
2. Poszukiwanie mutacji w innych, znanych genach związanych z fenotypem
•
MTND1-MTND6 -
LS
•
DGUOK, MPV17
- HC-MDS
3. W przyszłości dalsza analiza molekularna obejmująca geny nie badane lub nowo
odkryte.
podstawowy
skrining mtDNA

Zespół Leigha sprzężony z
deficytem COX i mutacjami w
genie
SURF1

S1
(COXI)
OXA1, COX10, COX15,
SURF1
Hem a, Hem a
3
COX11, COX17, SCO1, SCO2
Cu
B
COX11
COXIV
S2
SURF1
,
OXA1
COXII
COX17, SCO1, SCO2
Cu
A
COXIII, Va, Vb, VIb,
COXVIc, VIIb, VIIc, VIII
S3
COXVIa, VIIa
S4
Składanie kompleksu
COX
NH
2
61-79
274-290 COOH
1
300
TS N-TM C-TM
1
2
4
6
7
8
9
5
3
1 2
4
6
7
8
9
5
3
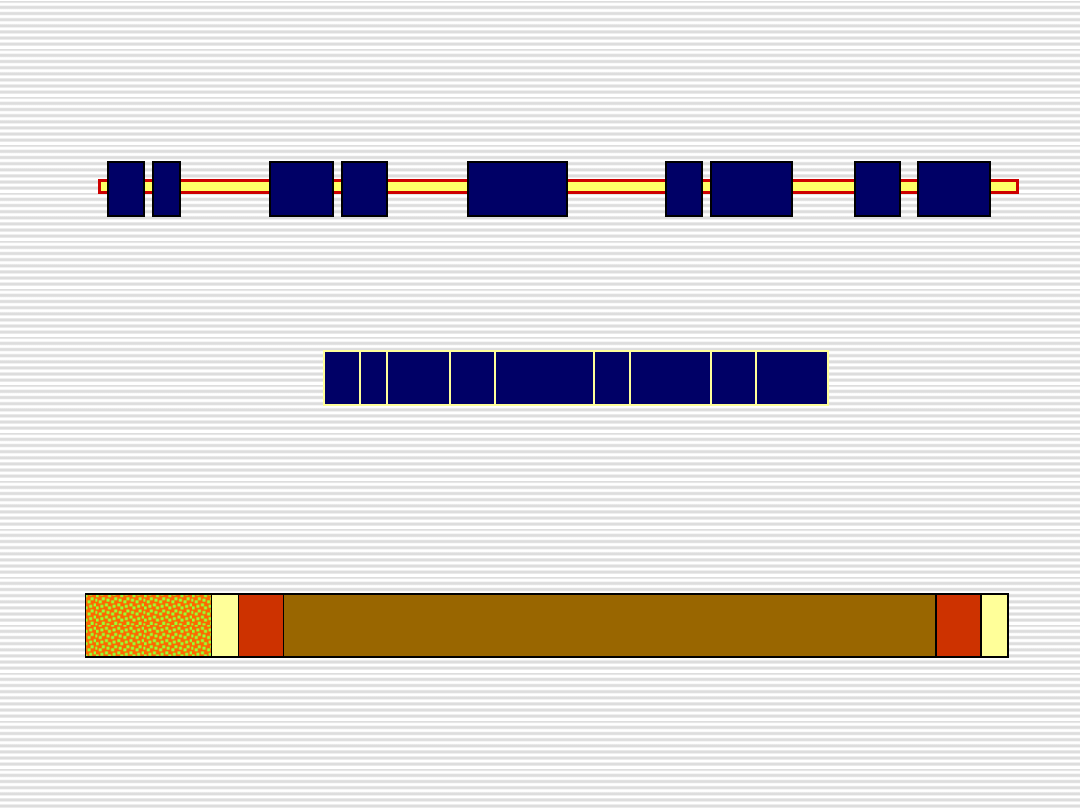
Gen
SURF1
9q34
4664 pz
965 pz
Białko SURF1
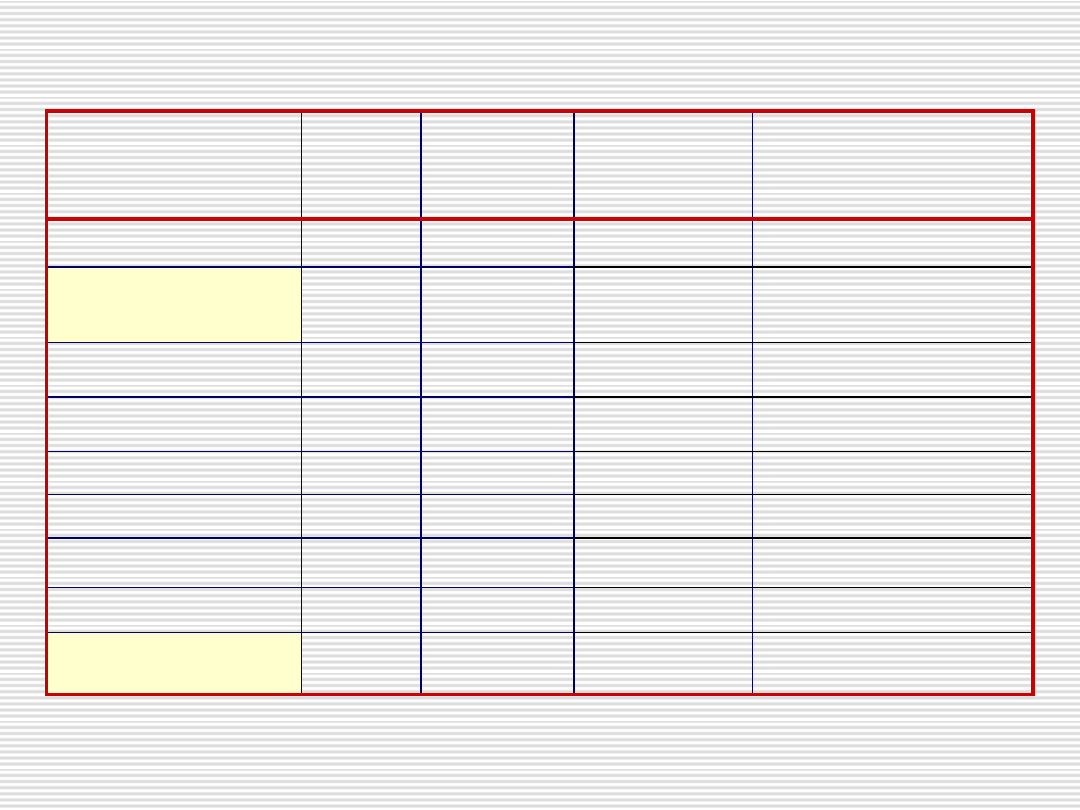
Mutacja
Liczba
alleli
Częstość
alleli (%)
Liczba
homozygot
Oczekiwana
zmiana w budowie
białka
c.39delG
1
1,4
0
p.Ala13AlafsX71
c.311_312insAT
312_321 del10
8
11,1
2
p.Pro104ProfsX105
c.688C>T
1
1,4
0
p.Arg230X
c.704T>C
1
1,4
0
p.Met235Thr
c.752-1G>C
4
5,5
0
spl int7
c.756_757delCA
1
1,4
0
p.Ser252SerfsX290
c.800_801insT
1
1,4
0
p.Leu267LeufsX290
c.821A>G
1
1,4
0
p.Tyr274Cys
c.845_846delCT
52
75
20
p.Leu281LeufsX290
44/36 pacjentów z LS
14
0
0
3
54
8
0
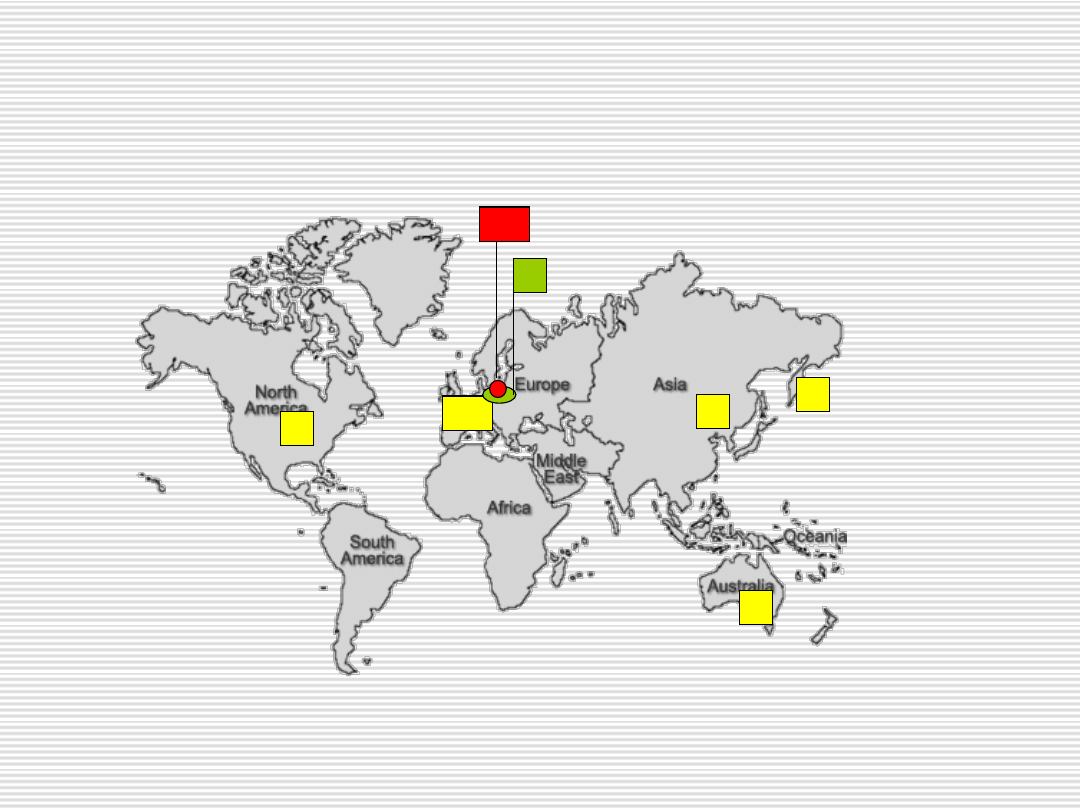
Częstość nosicielstwa powszechnej delecji w różnych populacjach

Badania populacyjne
Mutację c.845_846delCT w genie
SURF1
wykryto na 75% alleli
8 heterozygotycznych nosicieli/ 2890 noworodków
częstość nosicielstwa delecji 1: 357
(0.28%)
częstość nosicielstwa choroby 1: 277
(0.36%)
częstość choroby w populacji polskiej
1: 306 400
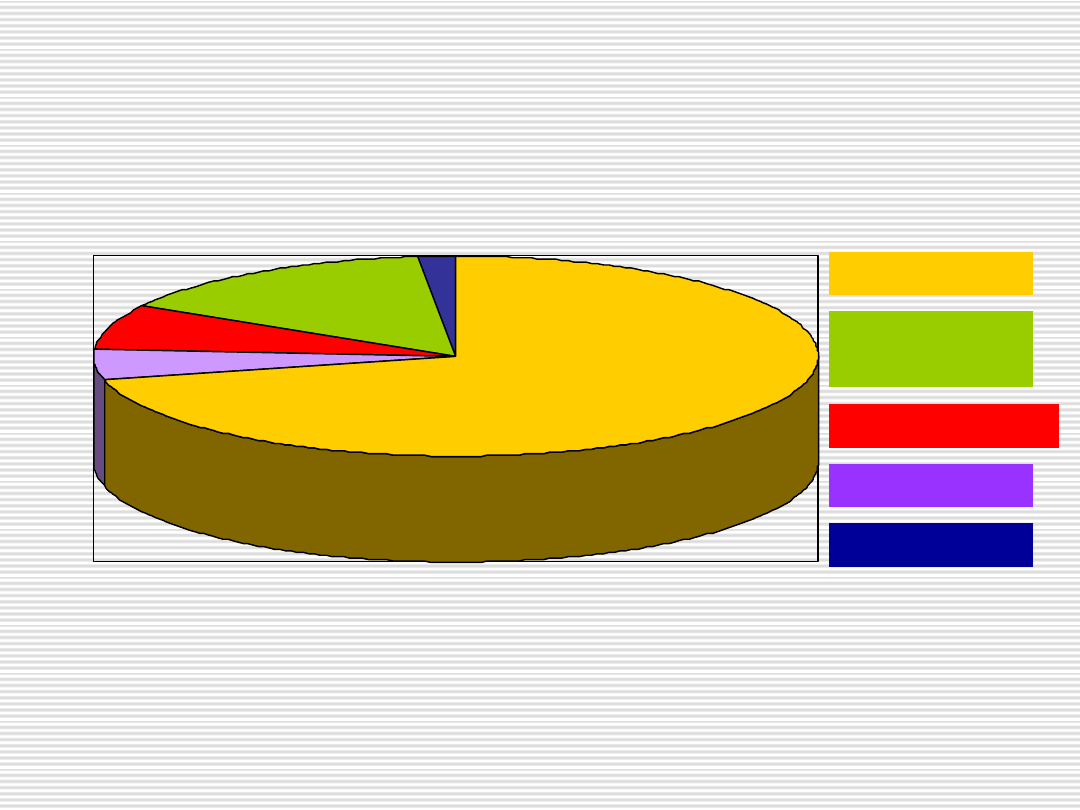
SURF1
69%
SCO2
4,9%
MTND1,3,5,6
14,7%
MTATP6
9,8%
MTTK
1,6%
Zespół Leigha w populacji polskiej

Schemat diagnostyki molekularnej w zespole
Leigha
1. Identyfikacja powszechnych mutacji w genie S
URF1
(c.845_846delCT,
c.311_312insAT312_321del10)
2. Identyfikacja mutacji w genie
MTATP6
(m.8993T>C/G)
3. Poszukiwanie mutacji w genach
MTND1-MTND6
4. Identyfikacja powszechnej mutacji w genie
SCO2
(g.1541G>A)
5. Identyfikacja mutacji za pomocą techniki MLPA: m.3243A>G (
MTTL1
),
m.8344A>G (
MTTK
), m.11778G>A (
MTND4
), m.3460G>A (
MTND1
),
m.14484T>C (
MTND6
)
6. W przyszłości dalsza analiza molekularna obejmująca geny nie badane lub nowo
odkryte, sekwencjonowanie całego genomu mitochondrialnego.

Mitochondrialny zespół deplecyjny (MDS)
(OMIM 251880)
heterogenna grupa chorób charakteryzującą się tkankowo
specyficznym obniżeniem ilości kopii mtDNA - deplecji (do poniżej
30%);
Wynika z zaburzeń replikacji mtDNA lub zaburzeń równowagi puli
nukleotydów, dostępnych do syntezy mtDNA;
odpowiada za ok. 50% złożonych deficytów OXPHOS u dzieci;
fenotypy: wątrobowo-mózgowy, mózgowo-mięśniowy, mięśniowy;
większość objawia się w okresie noworodkowym (wiotkość mięśni,
niewydolność wątroby, tubulopatia nerkowa, kwasica mleczanowa,
zgon przed ukończeniem 1 r.ż), lub we wczesnym dzieciństwie
(izolowana miopatia z regresją funkcji ruchowych lub wolno
postępująca encefalomiopatia);
objawy mogą początkowo ograniczać się do jednego narządu lub
tkanki, później dotyczą wielu narządów;
dziedziczony w sposób autosomalny recesywny;
w ok. 20-80% przypadków nie udaje się ustalić molekularnego
podłoża choroby.

Geny związane z MDS
TK2
RRM2B
f. mięśniowa
SUCLA2
TYMP
f. mózgowo-mięśniowa
SUCLG1
DGUOK
MPV17
f. wątrobowo-mięśniowa
C10orf2
POLG1
Najczęstsza przyczyna chorób mitochondrialnych u człowieka
adPEO i arPEO, parkinsonizm, SANDO (zespół zaburzeń
czuciowo-ruchowych), męska niepłodność

Postać wątrobowo-mózgowa (OMIM #251880)
Objawy:
od urodzenia do 6 mies. ż.
Początkowo – powiększenie wątroby i postępująca niewydolność, wymioty,
zaburzenia wzrastania, hipotonia niewielkiego stopnia;
koagulopatia, hipoglikemia, poziom całkowitej i związanej bilirubiny, AlAT i
AspAT, poziom tyrozyny i fenyloalaniny w osoczu;
później – odjawy neurologiczne – ataksja, drżenia, dystonia, neuropatia,
oczopląs, ciężkie upośledzenie rozwoju;
kwasica mleczanowa, znaczny wzrost stężenia AFP,
zgon przed końcem 1 r.ż.
Aktywność kompleksów łańcucha oddechowego obniżona (z wyjątkiem
kompleksu II)
Histopatologia: wielko- i drobnopęcherzykowate stłuszczenie, cholestaza
hepatocytarna i kanalikowa, włóknienie, odkładanie żelaza
Z. Alpersa
Padaczka oporna na leczenie, regresja rozwoju psychoruchowego, dysfunkcja
wątroby; przebieg postępujący, rokowanie niekorzystne.
W 50% przypadków czynnikiem inicjującym uszkodzenie wątroby jest podanie
kwasu walproinowego
Diagnostyka prenatalna
W rodzinach z defektem jądrowego DNA TAK
W rodzinach z defektem mtDNA ???
Diagnostyka preimplantacyjna, selekcja płci ???
m.8993 heteroplazmia <50%
prawdopodobnie zdrowe, >80%
chore
m.3243 <15%
prawdopodobnie zdrowe
<18%
zdrowe z prawdopodobieństwem 85%
Ścisła korelacja między poziomem mutacji a ciężkością objawów
Równy poziom mutacji w różnych tkankach
Brak zmian poziomu mutacji w czasie
OBIETNICA POSIADANIA
ZDROWEGO DZIECKA
ZMNIEJSZENIE RYZYKA URODZENIA
CHOREGO DZIECKA

Mitochondria w sieci
Kliniczne i molekularne informacje dla lekarzy i badaczy
On-line Mendelian Inheritance in Man www.ncbi.nlm.nih.gov
Mitomap www.mitomap.org
Geneclinics www.geneclinics.org
Dane molekularne Uppsala http://www.genpat.uu.se/mtDB/index.html
Dane biochemiczne i molekularne
Mitop mips.gsf.de/proj/medgen/mitop
Mitodat http://www-lecb.ncifcrf.gov/mitoDat/
Informacje dla pacjentów
United Mitochondrial Diseases Foundation (USA) www.umdf.org
Mitolinks (UK) http://www.communigate.co.uk/ne/
mitolinks/index.phtml

DZIĘKUJĘ ZA UWAGĘ
Wyszukiwarka
Podobne podstrony:
choroby mitochondrium peroksysomy lizosomy
2 Genom mitochondrialny, choroby mitochondrialne
Biochemia.Choroby mitochondrialne, Dietetyka CM UMK, Biochemia
Genom mitochondrialny. Choroby mitochondrialne(1), Immunologia
choroby mitochondrium peroksysomy lizosomy
CHOROBY MITOCHONDRIALNE I METABOLICZNE
MITOCHONDRIALNE CHOROBY
CHOROBY DZIEDZICZONE MITOCHONDRIALNIE
choroby naczyn i serca(1)
ŻYWIENIE A CHOROBY 4b
Choroby układu nerwowego ppt
Produkty przeciwwskazane w chorobach jelit II
Choroba niedokrwienna serca
CZLOWIEK I CHOROBA – PODSTAWOWE REAKCJE NA
Choroby nerwów czaszkowych
więcej podobnych podstron