21. H Y D R O K S Y K W A S Y i H A L O G E N O K W A S Y
Hydroksykwasy są to związki organiczne, których cząsteczki zawierają zarówno grupę karboksylową, jak i hydroksylową. Wzajemne ułożenie tych grup może być 1,2, 1,3, 1,4, 1,5, itd., mówimy wówczas odpowiednio o -, -, γ-, δ- i tym podobnych hydroksykwasach.
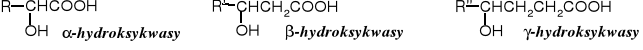
21.1 Występowanie
Hydroksykwasy pełnią ważna rolę biochemiczną i z tego powodu należą do popularnych związków naturalnych. Spotyka się zarówno hydroksykwasy alifatyczne, aromatyczne, jak i alifatyczno-aromatyczne. Do najbardziej znanych należą kwasy: glikolowy, mlekowy, winowy, jabłkowy, cytrynowy, askorbinowy, salicylowy i migdałowy.
Kwas glikolowy, czyli hydroksyetanowy znajduje się w niedojrzałych winogronach, w liściach dzikiej winorośli, burakach i innych roślinach.
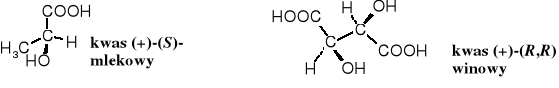
Kwas mlekowy, systematycznie - 2-hydroksypropanowy występuje zarówno w formie D-, L-, jak i racemicznej. Powstaje głównie w procesie fermentacji cukrów, np. laktozy zawartej w mleku, a także powszechnie występującej glukozy. Znajduje się we krwi, mięśniach, żółci, nerkach i w innych częściach ciała zwierząt i ludzi. Jego zawartość w mięśniach wzrasta w trakcie wysiłku fizycznego; jest odpowiedzialny za tzw. zakwasy w mięśniach. Znalazł praktyczne zastosowanie jako środek antyseptyczny, do ochrony przeciwdrobnoustrojowej leków i produktów spożywczych, a także w przemyśle do kompleksowania jonów niektórych metali. Jest produkowany na skalę przemysłową.
Kwas (-)-(S,S)-winowy (2,3-dihydroksybutanodiowy) rzadko występuje w naturze, natomiast jego stereoizomer (+)-(R,R)- jest szeroko rozpowszechniony w roślinach i owocach. Racemiczny kwas winowy powstaje w trakcie fermentacji moszczu winogronowego - osadza się na dnie naczyń fermentacyjnych w postaci kamienia winnego.
Kwas (-)-(S)-jabłkowy, składnik wielu owocach został wyizolowany z soku jabłkowego już w 1785 r. Stanowi produkt pośredni w niektórych procesach biochemicznych, np. w cyklu kwasu cytrynowego. Służy do impregnacji materiałów (papieru) do pakowania żywności.
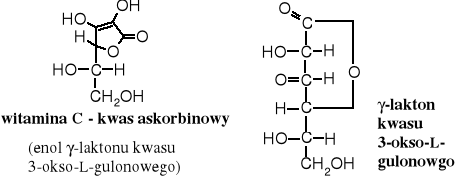
Kwas L-askorbinowy (witamina C) jest produkowany powszechnie przez rośliny wyższe i wiele organizmów zwierzęcych. Organizm ludzki go nie wytwarza, dlatego musi być dostarczany wraz z pożywieniem jako witamina. Jego brak lub niedobór wywołuje szkorbut i inne dolegliwości. W dużym stężeniu znajduje się w soku owoców cytrusowych, owocach dzikiej róży, jagodach i naci pietruszki. Rozkłada się pod wpływem światła, łatwo ulega autooksydacji, szczególnie w obecności śladów jonów metali ciężkich, np. żelaza. Syntetyczny izomer D- jest biologicznie nieaktywny.

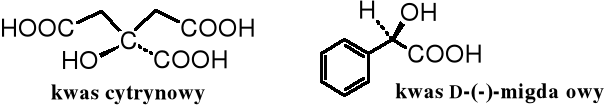
Kwas cytrynowy (kwas 2-hydroksypropano-1,2,3-trikarboksylowy) występuje w każdym organizmie jako produkt pośredni cyklu kwasu cytrynowego. Organizm dorosłego człowieka wytwarza go około 2 kg dziennie. Jest szeroko rozpowszechniony w świecie roślin, w soku cytrynowym jego stężenie dochodzi do 5-7%. Wytwarzany jest na skalę przemysłową metodą fermentacyjną z cukrów. Służy do zakwaszania napojów orzeźwiających, jako antykoagulant produktów emulsyjnych, a także w procesach galwanizacyjnych.
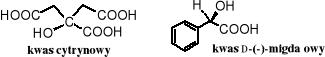
Kwas D-(-)-migdałowy [kwas (R)-2-hydroksy-fenylooctowy] w postaci nitrylu jest składnikiem amigdaliny, glikozydu obecnego w gorzkich migdałach. Oba jego enancjomery służą do rozdzielania racemicznych amin i alkoholi.
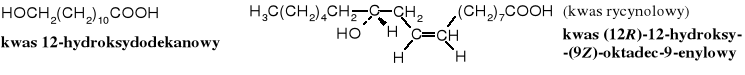
Znanych jest kilka popularnych hydroksyaminokwasów (np., seryna, hydroksylizyna, hydroksyprolina, tyrozyna i inne). Pośród naturalnych hydroksykwasów należy wymienić także kwasy hydroksytłuszczowe (np. kwas rycynolowy - (R)-12-hydroksy-(Z)-oktadec-9-enowy - składnik triglicerydów oleju rycynowego), hydroksykwasy w woskach, kwasy mykolinowe - składniki ściany komórkowej bakterii i wiele innych.
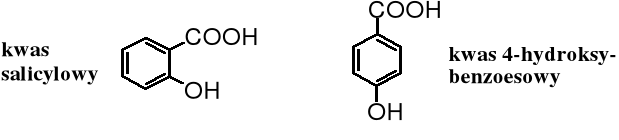
Kwas salicylowy (2-hydroksybenzoesowy) i 4-hydroksybenzoesowy są przedstawicielami naturalnych hydroksykwasów aromatycznych. Kwas salicylowy jest składnikiem wielu ziół, jego estry znajdują się w olejkach eterycznych i w korze niektórych drzew, np. wierzby, a jego pochodna - kwas O-acetylosalicylowy, popularny lek - aspiryna - powinien znajdować się w każdej apteczce. Kwas 4-hydroksybenzoesowy jest istotnym prekursorem ubichinonów (witamin K) oraz niektórych pigmentów bakteryjnych.
Do popularnych hydroksykwasów należą pochodne cukrów: kwasy onowe (inaczej aldonowe), uronowe i arowe. Kwasy onowe i arowe są kwasami syntetycznymi, natomiast kwas glukuronowy (uronowy) bierze czynny udział w oczyszczaniu organizmu z niektórych toksyn. Tworzy z nimi rozpuszczalne w wodzie pochodne, a przez to możliwe do usunięcia wraz z moczem. Nazwa uronowe wywodzi się od tego, że kwasy te zostały wykryte w moczu (urynie). Kwas galakturonowy jest składnikiem pektyn i agaru (policukrów).
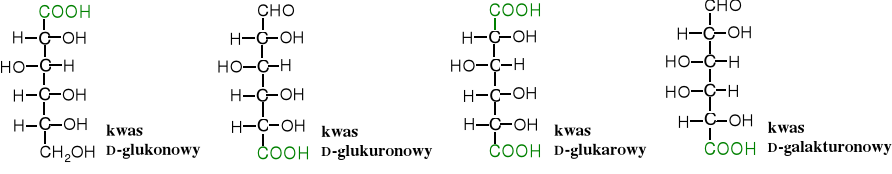
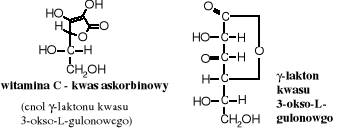
Do pochodnych cukrów należy też wspomniany wyżej kwas askorbinowy, czyli witamina C.
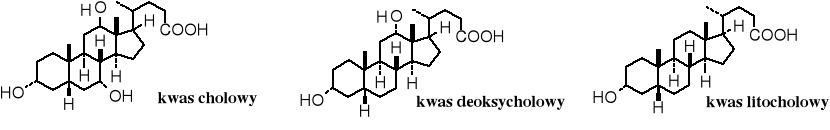
Kwasy żółciowe to także hydroksykwasy, pochodne sterydów. Ich przedstawicielami są kwas cholowy, deoksycholowy i litocholowy.
21.2 Nomenklatura
Wiele zwyczajowych nazw naturalnych hydroksykwasów jest dopuszczone do nazewnictwa systematycznego. Zalecane jest nazywanie hydroksykwasów sposobem podstawnikowym, polegającym na dołączeniu do rdzenia nazwy przedrostka hydroksy- wraz z odpowiednim lokantem.
21.3 Otrzymywanie
21.3.1 Hydroliza halogenokwasów
Substratem w tej metodzie są kwasy karboksylowe, które w reakcji Hella-Volharda-Zielinskiego przeprowadza się w -halogenokwasy, a następnie hydrolizuje do -hydroksykwasów.
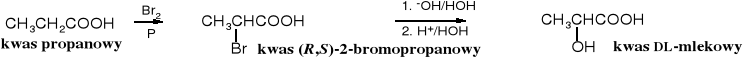
21.3.2 Reakcja Reformackiego
Z -bromoestrów i związków karbonylowych (aldehydów lub ketonów) w obecności cynku powstają -hydroksyestry, a ich ostrożna hydroliza prowadzi do kwasów -hydroksykarboksylowych.
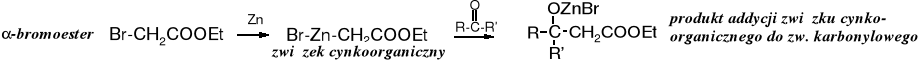
Tworzący się przejściowo związek cynkoorganiczny (np. BrZnCH2COOEt), przypomina związek Grignarda, jest jednak od niego mniej reaktywny - reaguje z aldehydami i ketonami, ale nie z estrami. Produkt addycji związku cynkoorganicznego do aldehydu lub ketonu po zakwaszeniu zostaje przekształcony w -hydroksyester, a z niego po hydrolizie powstaje -hydroksykwas.

Produkty reakcji Reformackiego mogą być przekształcane w inne związki, w tym nienasycone i nasycone estry, a z nich oczywiście w odpowiednie kwasy.
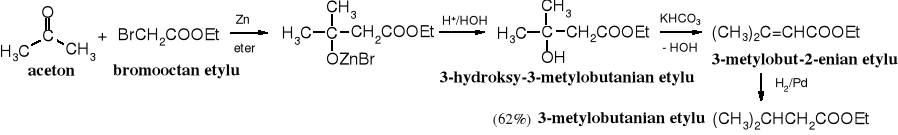
W reakcji Reformackiego biorą udział jedynie -halogenokwasy (nie - i dalsze).

21.3.3 Reakcja cyjanohydrynowa i hydroliza cyjanohydryn
Aldehydy i ketony w reakcji z cyjanowodorem zostają przekształcane w cyjanohydryny (-hydroksynitryle), z których po hydrolizie tworzą się -hydroksykwasy.
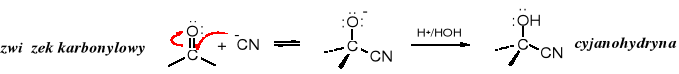
Poprzez odpowiednią cyjanohydrynę otrzymuje się racemiczny kwas migdałowy lub jego analogi.
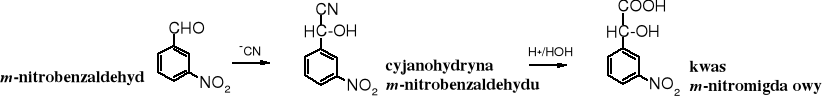
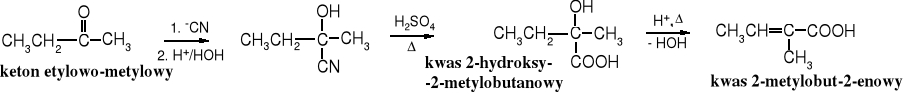
Również rozgałęzione -hydroksykwasy można otrzymać z cyjanohydryn. Kwasy -hydroksykarboksylowe znacznie trudniej ulegają dehydratacji niż -hydroksykwasy czy inne związki -hydroksykarbonylowe, ale w odpowiednio drastycznych warunkach można taką operację przeprowadzić. Reakcja prowadzi do kwasów ,-nienasyconych.
Po hydrolizie produktu reakcji metanalu z cyjanowodorem powstaje kwas glikolowy.
![]()
Ten sam kwas można otrzymać z kwasu octowego poprzez chlorowanie i hydrolizę kwasu chlorooctowego.

21.3.4 Z aminokwasów w reakcji z kwasem azotowym (III)
-Aminokwasy pod wpływem HNO2 ulegają przekształceniu w -hydroksykwasy. Produktem pośrednim jest odpowiedni kwas diazokarboksylowy. Reakcja przypomina syntezę fenoli z amin aromatycznych. Kwasy diazokarboksylowe są stosunkowo trwałe, np. diazooctan etylu powstający z estru glicyny pod wpływem HNO2 może być wyizolowany. Takiej reakcji nie ulegają zwykłe aminy alifatyczne.

21.3.5 Redukcja oksokwasów
Chemiczną redukcję oksokwasów do hydroksykwasów można przeprowadzić selektywnie, chociażby za pomocą tetrahydroboranu sodu.
![]()
Podczas redukcji chemicznej oksokwasu bez udziału czynników chiralnych powstają wyłącznie hydroksykwasy racemiczne. Natomiast enzymatyczna redukcja oksokwasów prowadzi do określonego enancjomeru.
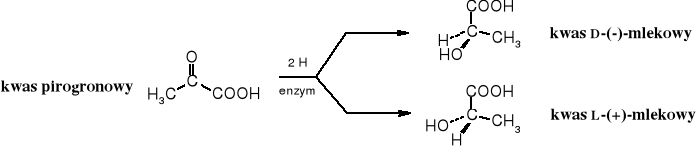
Konfiguracja produktu zależy od enzymu biorącego udział w reakcji. W naturze występują oba izomery kwasu mlekowego.
Racemiczny kwas mlekowy, podobnie jak i inne racemiczne chiralne kwasy można rozdzielić na enancjomery poprzez krystalizację ich soli z chiralnymi aminami (zasadami). Sole kwasów racemicznych z zasadami achiralnymi są nieprzydatne do tego celu.
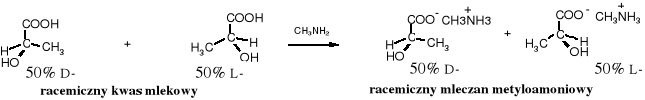
Oba składniki racemicznego mleczanu metyloamoniowego mają w środowisku achiralnym te same właściwości chemiczne i fizyczne, a tym samym nie są podatne na rozdzielenie za pomocą zwykłych sposobów fizycznych. Natomiast kwas racemiczny z enancjomeryczną zasadą tworzy sole distereoizomeryczne, które zwykle różnią się, np. rozpuszczalnością w odpowiednio dobranym rozpuszczalniku i można je rozdzielić przez krystalizację.
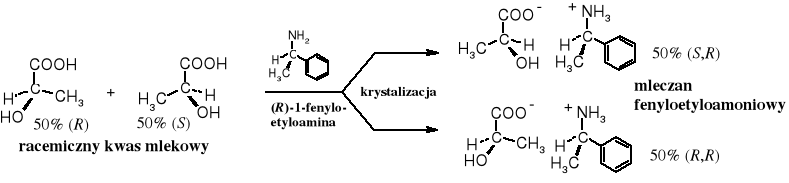
Znane są również inne sposoby rozdzielania racemicznych związków, np. poprzez:
- chromatografię chiralną;
- elektroforezę chiralną;
- tworzenie i rozdzielanie diastereoizomerycznych pochodnych;
- przemiany enzymatyczne lub
- krystalizację spontaniczną
21.4 Początki stereochemii
Odkrycie enancjomerów i tym samym stworzenie podstaw stereochemii przypisuje się L. Pasteurowi.
Louis Pasteur (1822-1895); ur. w Dôle, Francja; studia w Arbois, Besancon, prof. w Dijon, Strasbourgu, Lille i École Normale Supěrieure.
21.4.1 Odkrycia Pasteura
Pasteur ukończył studia chemiczne ze słaba oceną, ponieważ miał bardzo rozległe zainteresowania i nie był w stanie skupić się na określonym kierunku. Jedną z dziedzin, która go pasjonowała była krystalografia. Krystalizował między innymi sole kwasu winowego. Podczas rekrystalizacji winianu sodowo-amonowego z roztworu wodnego, w temperaturze zbliżonej do pokojowej, zauważył w otrzymanym osadzie dwa rodzaje bardzo podobnych do siebie kryształów. Różniły się jedynie tak, jak różni się przedmiot od swojego odbicia w lustrze. Ich wzajemne relacje przypominały dwie dłonie - lewą i prawą. Pasteur rozdzielił te kryształy od siebie za pomocą pęsety i stwierdził, że ich skład chemiczny, a także właściwości fizykochemiczne były identyczne. Różniły się jedynie znakiem skręcalności właściwej.
Roztwór o tym samym stężeniu sporządzony z kryształów jednego rodzaju skręcał światło spolaryzowane w polarymetrze o ten sam kąt, lecz w przeciwną stronę w porównaniu do roztworu sporządzonego z kryształków drugiego rodzaju. Jedna odmiana winianu sodowo-amonowego skręcała zatem, płaszczyznę światła spolaryzowanego w prawo (+), a druga w lewo (-). Zostały one nazwane prawoskrętną i lewoskrętna odmianą. Obie odmiany zmieszane razem traciły zdolność skręcania płaszczyzny światła spolaryzowanego i podobnie jak przed rozdzieleniem stawały się optycznie nieczynne. Taką mieszaninę określono mieszaniną racemiczną. Ponieważ obie odmiany soli miały identyczny skład chemiczny, Pasteur upatrywał ich odmienne właściwości optyczne w odmiennym ułożeniem w przestrzeni czterech różnych podstawników przy czterowiązalnym atomie węgla. Tą hipotezą znacznie wyprzedził poglądy współczesnych mu uczonych.
Samoistne rozdzielenie mieszaniny racemicznej, czyli to, co udało się Pasteurowi jest niezwykle rzadkim zjawiskiem. Zwykle dochodzi do tego przy współudziale czynnika chiralnego.
Znacznie później udowodniono, że odmienne właściwości optyczne izomerów winianu sodowo-amonowego wynikają z różnic w budowie przestrzennej cząsteczki kwasu winowego. W wyniku innego przestrzennego ułożenia podstawników przy C2 i C3 kwasu winowego powstają trzy jego odmiany: lewoskrętna (-), prawoskrętna (+) i optycznie nieczynna (±) zwana kwasem mezo-winowym.
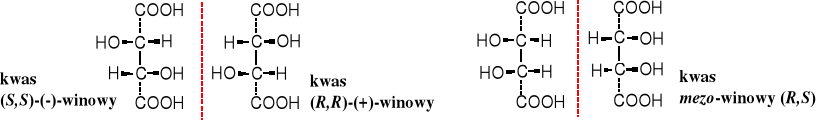
21.4.2 Właściwości fizykochemiczne
Właściwości fizykochemiczne (oprócz kierunku skręcalności światła spolaryzowanego - []D) kwasów (-) i (+)-winowego są identyczne, kwas mezo-winowy różni się od nich, inne właściwości ma też mieszanina racemiczna (±), pomimo tego, że składa się z jednakowych ilości kwasów (-) i (+). Ta mieszanina racemiczna zachowuje się, zatem jakby była odmiennym, jednorodnym związkiem chemicznym, a nie mieszanką dwóch związków. Z tego powodu tego typu mieszaninę nazywa się często krótko racematem. Nie zawsze jest tak, czasami mieszanina racemiczna zachowuje się faktycznie, jakby była mieszaniną dwóch różnych związków. Świadczy o tym chociażby obniżenie jej temperatury topnienia w porównaniu z tt. czystych enancjomerów.
Wybrane właściwości stereoizomerów kwasu winowego Tabela 20.1
Stereoizomer |
tt. [oC] |
Skręcalność []D |
Gęstość [g/cm3] |
Rozpuszczalność [g/100 ml wody] w 20oC |
(S,S)-(+) |
168-170 |
+12 |
1,7598 |
139,0 |
(R,R)-(-) |
168-170 |
-12 |
1,7598 |
139,0 |
(R,S)-mezo |
146-148 |
0 |
1,6660 |
125,0 |
(R,R/S,S)-(±) |
206 |
0 |
1,7880 |
20,6 |
Przypomnienie: symbole (R) i (S) oznaczają tzw. konfigurację absolutną, oznaczoną wg reguły Cahna-Ingolda-Preloga, zaś D- i L- konfigurację względną (względem aldehydu glicerynowego) określaną wg reguły Fischera. Natomiast symbole (+) i (-) wskazują kierunek kąta skręcenia płaszczyzny światła spolaryzowanego.
Enancjomery są stereoizomerami, których cząsteczki mają się do siebie jak odbicia lustrzane. Distereoizomery zaś są też stereoizomerami, ale ich cząsteczki nie odpowiadają odbiciom lustrzanym.
Kierunek skręcalności płaszczyzny światła spolaryzowanego zależy od właściwości podstawników wokół chiralnego atomu węgla, a nie od konfiguracji. Skręcalność właściwa []D zarówno dla enancjomerów (R) i (S), podobnie jak D- i L- może przyjmować wartości dodatnie lub ujemne.
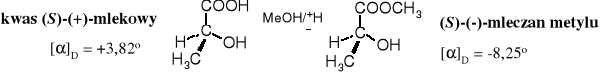
Estryfikacja kwasu (+)-mlekowego metanolem nie zmieniła konfiguracji związku, natomiast kierunek skręcalności właściwej []D estru przyjął znak przeciwny - (-).
Kierunek i wartość bezwzględna skręcalności właściwej jest stałą fizyczną, charakterystyczną dla określonego związku; nie ma prostej zależności pomiędzy skręcalnością właściwą a konfiguracją, poza przeciwnymi znakami skręcalności właściwej enancjomerów.
Reakcjom związków chiralnych może towarzyszyć retencja (zostaje zachowana konfiguracja), inwersja (produkt ma zmienioną konfigurację w stosunku do substratu) lub racemizacja (połowa produktu przyjmuje konfigurację przeciwną do konfiguracji substratu). Zwykle reakcje, które wywołują zmiany konstytucyjne z dala od centrum chiralnego nie mają wpływu na konfigurację. Reakcje biegnące na centrum chiralnym wg mechanizmu SN2 zachodzą z inwersją konfiguracji, a mechanizm SN1 często bywa przyczyną racemizacji.
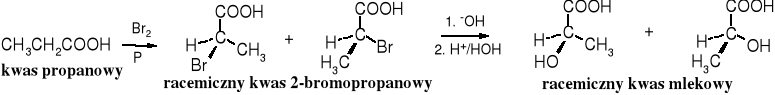
W reakcjach, w których tworzy się nowe centrum chiralne bez udziału czynników chiralnych (chiralnych substratów, chiralnych katalizatorów, czy chiralnego rozpuszczalnika) powstaje produkt racemiczny. Bromowanie kwasu propanowego prowadzi do racemicznego kwasu 2-bromopropanowego, a z niego w wyniku hydrolizy tworzy się racemiczny kwas mlekowy.
Podobnie racemiczny produkt powstaje w wyniku katalitycznej redukcji kwasu pirogronowego.
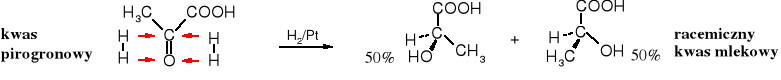
Pasteur postawił hipotezę wyjaśniająca przyczynę przeciwnych znaków skręcalności właściwej enancjomerów. Zjawisko to tłumaczył różnym ułożeniem w przestrzenni podstawników wokół chiralnego atomu węgla. Jego hipoteza znacznie wyprzedzała epokę, w której żył - dla współczesnych mu uczonych była niezrozumiała. Dużo czasu upłynęło zanim została powszechnie zaakceptowana, ale nadal nie znano sposobu ustalenia i zapisywania ułożenia atomów (podstawników) w przestrzeni wokół chiralnego atomu węgla.
21.4.3 Konfiguracja względna
W 1891 r. E. Fischer zaproponował, żeby umownie przyjąć konfigurację (ułożenie w przestrzeni) dodatnio skręcającego aldehydu glicerynowego jako D-, a aldehydu (-)-glicerynowego jako L-. W ten sposób można było zapisać za pomocą płaskich wzorów przestrzennie zróżnicowane cząsteczki - aldehydu D-(+)-glicerynowego i L-(-)-glicerynowego.

Konfigurację innych związków chiralnych określano na podstawie porównania ich konfiguracji z konfiguracją aldehydu glicerynowego. Z tego powodu, tak oznaczana konfiguracja była konfiguracją względną (względem aldehydu glicerynowego). Określanie konfiguracji innych związków dokonywano za pomocą reakcji o znanej stereochemii (biegnących z retencją albo z inwersją) prowadzących z aldehydu glicerynowego do porównywanego związku lub odwrotnie - z badanego związku otrzymywano aldehyd glicerynowy. Ten sposób oznaczania konfiguracji względnej nosił nazwę korelacji konfiguracji.
Fischer przyjął arbitralnie, że aldehyd D-glicerynowy, ma grupę hydroksylową po prawej stronie, jak na projekcji powyżej, a enancjomer L- po lewej stronie. Sposób zobrazowania cząsteczki, nazywany projekcją Fischera, polega na tym, że na powierzchnię kartki (ekranu, tablicy) robiony jest rzut cząsteczki ułożonej w ten sposób, że najbardziej utleniony atom węgla znajduje się u góry, a pozostałe atomy C w jednej płaszczyźnie pod nim. Należy sobie zdawać sprawę z tego, że na tym rzucie atomy (podstawniki) ułożone poziomo w cząsteczce realnej znajdują się nad płaszczyzną w stosunku do atomu chiralnego, a ułożone pionowo pod nim.
21.4.4 Konfiguracja absolutna
W roku 1951 okazało się, że założenia Fischera, co do rozmieszczenia podstawników wokół chiralnego atomu węgla odpowiadają rzeczywistej cząsteczce aldehydu glicerynowego. Od tego czasu mówi się o konfiguracji absolutnej, czyli takiej, jaka występuje w rzeczywistości. Prawdopodobieństwo, że założenie Fischera będzie zgodne z rzeczywistością wynosiło 50%.
21.4.5 Korelacja konfiguracji
Konfigurację względną kwasów winowych określono w wyniku szeregu reakcji prowadzących od aldehydu D-glicerynowego poprzez odpowiednią cyjanohydrynę do odpowiedniego kwasu winowego.
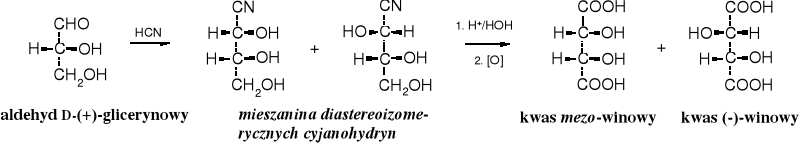
Konfiguracja kwasu mlekowego została określona też za pomocą korelacji do konfiguracji aldehydu D-glicerynowego.
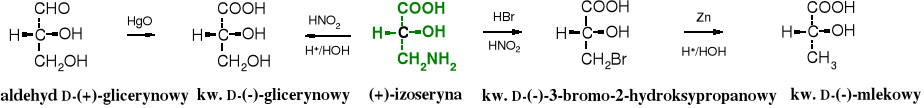
Od 1951 r. istnieje możliwość oznaczania rzeczywistego ułożenia podstawników wokół chiralnego atomu węgla, czyli konfiguracji bezwględnej. W tym właśnie roku J.M. Bijvoet stwierdził za pomocą rentgenografii, że aldehyd D-glicerynowy faktycznie ma taką konfigurację, jaką zaproponował E. Fischer. Tak więc, uprzednio określone konfiguracje względne innych związków poprzez korelacje z konfiguracją aldehydu D-glicerynowego odpowiadają konfiguracji bezwzględnej.
21.5 Właściwości fizykochemiczne hydroksykwasów
Hydroksykwasy zwykle są substancjami krystalicznymi. Kwas glikolowy (HOCH2COOH) topnieje w temperaturze 75-80oC, natomiast t.t. enancjomerów kwasu mlekowego wynosi 25-26oC, a kwasu (R)-jabłkowego 101oC, zaś jego racematu 131-132oC. Komercyjny kwas mlekowy dostępny jest w postaci stężonych (np. 90%) roztworów wodnych. Niższe hydroksykwasy są dobrze rozpuszczalne w wodzie. Dzięki obecności grupy hydroksylowej hydroksykwasy są lepiej rozpuszczalne w wodzie niż kwasy alkano- czy arenokarboksylowe. Hydroksykwasy alifatyczne są silniejszymi kwasami niż kwasy alkanowe, np. Ka kwasu glikolowego wynosi 1,5.10-4, w porównaniu do Ka kwasu octowego - 1,76.10-5. Wzrost kwasowości wywołany jest efektem indukcyjnym -I funkcji hydroksylowej. Natomiast efekt mezomeryczny +M tej funkcji osłabia moc kwasów hydroksyarenowych; Ka kwasu p-hydroksybenzoesowego wynosi 3,3.10-5, a benzoesowego 6,5.10-5. Zapach niższych hydroksykwasów jest ostry, drażniący.
21.6 Właściwości chemiczne
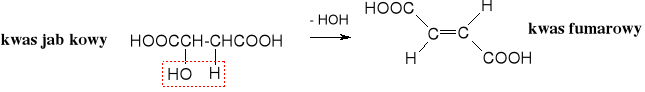
Reaktywność hydroksykwasów wynika z obecności dwóch grup funkcyjnych - hydroksylowej i karboksylowej. Z tego powodu z kwasami mogą tworzyć estry na grupie hydroksylowej, a z alkoholami - estry na grupie karboksylowej. Ich właściwości chemiczne zależą od wzajemnego położenia obu grup funkcyjnych. Charakterystyczną reakcją dla -hydroksykwasów jest tworzenie cyklicznych pochodnych zwanych laktydami, -hydroksykwasy ulegają dehydratacji do ,-nienasyconych kwasów, zaś γ- i δ- łatwo przekształcają się w wewnętrzne estry, czyli laktony.
Powolna destylacja kwasu mlekowego pod zmniejszonym ciśnieniem, przedstawiciela -hydroksykwasów, prowadzi do sześcioczłonowego dilaktonu, który został nazwany laktydem. Powstaje on z połączenia dwóch cząsteczek -hydroksykwasu i wydzielenia dwóch cząsteczek wody. Jest to reakcja estryfikacji.
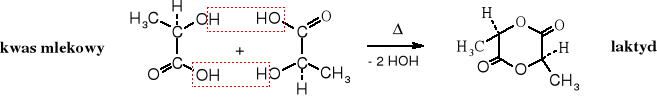
Kwasy -hydroksykarboksylowe, zarówno kwas glikolowy, jak i mlekowy odwadniane w temperaturze pokojowej tracą wodę przechodząc polimeryczne estry liniowe.
![]()
Ogrzewanie -hydroksykwasów powoduje ich dehydratację, prowadzącą do ,-nienasycownych kwasów karboksylowych.
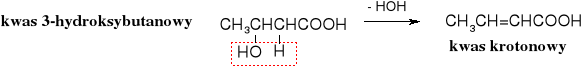
Z kwasów γ- i δ-hydroksykarboksylowych podczas ogrzewania powstają odpowiednio pięcio- lub sześcioczłonowe estry cykliczne zwane laktonami.
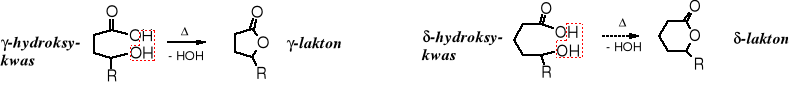
Utlenianie hydroksykwasów prowadzi do oksokwasów. Z -hydroksykwasów powstają kwasy dikarboksylowe.

21.7 HALOGENOKWASY
Halogenokwasy, to kwasy zawierające atom halogenu w łańcuchu bocznym, w odróżnieniu od halogenków kwasowych, w których halogen zastępuje grupę -OH w funkcji karboksylowej. Z uwagi na wzajemne położenie halogenu i funkcji karboksylowej rozróżnianie są -, - γ-halogenokwasy i inne.
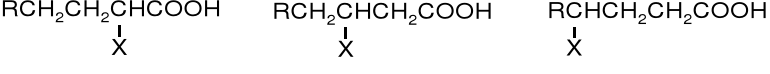
-halogenokwasy -halogenokwasy γ-halogenokwasy
Właściwości halogenokwasów zależą od tego wzajemnego ułożenia obu podstawników. Najbardziej reaktywne w reakcjach SN są -halogenokwasy. Atomy halogenów oddziałują na zasadzie indukcji na grupę karboksylową zwiększając jej kwasowość - najbardziej kwaśne są również -halogenokwasy.
21.7.1 Otrzymywanie
Chloro- i bromokwasy zawierające atom halogenu w położeniu otrzymuje się w reakcji halogenowania kwasów karboksylowych w obecności czerwonego fosforu, w przemianie zwanej reakcją Hella-Volharda-Zielinskiego.
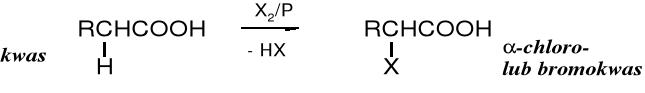
X: Cl; Br
Jodokwasy najłatwiej powstają poprzez przekształcenie chlorokwasów za pomocą jodku potasu w acetonie. Jodek potasu jest rozpuszczalny w acetonie, natomiast chlorek potasu nie. Dochodzi do wymiany chloru na jod, a wytrącanie się z roztworu nierozpuszczalnego chlorku potasu przesuwa równowagę na korzyść jodokwasu.

21.7.2 Właściwości chemiczne
Halogenokwasy (za wyjątkiem fluorokwasów, które są chemicznie bardzo inertne) ulegają hydrolizie do hydroksykwasów.
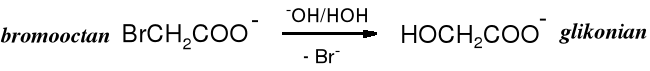
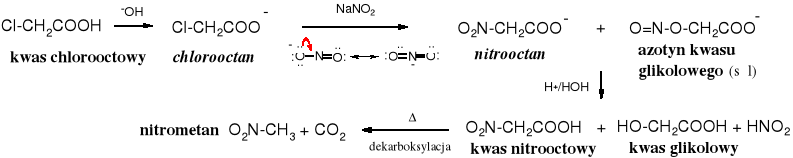
Atom halogenu w halogenokwasach można wymienić na grupę -NO2 otrzymując nitrokwasy obok azotynów hydroksykwasów [estrów kwasu azotowego (III)]. Jon NO2- jest odczynnikiem ambidentnym, tzn. że ma dwa centra aktywne i do reakcji dochodzi na atomie N lub/i O.
21.7.2.1 Reakcja Reformackiego
Służy do otrzymywania -hydroksyestrów z -halogenoestrów i związków karbonylowych w obecności cynku. Biegnie ona poprzez związki cynkoorganiczne. Reakcja Reformackiego została przedstawiona w podrozdziale dotyczącym otrzymywania hydroksykwasów.
21.7.2.2 Reakcje SE halogenokwasów
Reakcje substytucji nukleofilowej halogenokwasów prowadzą jeszcze do wielu innych pochodnych kwasów. Na poniższym schemacie przedstawione zostały produkty, jakie można otrzymać z halogenoestrów.
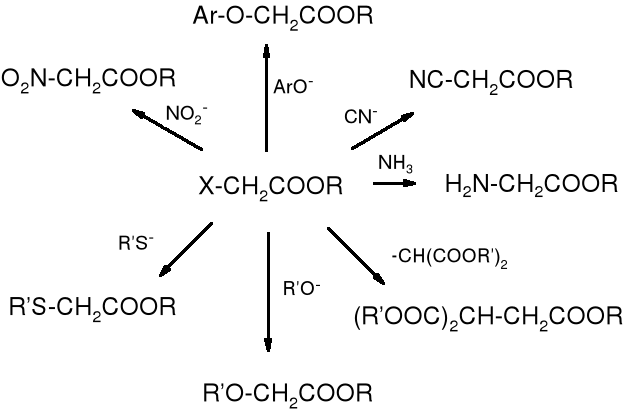
Zadanie: nazwij produkty powstające z halogenokwasów w reakcji substytucji nukleofilowej.
1
Wyszukiwarka
Podobne podstrony:
24. Cukry, Studia, Technologia chemiczna, Wykład I
Podstawy chemii organicznej - wstęp, Studia, Technologia chemiczna, Wykład I
11. Tiole i sulfidy, Studia, Technologia chemiczna, Wykład I
23. Kwas weglowy, Studia, Technologia chemiczna, Wykład I
12. Fenole (2), Studia, Technologia chemiczna, Wykład I
12. Fenole (1), Studia, Technologia chemiczna, Wykład I
Otrzymywanie wapna palonego, materiały naukowe do szkół i na studia, technologia chemiczna sprawozda
20. Oznaczanie zawartosci wody w cialach stalych i cieczach, materiały naukowe do szkół i na studia,
pytania na zaliczenie-zagrożenia ekologiczne, Studia, Technologia Chemiczna, I stopień, PK, II semes
HYDROKOLOIDY nowe, Studia, Technologia żywności
wykaz cwiczen, materiały naukowe do szkół i na studia, technologia chemiczna sprawozdania
16. Oznaczanie zawartosci tluszczu w nasionach oleistych, materiały naukowe do szkół i na studia, te
otrzymanie żywicy fenolowo-formaldehydowej, materiały naukowe do szkół i na studia, technologia chem
15. Otrzymywanie polistyrenu metoda perelkowa, materiały naukowe do szkół i na studia, technologia c
Węgle aktywne, Studia, Technologia chemiczna, Różne
Wnioski wapno palone, materiały naukowe do szkół i na studia, technologia chemiczna sprawozdania, wa
więcej podobnych podstron