22. A M I N O K W A S Y i P E P T Y D Y
Aleksander Kołodziejczyk 2007 grudzień
AMINOKWASY
Jak wynika z nazwy, aminokwasy są związkami organicznymi zawierającymi przynajmniej jedną funkcję aminową i jedną karboksylową. Należą do najważniejszych związków organicznych z uwagi na rolę, jaka pełnią w funkcjonowaniu organizmów żywych.
Z uwagi na ułożenie grupy aminowej w stosunku do grupy karboksylowej rozróżniamy -, -, γ- i kolejne aminokwasy.
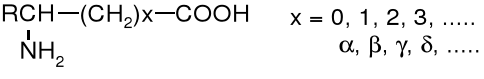
W zależności od liczby podstawników na atomie azotu aminokwasy dzielą się na takie, które zawierają grupę aminową 1o, 2o, 3o i czwartorzędową amoniową; te ostatnie noszą nazwę betain.
Występowanie
Aminokwasy należą do najpopularniejszych związków naturalnych, są bowiem składnikami białek - substancji stanowiących podstawowy budulec drobnoustrojów i organizmów zwierzęcych. Wchodzą w skład peptydów, czyli kopolimerów aminokwasów o mniejszej masie cząsteczkowej niż białka, pełnią również ważną rolę, jako wolne związki. Stanowią substraty wielu innych związków biologicznie czynnych, w tym neuroprzekaźników i alkaloidów.
Organizm wykorzystuje jedynie 23 aminokwasy do syntezy białek; są to tzw. aminokwasy kodowane, czyli rozpoznawalne przez kod genetyczny; najczęściej w białku występuje 20 z nich. W białkach, obok aminokwasów kodowanych stwierdzono obecność jeszcze kilkudziesięciu innych aminokwasów; razem nazwano je aminokwasami białkowymi. Aminokwasy białkowe, inne niż aminokwasy kodowane powstają w wyniku procesów biochemicznych, w tzw. postrybosomalnej modyfikacji reszt aminokwasów kodowanych wbudowanych w łańcuch białkowy. Pojawiają się one dopiero po utworzeniu białek na rybosomach w wyniku takich modyfikacji, jak N-alkilowanie, C-hydroksylowanie, halogenowanie, redukcja, tworzenie wiązań disulfidowych, wiązań amidowych i innych reakcji.
Dla ułatwienia zapisu długich wzorów peptydów i białek wprowadzono międzynarodowe kody aminokwasów: kody trójliterowe i jednoliterowe (te drugie tylko dla aminokwasów kodowanych).
Najpopularniejsze aminokwasy białkowe Tabela 21. 1
Wzór |
Nazwa |
Kod |
Punkt izoelek-tryczny (pHi) |
||
|
|
trójliterowy |
jednoliterowy |
|
|
aminokwasy alifatyczne |
|||||
|
glicyna |
Gly |
G |
5,97 |
|
|
alanina |
Ala |
A |
6,02 |
|
|
walina |
Val |
V |
5,97 |
|
|
leucyna |
Leu |
L |
5,98 |
|
|
izoleucyna |
Ile |
I |
6,02 |
|
|
prolina |
Pro |
P |
6,10 |
|
hydroksyaminokwasy |
|||||
|
seryna |
Ser |
S |
5,68 |
|
|
treonina |
Thr |
T |
6,53 |
|
|
4-hydroksyprolina |
Hyp |
- |
5,71 |
|
aminokwasy siarkowe |
|||||
|
cysteina |
Cys |
C |
5,02 |
|
|
metionina |
Met |
M |
5,75 |
|
|
cystyna |
(Cys)2 |
- |
|
|
aminokwasy aromatyczne |
|||||
|
fenyloalanina |
Phe |
F |
5,98 |
|
|
tyrozyna |
Tyr |
Y |
5,65 |
|
|
tryptofan |
Trp |
W |
5,88 |
|
aminokwasy kwaśne i ich amidy |
|||||
|
kwas asparaginowy |
Asp |
D |
2,87 |
|
|
asparagina |
Asn |
N |
5,41 |
|
|
kwas glutaminowy |
Glu |
E |
3,22 |
|
|
glutamina |
Gln |
Q |
5,65 |
|
aminokwasy zasadowe |
|||||
|
lizyna |
Lys |
K |
9,74 |
|
|
arginina |
Arg |
R |
10,8 |
|
|
histydyna |
His |
H |
7,64 |
|
|
hydroksylizyna |
Hly |
- |
|
|
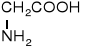
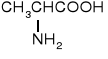
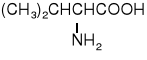
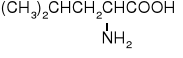
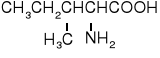
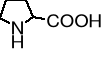
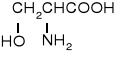
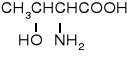
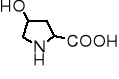
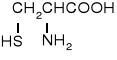
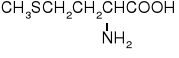
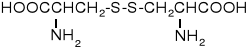
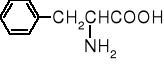
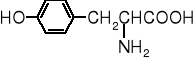
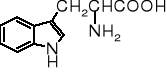
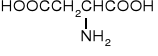
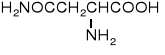
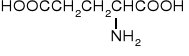
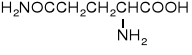
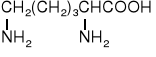
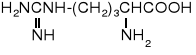
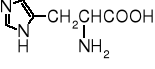
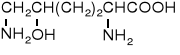
Oprócz glicyny wszystkie aminokwasy białkowe są chiralne i należą do L-aminokwasów, tzn. przy C mają konfigurację L. W niektórych aminokwasach białkowych znajdują się dwa centra chiralne.
Zadanie: wskaż, które aminokwasy zebrane w powyższej tabeli zawierają dwa centra chiralne.
Aminokwasy występujące w naturze, łącznie z aminokwasami białkowymi nazywane są aminokwasami naturalnymi, w odróżnieniu od aminokwasów syntetycznych, które zostały otrzymane na drodze syntezy chemicznej, a dotychczas nie znalezione pośród produktów naturalnych. Znanych jest ponad 1000 aminokwasów naturalnych. Aminokwasy naturalne mogą być białkowe i niebiałkowe.
Pośród niebiałkowych aminokwasów naturalnych znajdują się aminokwasy zarówno o konfiguracji L, jak i D, mogą one występować także w formie racemicznej.
Przykłady niebiałkowych aminokwasów naturalnych
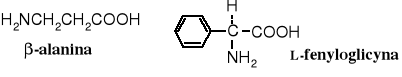
-Alanina jest składnikiem koenzymu A, zaś L-fenyloglicyna występuje w antybiotykach -laktamowych. Homocysteina jest powszechnie spotykanym aminokwasem, służy w biosyntezie metioniny. Produkowany przez rośliny kwas 1-aminocyklopropanokarboksylowy jest prekursorem etenu, hormonu roślinnego, przyspieszającego dojrzewanie owoców.
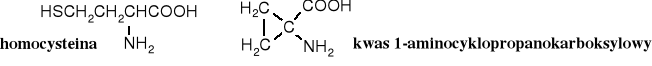
Znane są aminokwasy o szkodliwym działaniu, np. mimozyna ma właściwości depilatora; powoduje wypadanie sierści zwierząt karmionych roślinami produkującymi ten związek, a homarin wytwarzany przez niektóre ślimaki należy do najsilniejszych trucizn.
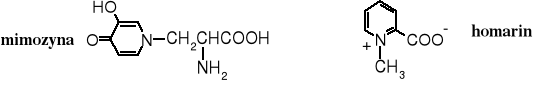
Toksyczny jest też kwas azetydyno-2-karboksylowy, występujący w konwaliach. Kod genetyczny nie odróżnia go od proliny, przez co zostaje wbudowywany w łańcuch białkowy, zmieniając funkcję takiego białka. Natomiast L-tyroksyna jest konieczna do prawidłowego funkcjonowania organizmu człowieka, ułatwia wychwyt jodu przez tarczycę.

Nomenklatura
Aminokwasom białkowym zostały nadane nazwy zwyczajowe i są one w powszechnym użyciu. Dla innych aminokwasów zaleca się stosowanie nazewnictwa wg uprzednio poznanych reguł IUPAC, tzn. traktuje się je jako kwasy z grupą aminową i innymi jako podstawnikami.
Zadanie: nazwij systematycznie tyroksynę.
Aminokwasy chiralne powinny mieć zaznaczoną konfigurację. Dla aminokwasów białkowych najczęściej podaje się symbole L i D. (rzadziej R czy S). Trójliterowe i jednoliterowe kody aminokwasów bez podania symbolu konfiguracji oznaczają konfiguracje naturalną aminokwasów białkowych czyli L, np. Leu oznacza L-leucynę. Konfigurację D lub DL trzeba zaznaczyć odpowiednim symbolem przed wzorem, np. D-Ala, czy DL-Val. Warto zauważyć, że symbole D i L są pisane mniejszą czcionką niż inne litery tekstu. Przy pełnej nazwie aminokwasu należy podać jego konfigurację.
Otrzymywanie
1. Z hydrolizatów białkowych
Wiele aminokwasów białkowych otrzymuje się z hydrolizatów białek. Jest to praktycznie użyteczny sposób ich pozyskiwania jedynie w tych przypadkach, kiedy aminokwas jest łatwy do wydzielenie z mieszaniny innych aminokwasów. Ten warunek spełnia, np. trudno rozpuszczalna w wodzie cystyna, która krystalizuje z hydrolizatu włosów. Innym aminokwasem łatwo krystalizującym jest tyrozyna. Aminokwasy kwaśne i zasadowe można izolować za pomocą jonitów. Aminokwasy aromatyczne wyjątkowo mocno adsorbują się na węglu aktywnym. W ten sposób usuwa się fenyloalaninę z hydrolizatu białkowego w procesie przygotowywania pożywek dla dzieci cierpiących na fenyloketonurię.
2. Synteza chemiczna
2.1 Amonoliza halogenokwasów
Najstarszy i nadal stosowany chemiczny sposób otrzymywania aminokwasów polega na amonolizie halogenokwasów, które są łatwo dostępne jako produkty reakcji Hella-Volharda-Zielinskiego.
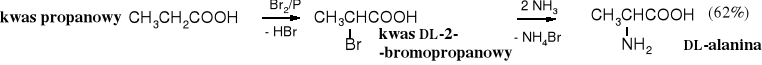
W reakcji tej powstają recemiczne -aminokwasy. Nadmiar amoniaku służy nie tylko do wiązania wydzielającego się bromowodoru, ale wielokrotny nadmiar amoniaku (powyżej 10x) zmniejsza wydajność niepożądanych 2o i 3o amin. Ten sam problem występował w syntezie amin poprzez alkilowanie amoniaku. Zamiast amoniaku można używać węglanu amonu, mieszaniny węglanu amonu z amoniakiem lub karbaminianu amonu. Ten ostatni tworzy z aminokwasem pochodne karbaminowe uniemożliwiające dalsze podstawianie.
2.2 Synteza Gabriela
Innym sposobem na uniemożliwienie tworzenia się 2o i 3o amin poprzez podstawianie atomów wodoru w amoniaku jest synteza Gabriela, polegająca na alkilowaniu ftalimidku potasu. Produktem tej reakcji jest aminokwas, który na grupie aminowej ma osłonę ftalilową. Można go w tej postaci używać do dalszych reakcji lub usunąć resztę ftalilową, najlepiej za pomocą hydrazynolizy.
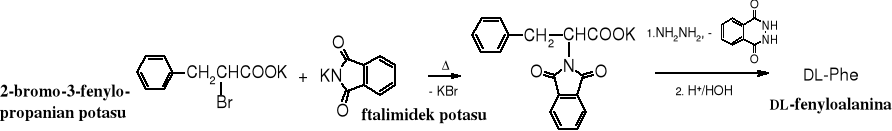
2.3 Synteza malonowa
Z malonianu dietylu łatwo otrzymuje się chronioną pochodną aminową, np. acetyloaminomalonian etylu, do której dołącza się resztę organiczną, tak jak w niepodstawiony ester malonowy. Można to osiągnąć poprzez alkilowanie pochodnej sodowej lub w reakcji Michaela. Po rozbudowie szkieletu węglowego produkt poddaje się hydrolizie i dekarboksylacji.

(izonitrozomalonian dietylu)

acetamidopropylomalonian dietylu kwas 2-aminopentanowy
Otrzymywanie kwasu asparaginowego
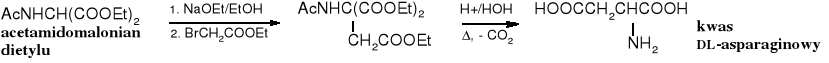
Z acetamidomalonianu dietylu i metanalu w reakcji Michaela można zsyntezować racemiczną serynę.
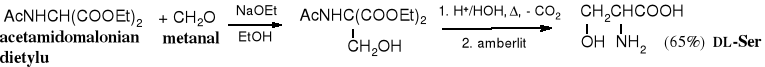
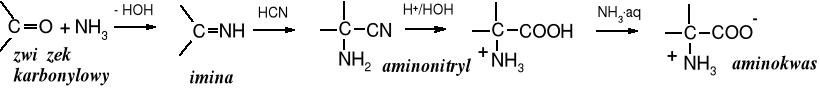
2.4 Synteza Streckera
Synteza aminokwasów z aldehydów i cyjanku sodu lub potasu w obecności amoniaku, nosząca nazwę syntezy Steckera, jest popularną metodą otrzymywania tych związków. Pośrednio tworzy imina, z niej powstaje aminonitryl, który w wyniku hydrolizy przekształca się w aminokwas.
Przykłady:
Fenyloglicyna jest ważnym surowcem w otrzymywaniu półsyntetycznych penicylin. Substratem dla niej jest aldehyd benzoesowy.
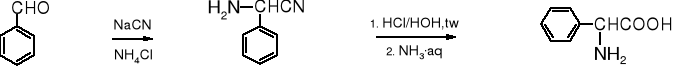
aldehyd benzoesowy cyjanohydryna aldehydu benzoesowego DL-fenyloglicyna
Do syntezy tyrozyny potrzebny jest aldehyd p-metoksyfenylooctowy.
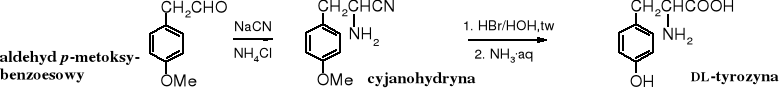
2.5 Redukcyjne aminowanie -oksokwasów
Oksokwasy redukowane w obecności amoniaku zostają przekształcone w aminokwasy. Jest to reakcja analogiczna do otrzymywania amin z aldehydów lub ketonów.
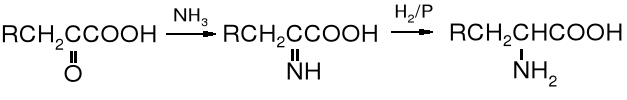
oksokwas iminokwas aminokwas
Otrzymywanie aminokwasów chiralnie czystych
Reakcje chemiczne z achiralnych substratów i bez udziału chiralnych czynników prowadzą do produktów racemicznych. Natomiast, zarówno do celów farmakologicznych, wzbogacania produktów żywnościowych, jak i do badań chemicznych zwykle potrzebne są czyste stereoizomery. Otrzymuje się je w reakcjach z chiralnych substratów lub w obecności chiralnych katalizatorów, w tym enzymów lub mikroorganizmów. Racemiczne aminokwasy można również rozdzielać na enancjomery.
Rozdzielanie racematów
Racematy można rozdzielić na enancjomery za pomocą soli diastereoizomerycznych, poprzez diastereoizomeryczne pochodne, reakcje enzymatyczne, wykorzystanie mikroorganizmów, chromatografię chiralną lub krystalizację spontaniczną. Rozdzielanie racematów na enancjomery wybieramy zamiast syntezy chiralnej wówczas, kiedy
- potrzebne są oba enancjomery;
- drugi niepotrzebny enancjomer można łatwo zracemizować i racemat zawrócić do rozdzielania;
- lub są inne przyczyny uzasadniające tę drogę.
Krystalizacja diastereoizomerycznych soli
Aminokwasy lub ich pochodne tworzą z chiralnymi aminami lub chiralnymi kwasami diastereoizomeryczne sole, które zwykle różnią się właściwościami, w tym rozpuszczalnością w odpowiednim rozpuszczalniku, co sprzyja rozdzielaniu ich poprzez krystalizację. Pośród chiralnych amin stosowanych do rozdzielania racemicznych aminokwasów stosuje się naturalne alkaloidy (np. brucynę, chininę, efedrynę, strychninę) lub aminy syntetyczne [np. 1-fenyloetyloaminę, 1-(p-nitrofenylo)etyloaminę, - i -1-naftyloetyloaminę], zaś jako chiralne kwasy używa się kwasy winowe, dibenzoilowinowe, glutaminowy, migdałowy, kamforosulfonowe, jabłkowy i N-chronione aminokwasy.
Formylo-DL-fenyloalaninę można rozdzielić na enancjomery poprzez jej sól z brucyną, wg niżej podane schematu.
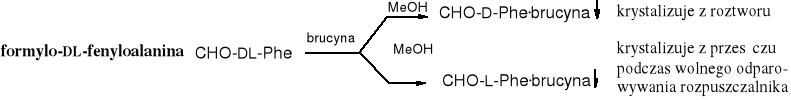
Rozdzielanie za pomocą enzymów
W tej metodzie stosuje się takie enzymy, jak np. acylazy, proteazy, syntetazy czy oksydazy. Enzymy te katalizują reakcję na określonym centrum sterycznym (najczęściej L), pozostawiając drugie centrum bez zmian. Produkty tych reakcji zwykle różnią się znacznie właściwościami fizycznymi i chemicznymi, co ułatwia ich separację.
Acylazy hydrolizują N-acylowane L-aminokwasy.
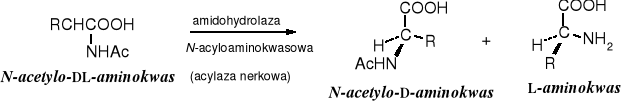
Proteazy są w stanie stereoselektywnie hydrolizować ugrupowania estrowe. Za pomocą proteazy w odpowiednio przygotowanej pochodnej kwasu diaminoheptanodionowego (diaminopimelinowego) można selektywnie zhydrolizować grupę estrową na centrum S.

Syntezy chiralnych aminokwasów
Jest wiele sposobów na otrzymywanie aminokwasów o pożądanej chiralności. Dla aminokwasów L najbardziej wydajne są metody wykorzystujące mikroorganizmy lub enzymy. Tak w przemyśle na wielką skalę produkuje się aminokwasy służące jako dodatki podwyższające wartość odżywczą produktów spożywczych (np. L-lizynę do mąki), polepszacze smaku (kwas L-glutaminowy), Phe i Asp do wytwarzania Aspartanu - słodzika czy farmaceutyki (Arg, Leu, Trp). Najczęściej stosowanym surowcem do ich produkcji jest glukoza lub cukry odpadowe (np. melasa).
Lys, podobnie jak i kilka innych L-aminokwasów jest produkowana za pomocą bakterii Corynebacterium glutamicum.
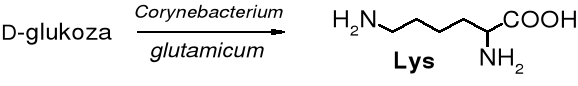
Kwas L-asparaginowy stosowany głównie do otrzymywania Aspartanu - peptydowego słodzika - produkowany jest metodą enzymatyczną. Enzym aspartaza osadzony na specjalnym podłożu jest bardzo trwały (półokres trwałości wynosi 2 lata) i zapewnia otrzymanie 220 ton aminokwasu z 1 kg tego biokatalizatora.
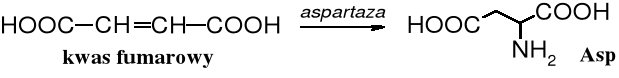
Właściwości fizyczne i fizjologiczne
Aminokwasy jako sole wewnętrzne są związkami bardzo polarnymi i podobnie, jak zwykłe sole charakteryzują się wysokimi temperaturami topnienia (200-300oC). Większość z nich rozpuszcza się w wodzie, w kwaśnych roztworach (wodnych, alkoholowych, w kwasie mrówkowym i octowym) i wodnych roztworach wodorotlenków alkalicznych. Spośród aminokwasów kodowanych jedynie prolina jest rozpuszczalna w etanolu. Inne rozpuszczalniki organiczne nie rozpuszczają tych aminokwasów. Kilka aminokwasów białkowych należy do trudno rozpuszczalnych nawet w wodzie, są to (Cys)2, Tyr, His, Asp i Glu. N-acylowane aminokwasy wykazują podobne właściwości jak kwasy karboksylowe, np. są dobrze rozpuszczalne w rozpuszczalnikach organicznych, zaś estry aminokwasów stają się aminami.
Właściwości fizjologiczne
Większość aminokwasów białkowych jest pozbawiona zapachu. Aminokwasy siarkowe (Cys i Met) charakteryzują się nieprzyjemną tiolową wonią. Kwas L-glutaminowy, aromatyczny składnik przypraw do zup typu maggi czy sosu sojowego, ma zapach pobudzający apetyt, określany zapachem rosołu (hydrolizatu białkowego). Monoglutaminian sodu jest znaną i ceniona przyprawą kuchenną, typu polepszacza smaku (umami).
Smak większości aminokwasów białkowych jest określany jako słodki lub gorzki, ale występują różnice w jego ocenie, z uwagi na subiektywne odczuwanie smaku. Słodkimi aminokwasami są Gly (stąd jej nazwa), Ala, Ser i Thr. Natomiast Lys, Trp, Asp i Asn mają smak obojętny (są bez smaku).
Niektóre aminokwasy spożywane w nadmiarze mogą powodować poważne dolegliwości. Najbardziej niebezpieczne są aminokwasy hydrofobowe, a pośród nich Leu, Tyr, Phe i Trp. Nadmiar Leu wywołuje pelagrę, Tyr jest neurotoksyczna, duża dawka Phe daje podobne objawy do fenyloketonurii. Groźny jest także nadmiar Cys i Met - prowadzi do nekrozy wątroby i nerek.
Aminokwasy hydrofilowe (Asp, Arg, Glu, Orn, i Lys) są nieszkodliwe nawet w kilkugramowych dawkach. Znalazły zastosowanie w chemioterapii.
Chiralność
Wszystkie aminokwasy , oprócz glicyny i jej N-pochodnych są chiralne, przy czym aminokwasy białkowe mają konfigurację L, inne aminokwasy naturalne występują w postaci L, D lub racemicznej. Syntetycznie można otrzymać każdy stereoizomer. Aminokwasy zawierające rozgałęzione łańcuchy lub dodatkowe grupy funkcyjne mogą mieć kilka centrów chiralnych.

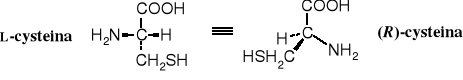
Wszystkie aminokwasy kodowane (oprócz G i C) mają konfigurację L na centrum , odpowiada ona konfiguracji absolutnej S; L-cysteina z konfiguracją R na centrum stanowi wyjątek.
Przykłady:
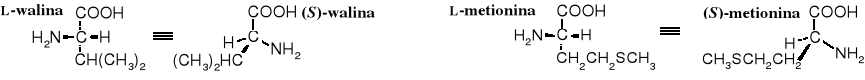
Dwa aminokwasy kodowane zawierają po dwa centra chiralne: izoleucyna [kwas (2S,3S)-2-amino-3-metylopentanowy] i treonina [kwas (2S,3R)-2-amino-3-hydroksybutanowy].
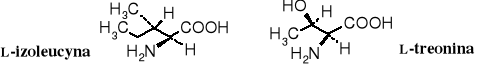
Właściwości chemiczne aminokwasów
1. Właściwości kwasowo-zasadowe
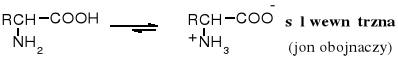
Obie główne grupy funkcyjne aminokwasów aminowa (zasadowa) i karboksylowa (kwasowa) reagują z sobą wewnątrzcząsteczkowo, dlatego aminokwasy występują w formie soli wewnętrznej, zwanej jonem obojnaczym (niem. zwitterion).
Jon obojnaczy w zależności od pH środowiska może ulegać protonowaniu i przekształcać się w kation lub odszczepiać proton z grupy NH3+ by stać się anionem.
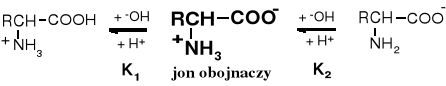
W środowisku silnie kwaśnym aminokwas występuje w postaci kationu, a w silnie zasadowym w postaci anionu, w pośrednim natomiast tworzy sól wewnętrzną. Wartość pH, przy którym w stężenie kationów aminokwasu jest takie same jak anionów nazywa się punktem izoelektrycznym; nadano mu symbol pHi. Punkt ten wyznacza się za pomocą miareczkowania. Wartość pHi dla aminokwasów obojętnych i kwaśnych równa się średniej arytmetycznej wartości pK1 i pK2, które opisują o stałe równowagi przejścia jonu obojnaczego dpowiedniow kation i anion.
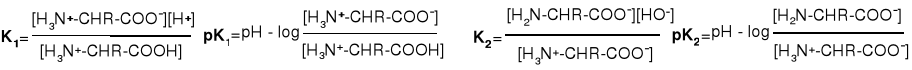
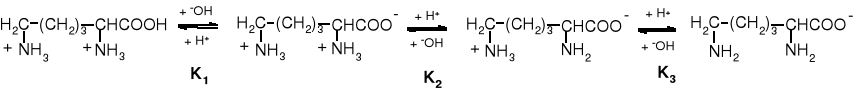
Dla aminokwasów trójfunkcyjnych podawane jest jeszcze stała K3. Opisuje ona dysocjację przy najwyższych wartościach pH, tzn. tworzenie anionu karboksylanowego. Dla aminokwasów kwaśnych K1 i K2 są to stałe równowagi dysocjacji grup karboksylowych, a dla cysteiny K2 jest stałą dysocjacji grupy -SH, zaś dla aminokwasów zasadowych K2 jest stałą odszczepienia protonu z jednej z protonowanych grup zasadowych.
Schemat dysocjacji aminokwasu zasadowego:
Stałe dysocjacji kilku wybranych aminokwasów kodowanych Tabela 21.2
Aminokwas |
pK1 |
pK2 |
pK3 |
pHi |
Gly |
2.3 |
9,6 |
- |
5,97 |
Ala |
2,3 |
9,7 |
- |
6,01 |
Ser |
2,2 |
9,2 |
- |
5,68 |
Cys |
1,7 |
8,3 |
10,8 |
5,02 |
Lys |
2,2 |
9,1 |
10,5 |
9,82 |
Arg |
2,2 |
9,0 |
12,8 |
10,8 |
Asp |
1,9 |
3,7 |
9,6 |
2,77 |
Glu |
2,2 |
4,3 |
10,0 |
3,24 |
Dla aminokwasów obojętnych wartość pHi zawarta jest w granicach 6, np. dla Gly = 5,97, a dla Ala = 6,01. Dla aminokwasów kwaśnych jest niższa (dla Asp wynosi 2,77, a Glu 3,24), zaś dla aminokwasów zasadowych jest wyższa, odpowiednio - 9,82 dla Lys i 10,76 dla Arg.
W stanie krystalicznym aminokwasy występują w formie jonów obojnaczych.
Reakcja aminokwasów z kwasami
Pod wpływem silnych kwasów aminokwasy ulegają protonowaniu tworząc sole amoniowe. Kwasy słabe i średniej mocy nie reagują z aminokwasami.
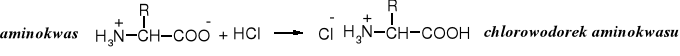
Z silnymi zasadami aminokwasy tworzą sole karboksylanowe. Amoniak i aminy nie reagują z aminokwasami.
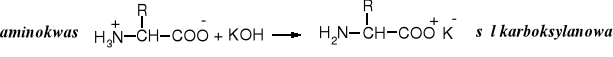
2. Reakcja z kwasem azotowym (III)
Kwas azotawy przekształca aminokwasy w hydroksykwasy. Reakcja ta została wykorzystana do ustalaniu korelacji konfiguracji aminokwasów względem aldehydu glicerynowego.
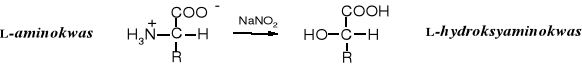
3. Acylowanie aminokwasów
Grupa aminowa aminokwasów ulega acylowaniu pod wpływem halogenków kwasowych lub bezwodników. Powstają N-acyloaminokwasy. Acylowanie prowadzi się najczęściej w środowisku zasadowym.
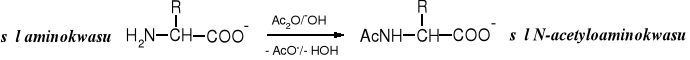
Formylowe pochodne aminokwasów powstają pod wpływem kwasu mrówkowego.
![]()
Acylowane aminokwasy tracą zdolności tworzenia soli wewnętrznej, stają się typowymi kwasami karboksylowymi. Ze względu na obecność grupy amidowej w położeniu ich moc jest zwykle większa niż, np. kwasu octowego.
4. Estryfikacja grupy karboksylowej
Grupę karboksylową aminokwasu można estryfikować typowymi metodami, np. działając na nie alkoholami w bezwodnym środowisku, w obecności kwasów. Produktami są sole amoniowe aminoestrów. Chlorowodorki aminoestrów są zwykle trudno rozpuszczalne w alkoholach.
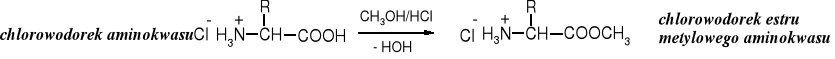
Dogodnym sposobem wytwarzania bezwodnego chlorowodoru in situ jest reakcja chlorku tionylu z alkoholem.

Chlorowodorki estrów metylowych lub etylowych aminokwasów otrzymuje się przez ostrożne wkroplenie chlorku tionylu do zimnego alkoholu. Następnie do tak otrzymanego roztworu chlorowodoru dodaje się aminokwas i pozostawia mieszaninę w niskiej temperaturze. W trakcie reakcji obserwuje się następujące zmiany: nierozpuszczalny w alkoholu aminokwas przechodzi w rozpuszczalny chlorowodorek aminokwasu, po czym zwykle po chwili zaczyna krystalizować trudno rozpuszczalny chlorowodorek estru.
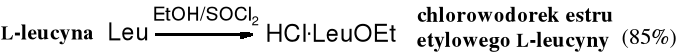
Warto zdawać sobie sprawę z tego, że większość wolnych aminoestrów należy do związków nietrwałych. Szybko ulegają międzycząsteczkowej aminolizie do cyklicznych dioksopiperazyn.
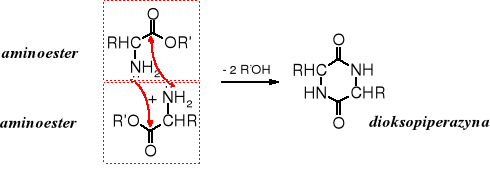
Jedynie estry t-butylowe są trwałe z uwagi na utrudnioną aminolizę, powodowaną obecnością rozbudowanej przestrzennie grupy R' reszty alkoksylowej.
PEPTYDY
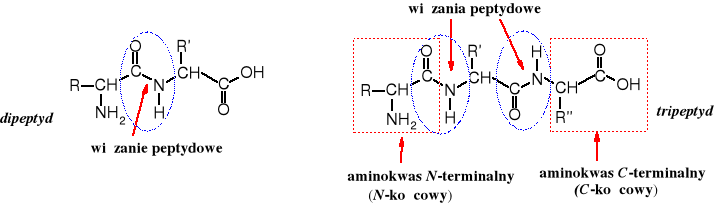
Amidy powstałe z aminokwasów noszą nazwą peptydów. Powstają one w wyniku utworzenia wiązania amidowego, zwanego w tym przypadku wiązaniem peptydowym, pomiędzy grupą karboksylową jednego aminokwasu, a grupą aminową drugiego aminokwasu. Peptydy mogą zawierać reszty tych samych aminokwasów (są to tzw. homopeptydy), np. polilizyna. Częściej spotyka się jednak peptydy zbudowane z reszt różnych aminokwasów.
Peptyd złożony z dwóch reszt aminokwasów nazywa się dipeptydem, z trzech tripeptydem i tak dalej. Peptydy, w których liczba reszt aminokwasowych nie przekracza 10 określane są oligopeptydami, a powyżej 100 reszt AA białkami. Liczbę reszt AA w peptydzie podaje się za pomocą liczebników łacińskich: dipeptyd, tripeptyd, ...... dekapeptyd. Dla ułatwienia określenia wielkości peptydów zawierających ponad 10 reszt aminokwasowych podaje się tę informację używając polskich liczebników, np. mówi się jedenastopeptyd, dwudziestopeptyd, piędziesięciojedenpeptyd, a zapisuje odpowiednio 11-peptyd, 20-peptyd czy 51-peptyd.
Wiązanie peptydowe
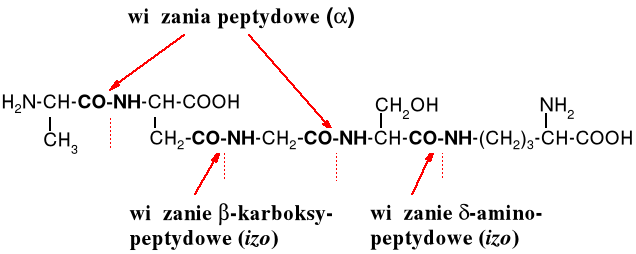
Za wiązanie peptydowe uważa się wiązanie utworzone przez grupę -karboksylową z grupą -aminową. Jeżeli przynajmniej jedna z tych grup nie jest , to takie wiązanie nazywa się izopeptydowym.
Zadanie: zidentyfikuj aminokwasy tworzące powyższy peptyd.
W peptydach ważna jest sekwencja, tzn. kolejność powiązania ze sobą aminokwasów. Z trzech aminokwasów, np. Gly, Val i Tyr można utworzyć kilka izomerycznych tripeptydów, różniących się właściwościami fizykochemicznymi, chemicznymi i co bardzo ważne biologicznymi: Gly-Val-Tyr, Gly-Tyr-Val, Val-Gly-Tyr, Val-Tyr-Gly, Tyr-Gly-Val i Tyr-Val-Gly. Warto zwrócić uwagę, że trójliterowe symbole tworzące zapis peptydu mają różne znaczenie w zależności od miejsca, jakie zajmują w peptydzie. Wszystkie symbole aminokwasów nie będące aminokwasami N- ani C-terminalnymi oznaczają resztę pozbawioną jednego atomu wodoru przy grupie aminowej i funkcji -OH przy grupie karboksylowej, czyli symbolizują następujące ugupowanie: -HN-CHR-CO-. Symbol reszty N-terminalnego aminokwasu to H2N-CHR-CO- (aminokwas pozbawiony -OH), w C-końcowym aminokwasie brakuje jedynie jednego wodoru: -HN-CHR-COOH.
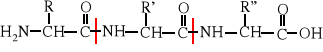
Tripeptyd Gly-Val-Tyr można zapisać też jako H-Gly-Val-Tyr-OH. Wówczas trójliterowe symbole oznaczają to samo - reszty aminokwasów bez atomu wodoru przy grupie aminowej i bez -OH przy grupie karboksylowej. Najczęściej, dla uproszczenia zapisu peptydu za pmocą symboli pomijamy atom wodoru przy aminokwasie N-terminalnym i -OH przy aminokwasie C-terminalnym.
Tylko w wyjątkowych wypadkach peptyd przedstawiamy całym wzorem strukturalnym. Tripeptyd Gly-Val-Tyr w takim zapisie wygląda jak wzór A.
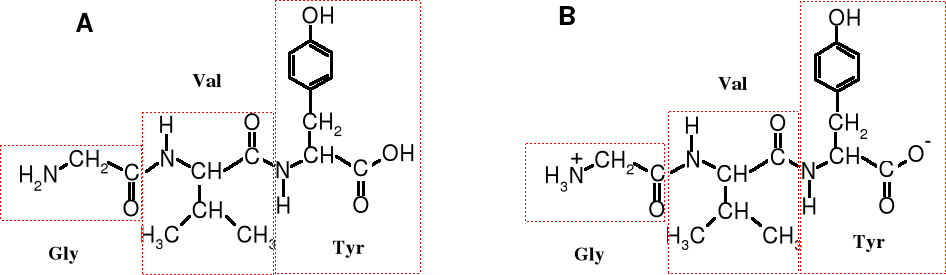
Warto też sobie uzmysłowić, że peptyd zawierający wolną grupę aminową na N-końcu i wolną grupę karboksylową na C-końcu tworzy, podobnie jak aminokwas sól wewnętrzną. W praktyce, dla uproszczenia, rzadko zapisujemy peptyd z rozdzielonymi ładunkami (wzór B).
Pełna nazwa peptydów zawiera nazwy reszt aminokwasów, z jakich jest zbudowany. Tworzymy ją w ten sposób, że aminokwas poprzedzający traktowany jako reszta acylująca, a nazwa C-końcowego aminokwasu pozostaje bez zmian. W ten sposób tripeptyd Gly-Val-Tyr otrzymuje nazwę: glicylowalilotyrozyna, zaś Val-Gly-Tyr to waliloglicylotyrozyna, a Tyr-Val-Gly - tyrozylowaliloglicyna, itp.
Peptydy można też zapisywać symbolami jednoliterowymi, np: GVT, VGT czy TVG. Taki zapis preferują biochemicy i biolodzy do przedstawiania cząsteczek dużych peptydów i białek.
Sposób oznaczania aminokwasu N-terminalnego w peptydach i białkach
Jedną z pierwszych czynności rozpoczynających procedurę prowadzącą do rozpoznania nieznanego białka jest oznaczenie aminokwasów końcowych. Aminokwas N-końcowy identyfikuje się poprzez N-arylowanie niepodstawionej grupy aminowej końcowego aminokwasu, najczęściej za pomocą 2,4-dinitrofluorobenzenu. Reszta 2,4-dinitrofenylowa przyłącza się do grupy aminowej aminokwasu N-terminalnego, podobnie jak i do innych wolnych grup aminowych łańcuchów bocznych aminokwasów zasadowych. Następnie arylowany peptyd poddaje się hydrolizie i za pomocą HPLC (wysokosprawnej chromatografii cieczowej) poszukuje się aminokwasu zawierającego resztę arylowa na grupie -aminowej. Reszta arylowa nie tylko ułatwia zidentyfikowanie N-końcowego aminokwasu, ale również obniża jego próg wykrywalności przy użyciu detektora UV.
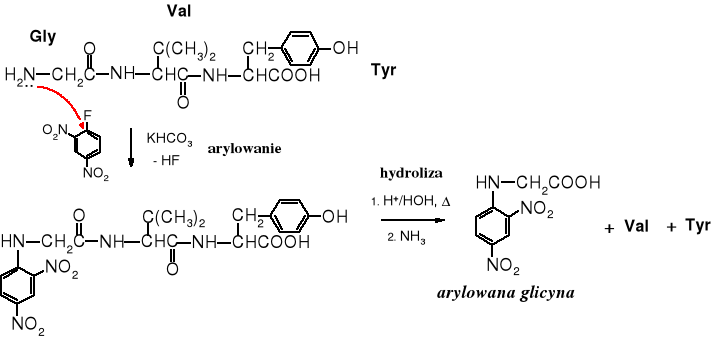
Za pomocą wzorców lub porównania widm UV można zidentyfikować aminokwas z arylowaną grupą aminową , czyli N-terminalny.
Sposób oznaczania aminokwasu C-terminalnego w peptydach i białkach
Najprostszym sposobem identyfikowania aminokwasu C-terminalnego jest poddanie peptydu (białka) hydrazynolizie. W wyniku tej reakcji reszty wszystkich aminokwasów tworzących łańcuch peptydowy, oprócz aminokwasu C-terminalnego, zostajną przekształcone w hydrazydy, a aminokwas z wolna grupą karboksylową tworzy z nią sól.
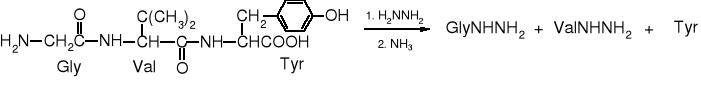
Za pomocą HPLC nietrudno odróżnić wolny aminokwas od hydrazydów aminokwasów.
Skład aminokwasowy i sekwencja aminokwasów w peptydach i białkach
Skład aminokwasowy peptydów i białek oznacza się za pomocą HPLC po hydrolizie wszystkich wiązań peptydowych. Hydrolizę prowadzi się najczęściej 6N kwasem solnym w temperaturze 120oC przez 12 godzin. W tych warunkach niektóre aminokwasy, przede wszystkim Cys ulegają rozkładowi. Następuje również hydroliza Asn i Gln do Asp i Glu. Wykrycie aminokwasów ulegających rozkładowi ułatwiają inne sposoby hydrolizy, np. enzymatyczna.
Popularnym sposobem oznaczania sekwencji, czyli kolejności ułożenia aminokwasów w łańcuchu peptydowym była degradacji Edmana. Pod wpływem fenyloizotiocyjanianu N-końcowy aminokwas ulega odszczepieniu i przekształceniu w odpowiednią pochodną 3-fenylo-2-tiohydantoiny, którą można zidentyfikować chromatograficznie. Następna porcja fenyloizotiocyjanianu odszczepia drugi aminokwas od N-końca, który stał się aminokwasem N-terminalnym po odszczepieniu pierwszego aminokwasu.
Postępując w ten sposób można poznać sekwencję peptydu zawierającego do 10 reszt aminokwasowych. Sekwencja dłuższych peptydów jest trudna do wykonania tym sposobem, ponieważ reakcje odszczepienia aminokwasów nie biegną ze 100% wydajnością i po kilku operacjach wzrasta stężenie zanieczyszczeń i wynik staje się niepewny. Sekwencję dużych peptydów i białek oznacza się na podstawie sekwencji ich fragmentów, otrzymywanych za pomocą częściowej hydrolizy chemicznej lub enzymatycznej. Zastosowanie automatycznych sekwentatorów umożliwia zrobienie sekwencji nawet 100-peptydu.
Sekwencję aminokwasów w białkach coraz częściej oznacza się za pomocą spektrometrii mas. Można ją też określić po oznaczeniu sekwencji nukleotydów genu kodującego dane białko. Jest to jednak sekwencja białka „surowego”, to znaczy takiego, jakie powstało w trakcie jego syntezy na rybosomach, bez późniejszych modyfikacji.
Synteza peptydów
Synteza peptydów sprawiała chemikom poważne trudności przez długi czas, ponieważ zmieszanie dwóch aminokwasów i doprowadzenie jakimś sposobem do utworzenia wiązania peptydowego prowadzi do powstania mieszaniny wielu produktów: dipeptydów, tripeptydów i wyższych peptydów, o różnej sekwencji. Dzieje się tak, ponieważ w obu aminokwasach obie grupy są reaktywne (aminowa i karboksylowa), a także tworzące się peptydy wchodzą w dalsze reakcje.
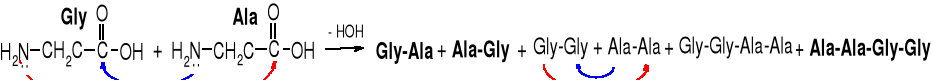
Dipeptydy Gly-Ala i Ala-Gly, a także Gly-Gly i Ala-Ala reagują zarówno między sobą tworząc tetratpeptydy, jak i z aminokwasami dając tripeptydy.
![]()
Tripeptydy i tetrapeptydy mogą dalej reagować zarówno z sobą, jak i z wszystkimi innymi peptydami oraz aminokwasami znajdującymi się w środowisku reakcji. W rezultacie powstaje skomplikowana mieszanina, z której trudno wydobyć poszczególne składniki. W celu otrzymania jednorodnego produktu, należy czasowo osłonić funkcję aminową aminokwasu acylującego i karboksylową aminokwasu acylowanego.
Do osłony funkcji aminowej najczęściej stosuje się osłonę benzyloksykarbonylową (w skrócie Z) lub t-butoksyksykarbonylową (w skrócie Boc). Z-aminokwasy otrzymuje się w reakcji aminokwasu z chloromrówczanem benzylu, a Boc-aminokwasy powstają pod wpływem pirowęglanu t-butylu.

Funkcję karboksylową aminokwasu acylowanego chroni się najczęściej za pomocą estrów: metylowego, etylowego, benzylowego lub t-butylowego.

Do połączenia tak chronionych aminokwasów i otrzymania dipeptydu potrzebny jest odczynnik aktywujący grupę karboksylową aminokwasu acylującego. Takim odczynnikiem jest, np. DCC (dicykloheksylokarbodiid), można też aminokwas acylujący przeprowadzić w bezwodnik lub zastosować inną metodę aktywacji. Zasada (B), np. trietyloamina ma za zadanie uwolnić aminoestr, np. Ala-OBz z jego soli.
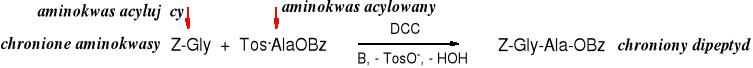
Mechanizm aktywacji grupy karboksylowej za pomocą DCC
W reakcji kwasu (w tym N-chronionego aminokwasu) z DCC powstaje O-acyloizomocznik; który ma właściwości acylujące; należy do tzw. „aktywnych estrów”. Powstający ubocznie jego izomer N-acylomocznik jest nieaktywny i obniża wydajność reakcji.
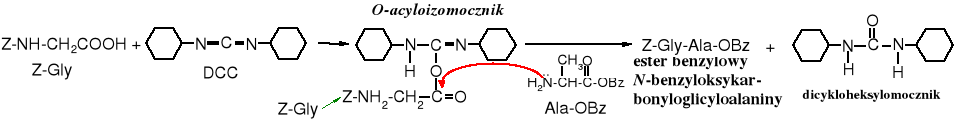
Zadanie: napisz wzór strukturalny N-acylomocznika.
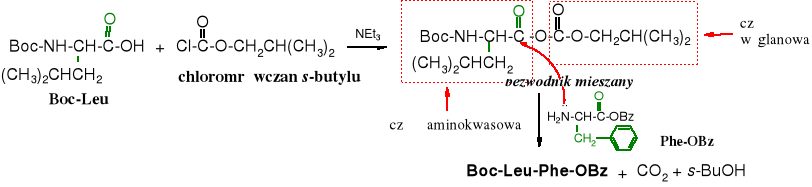
Aktywacja grupy karboksylowej za pomocą utworzenia mieszanego bezwodnika
Synteza peptydów metodą mieszanych bezwodników zaczyna się od reakcji N-chronionego aminokwasu z chloromrówczanem, najlepiej s-butylu. Tworzy się mieszany bezwodnik - aminokwasowo-węglanowy, który jest zdolny częścią aminokwasową acylować aminy, w tym aminoestry. Część węglanowa jest w nim mniej aktywna z uwagi na mniejszy ładunek dodatni na karbonylowym atomie węgla i zawadę przestrzenną reszty s-butylowej.
Reakcję tworzenia mieszanego bezwodnika prowadzi się w temperaturze poniżej -10oC przez kilkanaście minut, po czym dodaje się substrat przeznaczony do acylowania (w poniższym przykładzie ester fenyloalaniny).
Metoda estrów aktywnych
Niektóre estry aktywne popularnych N-chronionych aminokwasów są odczynnikami komercyjnymi, a przez to łatwodostępnymi. Inne przyrządza się in situ w prosty sposób. Rozpuszcza się N-chroniony aminokwas w odpowiednim rozpuszczalniku, dodaje się substratu, z którego ma powstać ester i DCC. Po 2-3 godzinach aktywny ester jest gotowy do użycia. Bez wyodrębniania miesza się go z aminoestrem i pozostawia do utworzenia peptydu.

Tripeptyd najczęściej otrzymuje się z dipeptydu poprzez usunięcie osłony grupy aminowej aminokwasu N-terminalnego i acylowanie go kolejnym N-chronionym aminokwasem. Czynności takie powtarza się aż do uzyskania peptydu o zaplanowanej wielkości i sekwencji. Taki sposób otrzymywania peptydów, nazywany krok po kroku (ang. step by step) jest bardzo pracochłonny. Dodatkowo zużywa się czas na oczyszczanie i identyfikowanie każdego pośredniego peptydu. Syntezę można usprawnić poprzez łączenie gotowych fragmentów, np. w celu uzyskania 10-peptydu poddaje się kondensacji 6-peptyd z 4-peptydem. Jednak te fragmenty trzeba uprzednio także przygotować. Daje się jednak syntezować je równocześnie i wtedy oszczędność w czasie jest istotna. Oba te sposoby należą do tzw. klasycznej metody syntezy peptydów w roztworze.
Duże peptydy najłatwiej otrzymuje się na fazie stałej, tzw. metodą Merifielda. Zaczyna się ona od przytwierdzenia C-końcowego aminokwasu do polimeru, który stanowi zarazem osłonę grupy karboksylowej. Następnie przyłącza się kolejne aminokwasy, aż do pożądanej wielkości. Usprawnienie polega na tym, że rosnący peptyd jest cały czas w jednym i tym samym naczyniu, do którego wprowadza się roztwory odpowiednich reagentów, a po reakcji rozpuszczalniki w celu wymycia nadmiaru reagentów i produktów ubocznych oraz towarzyszących. Po zakończeniu syntezy peptyd zdejmuje się z polimerycznego nośnika i przerabia wg potrzeb. Wszystkie czynności syntezy peptydu na fazie stałej można zautomatyzować i prowadzić ją za pomocą automatycznego syntezatora. Za opracowanie tej metody Meriefield otrzymał nagrodę Nobla.
Zdejmowanie osłon z grup funkcyjnych
Osłony grup funkcyjnych aminokwasów należy tak dobierać, żeby można je było, w zależności od potrzeb usuwać selektywnie lub równocześnie. Taka możliwość istnieje, ponieważ różne grupy usuwa się w różnych warunkach. Obie osłony zastosowane do syntezy powyższego peptydu, a więc Z i Bz można usunąć równocześnie poprzez hydrogenolizę, czyli działanie wodorem w obecności katalizatora (Pd lub Pt).
![]()
Na uwagę zasługuje nie tylko wysoka wydajność powyższej reakcji, ale i to, że obie osłony zostają zredukowane do niekłopotliwych produktów - toluenu i ditlenku węgla.
Odczynnikiem usuwającym większość osłon peptydowych jest ciekły fluorowodór.
Osłonę Z daje się usunąć selektywnie w obecności estru benzylowego poprzez acydolizę bromowodorem w kwasie octowym.
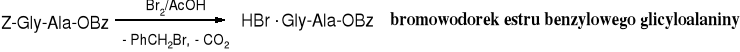
Estry metylowe, etylowe i benzylowe usuwa się poprzez hydrolizę zasadową w niskiej temperaturze. Nieostrożnie prowadzone hydrolityczne odszczepienie grup estrowych może spowodować reakcje uboczne, w tym racemizację.
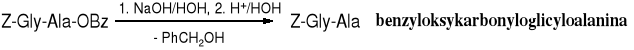
W najłagodniejszych warunkach usuwa się osłony zawierające ugrupowanie t-butylowe: Boc i ester t-butylowy; zostają one odszczepione na zimno już pod wpływem kwasu średniej mocy, jakim jest kwas trifluorooctowy.
![]()
Peptydy naturalne
Organizmy żywe wytwarzają różnorodne związki organiczne, w tym wiele peptydów. Pośród naturalnych peptydów znajdują się krótkie oligopeptydy, dłuższe zawierające kilkanaście do kilkudziesięciu reszt aminokwasowych, a także takie, w których liczba reszt aminokwasowych dochodzi do 100, czyli stają się podobne do białek. Peptydy naturalne zbudowane są zarówno z aminokwasów białkowych, jak i niebiałkowych. Mikroorganizmy wytwarzają peptydy, w których występują również D-aminokwasy. Znane są peptydy zbudowane nie tylko z aminokwasów, ale także z reszt innych związków, np. hydroksykwasów (takie peptydy nazywane są depsipeptydami), cukrów (glikopeptydy), lipidów (lipopeptydy). Peptydy mogą być estryfikowane kwasem fosforowym, powstają wówczas fosfopeptydy. Znane są peptydy nierozgałęzione, rozgałęzione i cykliczne.
Przykłady naturalnych peptydów
1. Glutation (GSH)
Glutation występuje prawie we wszystkich komórkach żywych organizmów. Jest tripeptydem, w którym aminokwas N-terminalny - kwas L-glutaminowy przyłączony jest do Cys grupą γ-karboksylową:
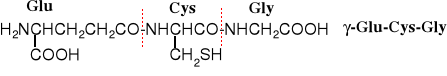
Glutation bierze udział w reakcjach redoks, przechodząc przy tym w dimer. Jest antyutleniaczem, aktywatorem pewnych enzymów, pełni rolę koenzymu.
Pomimo, prostej budowy (tripeptyd) chemiczna synteza glutationu z uwagi na jego skład aminokwasowy jest trudna. Szczególnej osłony wymaga funkcja tiolowa, a acylowanie grupą γ-karboksylową kwasu glutaminowego biegnie wolniej niż grupą . Dlatego glutation pozyskuje się z materiałów biologicznych, najczęściej z drożdży. Natomiast często nowe odczynniki kondensujące, jak i nowe grupy ochronne testuje się w reakcjach otrzymywania glutationu. Panuje opinia, że jeżeli sprawdzą się w syntezie glutationu, to będą przydatne i w otrzymywaniu innych peptydów.
Zsyntezowano wiele peptydów podobnych do glutationu (analogów glutationu), ale żaden z nich nie okazał się aktywniejszy od peptydu natywnego (naturalnego). Natomiast znanych jest kilka naturalnych analogów glutationu.
2. Tyreoliberyna (TRF)
Tyreoliberyna, tripeptyd występujący w mózgu (w podwzgórzu) uwalnia tyreotropinę - hormon regulujący czynność tarczycy, w tym wychwyt jodu. Stymuluje on też aktywność innych hormonów, np. prolaktyny, hormonu wzrostu, wazopresyny, insuliny, a także neuroprzekaźników. Jeden spośród syntetycznych analogów TRF - [1-Me-His2]TRF (dawniej [3-Me-His2]TRF) - jest aktywniejszy od peptydu natywnego. Oba te tripeptydy znalazły zastosowanie w diagnostyce medycznej i w terapii uaktywniania tarczycy.
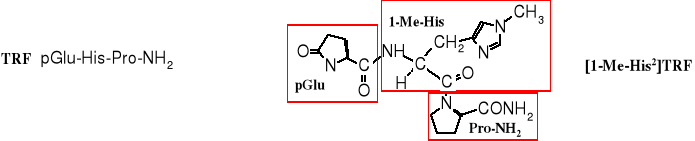
Symbol pGlu jest oznaczeniem kwasu piroglutaminowego; powstaje on przez cyklizację (w tym termiczną) kwasu glutaminowego.
Zadanie: napisz schemat przemiany Glu _→ pGlu.
3. Peptydy opioidowe
Nazwa peptydy opioidowe pochodzi od opium i oznacza, że ich aktywność biologiczna jest podobna do działania opium (opioidowe). Mają zbliżone właściwości biologiczne pomimo istotnej różnicy w budowie chemicznej. Z produktów naturalnych wyizolowano kilkanaście peptydów opioidowych, a kilka tysięcy ich analogów otrzymano syntetycznie. Duże zainteresowanie syntezą nowych analogów peptydów opioidowych wynikało z tego, że istniała uzasadniona nadzieja, iż uda się otrzymać peptyd opioidowy o działaniu przeciwbólowym, który byłby pozbawiony niekorzystnych cech morfiny. Jak na razie nie osiągnięto tego celu, pomimo że niektóre analogi enkefalin są znacznie aktywniejsze od morfiny w działaniu przeciwbólowym. Niestety, tak jak morfina wykazują właściwości narkotyczne, uszkadzają organy wewnętrzne, szkodliwie działają na układ oddechowy i krążenia.
Pośród naturalnych peptydów opioidowych do najbardziej znanych należą enkefaliny i endorfiny.
Enkefaliny
Obie naturalne enkefaliny są pentapeptydami, różnią się resztą aminokwasu C-terminalnego, jedna na C-końcu ma L-leucynę (Leu-enkefalina), a druga zawiera L-metioninę (Met-enkefalina).
Tyr-Gly-Gly-Phe-Leu Leu-enkefalina Tyr-Gly-Gly-Phe-Met Met-enkefalina
Enkefaliny odkryto początkowo w mózgu, stąd ich nazwa (gr. enkephalos = mózg), później okazało się, że występują prawie we wszystkich tkankach).
Endorfiny
Nazwa endorfiny wywodzi się od wyrażenia endogenne morfiny, co oznacza morfinopodobne substancje wytwarzane przez własny organizm. Występują w wielu tkankach, np. w mózgu, płynie mózgowo-rdzeniowym, nerkach, we krwi, w żołądku, jelitach, a nawet w łożysku.
Poznano kilka endorfin: , , γ i δ różnią się one długością łańcucha peptydowego (około 30 AA) i nieznacznie składem aminokwasowym. Sekwencja -endorfiny człowieka:
1 5 10 15
Tyr-Gly-Gly-Phe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val-
20 25 30
Thr-Leu-Phe-Lys-Asn-Ala-Ile-Ile-Lys-Asn-Ala-Tyr-Lys-Lys-Gly-Glu
Zadanie: co przypomina fragment 1-5 -endorfiny człowieka?
Wysiłek fizyczny, zadowolenie z osiągniętego sukcesu, akupunktura i stres zwiększają stężenie endorfin w organizmie; ich rolą jest polepszenie samopoczucia.
Przyjmowanie egzogennnych endorfin prowadzi do uzależnienia.
Neuropeptydy
Neropetydami nazywane są peptydy wytwarzane w tkance nerwowej, w tym w mózgu. Oprócz nich są jeszcze peptydowe hormony tkankowe (wytwarzane przez tkanki pełniące inne funkcje, znane np. w żołądku wydzielana jest gastryna) i hormony gruczołowe (produkowane przez specjalne gruczoły dokrewne, np. androgeny, estrogeny czy kortykosteroidy).
Oksytocyna i wazopresyna
Do hormonów neuropeptydowych zaliczane są cykliczne nonapeptydy: oksytocyna i wazopresyna.

Oba peptydy różnią się nieznacznie składem aminokwasowym. Oksytocyna jest jednakowa u wszystkich ssaków, natomiast skład wazopresyny jest nieznacznie gatunkowo zmienny. U ludzi w położeniu 8 zawiera argininę i dlatego często nazywana jest argininowazopresyną.
Oksytocyna ma aktywność wazopresorową, która polega na wywoływaniu skurczów mięśni gładkich ciężarnej macicy, a wraz z prolatyną stymuluje laktację - aktywność laktacyjna. Natomiast najważniejszym zadaniem wazopresyny jest regulowanie stężenia moczu. Brak lub zbyt mały poziom wazopresyny jest przyczyną moczówki prostej, dolegliwości polegającej na wydalaniu zbyt dużej ilości rozcieńczonego moczu. W czasie snu poziom wazopresyny rośnie, dzięki temu można przespać 8, i więcej godzin bez potrzeby opróżniania pęcherza.
Syntetyczne analogi zarówno oksytocyny, jak i wazopresyny znalazły zastosowanie w terapii. Oksytocyna i jej analogi służą w wspomaganiu akcji porodowej oraz do stymulowania laktacji, a analogi wazopresyny stosuje się w leczeniu niektórych nieprawidłowości w funkcjonowaniu układu moczowego.
Adrenokortykotropina (ACTH)
Adrenokortykotropina jest 39-peptydem produkowanym przez przedni płat przysadki mózgowej, a działa w nadnerczach jako stymulator syntezy kortykosterydów. Była jednym z najdawniej poznanych hormonów peptydowych i dzięki temu przeprowadzono na niej wiele badań. Odkryto, a później potwierdzono to i na innych biologicznie czynnych peptydach, że w łańcuchu peptydowym znajduję się fragmenty pełniące różne funkcje. Jest grupa aminokwasów odpowiedzialnych za połączenie się z receptorem, jest miejsce aktywne, w którym dochodzi do reakcji biochemicznej, jest adres, czyli receptory w organizmie, do którego hormon ma trafić, jest także fragment decydujący o specyficzności gatunkowej. Syntetyczny fragment ACTH (1-24) wykazuje pełną aktywność biologiczną peptydu natywnego (naturalnego). Znalazł on zastosowanie kliniczne.
Insulina
Insulina, 51-peptyd wytwarzany w trzustce należy do najpopularnieszych peptydów, ponieważ jest powszechnie stosowanym przez diabetyków lekiem. Wielu badaczy zalicza ją do białek, ponieważ zwykle występuje w postaci dimeru, czyli agregatu zawierającego ponad 100 reszt aminokwasowych. Została odkryta w 1921 r. a jej odkrywcy otrzymali nagrodę Nobla już w 1923 r.
Insulina zbudowana jest z dwóch łańcuchów: A, w którym jest 21 reszt aminokwasów i B, zawierającym 30 takich reszt. Oba łańcuchy połączone są dwoma mostkami disilfidowymi, a ponadto w łańcuchu A znajduje się trzeci mostek disulfidowy.

Do niedawna insulinę, lek ratujący życie, ekstrahowano z trzustek zwierząt rzeźnych, głównie wołowych i wieprzowych. Insulina zarówno wołowa jak wieprzowa różnią się nieznacznie składem aminokwasowym w porównaniu z insuliną ludzką (HI), ale ich aktywność w stosunku do ludzi jest podobna jak oryginalnej HI. Jednak insulina pochodzenia zwierzęcego może być przyczyną poważnych reakcji alergicznych. Dlatego, dla ludzi uczulonych na insulinę zwierzęcą podawanie HI jest sprawą życia i śmierci, zaś jej pozyskiwanie stanowiło nie lada problem. Nie można jej izolować z trzustek ludzkich, a produkcja tak dużego peptydu na drodze syntezy chemicznej jest nieopłacalna ekonomicznie. Na szczęście pod koniec XX w. opracowano syntezę HI w oparciu o inżynierię genetyczną. Bakterie E. coli, którym wszczepiono zrekombinowany gen odpowiedzialny za syntezę HI u ludzi wytwarzają ten peptyd na skalę przemysłową.
Ostatnio insulina stała się popularnym, acz niedozwolonym środkiem dopingującym. Jest szczególnie trudna do wykrycia, ponieważ występuje naturalnie w organizmie, a czas jej połowicznego rozkładu wynosi tylko kilka minut. Przyjmowana przez ludzi zdrowych powoduje poważne uszkodzenie organizmu. Daje jednak doraźne korzyści dla uprawiających sport. Przyspiesza rozrost mięśni, a przyjęta razem z glukozą bezpośrednio przed samymi zawodami znacznie zwiększa wydolność zawodników. Jest tania i ogólnie dostępna. To wszystko czyni z niej nadzwyczaj atrakcyjny, a zarazem bardzo niebezpieczny środek dopingujący.
Peptydy toksyczne
Bardzo wiele niezwykle groźnych toksyn naturalnych ma budowę peptydów. Należą do nich między innymi toksyny grzybów, jady węży, pająków, a także niektóre antybiotyki. Zawierają one często D-aminokwasy i -rozgałęzione aminokwasy, co utrudnia biodegradację i przez to przedłuża ich szkodliwe działanie. Na rysunku poniżej przedstawiona jest struktura amatoksyny, toksycznego peptydu muchomora sromotnikowego.
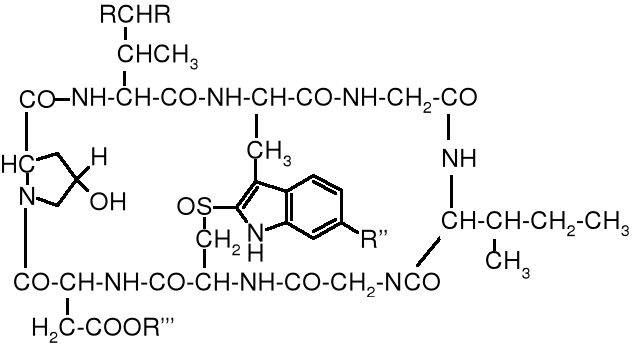
19
Wyszukiwarka
Podobne podstrony:
8 AMINOKWASY, PEPTYDY, BIA id 4 Nieznany (2)
Æwiczenie 7 Aminokwasy, peptydy i bia³ka
AMINOKWASY I PEPTYDY
01 Aminokwasy, peptydy, białka, enzymyid 3054 ppt
Aminokwasy, peptydy 2
Kopia 3 AMINOKWASY, PEPTYDY, BIAŁAKA
68 Aminokwasy peptydy i bialka(1)
AMINOKWASY I PEPTYDY protokól
68 Aminokwasy peptydy i bialka
aminokwasy i peptydy kapilarna elektroforeza
aminokwasy peptydy
Aminokwasy, peptydy 1
27. Aminokwasy i peptydy, ♦ różne, różne
aminokwasy peptydy, Ogrodnictwo, Ogrodnictwo UP Wro, ROK I, semestr II, biochemia, egzamin, poilkj,
Aminokwasy,peptydy,białka i inne
26. Aminokwasy i peptydy, Technologia Żywnośći UR, II rok, biochemia
3 AMINOKWASY, PEPTYDY, BIAŁAKA
Aminokwasy, peptydy, białka
więcej podobnych podstron