
CHOROBY MITOCHONDRIALNE

GENOM MITOCHONDRIALNY:
• KOLISTE DWUNICIOWE DNA
, ZASADY
REPLIKACJI PRZYPOMINAJĄ
REPLIKACJĘ BAKTERYJNĄ
• DNA
NIEZALEŻNE
OD DNA
JĄDROWEGO
• NIE POSIADA ENZYMÓW
REPARACYJNYCH
• KODUJE RÓŻNE PODJEDNOSTKI
BIAŁEK ODDECHOWYCH, tRNA, rRNA
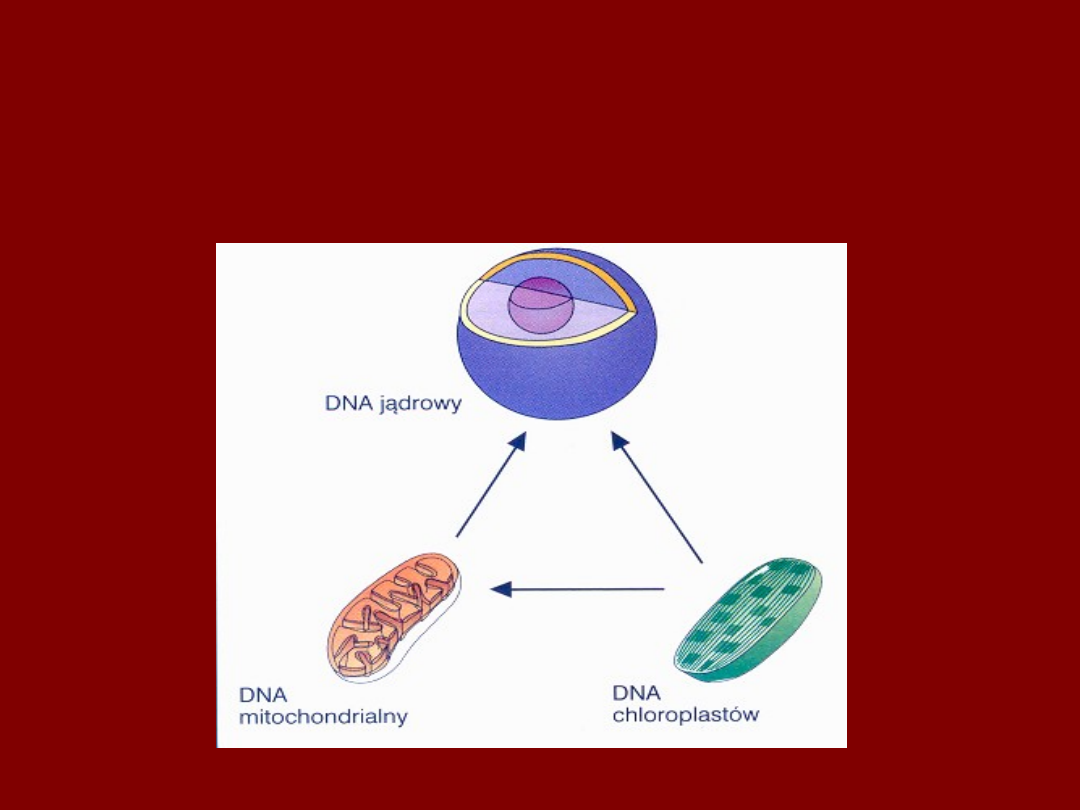
MITOCHONDRIA I CHLOROPLASTY BYŁY
KIEDYŚ BAKTERIAMI
SYMBIONTYCZNYMI
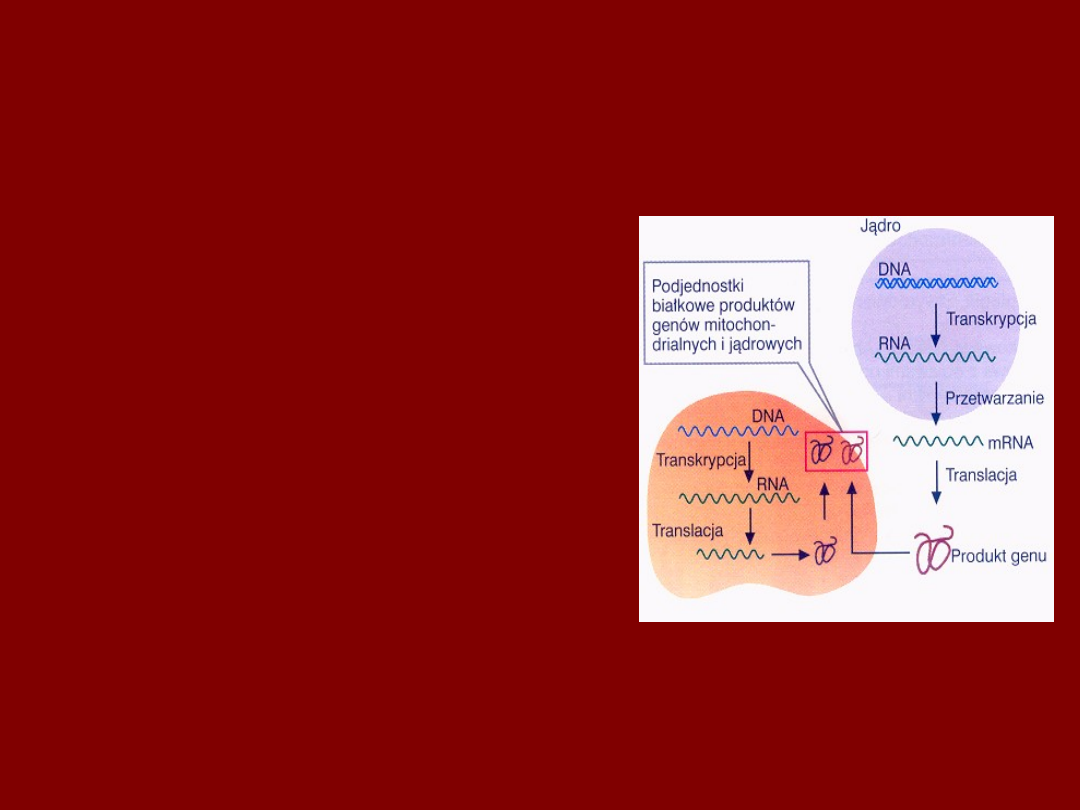
WSPÓŁPRACA m-DNA i DNA
JĄDROWEGO
• POSZCZEGÓLNE PODJEDNOSTKI
TYCH SAMYCH BIAŁEK
ODDECHOWYCH SĄ KODOWANE
m-DNA I DNA JĄDROWYM
• TAKI SAM KOŃCOWY DEFEKT KLINICZNY
MOŻE BYĆ WYWOŁANY NIEZALEŻNYMI
MUTACJAMI W OBU TYPACH DNA
• TAKI SAM FENOTYP CHOROBOWY
MOŻE MIEĆ ODMIENNE DZIEDZICZENIE
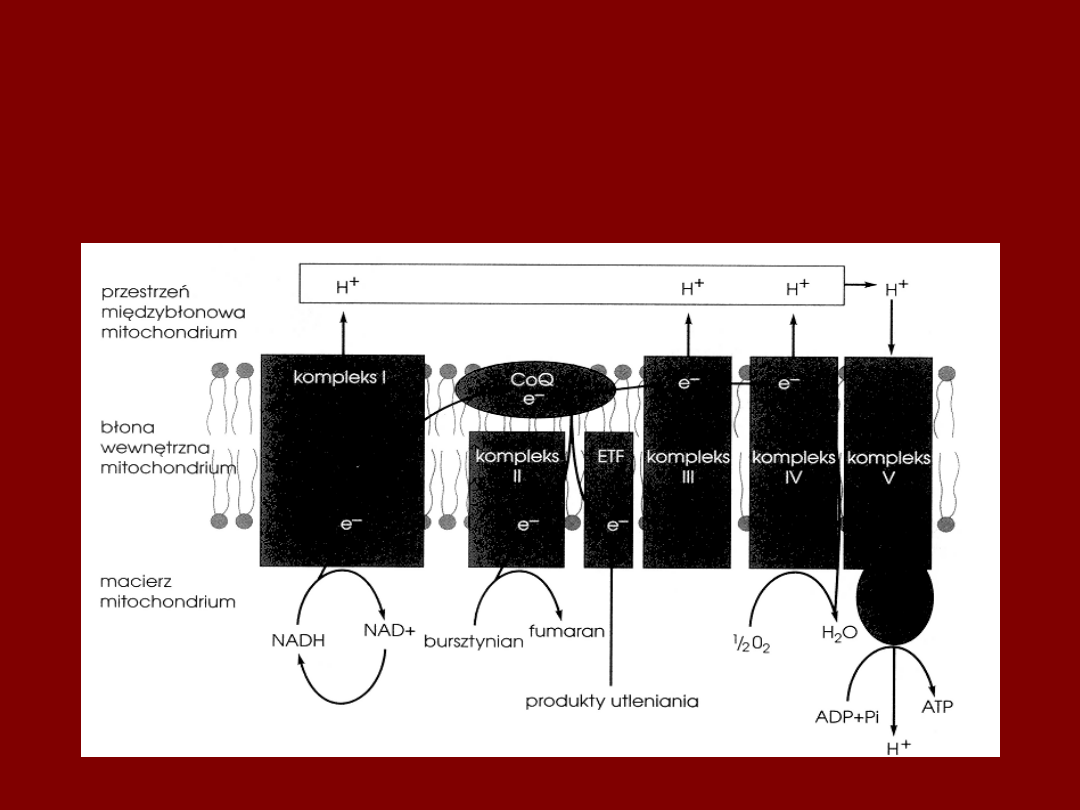
KOMPLEKSY FOSFORYLACJI
OKSYDACYJNEJ
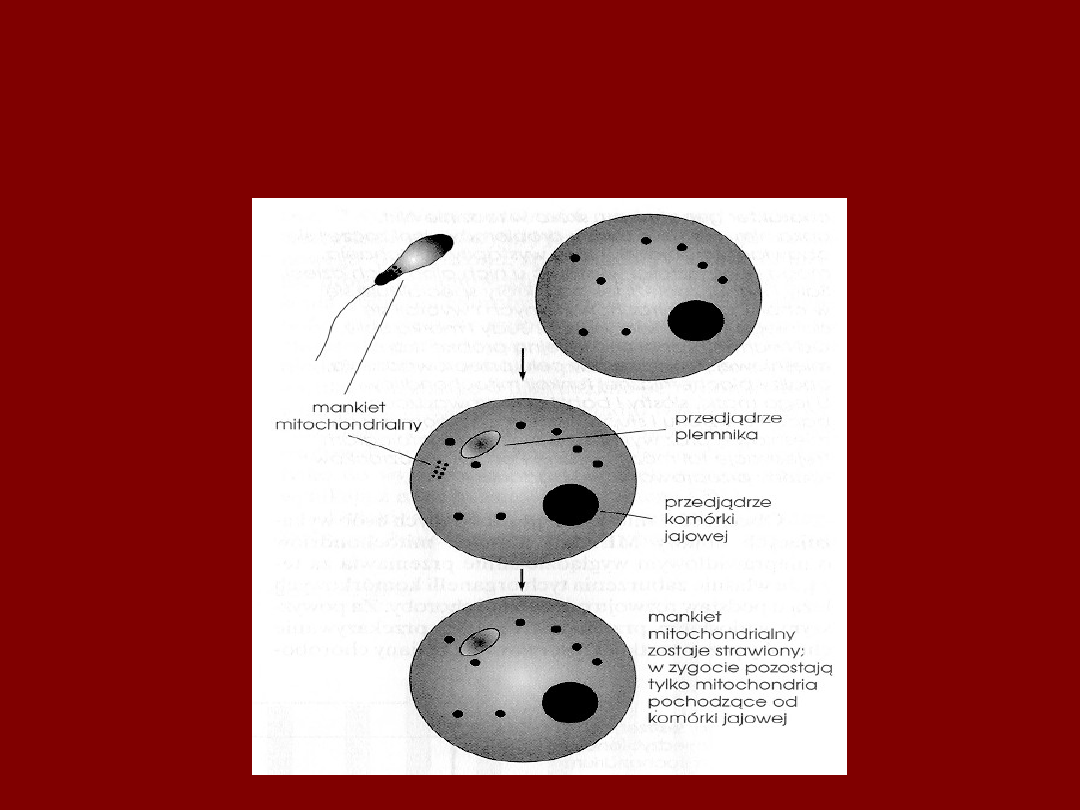
MITOCHONDRIA
DZIEDZICZYMY PO MATCE
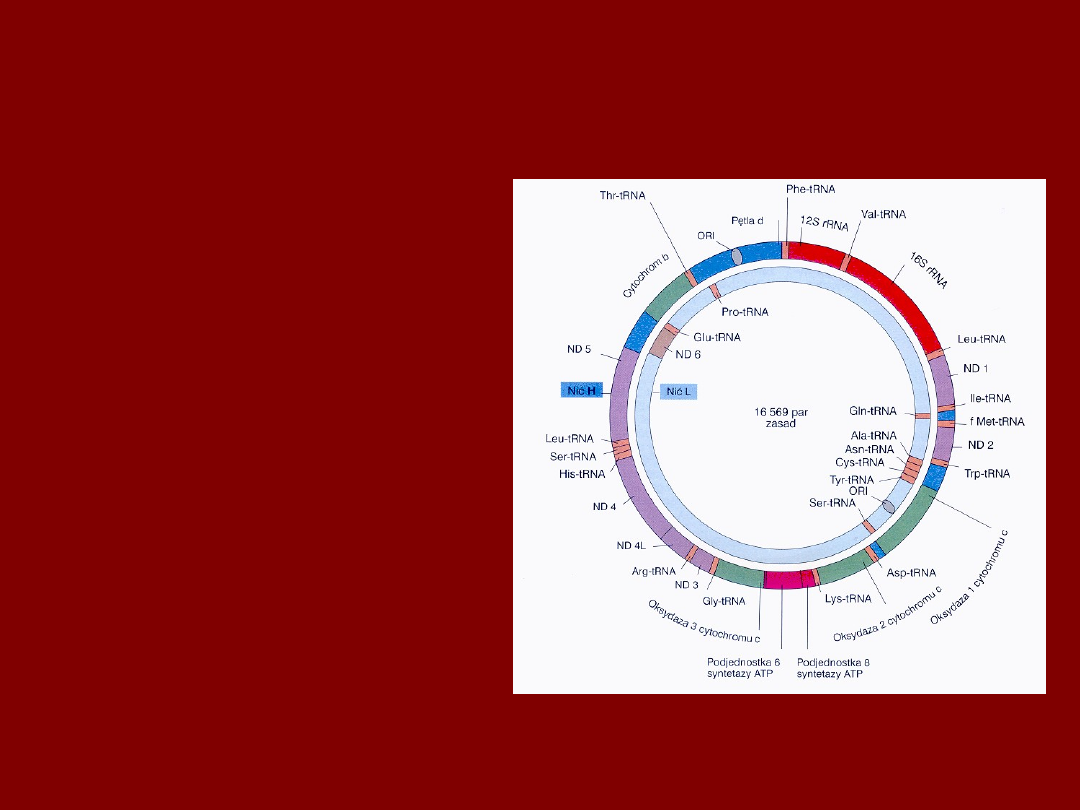
GENOM MITOCHONDRIALNY
•
KAŻDE MITOCHONDRIUM
ZAWIERA 2-10 KOLISTYCH DNA
•
M-DNA SKŁADA SIĘ Z:
13 REGIONÓW KODUJĄCYCH
- Podj. 1, 2, 3 OKSYDAZY CYT - C
- CYT-B
- Podj. 6, 8 ATP - azy
- Podj. ND 1, 2, 3, 4L, 4, 5, 6 NADH
- 7 Podj. NADH – Reduktazy
( 60 % pojemności kodującej)
•
ZBUDOWANY Z DWU NICI H i
L
-nić H to większość genów,
dwa traskrypty
-nić L to geny ND6, i 8 jedn. tRNA,
jeden transkrypt

HETEROPLAZATYCZNOŚĆ
• MITOCHONDRIA DELECYJNE
NAMNAŻAJĄ SIĘ SZYBCIEJ NIŻ
PRAWIDŁOWE
•
RÓŻNE PROPORCJE
USZKODZONYCH MITOCHONDRIÓW
W TEJ SAMEJ LINII KOMÓRKOWEJ
•
MOZAIKOWE RÓŻNICE
ZMIAN POMIEDZY TKANKAMI
A NAWET W TEJ SAMEJ
TKANCE
•
ZMIENNOŚĆ OBJAWÓW KLINICZNYCH
A.
CIĘZKIE ZMIANY W WIELU NARZĄDACH
B.
CIĘZKIE ZMIANY W POJEDYNCZM NARZĄDZIE
C.
PORONNE, LOKALNE, PÓZNE OBJAWY
D. KOMPLEMENTACJA MUTACJI – BEZ OBJAWÓW

DEFEKTY GENOMU
MITOCHONDRIALNEGO
• PRZEKAZYWANE TYLKO
W OOCYCIE
• DZIEDZICZĄ SIĘ
W LINII MATCZYNEJ
NIEZALEŻNIE OD PŁCI POTOMKA
• OBJAWY DOTYCZĄ
NARZĄDÓW NAJBARDZIEJ
ZALEŻNYCH OD METABOLIZMU TLENOWEGO
(OUN, SIATKÓWKA, MIĘŚNIE, NERKA itp.)
• BARDZO
ZMIENNA EKSPRESJA
( WIEK,
UMIEJSCOWIENIE, NASILENIE OBJAWÓW) –
czasem u dzieci wcześniej niż u rodziców
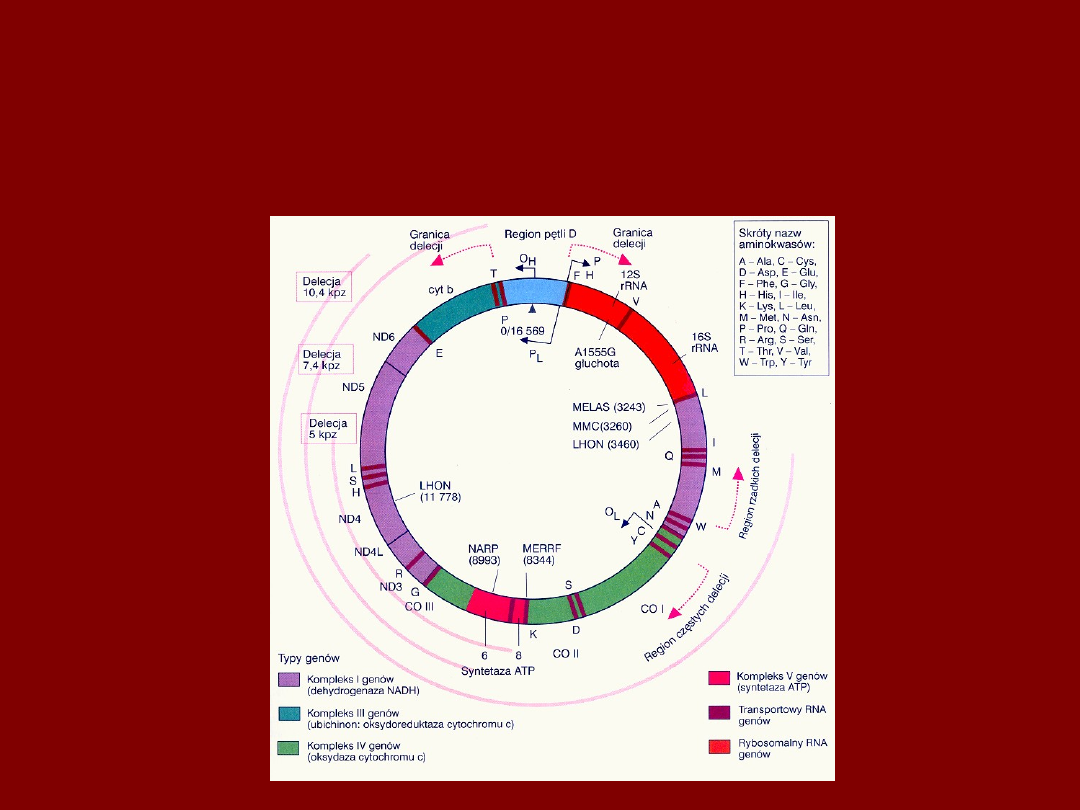
DEFEKTY GENOMU
MITOCHONDRIALNEGO

RÓŻNE DEFEKTY MITOCHONDRIALNE
MOGĄ MIEĆ PODOBNĄ KOŃCOWĄ
EKSPRESJĘ
N.
wzrokowy-LHON- zanik
Kora potyliczna
MELAS, udar
Siatkówka – retinopatia
Delecje, mutacja 3243,8993
Soczewka-zaćma
Różne
delecje
Rogówka –dystrofia
Delecje pojedynczych
zasad
Powieka, mięśnie
Różne mutacje
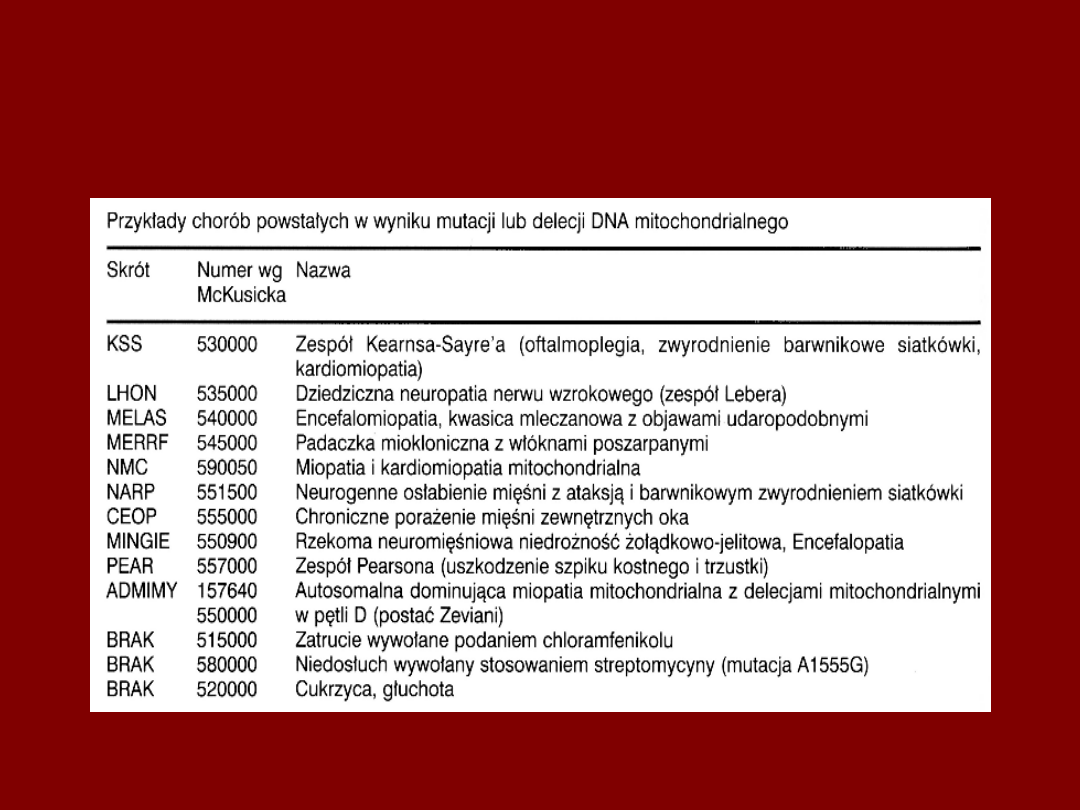
CHOROBY MITOCHONDRIALNE

MERF
• M
YOCLONIC
E
PILEPSY WITH
R
AGGED
R
ED
F
IBERS
• PADACZKA MIOKLONICZNA : napadowe skurcze
różnych grup mięśni, nagłe ruchy, ”grymasy”,
drgawki uogólnione, utrata siły i napięcia mięśni
• CZERWONE WŁÓKNA MIĘŚNIOWE POSTRZĘPIONE,
„SZMATOWATE”
• ATAKSJA, SPASTYCZNOŚĆ, DEMENCJA,
GŁUCHOTA, ŚLEPOTA.
• DEFEKT Z NIEDOBORU KOMPLEKSÓW
ODDECHOWYCH
I
i
IV
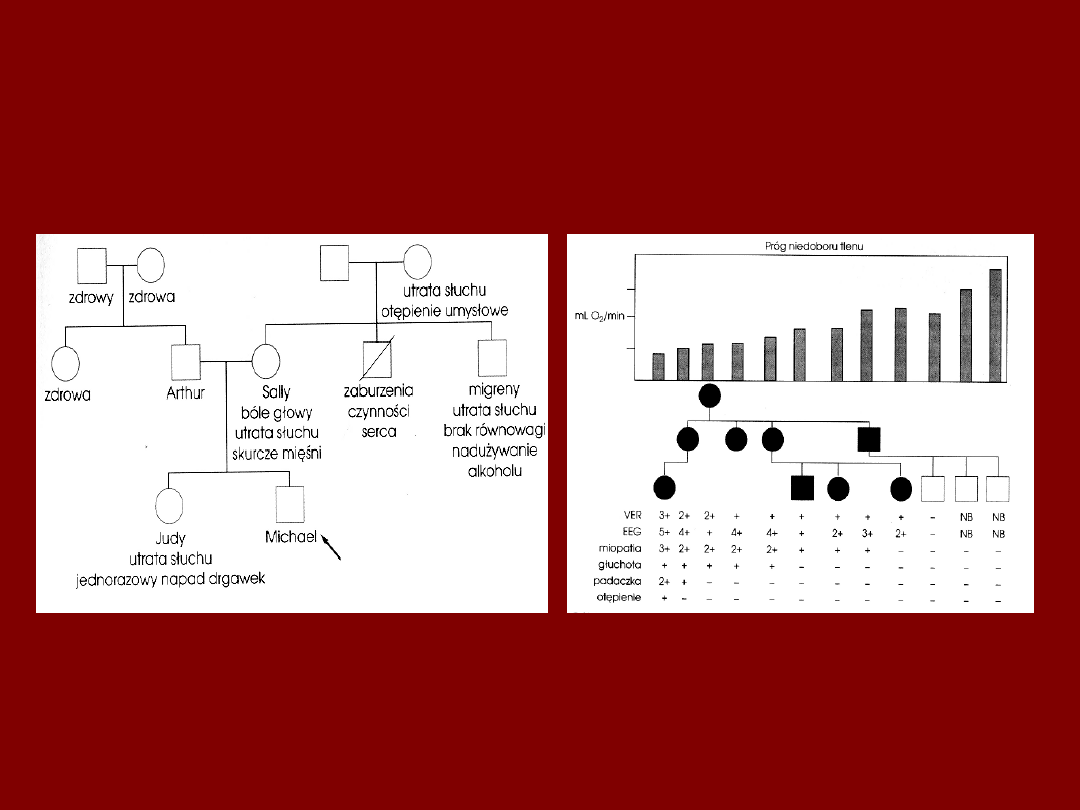
ZMIENNOŚĆ EKSPRESJI
KLINICZNEJ W MERF
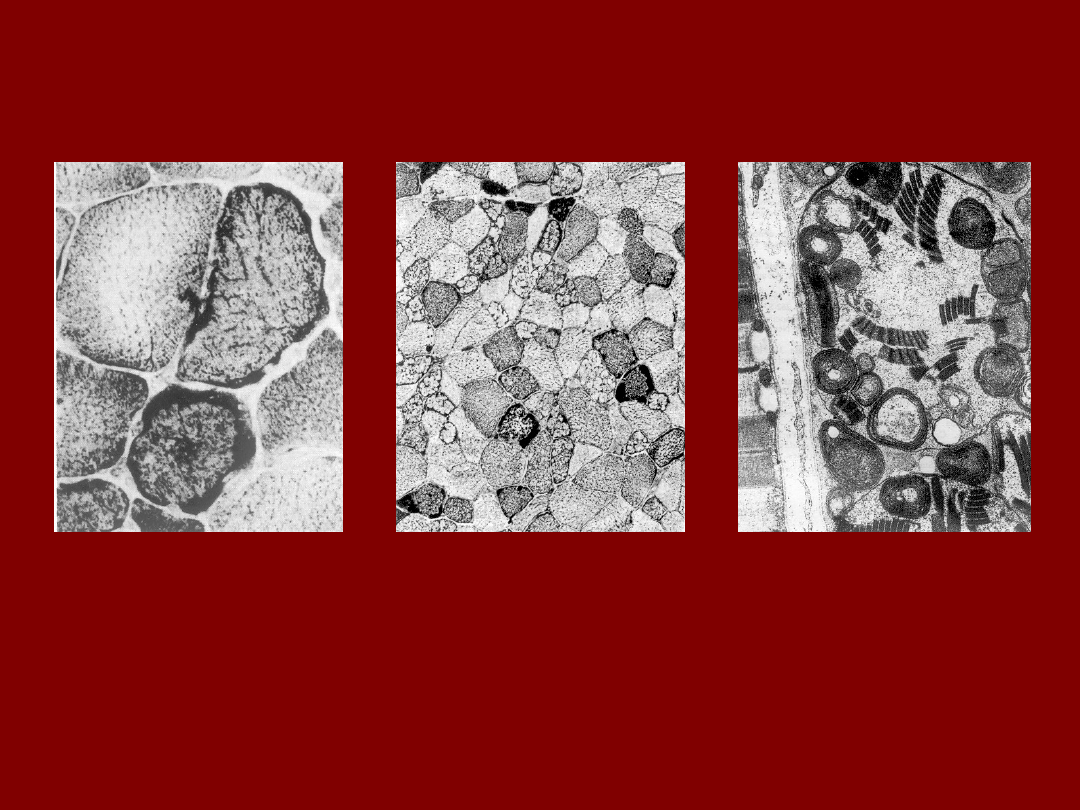
MIĘSIEŃ W MERF
a. Reduktaza NADH-terazoliowa : ciemno barwiące się peryferyjne agregaty
mitochondrialne.
b. Sudan Czarny: nieprawidłowe nagromadzenie lipidów obojętnych.
c. EM: struktury parakrystaliczne w mitochondriach.
Mechler et al..Austr.J.Med.1986,16, 185

MELAS
• ENCEFALOMIOPATIA
• DRGAWKI
• EPIZODY UDAROWE
• DYSFUNKCJE I ZMIANY
STRUKTURALNE MÓZGU
• KWASICA MLECZANOWA
• MUTACJE PUNKTOWE
np. 3443mt tRNAleu
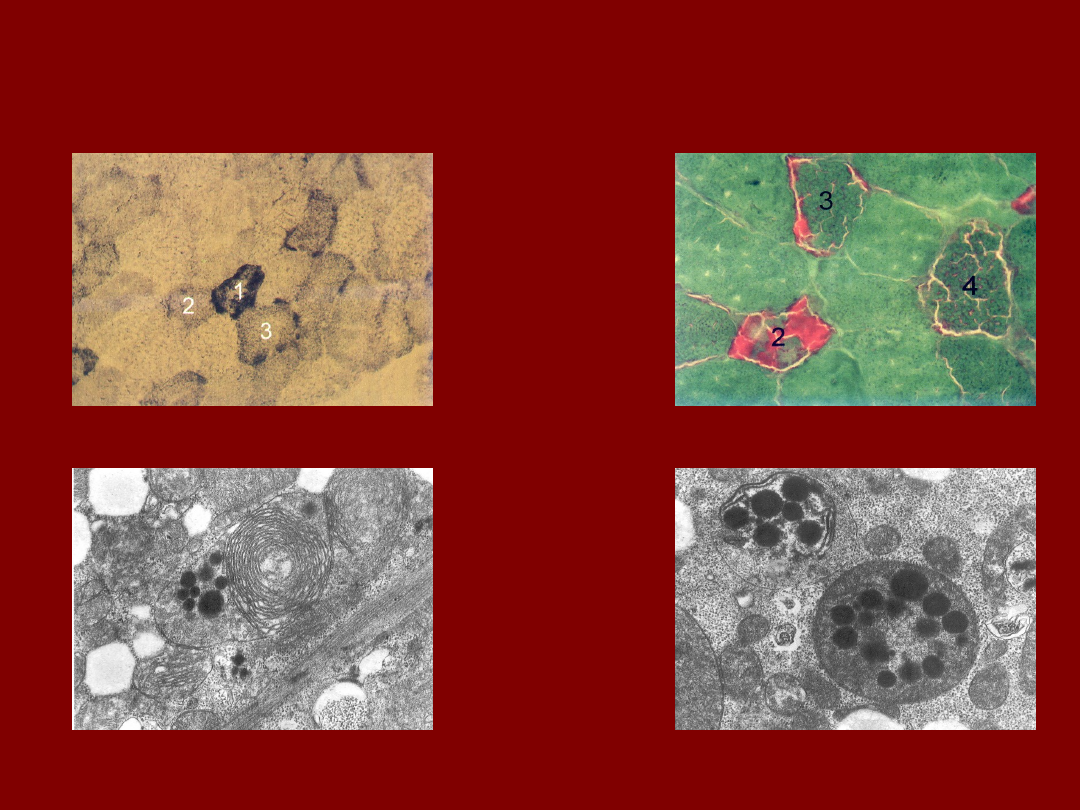
WŁÓKNA RRF W MELAS
a. Dodtnia reakcja SDH o róznym nasileniu b. Reakcja trichrom-Gomori
c. Zmiany strukturalne mitochondriów d. Mitochondria bez grzebieni, wtręty kuliste
doc. M. Pronicki, Zakład Patologii CZD, 2004

LHON=LEBER HEREDITARY
OPTIC NERVE ATROPHY
• PODOSTRA, PROGRESYWNA OBUSTRONNA UTRATA WZROKU
Z UBYTKAMI CENTRALNYMI, ZABURZENIAMI WIDZENIA
KOLORÓW, ZANIKIEM N. WZROKOWEGO
• DUŻE RÓŻNICE W WIEKU ZACHOROWANIA( ŚR. 23 R.Ż.)
• MUTACJE 9 RÓŻNYCH GENÓW m-DNA
• PALENIE, ALKOHOL WYRAŹNIE NASILAJĄ CIEŻKOŚĆ
• MUTACJE PRZYNAJMNIEJ 9 GENÓW KODUJĄCYCH
PODJEDNOSTKĘ IX OXFOS I GENU OKSYDAZY Cyt C
• WARIANTY: PODOSTRA LHON + DYSTONIA
LHON + MIELOPATIA

ZESPÓŁ LEIGHA
• SYMETRYCZNE OBUSTRONNE ZMIANY
PODKOROWE, PNIA, RDZENIA
PRZEDŁUŻONEGO
• OBWODOWA NEUROPATIA SENSORYCZNA
• ATAKSJA
• DRGAWKI
• RETINITIS PIGMENTOSA
• OPÓŹNIENIE PNR, DEMENCJA
• MUTACJE GENU SURF1 ( OKSYDAZA
CYTOCHROMOWA), ATP-azy 6

ZESPÓL KEARNS - SAYRE
• RETINOPATIA
• ATAKSJA
• ZABURZENIA PRZEWODZENIA W MS
• RRMF

ZESPÓŁ PEARSONA
• ANEMIA SIDEROBLASTYCZNA
• NIEWYDOLNOŚĆ TRZUSTKI, CUKRZYCA
• NIEWYDOLNOŚĆ WĄTROBY, KWASICA
MLECZANOWA
• OFTALMOPLEGIA
• MIOPATIA PROKSYMALNA Z MĘCZLIWOŚCIĄ
MIĘŚNI
• WCZESNY POCZĄTEK, ZGON OK. 3 R.Ż.

DEFEKTY m-DNA ZMIENIAJĄCE
REAKCJE NA LEKI
• USZKODZENIE SŁUCHU PO
GENTAMYCYNIE I STREPTOMYCYNIE
• APLAZJA SZPIKU
PO CHLORAMFENIKOLU –
MUTACJA 1555 tRNA mtDNA

OBJAWY SYSTEMOWE W ZESPOŁACH
MITOCHONDRIALNYCH
•
KARDIOMIOPATIA, ZABURZENIA PRZEWODNICTWA.
•
CUKRZYCA
•
RETINOPATIA BARWNIKOWA, OFTALMOPLEGIA, ZAĆMA
•
KWASICA MLECZANOWA
•
GLOMERULOPATIA, DYSFUNKCJA PROKSYMALNEJ CZ. NEFRONU
•
USZKODZENIE WĄTROBY, NAPADOWE NUDNOŚCI, W YMIOTY
•
EGZO – I ENDOKRYNOWA NIEWYDOLNOŚĆ TRZUSTKI
•
PANCYTOPENIA
•
UTRATA SŁUCHU
•
ZABURZENIA PSYCHICZNE ZWŁASZCA DEPRESJE

OBJAWY NEUROLOGICZNE DEFEKTÓW
MITOCHONDRIALNYCH
• OFTALMOPLEGIA, NEUROPATIA N.
WZROKOWEGO
• UDARY U MŁODYCH OSÓB
• DRGAWKI, MIOKLONIE, ATAKSJA,
ZMĘCZENIE PRZEWLEKŁE, NIETOLERANCJA
WYSIŁKU
• DEMENCJA, ZWAPNIENIA JĄDER PODSTAWY
• „NACZYNIOWE” BÓLE GŁOWY
• DYSTONIA NEUROWEGETATYWNA

DIAGNOSTYKA LABORATORYJNA
DEFEKTÓW MITOCHONDRIALNYCH
• BIOPSJA MIĘŚNIA ( Ragged Red Muscle Fibers) –
Gomori Trichrome, badania histochemiczne
• MLECZANY SUROWICY – podwyższone
• OCENA PRZEWODNICTWA NERWOWEGO
• AUDIOMETRIA
• EKG - ZAWSZE HOLTER
• MRI, CT – zaniki, zwapnienia jąder podstawy
• OCENA BIOCHEMICZNYCH PARAMETRÓW OXPHOS
• ANALIZY MOLEKULARNE – SEKWENCJONOWANIE
mtDNA

DIAGNOSTYKA MUTACJI
mtDNA
•
W TEJ SAMEJ CHOROBIE ( A NIEKIEDY TAKŻE TEJ SAMEJ RODZINIE)
SPOTYKAMY MUTACJE RÓŻNYCH SEKWENCJI
•
PRZY TAKIEJ SAMEJ MUTACJI ( A NIEKIEDY TAKŻE W TEJ SAMEJ RODZINIE)
SPOTYKAMY RÓŻNE CHOROBY MITOCHONDRIALNE
•
OBJAWY IDENTYCZNE JAK W CHOROBIE MITOCHONDRIALNEJ MOGĄ
WYWOŁYWAC ROWNIEZ MUTACJE JĄDROWEGO DNA
•
WIĘKSZOŚĆ MUTACJI BŁĘDNEGO SENSU BIAŁEK I O UMIARKOWANEJ
EKSPRESJI JEST HOMOPLAZMATYCZNA
•
W ENCEFALOPATIACH PRZEWAŻAJĄ MUTACJE tRNA
•
W LHON PRZEWAŻAJĄ MUTACJE GENÓW KODUJĄCYCH BIAŁKA
•
MUTACJE PUNKTOWE 12S RNA CZĘSTO KOJARZĄ SIĘ Z GŁUCHOTĄ
•
DUŻE POJEDYNCZE DELECJE DE NOVO SĄ CZĘSTE W PROGRESYWNYCH
OFTALMOPLEGIACH

PORADNICTWO GENETYCZNE W
CHOROBACH MITOCHONDRIALNYCH
• OCENIĆ JAKI TYP DZIEDZICZENIA
WYSTĘPUJE W DANEJ RODZINIE,
• POSZUKIWAĆ W RODZINIE TAKŻE OSÓB Z
ŁAGODNĄ EKSPRESJĄ
• KOBIETA – NOSICIELKA MUTACJI
PRZEKAZUJĄ JĄ WSZYSTKIM DZIECIOM,
• KAŻDE Z NICH MOŻE CHOROWAĆ W
INNYM CZASIE I Z INNĄ EKSPRESJĄ
• POSZUKIWANIE MUTACJI PROWADZIĆ W
RÓŻNYCH TKANKACH

INNE CHOROBY
MITOCHONDRIALNE
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
Wyszukiwarka
Podobne podstrony:
2 Genom mitochondrialny, choroby mitochondrialne
Genom mitochondrialny. Choroby mitochondrialne(1), Immunologia
choroby mitochondrium peroksysomy lizosomy
Biochemia.Choroby mitochondrialne, Dietetyka CM UMK, Biochemia
choroby mitochondrium peroksysomy lizosomy
CHOROBY DZIEDZICZONE MITOCHONDRIALNIE
Choroby mitoch2011
CHOROBY MITOCHONDRIALNE I METABOLICZNE
choroby naczyn i serca(1)
ŻYWIENIE A CHOROBY 4b
Choroby układu nerwowego ppt
Produkty przeciwwskazane w chorobach jelit II
Choroba niedokrwienna serca
CZLOWIEK I CHOROBA – PODSTAWOWE REAKCJE NA
Choroby nerwów czaszkowych
więcej podobnych podstron