Ilona Lewandowska, Tadeusz Trzmiel
Instytut Biochemii Technicznej PŁ
MIKROBIOLGICZNY PROCES DEGRADACJI WĘGLOWODRÓW AROMATYCZNYCH
1 WSTĘP
Na przestrzeni ostatnich kilkudziesięciu lat do wykazu ekologicznych zagrożeń doszło jeszcze jedno, możliwość zatrucia środowiska życia węglowodorami oraz ich związkami pochodnymi. W krajach o wysokiej kulturze ekologicznej, co najczęściej wiąże się z wysokim uprzemysłowieniem, coraz bardziej zaczyna się zwracać uwagę na zagrożenia środowiska takimi związkami jak: tlenki siarki, azotu, węglowodory aromatyczne itp. Aktualne badania naukowców potwierdzają, że emitowane są do atmosfery, wód śródlądowych i do gleby grupy związków chemicznych, których stężenie w emisji jest bardzo małe ale ze względu na niezwykle silne właściwości toksyczne, nawet na niskim poziomie stężeń są one bardzo niebezpieczne dla zdrowia ludzi i zwierząt.
Na szczególną uwagę zasługują różnego rodzaju węglowodory, m.in. monopierścieniowe i policykliczne. Wykazują one potencjał kancerogenny i genotoksyczny.
Wzrost zanieczyszczeń środowiska związkami organicznymi skłania do poszukiwań jak najbardziej skutecznych metod ich utylizacji. W warunkach naturalnych jest to proces długotrwały i dlatego człowiek dąży do jego przyspieszenia. Do tych zabiegów wykorzystuje się osiągnięcia w biotechnologii, gdzie drobnoustroje wykorzystują węglowodory jako źródło węgla i energii. Należy jednak znać warunki biochemicznego rozkładu węglowodorów jak i ich metabolitów pośrednich, które mogą powstawać w wyniku procesu biodegradacji, ponieważ taka niewiedza może spowodować straty finansowe i badawcze.
Duże ilości związków aromatycznych są wynikiem działalności człowieka. Obecne są również w środowisku naturalnym np. w trakcie erupcji wulkanów. Innym źródłem tych związków są rośliny. Przykładem takim jest lignina.
Reakcją obrony środowiska na zanieczyszczenie węglowodorami aromatycznymi są mikroorganizmy, które są wstanie je degradować. Są to:
bakterie: Aeromonas, Flavobakterium, Beneckea, Anabaena, Moraksella,
Achromobacter, Mycobacterium, Nocardia, Pseudomonas,
Chryzosporium, Micrococus, Alcaligenes, Vibrio, Acinetobakter,
Bacillus, Arthrobacter.
drożdże: Sacharomyces, Trichoderma.
grzyby nitkowate: Aspergillus, Penicylium, Mucor.
promieniowce: Actinomyces, Nocardia, Streptomyces.
cyjanobakterie i glony z rodzajów: Oscillatoria, Anabaena, Nostoc, Chlorella,
Chlamydomonas, Scenedesmus, Phormidium.
W niektórych procesach biodegradacji konieczne jest współdziałanie dwóch lub większej liczby szczepów, ponieważ pojedyńczo nie były by w stanie przeprowadzić podanej zmiany do końca.
W trakcie procesów biodegradacji oprócz rozkładu lub przemian niektórych związków mogą powstać produkty znacznie trwalsze, które mogą mieć działanie rakotwórcze lub mutagenne. Jako przykład mogą tu posłużyć wielopierścieniowe węglowodory aromatyczne. Stanowią one grupę związków które wraz ze wzrostem procesów przetwórczych i związanym z tym od lat niekontrolowanym zanieczyszczeniem środowiska towarzyszą człowiekowi praktycznie wszędzie. Wiele związków z tej grupy występuje w dymie tytoniowym, powietrzu, wodzie, pożywieniu, glebie, wodnych organizmach, olejach mineralnych i rafinowanych produktów naftowych.
Związki aromatyczne ulegają biodegradacji do jednego z pięciu podstawowych produktów: katecholu, kwasu protokatechowego, kwasu gentyzynowego, kwasu homokatechowego i kwasu homogantyzynowego jak również do kilku niekonwencjonalnych związków, takich jak: kwas salicylowy, kwas antranilowy i ortoaminofenol.
2. WĘGLOWODORY - PODZIAŁ, CHARAKTERYSTYKA I
WŁAŚCIWOŚCI CHEMICZNE
Węglowodory to grupa związków węgla i wodoru. Stanowią jedną z największych grup związków organicznych. W warunkach normalnych występują jako gazy (np. początkowe węglowodory szeregu alkanów), ciecze (np. nizsze węglowodory aromatyczne) lub ciała stałe (np. wyższe węglowodory aromatyczne). Węglowodory nasycone acykliczne i alicykliczne wykazują małą aktywność chemiczną i reakcje z ich udziałem zachodzą na ogół w wysokich temperaturach. Bardziej czynnie chemicznie są alkeny, które wykazują zdolność do reakcji przyłączania oraz do polimeryzacji. Jeszcze większą aktywność chemiczną wykazują alkiny, które oprócz reakcji przyłączania ulegają reakcji podstawiania. Dla węglowodorów aromatycznych najbardziej typowe są reakcje podstawiania. Wszystkie węglowodory są palne, mają wysokie ciepło spalania.
Bakterie wykorzystujące węglowodory są szeroko ropowszechnione. Można je wyizolować ze wszystkich gleb uprawnych, pastewnych i leśnych. Związki te są obecne w wielu organizmach i w sposób ciągły są syntetyzowane przez bakterie i rośliny; należą do nich np. woskowate substancje pokrywające liście wielu roślin. Węglowodory należy zatem uważać nie za skamieniałe relikty pierwotnej produkcji roślinnej z czasów prehistorycznych, lecz również za wtórne metabolity nadal syntetyzowane w znacznych ilościach przez rośliny zielone [Schlegel H.G. 1996].
Podział węglowodorów na różne grupy systematyczne przedstawia poniższy schemat
[Gałamon T. 1988].
WĘGLOWODORY
ALIFATYCZNE AROMATYCZNE
NASYCONE NIENASYCONE JEDNOPIERŚCIENIOWE
O PIERŚCIENIU
SKONDENSOWANYM
ALKANY ALKENY ALKINY
CYKLOALKANY CYKLOALKENY
2.1. WĘGLOWODORY AROMATYCZNE
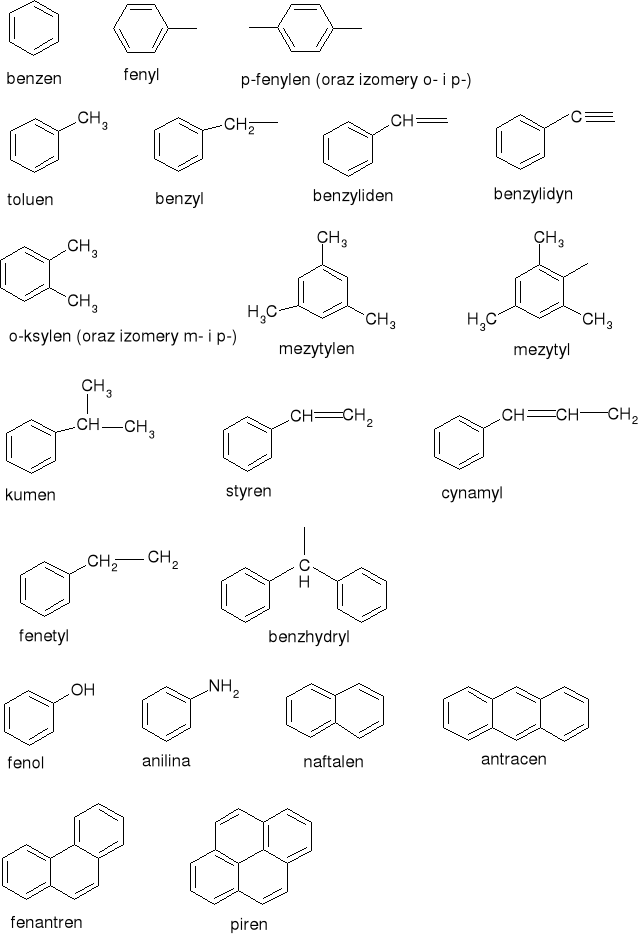
Węglowodory aromatyczne zostały odkryte w XIX wieku. Są to związki, które w cząsteczce mają szczególne ugrupowanie złożone z sześciu atomów węgla. Ich nazwa powstała w związku z tym, że pochodne benzenu były po raz pierwszy wyodrębnione z olejków eterycznych, balsamów i innych subsatancji odznaczających się przyjemnym zapachem. Nazwa ta służy do dziś, chociaż termin „aromatyczność” oznacza obecnie zespół właściwości i reakcji charakteryzujących pewną grupę związków.
Związki te cechuje wysoki stopień nasycenia a ich cechą charakterystyczną jest to, że posiadają dużą stabilność cząsteczki. Róznią się od alifatycznych mniejszą zawartością wodoru, większą łatwością sulfonowania i nitrowania oraz dużą odpornością na działanie czynników utleniających [Uzarewicz A. 1978]. Są nierozpuszczalne w wodzie, natomiast dobrze rozpuszczają się w polarnych rozpuszczalnikach organicznych. Palą się silnie kopcącym płomieniem.
Najprostszym przedstawicielem węglowodorów aromatycznych jest benzen C6H6.
Węglowodory aromatyczne możemy podzielić na dwie podstawowe grupy:
Węglowodory aromatyczne jednopierścieniowe, czyli substancje ciekłe lub stałe,
które tworzą szereg homologiczny, gdzie homologi różnią się między sobą grupą
CH2. Do tej grupy zaliczy jest benzen i jego pochodne.
2. Wielopierścieniowe węglowodory aromatyczne (WWA), czyli związki organiczne
składające się z paru pierścieni benzenowych (przy czym sąsiednie pierścienie
mają wspólne dwa atomy węgla), niekiedy też pierścieni niearomatycznych.
Pierścienie mogą być skondensowane: liniowo, kątowo lub zawierać wspólne
atomy węgla w trzech pierścieniach, np. naftalen, fenantren, antracen, piren
[Uzarewicz A.1978]. Policykliczne węglowodory aromatyczne mogą zawierać
podstawniki alkilowe w łańcuchach bocznych. W związkach heterocyklicznych
pierścień aromatyczny zawiera zamiast atomu węgla atom azotu, tlenu czy siarki.
Poniżej przedstawiono wzory chemicznie najważniejszych węglowodorów aromatycznych:
3. skażenie środowiska przez węglowodory
3.1. Skażenie środowiska glebowego przez węglowodory.
Intensywna działalność baz wojskowych, jednostek przemysłu obronnego, chemicznego, petrochemicznego oraz transportu spowodowała duże zniszczenie gleb węglowodorami oraz ich pochodnymi (oleje, smary, WWA itd.). Stopień zagrożenia zależy jednak od rodzaju gruntu. I tak (Obara K. I wsp. 2000):
grunt nieprzepuszczalny - we wstępnej fazie zanieczyszczeniu ulega wierzchnia warstwa gruntu;
grunt przepuszczalny - zanieczyszczenia wsiąkają w głąb i dzięki siłom grawitacji są transportowane aż do wód podziemnych.
Zanieczyszcenie gleb węglowodorami aromatycznymi, które np. pochodzą z ropy naftowej nie stanowią aż tak dużego niebezpieczeństwa. W glebie dobrze przewietrzonej zostają one w krótkim czasie całkowicie rozłożone. Zaś na znacznej głębokości, przy braku dostępu powietrza jest prawdopodobne, że utrzymają się w gruncie i przenikną do wody pitnej. Związki aromatyczne mogą równiż toksycznie oddziaływać na organizmy glebowe, ogniżać plonowanie roślin, powodować zniszczenie materiału siewnego, zmieniać warunki geotechniczne jak również powietrzno-wodne w glebie (Schlegel H. G. 1975, Muszyński 2000, Łebkowska 2003).
Obecność węglowodorów w środowisku naturalnym powoduje oddziaływania między nimi a otaczającymi je gruntami. Można zauważyć wówczas następujące procesy: adsorpcję na ziarnach, odparowywanie do powietrza gruntowego, rozpuszczanie w wodzie lub degradację chemiczną i biologiczną tych gleb.
Różne są obszary zanieczyszczeń gleb, ich skład chemiczny oraz czas oddziaływania na środowisko. Zróżnicowane muszą być także metody oddziaływania. Generalnie są one kosztowne i nie pozostają obojętne dla środowiska naturalnego.
3.2. Skażenie środowiska wodnego przez węglowodory.
Zanieczyszczenie wód gruntowych związkami organicznymi, głównie węglowodorami stanowi poważny problem. Wynika to m.in. z powszechności ich stosowania, szkodliwego wpływu na organizmy żywe oraz specyficznych możliwości w zakresie rozprzestrzeniania się i długotrwałego pozostawania w środowisku.
Obecność węglowodorów w wodach morskich i oceanicznych jest skutkiem wycieków ropy naftowej powstałych na skutek wypadków tankowców i katastrof morskich. Oszacowano, że około 0,1% ropy (czyli 35 mln ton rocznie) wpływa do morza (Badawcza Rada Naukowa 1985 rok). Natomiast przedostawanie się ropy do morza z naturalnych źródeł (głównie jest to przesączanie się z pokładów złóż ropy) wynosi około 0,5 mln ton rocznie. Ponieważ czas „życia” węglowodorów w morzu to dekady lub wieki zatem rozpuszczone w głębokich oceanach węglowodory krążą przez wiele lat skutkiem czego stanowią światowy problem [Mueller J.G.. i wsp. 1996], ponieważ ich obecność zagraża obniżeniu wydajności rybołówstwa, rozwoju terenów rekreacyjnych i wodnych.
Aby zapewnić rozwój rekreacji i wypoczynku w obrębie czystych zbiorników wodnych należy pamiętać o czystości wód gruntowych, a co za tym idzie i czystości wód pitnych. Problem ten ma istotne znaczenie ze względu na obecność w środowisku wodnym węglowodorów aromatycznych takich jak: benzen, toluen i ksylen. Związki te znalazły się na liście priorytetowych zanieczyszczeń Agencji Ochrony Środowiska [Tsao C.W 1998, Shuttleworth K.L 1995]. Ich pozycja tam wynika z:
szerokiej stosowalności w różnych przemysłach (jako składniki benzyn, rozpuszczalników),
stosunkowo wysokiej rozpuszczalności w wodzie i szybkiej możliwość skażenia roślinności wodnej i wód gruntowych
oraz ze względu na silne działanie kancerogenne [Tsao C.W 1998, Obara K. i wsp 2000].
W tabeli 1 przedstawiono dopuszczalne stężenia monocyklicznych węglowodorów aromatycznych (MWA) w wodach gruntowych [Obara K. i wsp 2000].
Tabela. 1 Dopuszczalne stężenie MWA w wodach podziemnych.
Związek |
obszar |
||
|
parki narodowe, rezerwaty, strefy ochronne źródeł i ujęć wód podziemnych |
tereny upraw rolniczych, tereny zabudowy mieszkaniowej, rekreacji, wypoczynku oraz obszary leśne |
zakłady przemysłowe, trasy komunikacyjne, lokomotywownie, poligony wojskowe, lotniska |
|
stężenie monocyklicznych węglowodorów aromatycznych [ug/dm3] |
||
Benzen |
0,2 |
1 |
5 |
Etylobenzen |
0,2 |
20 |
60 |
Toluen |
0,2 |
15 |
50 |
Ksylen |
0,2 |
20 |
60 |
całkowita ilość związków aromatycznych |
0,2 |
15 |
50 |
W przypadku wody do picia i na potrzeby gospodarcze spośród MWA normowany jest tylko benzen - jego dopuszczalna zawartość 0,01mg/dm3.
3.3. Skażenie atmosfery przez węglowodory.
Węglowodory aromatyczne obecne w atmosferze pochodzą ze źródeł antropogenicznych i naturalnych [Kluska M. 2000]. Naturalne źródło występowania tych związków związane jest z szeroko rozpowszechnionymi procesami wegetacyjnymi niektórych organizmów, procesami gnilnymi, pożarami lasów oraz obecnością ich jako podstawowych składników wosków liści, olei roślinnych, pancerzyka insektów (kutikuli), tłuszczy mikroorganizmów czy gazu ziemnego [Kluska M. 2000, Mueller J.G. 1996]. Natomiast za antropogennego źródła występowania węglowodorów aromatycznych uważa się niekompletne spalanie paliw kopalnych (elektrociepłownie, gospodarstwa domowe i pojazdy mechaniczne), złe przechowywanie paliw ciekłych, nieszczelności podczas tankowań, obróbki i transportu [Kluska M. 2000, Mueller J.G. 1996]. Szczególnie niebezpieczne dla powietrza są benzyny, tzn lekkie frakcje nafty, stanowiące mieszaninę węglowodorów nasyconych, naftalenowych, nienasyconych i aromatycznych. Benzyny wykazyją łatwość przechodzenia w stan pary w warunkach normalnych. Wielkość emisji węglowodorów do atmosfery zależy od wielu czynników:
rodzaju paliwa;
sposobu spalania;
zastosowania urządzeń oczyszczających powstałe po spaleniu produkty;
warunków atmosferycznych;
organizacji pracy.
Wśród najczęściej występujących w powietrzu węglowodorów wymienić należy związki alifatyczne, aromatyczne o małej masie, a także policykliczne węglowodory aromatyczne tworzące się w wyniku polikondensacji. Uwolnione do atmosfery węglowodory występują w postaci par lub adsorbują się na powierzchni pyłów ( sadza, popioły).
4. Drobnoustroje degradujące węglowodory
Proces samooczyszczania środowiska naturalnego ze zgromadzonych w nim węglowodorów trwa długo i przebiega powoli. Trwają badania nad przyśpieszeniem ich biodegradacji na drodze procesów biotechnologicznych. Wykonuje się to przy pomocy aktywnych i czystych kultur uprzednio wyizolowanych ze skażonych środowisk. Dlatego zwrócono szczególną uwagę na rozmieszczenie bakterii zdolnych do utleniania węglowodorów, z powodu możliwości wykorzystania ich potencjału degradacyjnego przy likwidacji zanieczyszczeń powstałych w wyniku wycieków ropy [Rosenberg E. 1996 ].
Węglowodory są obecne w wielu organizmach i w sposób ciągły są syntetyzowane przez bakterie i rośliny w postaci woskowatych substancji pokrywających liście. Dlatego też węglowodory należy uważać za wtórne metabolity syntetyzowane w dużych ilościach przez rośliny zielone [Schlegel H.G. 1996].
Zdolność drobnoustrojów do biodegradacji węglowodorów uzależniona jest od [Kwapisz E. 1999]:
funkcjonowania odpowiednich systemów ich transportu do komórki;
posiadania przez drobnoustroje potencjału genetycznego umożliwiającego wprowadzenie tlenu do cząsteczki węglowodoru;
specyficzności substratowej oksygenaz;
funkcjonowania mechanizmu indukowania enzymów takich jak: dehydrogenazy,
hydrolazy czy dekarboksylazy.
Pobieranie węglowodorów przez komórki mikroorganizmów może zachodzić w drodze [Łebkowska M, Klimiuk E. 2003]:
wprowadzania mikrokropli o wymiarach mniejszych niż wielkość komórki;
transportu makrokropel;
pobierana składników rozpuszczonych lub wolnych, przy czym w wodzie ulega rozpuszczeniu niewielka ilość węglowodorów charakteryzujących się małym ciężarem cząsteczkowym.
Środowisko naturalne obfituje w liczne gatunki tlenowych i beztlenowych drobnoustrojów, z których niektóre wykazują uzdolnienia do wykorzystywania węglowodorów jako źródła węgla i energii a tym samym do ich degradacji. Świat drobnoustrojów obfituje w liczne ich grupy. Wśród nich można wyróżnić zarówno bakterie, drożdże, glony i grzyby strzępkowe. Tabela 2 przedstawia przegląd tych drobnoustrojów, u gatunków zaznaczonych wytłuszczonym drukiem udokumentowano zdolność do degradowania węglowodorów aromatycznych [Watkinson R.J. 1990, Łebkowska M. 2003].
Tabela. 2 Drobnoustroje degradujące węglowodory.
BAKTERIE |
DROŻDŻE |
GRZYBY NITKOWATE |
GLONY |
PROMIENIOWCE |
Acetobacter |
Candida |
Aspergillus |
Anabaena |
Actinomyces |
Achromobacter |
Cryptococcus |
Cladosporium |
Chlamydomonas |
Nocardia |
Acinetobacter |
Debaryomyces |
Corollaspora |
Chlorella |
Streptomyces |
Aeromonas |
Hansenula |
Cunninghamella |
Nostoc |
|
Alcaligenes |
Pichia |
Dendryphiella |
Oscilatoria |
|
Arthrobacter |
Rhodotorula |
Glicadium |
Scenedesmus |
|
Bacillus |
Sacharomyces |
Lulworthia |
Phormidium |
|
Beijerinckia |
Sporobolomyces |
Mucor |
|
|
Beneckea |
Torulopsis |
Penicillium |
|
|
Chrysosporium |
Trichoderma |
Varicospora |
|
|
Corynebacterium |
Trichospora |
|
|
|
Flavobacterium |
|
|
|
|
Micrococcus |
|
|
|
|
Moraksella |
|
|
|
|
Mycobacterium |
|
|
|
|
Pseudomonas |
|
|
|
|
Vibrio |
|
|
|
|
Xentomonas |
|
|
|
|
Spośród dużej grupy bakterii o uzdolnieniach do degradacji węglowodorów najlepiej zbadanymi i poznanymi są bakterie z rodzaju Pseudomonas, które utleniają katechol i pokrewne związki za pomocą dioksygenaz.
Postępujące zanieczyszczenie środowiska, świadomość istniejącego zagrożenia oraz rygory prawne i administracyjne powodują, że wykorzystanie wyspecjalizowanych mikroorganizmów do remediacji gruntów, jako metody dość taniej i stosunkowo mało skomplikowanej, wydaje się użyteczne. Namnożenie ich, a następnie immobilizacja na nośniku stałym to jeden ze sposobów uzyskania odpowiednio dużej ilości biomasy aktywnych drobnoustrojów. Uzyskana w ten sposób biomasa może być stosowana zarówno „in situ” jak i „ex situ” w różnego rodzaju bioreaktorach [Muszyński A. i wsp 1996, Mueller J.G. 1996]. Nie należy jednak zapominać o kontroli mikrobiologicznej procesu remediacji gruntów. Powinna ona obejmować ocenę zmian ilościowych bakterii w celu zapewnienia co najmniej 105 komórek w 1g gleby. Niezbędna jest także okresowa identyfikacja mikroorganizmów celem stwierdzenia braku rozwoju bakterii o cechach chorobotwórczych w obrębie biomasy wprowadzanej do środowiska [Muszyński A. i wsp 1996].
Naturalny proces biodegradacji zachodzi stosunkowo powoli i aby zwiększyć szybkość i skuteczność rozkładu korzystne jest stosowanie biopreparatów. Obecnie coraz częściej do remediacji gruntów wykorzystuje się biopreparaty zawierające autochtoniczne mikroorganizmy zdolne do biodegradacji węglowodorów oraz składniki mineralne, głównie związki azotu i fosforu. Zasiedlenie ekosystemu glebowego przez obce środowisku szczepy drobnoustrojów może niekiedy doprowadzić do zachwiania naturalnej równowagi biologicznej. Dlatego też lepszym rozwiązaniem jest stosowanie mikroorganizmów autochtonicznych pochodzących z zanieczyszczonego środowiska [Łebkowska M. i wsp. 2003]. Wykorzystanie biopreparatów, zawierających obce środowisku glebowemu mikroorganizmy, może być dopuszczone do oczyszczania gruntu jeśli badania laboratoryjne wykazały ich skuteczność. Istotna jest modyfikacja warunków panujących w glebie, tak aby stały się one optymalne dla rozwoju biomasy mikroorganizmów [Muszyński A. i wsp. 1996].
5. warunki biodegradacji
Proces biodegradacji w warunkach naturalnych zachodzi stosunkowo powoli i przy udziale bytującej w danym środowisku mikroflory. Zadaniem kierowanych procesów biotechnologicznych jest likwidacja skażeń przez wzbogacenie autochtonicznej mikroflory wyselekcjowanymi, wysokoaktywnymi mikroorganizmami przy jednoczesnym niedopuszczeniu do zachwiania naturalnej równowagi biologicznej. Celem prac biotechnologa jest również stworzenie optymalnych warunków do rozwoju tychże drobnoustrojów. Przebieg bioremediacji gruntów zanieczyszczonych paliwami i smarami zależy od wielu czynników, które należy uwzględnić aby proces oczyszczania przebiegał w sposób prawidłowy. Do podstawowych czynników mających wpływ na szybkość procesu biodegradacji węglowodorów, w tym aromatycznych należą:
Stan fizyczny produktów naftowych.
Węglowodory naftowe mogą występować w ziemi w wielu fazach:[Surygała J. 2000];
faza stała (asfalt, bituminy, żywice, woski naftowe - do momentu gdy temperatura otoczenia nie wzrośnie powyżej punktu topnienia lub mięknięcia);
faza ciekła (jako błonka zaadsorbowana na powierzchni ziaren mineralnych dzięki siłom Van der Waalsa; zaadsorbowanie przez ziarna w wyniku chemisorpcji, uwięzione w szczelinach i porach ziaren);
faza ciekła wolnych płynów;
w stanie rozpuszczonym (w infiltrującej wodzie strefy areacji, w otoczce wodnej pokrywającej ziarna mineralne, w wodzie porowej, w wodach gruntowych);
faza parowa
Budowa chemiczna i właściwości węglowodorów.
Podatność węglowodorów na biodegradację związana jest z [Łebkowska M. 2003]:
długością łańcucha;
ilością rozgałęzień;
obecnością pierścieni aromatycznych i podstawników;
budową alicykliczną.
Do związków trudno degradowanych należą policykliczne węglowodory aromatyczne, długołańcuchowe alifatyczne, alicykliczne. Dlatego też węglowodory aromatyczne są trudniej degradowlne niż alifatyczne.
3. Rozpuszczalność w wodzie.
Związki rozpuszczalne w wodzie są łatwo dostępne dla drobnoustrojów, te z nich, które w niej się nie rozpuszczają mogą być wykorzystywane w stanie zdyspergowanym [Łebkowska M. 2003]. Jedynie węglowodory o niskiej masie cząsteczkowej łatwo ulegają rozpuszczeniu w wodzie. Aby możliwe było przyswojenie przez drobnoustroje węglowodorów wyższych, które są trudniej rozpuszczalne, konieczne jest rozproszenie ich w formie makro- czy mikrokropli. Sprzyja temu produkcja biosurfaktantów poprzedzająca proces degradacji węglowodorów.[Watkinson R.J. 1990] Wiele mikroorganizmów zdolnych jest do wytwarzania substancji powierzchniowo czynnych (SPC) o właściwościach emulgacyjnych [Łebkowska M. 2003]. Związki o charakterze substancji powierzchniowo czynnych powodują zwiększanie rozpuszczalności węglowodorów w fazie wodnej oraz utworzenie węglowodorowej emulsji, a tym samym zwiększenie powierzchni między fazami.
4. Stężenie węglowodorów i ich toksyczność w stosunku do mikroflory.
Coraz częściej oznaczenie toksyczności jest włączane do badań nad biodegradacją. Jest to szczególnie ważne, ponieważ potrzeba bioremediacji bazuje na potrzebie zmniejszenia ilości toksycznych związków w terenie. Stężenie węglowodorów zawartych w produktach naftowych zasadniczo wpływa na efekt biodegradacji. Związane jest to ze stopniem toksyczności węglowodorów (LC50), która waha się zależnie od ich rodzaju. Na przykład wartość LC50 benzenu dla bakterii wynosi 100mg/l, toluenu 20mg/l a ksylenu 6mg/l [Łebkowska M. 2003, Shuttleworth K.L. 1995, Persoone G. 1993]. Wiele węglowodorów ulega biodegradacji po okresie adaptacji drobnoustrojów, w którym wytwarzają one enzymy indukcyjne. Na proces ten ma wpływ stężenie węglowodorów jako substancji pokarmowych - zbyt niskie stężenie ogranicza wytwarzanie enzymów indukcyjnych [Łebkowska M. 2003].
5. Procesy sorpcji węglowodorów w glebie.
Niektóre badania [Maliszewska B. 1991] wykazały, że nawożenie gleb nawozami naturalnymi i kompostami może opóźnić rozkład wielopierścieniowych węglowodorów aromatycznych (WWA). Jako związki hydrofobowe i niepolarne, WWA adsorbują się w huminach i kwasach huminowych. Natomiast sorbcja policyklicznych węglowodorów aromatycznych do materii organicznej w glebach i osadach oraz szybkość ich desorpcji silnie wpływa na szybkość degradacji zanieczyszczeń przez mikroorganizmy. Czas trwania kontaktu pomiędzy WWA a glebą może mieć znaczący wpływ na dostępność WWA. Uzyskiwanie rzeczywistej równowagi pomiędzy WWA a glebą może być procesem powolnym. Nie do końca zrozumiany jest mechanizm, który powoduje, że WWA stają się bardziej odporne na degradację wraz z upływem czasu. Wiadomo, że sorpcja WWA do organicznej materii gleby jest czynnikiem krytycznym przy określaniu ich biodostępności.
6. Zawartość związków biogennych w tym azotu i fosforu.
Węglowodory nie zawierają azotu i fosforu, które są niezbędnymi pierwiastkami do budowy biomasy i do ich procesów fizjologicznych. Dlatego w procesach remediacji należy uzupełnić glebę o te biogeny. Związki azotowe i fosforowe dostarczane są w postaci nawozów sztucznych [Łebkowska M. 2003]. W naturalnych środowiskach proporcje występowania wyżej wymienionych składników są zazwyczaj bliskie wymaganiom bytującej w nim mikroflory. Podczas skażenia węglowodorami proporcje te ulegają zachwianiu w skutek zwiększenia stężenia źródła węgla [Rashiah V. 1991, 1992].Jest wiele publikacji dotyczących optymalnego stosunku węgla do azotu i fosforu; najczęściej uznawany za odpowiedni jest stosunek 100 : 9 : 2 lub 100 : 10 : 1 [Morgan P. i Watkinson R.J. 1990]. Mimo obfitości źródła węgla drobnoustroje nie są w stanie go wykorzystać, ponieważ w podłożu występuje duży niedostatek azotu i fosforu uniemożliwiający prawidłowy rozwój drobnoustrojów. Jednak w pewnych przypadkach, jeśli w glebie dominuje mikroflora oligotroficzna, nie jest wskazane nawożenie mineralne, gdyż nadmiar azotu i fosforu może hamować usuwanie węglowodorów [Łebkowska M. 2003].
Ilość i jakość mikroorganizmów.
Wiele mikroorganizmów zdolnych jest do rozkładu węglowodorów Często reakcje rozkładu zachodzą sekwencyjnie przy udziale wielu rodzajów mikroorganizmów synergistycznie zależnych od siebie [Łebkowska M. 2003]. Podkreślić należy, że do bezpośredniej remediacji gleb powinno się stosować aktywne szczepy bakterii pochodzących z danego zanieczyszczonego środowiska. Szczepy te po namnożeniu można wprowadzać ponownie do gruntu. Wykorzystanie biopreparatów, zawierających obce środowisku glebowemu mikroorganizmy, może być dopuszczone do oczyszczania gruntu jeśli badania laboratoryjne wykazały ich skuteczność. Efektywność procesu biodegradacji węglowodorów zależy od ilości mikroflory zdolnej do rozkładu zanieczyszczeń naftowych. Na ogół stężenie tych mikroorganizmów stanowi około 0.01 - 1% ogólnej liczby drobnoustrojów glebowych. W procesach biologicznej remediacji gleb stosuje się metody z wykorzystaniem:
naturalnej mikroflory, której aktywność biochemiczną reguluje się zabiegami technologicznymi, jak napowietrzanie, uzupełnienie biogenów;
naturalnej mikroflory, wyizolowanej z gruntu, namnożonej i ponownie wprowadzonej do gleby w postaci komórek wolnych lub zimobilizowanych na nośnikach [Łebkowska M. 2003].
8. Odczyn pH.
Prawidłowy rozwój i funkcjonowanie wszystkich organizmów zależy od odpowiedniego pH. W zależności od grupy taksonomicznej drobnoustroje degradujące węglowodory wymagają różnych wartości pH środowiska:
bakterie -optimum pH 6.0 -8.5;
grzyby - środowisko lekko kwaśne o pH 4.5 -6.0
bakterie glebowe - optymalne pH 5 - 7.8.
Jak widać z powyższych wielkości pH odczyn środowiska powinien zawierać się w granicach 6-8,5. Często zaleca się dodawanie wapna w iości około 20 - 30% do gleb kwaśnych dla podwyższenia odczynu, a także aby zneutralizowac kwasy powstające podczas biodegradacji. Nie zawsze jednak jest to korzystne, ponieważ może spowodować wzrost trwałości węglowodorów aromatycznych [Łebkowska M. 2003].
Uwodnienie środowiska.
Woda jest czynnikiem, który umożliwia procesy metaboliczne mikroorganizmów oraz warunkuje możliwość rozpuszczania pewnej ilości węglowodorów o małej wartości log wspóczynnika podziału oktanol/woda. Nadmiar wody prowadzi do powstania warunków beztlenowych, które opóźniają biodegradację. Dlatego optymalna wilgotność gruntu powinna wynosić około 80% pojemności wodnej [Morgan P. i Watkinson R.J. 1989], a wilgotność względna nie powinna być mniejsza niż 15% [Cain R.B.][Łebkowska M]. Drożdże i pleśnie wykazują większą tolerancję na zmniejszoną dostępność wody niż bakterie.
Temperatura.
Temperatura jest czynnikiem silnie wpływającym na aktywność drobnoustrojów, czyli również na intensywność biodegradacji węglowodorów. Optimum temperaturowe dla flory bakteryjnej degradującej węglowodory wynosi zwykle 22 - 30°C. Utrzymanie temperatury na tym poziomie w warunkach naturalnych możliwe jest jedynie latem, więc złoża do biologicznego rozkładu są poddawane procesowi podgrzania do odpowiedniej temperatury.
Zawartość tlenu w środowisku.
Do pełnej biodegradacji węglowodorów niezbędny jest tlen. Warunkuje on szybkość wzrostu bakterii, ich fizjologię oraz wpływa na szybkość degradacji związków ropopochodnych. Najczęściej drobnoustroje włączają tlen cząsteczkowy bezpośrednio wiążąc go przy udziale oksygenaz. U organizmów o mniej typowym metabolizmie tlen może być pobierany z substratów organicznych, tj. wybranych jonów (SO42-, NO3-), oraz CO2, H2O [Hartmans S. 1989, Zayer J. 1990]. Silne natlenienie środowiska jest szczególnie ważne w przypadku występowania węglowodorów jako źródła węgla, gdyż są one bardzo ubogie w tlen. Prowadzone są również badania mające na celu wykorzystanie tlenu doprowadzonego do podłoża pochodzącego z H2O2 jako jego źródła [Chmiel A. 1991]. Najefektywniejszym jednak sposobem natleniania jest doprowadzenie powietrza do warstwy oczyszczonego gruntu [Łebkowska M. 2003].
Obecność innych, aniżeli węglowodory, źródeł węgla dla drobnoustrojów oraz związków toksycznych.
Dostępność innych niż węglowodory źródeł wegla może wudłużyć czas bioremediacji, ponieważ drobnoustroju głównie wykorzystują łatwo rozkładalne substraty organiczne. Nawożenie organiczne gleb torfem lub kompostem powoduje zwiększenie sorpcji i trwałość niektórych węglowodorów, ale także wykorzystanie łatwo przyswajalnych źródeł węgla przed trudniej rozkładalnymi węglowodorami. Obecność związków toksycznych, takich jak metale ciężkie, czy pestycydy, może zahamować mikrobiologiczne procesy degradacji produktów naftowych. W pewnych przypadkach stosuje się zabiegi technologiczne do usunięcia związków toksycznych [Łebkowska M. 2003].
6. BIODEGRADACJA WĘGLOWODORÓW AROMATYCZNYCH
W środowisku obecne są naturalne związki aromatyczne jak i sztucznie otrzymane przez człowieka. Działanie węglowodorów aromatycznych w środowisku naturalnym zależy od wielu czynników, które determinują szybkość procesu biodegradacji oraz stopień metabolizmu. Wśród nich wyróżniamy czynniki: fizykochemiczne, mikrobiologiczne i środowiskowe. Pierwszą grupę stanowią: wielkość cząsteczki węglowodorowej i rozpuszczalność jej w wodzie, charakter lipofilowy, lotność, stężenie w środowisku oraz obecność wielu podstawników i grup funkcyjnych. Druga grupa czynników to: typ populacji, jej rozpowszechnienie oraz dostępność mikroflory do węglowodorów. Zaś czynniki środowiskowe tworzą: temperatura otoczenia, pH, stężenie tlenu, zasolenie, pora roku, intensywność nasłonecznienia, typ osadu oraz obecność kosubstratów i warunki odżywcze [Carnigilia C.E. 1984]. Bakterie, drożdże i grzyby strzępkowe wykazują możliwość wykorzystania węglowodorów jako źródła węgla i energii a tym samym do ich degradacji.
Związki aromatyczne mogą być degradowane jeżeli ich pierścień ulegnie rozszczepieniu. Daje to możliwość dalszej asymilacji tych związków. Część organizmów prokariotycznych i eukariotycznych posiada zdolność rozszczepienia pierścienia aromatycznego. Istnieją główne różnice między szybkością rozkładu węglowodorów aromatycznych w glebie i systemach wodnych (w tych ostatnich przebiega on wolniej), oraz stanie się węglowodorów związkami bardziej opornymi na działanie degradującej mikroflory w zależności od ilości pierścieni benzenowych w cząsteczce. Potencjał enzymatyczny tych drobnoustrojów pozwala na utlenienie rozpatrywanych związków według wielkości pierścienia. I tak kolejno degradowane są: benzen, toluen, ksylen, naftalen, antracen, fenantren, benzoapiren, benzoantracen [Rosenberg E. 1996].
Metabolizm związków aromatycznych przez mikroorganizmy zależny jest od:
ilości pierścieni aromatycznych w cząsteczce węglowodoru,
wstępnych reakcji prowadzących do rozszczepienia pierścienia;
oraz sposobu jego rozszczepienia.
6.1. ogólny schemat biodegradacji wybranych węglowodorów aromatycznych
Rośliny produkują wiele związków organicznych zawierających pierścienie aromatyczne. Pod względem ilościowym dominuje lignina, która stanowi 20% (wagowych) drewna. Pierścień aromatyczny rozszczepia wiele gatunków bakterii i grzybów. Niektóre bakterie z rodzaju Pseudomonas rosną szybciej w obecności benzoesanu niż cukrów, jednakże warunkiem szybkiej degradacji związków aromatycznych jest obecność tlenu [Schlegel H.G. 2003].
Rozszczepienie pierścienia aromatycznego często, lecz nie zawsze, jest poprzedzane usunięciem z niego podstawników. Grupy chlorowe, nitrowe i sulfonowe są zastępowane grupami hydroksylowymi. Eliminacja halogenów jest bardzo ważna ponieważ, związki aromatyczne z podstawnikiem są bardzo wolno rozkładane [Rehm J. 1996].
Alifatyczne łańcuchy boczne mogą być różnie modyfikowane i skracane lub mogą pozostawać nietknięte [Schlegel H.G. 2003, Rehm J. 1996, Evans W.C.1963]. Np.: m-krezol i p-krezol może być przekształcany do hydroksymetylowej pochodnej lub bezpośrednio hydroksylowany do 3-metylokatecholu i 4-metylokatecholu.
Większość spośród związków aromatycznych występujących w przyrodzie ulega degradacji przez bakterie do katecholu i kwasu protokatechowego. Oba te związki posiadają dwa typy szlaków biodegradacji:
szlak intradiolowy - rozszczepienie pierścienia między dwoma sąsiadującymi
atomami węgla, co prowadzi do powstania kwasów dikarboksylowych;
szlak ekstradiolowy - rozszczepienie pierścienia między hydroksylowym i
niehydroksylowym atomem węgla.
Benzen, toluen, fenantren i antracen w pierwszej fazie procesu biodegradacji są przekształcane do katecholu (rys.1) [Rehm J. 1996].
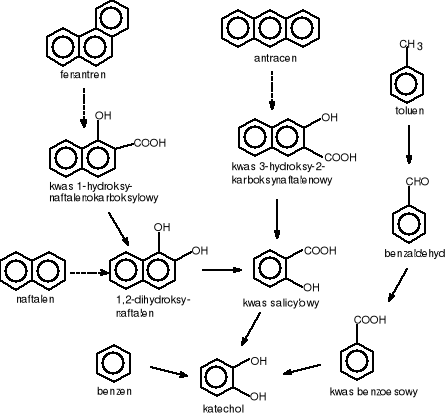
Rysunek 1 Degradacja związków aromatycznych do katecholu
Do kwasu protokatechowego są przekształcane pierścienie aromatyczne podwójnie podstawione w pozycjach 1,2-, 1,3- i 1,4- np.: w m-krezolu, p-krezolu, jak również pierścienie podstawione wielokrotnie. We wszystkich przyoadkach do pierścienia włączają się grupy hydroksylowe (rys.2.) [Schlegel H.G. 2003, Rehm J. 1996].
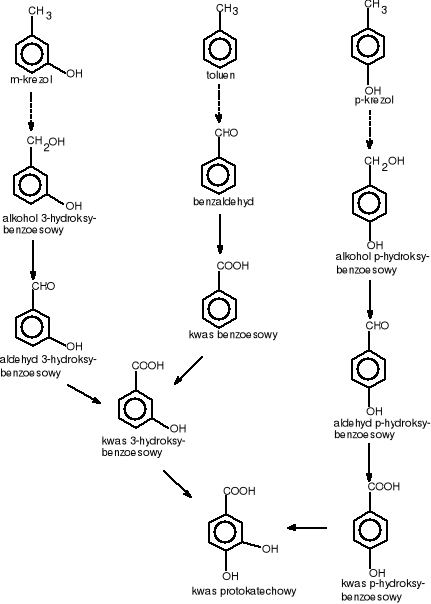
Rysunek 2 Degradacja związków aromatycznych do kwasu protokatechowego
6.2. BIODEGRADACJA WĘGLOWODORÓW AROMATYCZNYCH W
WARUNKACH TLENOWYCH
Jednym z czynników, który sprzyja szybkiej degradacji związków aromatycznych jest tlen. Metabolizm tlenowy charakteryzowany jest przez dwa podstawowe procesy związane z obecnością cząsteczki tlenu, [Rehm J. 1996]:
przeniesieniem elektronów z substratu organicznego na tlen wiążące się z wytworzeniem energii dla komórki;
wprowadzeniem cząsteczki tlenu do substratu organicznego - aktywacja do dalszego metabolizmu.
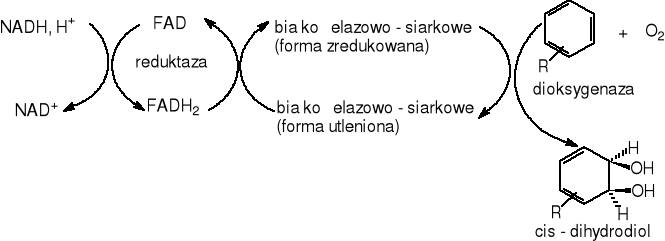
Wiele gatunków bakterii rozpoczyna degradację związków aromatycznych od przyłączenia dwu atomów z cząsteczki tlenu do pierścienia aromatycznego w wyniku czego tworzy się cis-dihydrodiol [Rosenberg E. 1996, Schlegel H.G. 2003]. Wprowadzenie tlenu następuje przy wykorzystaniu systemu dioksygenaz. W następnym etapie odpowiednie dehydrogenazy odrywają dwa atomy wodoru, przez co przywracają aromatyczny charakter pierścienia. W wyniku tych reakcji węglowodory aromatyczne przekształcane są do katecholu lub kwasu protokatechowego, które traktowane są jako metabolity pośrednie tych przemian.W dalszych etapach pierścień jest rozrywany między dwiema sąsiadującymi grupami hydroksylowymi, albo między hydroksylowanym i sąsiadującym niehydroksylowanym atomem węgla. Najważniejszymi typami otwarcia pierścienia aromatycznego są: rozszczepienie typu „orto” i typu „meta”[ Rosenberg E. 1996, Schlegel H.G. 2003, Rehm J. 1996].
6.2.1. Intradiolowe rozszcepienie pierścienia aromatycznego ( typu „orto”).
Rozszczepienie pierścienia między dwoma sąsiadującymi hydroksylowanymi atomami węgla (rys.3) prowadzi do powstania kwasów dikarboksylowych. Przypuszczalnie w reakcji tej zachodzi pierwotne przyłączenie cząsteczki O2 do atomów węgla obok grupy hydroksylowej z wytworzeniem cyklicznego nadtlenku. Z kolei, wewnątrzcząsteczkowe przekształcenia prowadzą do zerwania wiązania C-C z wytworzeniem kwasu cis,cis-mukonowego. Katechol jest rozszczepiany przez 1,2-dioksygenazę katecholową, a kwas protokatechowy przez 3,4-dioksygenazę
x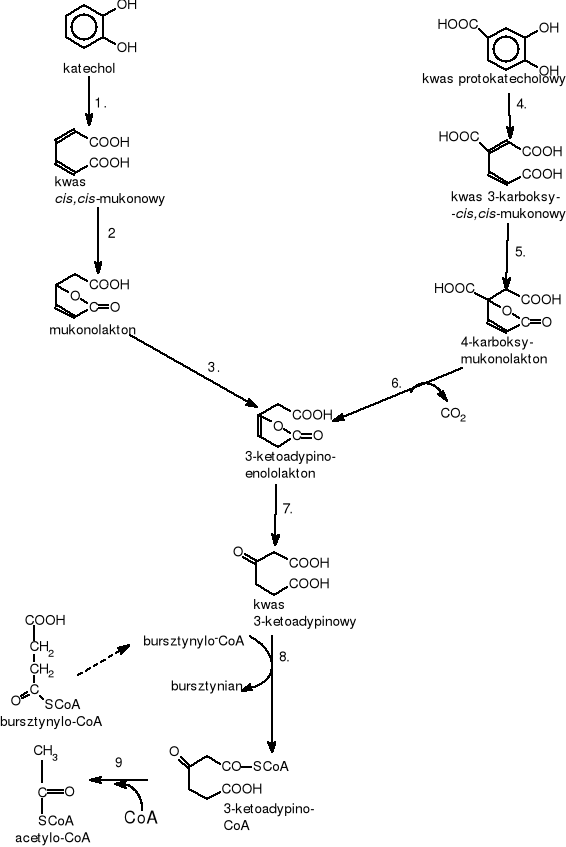
gdzie: 1.EC.1.13.11.1. - 1,2 dioksygenaza katecholowa (pirokatechaza); 2.EC. 5.5.1.1. - cykloizomeraza mukonianowa; 3.EC.5.3.3.4. - izomeraza mukonolaktonowa; 4.EC.1.13.11.3. - 3,4 dioksygenaza protokatecholowa; 5.EC.5.5.1.2. - cykloizomeraza 3-karboksymukonianowa; 6.EC.4.1.1.44. - dekarboksylaza 4-karboksymukonolaktonowa; 7.EC.3.1.1.24. - hydrolaza 4-ketoadypinolaktonowa; 8.EC.2.8.3.6.-transferaza 3-ketoadypino-bursztynylo-CoA; 9.EC.2.3.1.16. - tiolaza 3-ketoadypinylo-CoA
Rysunek 3 Schemat rozszczepienia pierścienia aromatycznego typu „orto”
protokatechową. Produkty tych dwóch reakcji enzymatycznych (cis,cis-mukonian i 3-karboksy-cis,cis-mukonian) ulegają dalszym przemianom poprzez ten sam związek pośredni tj. kwas 3-ketoadypinowy. Związek ten jest aktywowany przez transferazę-CoA i zostaje rozszczepiony do bursztynylo-CoA i aetylo-CoA, które są włączane do cyklu Krebsa [Schlegel H.G. 2003, Parales R.E. 2000, Rehm J. 1996, Chodkowski J. 1996].
6.2.2. Ekstradiolowe roszczepienie pierścienia aromatyczne go (typu „meta”)
Rozszczepienie pierścienia między hydroksylowanym i niehydroksylowanym atomem węgla jest katalizowane przez 2,3-dioksygenazę katecholową lub 4,5-dioksygenazę protokatechową (rys.4) [Schlegel H.G. 2003, Chodkowski J. 1996, Rehm J. 1996, Sala-Trepa J.M. 1971]. W wyniku reakcji powstają semialdehydy kwasu 2-hydroksymukonowego (semialdehydy kwasu 2-hydroksy-4-karboksymukonowego), które następnie wchodzą w szlaki metabolizmu pośredniego poprzez pirogronian, aldehyd octowy, szczawiooctan, fumaran i inne produkty pośrednie (zależnie od typu podstawienia powstałych kwasów alifatycznych).
Na uwagę zasługuje fakt, iż dla różnych drobnoustrojów szlaki kataboliczne związków aromatycznych znacznie różnią się zarówno sposobami rozszczepienia pierścienia, jak i reakcjami wstępnymi prowadzącymi do jego rozszczepienia (jak ma to miejsce w przypadku toluenu gdzie w pierwszym etapie utleniona zostaje grupa metylowa). W przypadku niektórych bakterii również faza wzrostu i warunki hodowli mogą decydować o tym, czy postają enzymy katalizujące rozszczepienie typu „orto” czy typu „meta”, np.: u bakterii z rodzaju Pseudomonas związki aromatyczne katabolizowane poprzez katechol ulegają rozszczepieniu typu „orto”, podczas gdy katabolizowane poprzez kwas protokatechowy ulegają rozszczepieniu typu „meta” [Schlegel H.G. 2003].
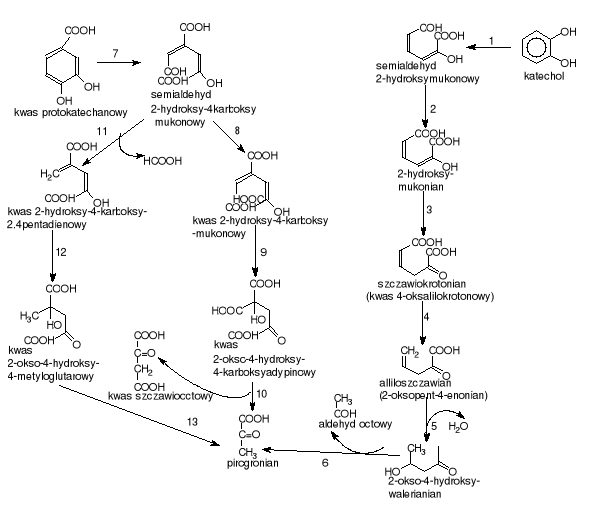
gdzie: 1.EC.1.13.11.2. - 2,3-dioksygenaza katecholowa; 2.EC.1.2.1.32. - dehydrogenaza aldehydowa; 3. EC.5.3.3.? - tutomeraza; 4.EC.4.1.1.77. - dekarboksylaza oksalilokrotonianowa; 5.EC. 4.2.1.80. - hydrataza 2-oksopent-4-enonianowa ; 6.EC.4.2.1.18. - aldolaza 2-keto-3-deoksypentozowa, 7.EC.1.13.11.8. - 4,5-dioksygenaza protokatechanowa, 8.EC.1.2.1.45. - dehydrogenaza semialdehydu 2-hydroksy4-karboksy mukonowego, 9.EC.4.2.1.?. - 10.EC.4.1.3.17 - aldolaza 2-okso-4-hydroksy-4-karboksyadypinowa; 11.EC.3.7.1.9. - hydrolaza semialdehydu 2-hydroksymukonowego; 12.EC.4.2.1.? - 13.EC.4.1.3.17. - aldolaza 2-okso-4-hydroksy-4-metyloglutarowa
Rysunek 4. Schemat rozszczepienia pierścienia aromatycznego typu „meta”
6.3. biodegradacja węglowodorów aromatycznych w warunkach beztlenowych
Zwiazki aromatyczne są również degradowane beztlenowo, jednak dotychczasowa wiedza na ten temat jest niewielka, ponieważ w ostatnich latach większą uwagę poświęcono beztlenowej degradacji monoaromatycznych i policykliczych węglowodorów [Parales R.E. 2000, Rooney-Varga J.N. 1999, Coates J.D. 1997, Żiriakow W.G. 1981]. Warto zauważyć, iż węglowodory wielopierścieniowe mogą być degradowane w warunkach nie całkowicie beztlenowych, bowiem badano ich wykorzystanie przez mikroorganizmy w obecności związków azotowych jako akceptorów elektronów [ Coates J.D. 1997].
We wstępnych etapach rozszczepienia pierścienia aromatycznego bierze udział tlen cząsteczkowy. Jeszcze niedawno sądzono, że benzen, kwas benzoesowy, kwas 4-hydroksylobenzoesowy i wiele innych związków nie mogą być degradowane w warunkach beztlenowych. Okazało się to jednak nie prawdziwe, ponieważ fenol, kwas benzoesowy, czy nawet alkohol benzylowy są atakowane również w warunkach beztlenowych. Warunkiem jest obecność takich akceptorów wodoru, jak azotan, siarczan, CO2, które pozwalają na oddychanie beztlenowe, lub dostęp światła. Zjawiska takie obserwowano od lat. Już w 1932 oku Van Niel [Schlegel H.G. 2003] w ramach swoich badań wyizolował z hodowli w obecności benzenu i wystawionej na działanie światła szczep Rhodopseudomonas palustris, który degradował kwas benzoesowy do kwasu pimelinowego.
Od tego czasu stwierdzono, że we wczesnych etapach aktywacji benzoesanu uczestniczy koenzym-A i powstaje związek pośredni (benzyloCoA), który ulega hydratacji do pochodnej cykloheksanu, a następnie degradacji z uwolnieniem acetyloCoA. Beztlenowy rozkład podstawionego benzenu jest szlakiem reduktywnym [Schlegel H.G.. 2003]. Van Niel [Rehm J. 1996] zaproponował następujący szlak biodegradacji kwasu benzoesowego (rys.5): podczas, którego w pierwszym etapie następuje redukcja pierścienia benzenowego przez przyłączenie 3 cząsteczek wodoru, dalej zachodzi β-oksydacja. Rhodopseudomonas palustris degraduje kwas benzoesowy do kwasu pimelinowego, natomiast Moraxella sp. rozkłada ten sam substrat do kwasu adypinowego.
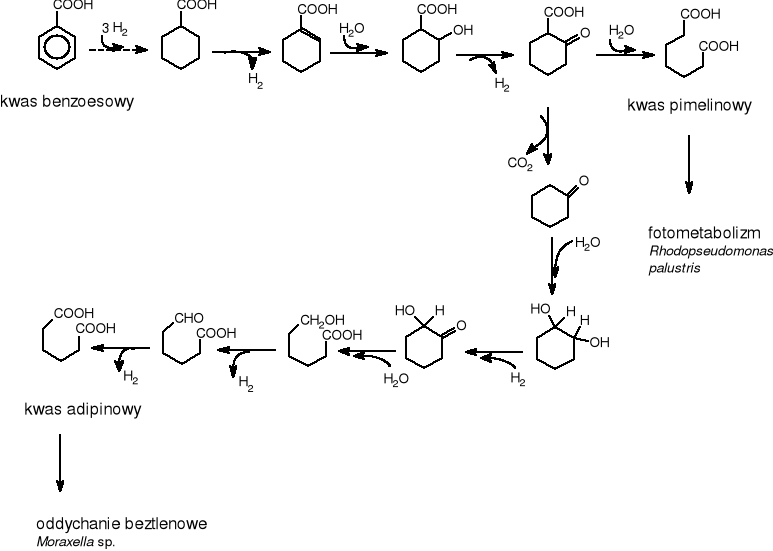
Rysunek 5 Beztlenowa biodegradacja kwasu benzoesowego przez Rhodopseudomonas palustris i Moraxella sp.
Tarvin i Buswell [Rehm J. 1996] w 1934 roku opisali degradacje związków aromatycznych podczas metanowej fermentacji. Należy pamiętać, że badali oni tylko rozerwanie pierścienia benzenu w warunkach beztlenowych wobec egzystujacych szczepów bakteryjnych. Rozerwanie pierścienia najprawdopodobniej miało miejsce podczas fotometabolizmu - fermentacji metanowej. Proces ten polega na redukcji jądra benzenu, a następnie rozerwaniu pierścienia w wyniku hydrolizy, co prowadzi do powstania kwasów o budowie łańcuchowej.
6.4. biodegradacja policyklicznych węglowodorów aromatycznych na drodze metabolizmu mikrobiologicznyego
Bakterie, grzyby i algi odgrywają znaczącą rolę w przemianie policyklicznych węglowodorów aromatycznych w środowiskach wodnych i lądowych. Gibson [Carnigilia C.E. 1984] stworzył fundamenty ogólnych zasad metabolizmu węglowodorów aromatycznych przez bakterie. W 1968 roku wyizolował on szczep bakterii Pseudomonas putidia mającej zdolność do wzrostu wobec etylobenzenu jako jedynego źródła węgla i energii, szczep ten mógł rozwijać się również na benzenie i toluenie. Dalsze badania przeprowadzone na szczepie Pseudomonas putidia 39/D rosnącym na glukozie wobec benzenu wykazały zgromadzenie w komórkach bakteryjnych cis-1,2-dihydroksy-1,2-dihydrodiolu. Zastosowanie O18 dowiodło, że oba atomy tlenu w produkcie przemiany benzenu (cis - dihydrodiolu) pochodzą z tlenu cząsteczkowego. Bakterie wstępnie utleniają węglowodory do cis-dihydrodioli dzięki wieloskładnikowemu systemowi dioksygenaz, następnie dzięki dehydrogenazie cis-dihydrodiolowej do pochodnej pozbawionej grup hydroksylowych. Dalsze utlenienie prowadzi do wytworzenia katecholi, które są substratami dla innych dioksygenaz rozszczepiających pierścień aromatyczny. Rozszczepienie pierścienia następuje zgodnie z zasadami podanymi w punkcie 2.5.2.
W przeciwieństwie do bakterii grzyby utleniają węglowodory aromatyczne wykorzystując system monooksygenaz sprzężonych z cytochromem P-450 na drodze reakcji epoksydowych do trans - dihydrodioli [Carnigilia C.E. 1984, Mueler J.G. 1996]. Metabolizm biodegradacji policyklicznych węglowodorów aromatycznych przez organizmy prokariotyczne i eukariotyczne przedstawia rys.6
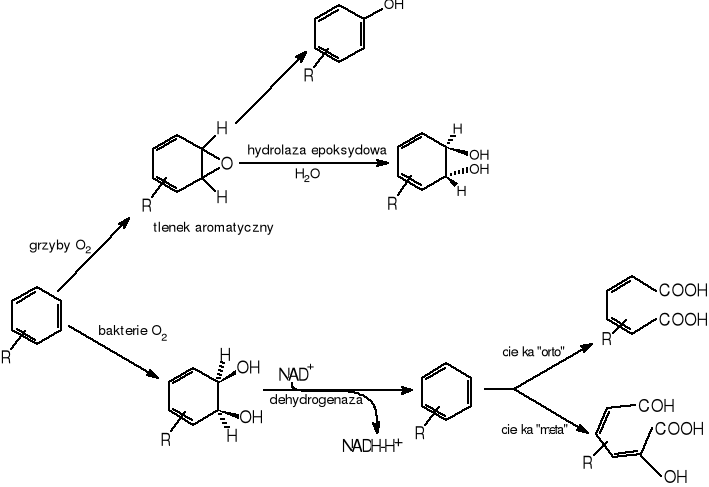
Rysunek 6 Mechanizm biodegradacji aromatycznych węglowodorów policyklicznych przez mikroorganizmy [11]
7.MECHANZMY BIODEGRADACJI WĘGLOWODORÓW AROMATYCZNYCH
7.1. węglowodory aromatyczne jednopierścieniowe
BENZEN
Benzen to najprostszy węglowodór aromatyczny. Jest bezbarwną cieczą o charakterystycznym zapachu. Związek prawie nierozpuszczalny w wodzie, natomiast nieorganicznie rozpuszczalny w alkoholu, eterze. Jest łatwopalny i toksyczny. Benzen otrzymuje się z lekkich frakcji smoły pogazowej, z niektórych gatunków ropy naftoweji i z produktów przerobu benzyn, stanowiących obecnie główne jego źródło. Stosowany jest w synyezie chemicznej jako surowiec,rozpuszczalnik i jako składnik paliw silnikowych.
Inicjacja biodegradacji benzenu zachodzi pod wpływem 1,2-dioksygenazy benzenowej, które wprowadza w jego strukturę całą cząsteczkę tlenu. Mechanizm działania enzymu nie jest do końca wyjaśniony. Sądzi się, że w swej cząsteczce dioksygenazy zawierają jon metalu, który powoduje aktywację cząsteczki tlenu i substratu, co umożliwia połączenie obu substancji. W wyniku czego benzen ulega hydroksylacji, z jednoczesną utratą właściwości aromatycznych, do cis-1,2-dihydro-1,2-dihydroksybenzenu, który następnie ulega dehydratacji (ponowna aromatyzacja) do katecholu. Powstały katechol ulega dalszej degradacji szlakiem intradiolowym „orto” lub ekstradiolowym „meta”, które zostały opisane w pkt. 6.3. i 6.4.
Inicjację mirobilogicznej degradacji benzenu przez 1,2-dioksygenazę benzenową pokazuje rys.7.
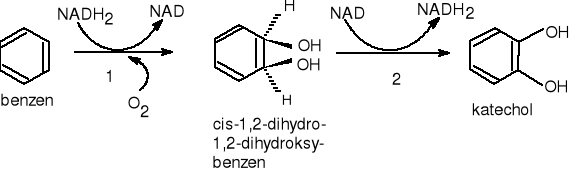
gdzie: 1.EC.1.14.12.3. - 1,2-dioksygenaza benzenowa
2.EC.1.3.1.19. - dehydrogenaza cis-dihydrokatecholowa
Rysunek 7 Degradacja benzenu przy udziale dioksygenaz
TOLUEN
Jest homologiem benzenu. Związek ten to bezbarwna ciecz o charakterystycznym zapachu, lżejsza od wody. Nierozpuszczalny w wodzie ale dobrze rozpuszczalny w alkoholu, benzenie i eterze. Otrzymywany jest w wyniku katalitycznego reformowania ropy naftowej lub frakcyjnej destylacji smoły pogazowej. Znajduje zastosowanie jako surowiec i rozpuszczalnik w wielu dziedzinach przemysłu organicznego, farmaceutycznego, tworzyw sztucznych, do produkcji trotylu, barwników, detergentów i substancji zapachowych oraz jako składnik wysokooktanowych paliw lotniczych.
Związk ten ulega wstępnej degradacji w trojaki sposób[Oh D.J. 2001, Weiner J.M. 1998]. Po pierwsze (1), na drodze dihydroksylacji pierścienia, podobnie jak beenzen. Reakcję tą katalizuje dioksygenaza toluenowa, która składa się z trzech białkowych komponentów. Dwa elektrony ze zredukwanego NADH2 są przenoszone na flawoproteinę, którą nazwano reduktazą ferrodoxinTOL. Dalej te dwa elektrony są przenoszone ze zredukowanej flawoproteiny na małe żelazowo-siarkowe białko- FERRODOXINTOL, które z kolei redukuje końcowy składnik dioksygenazy: duże diżelazowo-siarkowe białko nazwane ISPTOL. Ten ostatni element systemu w zredukowanej formie wykazuje zdolność do aktywowania tlenu cząsteczkowego i wbudowywania go do pierścienia toluenu przy 2 i 3 atomie węgla.
Rysunek 8 Budowa dioksygenazy toluenowej
Drugi sposób inicjacji degradacji toluenu (2) oparty jest na ataku monooksygenazy metanowej (EC 1.14.13.25) na grupę metylową łańcucha boczngo.
Trzecia możliwość poznana w trzech wariantach (3,4,5) to przyłączenie jednego atomu tlenu cząsteczkowego do któregoś z węgli pierścienia przez odpowiednie monooksygenazy w wyniku tego pierścień toluenu ulega hydroksylacji. Drugi atom tlenu jest redukowany do wody, a jako donory wodoru mogą służyć nukleotydy pirydynowe. Na rysunku 9 przedstawiono poznane dotychczas drogi mikrobiologicznej degradacji toluenu w warunkach tlenowych [Schlegel H.G. 2003,Carnigilia C.E. 1984, Rehm J. 1996].
Rysunek 9 Schemat wstępnych przekształceń toluenu
KSYLEN
Ksylen jest łatwo palną cieczą o charakterystycznym zapachu, nie mieszajacą się z wodą. Razem z benzenem, toluenem i etylobenzenem wchodzi w skład benzyn i zanieczyszcza wody podziemne. Możemy wyróżnuić trzy jego izomery: o-, m- i p-ksylen. Każdy z tych związków ulega degradacji w odmienny sposób [Oh D.J. 2001, Hyatt B.A 2001, Jeon M. 2001].
o-ksylen jest głównie używany do produkcji bezwodnika ftalowego, a niewielkie jego ilości używane są jako rozpuszczalnik do farb.
m- ksylen znaleziony został w paliwach kopalnych i używany jest jako rozpuszczalnik. Do atmosfery dostaje się w wyniku spalania benzyn, z kolei wycieki benzyn i olejów ropy naftowej do gleby powodują jej skażenie.
Każdy z tych izomerów ulega degradacji w odmienny sposób [Oh D.J.2001, Hyatt B.A. 2001, Jeon M. 2001] ale do tego samego zwiazku pośredniego jakim jest 3-metylokatechol
(rys. 10).
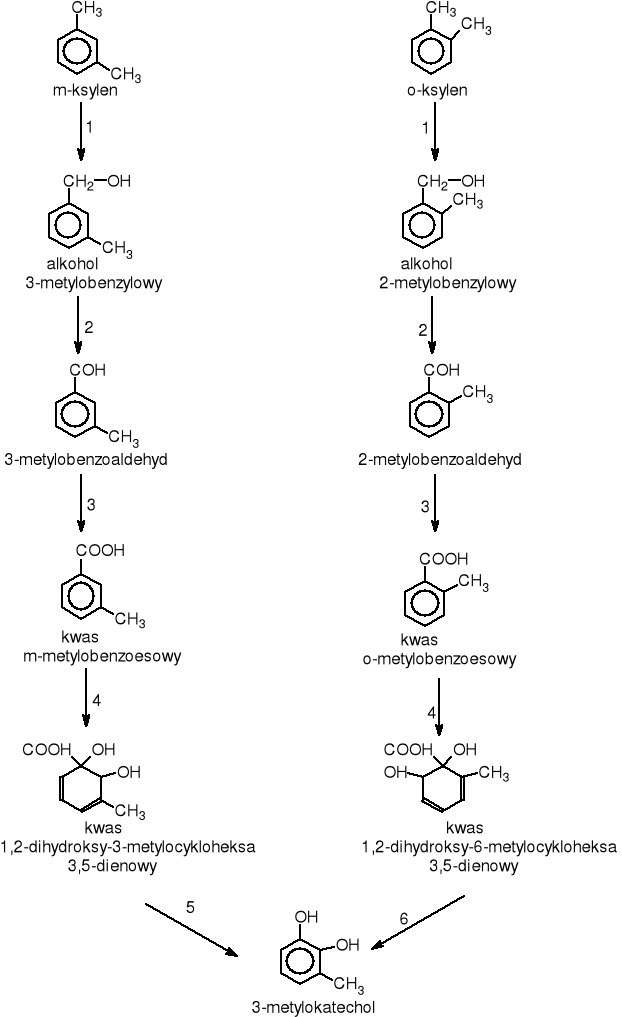
gdzie: 1.EC.1.14.13.?. - monooksygenaza ksylenowa, 2.EC.1.1.1.90. - dehydrogenaza benzyloalkoholowa, 3.EC.1.2.1.7. - dehydrogenaza benzyloaldehydowa, 4EC.1.14.12.? - dioksygenaza kwasu m-metylobenzoesowego, 5.EC.1.3.1.59. - dehydrogenaza kwasu 1,2-dihydroksy 3-metyloheksa-3,5-dienowego, 6.EC.1.3.1.68. - dehydrogenaza kwasu 1,2-dihydroksy 6-metylocykloheksa 3,5-dienowego
Rysunek 10 Szlak biodegradacji o- i m-ksylenu
p-ksylen otrzymywany jest z ropy naftowej w Stanach Zjednoczonych, wykorzystuje się je do produkcji kwasu tereftalowego (kwas benzeno 1,4-dikarboksylowy), który z kolei używany jesy do produkcji włókna poliestrowego, żywic, lakierów i filmów.
Schemat degradacji p-ksylenu przedstawia rys. 11.
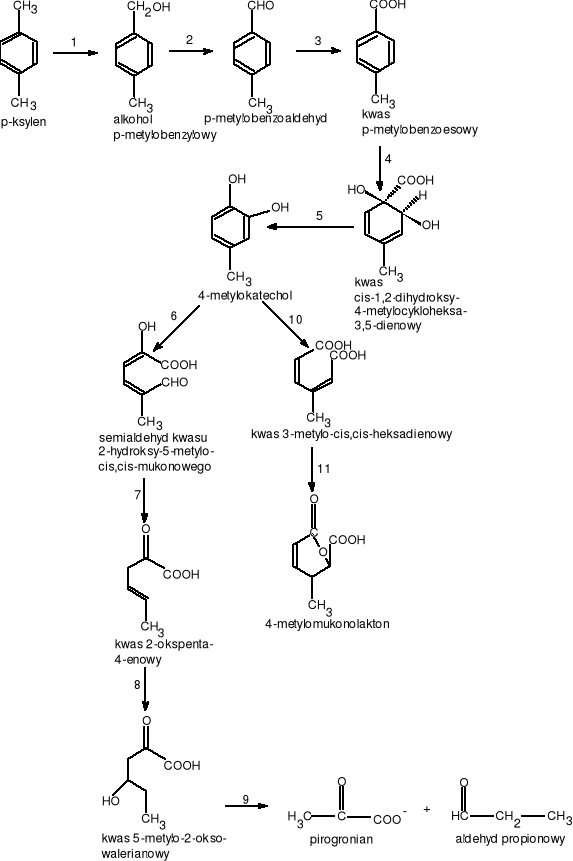
gdzie:1.EC.1.14.13.? - monooksygenaza ksylenowa, 2.EC.1.1.1.90. - dehydrogenaza benzyloalkoholowa, 3.EC.1.2.1.7. - dehydrogenaza benzyloaldehydowa, 4.EC.1.14.12.10. - 1,2 -dioksygenaza kwasu benzoesowego, 5.EC.1.3.1.67. - dehydrogenaza kwasu 1,2-dihydroksy 4-metylocykloheksa-3,5-dienowego, 6.EC.1.13.11.2. - 2,3-dioksygenaza katecholowa, 7.EC.3.7.1.9. hydrolaza semialdehydu 2-hydroksymukonowego, 8.EC.4.2.1.80. - hydrataza kwasu 2-oksopenta -4-enowego, 9.EC.4.1.2.18. -aldolasa 4-hydroksy - oksowalerianowa, 10.EC.1.13.11.1. - 1,2-dioksygenaza katecholowa, 11.EC.5.5.1.1. cukloizomeraza kwasu mukonowego
Rysunek 11 Schemat degradacji p-ksylenu
7.2. węglowodory aromatyczne o pierścieniu skondensowanym
naftalen
Naftalen to węglowodór o dwóch skondenowanych pierścieniach benzenowych. Występuje w postaci białych krystalicznych płatków. Występuje w smole węglowej, ropie naftowej i olejkach eterycznych, jest domieszką gazu świetlnego, zwiększającą jego siłę świecenia. Ma charakterystyczny zapach. Jest stosowany do syntezy różnych chemikaliów, barwników ftaleinowych, indygo, środków owadobójczych i wybuchowych, rozpuszczalników, żywic syntetycznych i garbników.
Naftalen oraz naftalen z podstawnikami alkilowymi są najbardziej toksycznymi składnikami frakcji olejowych rozpuszczalnych w wodzie (Boylan [Carnigilia C.E. 1984] i Tripp [Carnigilia C.E. 1984] 1971). Związek ten został sklasyfikowany przez Agencje Ochrony Środowiska Stanów Zjednoczonych (EPA) jako „priority toxic polutant” czyli „priorytetowy toksyczny polutant” [US-EPA 1980, 1986].
Po raz pierwszy szlak metaboliczny oraz reakcje enzymatyczne prowadzące do rozkładu naftalenu zostały opisane w 1964 roku. Najnowsze badania wykazały, że bakterie wstępnie utleniają naftalen przez włączenie dwu atomów tlenu cząsteczkowego do pierścienia aromatycznego z wytworzeniem cis-1,2-dihydroksy-1,2- dihdronaftalenu (Cattevall [Carnigilia C.E. 1984] i Jerina [Carnigilia C.E. 1984] 1971). Drogę degradacji naftalenu u bakterii pokazano na rys.12
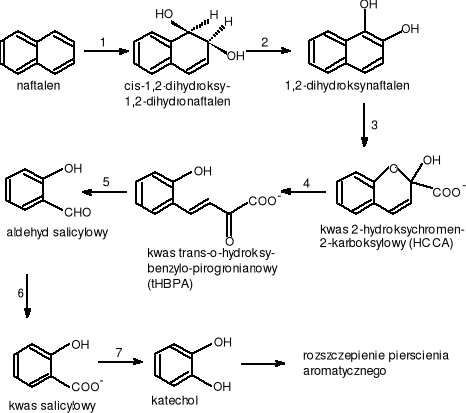
gdzie: 1.EC.1.14.12.12. - 1,2-dioksygenaza naftalenowa; 2. EC.1.3.1.29. - dehydrogenaza cis-1,2-dihydroksy-1,2-dihydronaftalenu; 3.EC.1.13.?.?. - dioksygenaza 1,2-dihydroksynaftalenu; 4.EC.5.3.99.?. - izomeraza kwasu 2-hydroksychromen-2-karboksylowego; 5.EC.4.2.1.?. - aldolaza kwasu trans-o-hydroksybenzylopirogronianowego (tHBPA); 6.EC.1.2.1.65. - dehydrogenaza aldehydu salicylowego; 7.EC.1.14.13.1. - hydroksylaza kwasu salicylowego.
Rysunek 12 Drogi biodegradacji naftalenu przez bakterie
W 1975 roku Jeffrey [Carnigilia C.E. 1984] sprecyzował przestrzenną formę powstającego cis-1,2-dihydroksy-1,2-dihydronaftalenu jako formę (+)(1R,2S), natomiast Ensley [Carnigilia C.E. 1984] w 1982 roku scharakteryzował dioksygenazę naftalenową wyizolowaną ze szczepu Pseudomonas sp.NCIB 9816 (rys.13). Enzym ten jest odpowiedzialny za wprowadzenie cząsteczki tlenu i wytworzenie (+) cis -(1R,2S) -1,2 - dihydroksy - 1,2- dihydronaftalenu. Enzym ten jest bardzo niestabilny i musi być szybko oczyszczany w obecności ditiotheritolu (mieszanina 10% v/v etanolu i 10% v/v glicerolu w buforze trichydrochlorowym ) dla zapewnienia aktywności enzymatycznej otrzymanego preparatu (Ensley w 1982r [Carnigilia C.E. 1984]).
Rysunek13 Schemat budowy bakteryjnej dioksygenay naftalenowej
Dalszym krokiem utlenianie naftalenu przez bakterie jest przemiana cis-1,2-dihydroksy-1,2-dihydronaftalenu w 1,2-dihydroksynaftalen. Reakcja ta jest katalizowana przez dehydroksynazę (+) - cis - dihydrodiolową naftalenu zależną od NAD, jako akceptora elektronów (Patel i Gibson w 1974r [Carnigilia C.E. 1984]). Dehydrogenaza ta jest wysoce stereoselektywnym enzymem metabolizującym tylko substraty typy „cis”. Następnymi etapami przemiany naftalenu jest przekształcenie cis-1,2- dihydrohsynaftalenu do cis-2'-hydroksybenzylopirogronianu. Ten z kolei przez serię przemian enzymatycznych przy udziale dioksygenaz przechodzi do salicylanu i pirogronianu. Salicylan dzięki hydroksylazie salicylanowej utleniany jest do katecholu, który podlega rozszczepieniu pierścienia aromatycznego na drodze „orto” lub „meta”. Prowadzono również wiele badań nad metabolizmem naftalenu przez grzyby. Okazało się, że grzyby są wstanie degradować naftalen ale zupełnie inaczej niż miało to miejsce u bakterii. W odróżnieniu od bakterii grzyby przyłączają tylko jeden atom tlenu cząsteczkowego do naftalenu przy pomocy monooksygenazy zależnej od cylochromu P-450. W wyniku tej reakcji powstaje 1,2 -tlenek naftalenu (rys. 14).
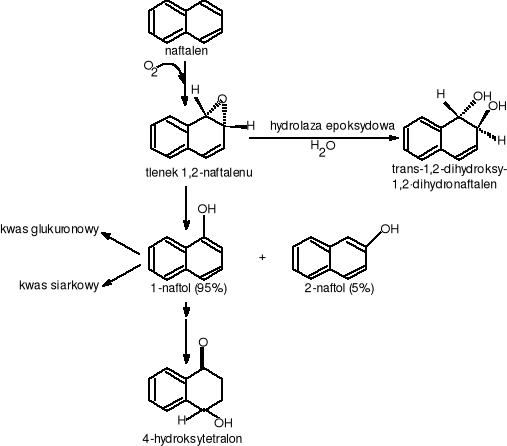
Rysunek 14 Drogi biodegradacji naftalenu przez grzyby
Utworzony tlenek aromatyczny jest formą bardzo nietrwałą. Może on podlegać:
reakcji głównej (przegrupowaniu elektronów i utworzeniu 1- naftolu);
reakcji pobocznej z wytworzeniem 2 - naftolu dzięki istnieniu mechanizmu przesunięcia NIH;
reakcji enzymatycznego uwodnienia katalizowanego przez hydroksylaę epoksydową prowadzącą do utworzenia (+) trans -(1S.2S) - dihydroksy - 1,2- dihydronaftalenu (Cerniglia i Gibson w 1978 i 1983 [Carnigilia C.E. 1984]).
Grzyby wykazują również zdolność do tworzenia glukuronowych i siarkowych soli naftolowych węglowodorów aromatycznych. W momencie odkrycia toksycznego charakteru 1-naftolu zaczęto baczniej interesować się owymi solami, sądząc, że takie połączenia są ważne w odtruwaniu i eliminacji ksenobiotyków przez grzyby.
W 1998 roku Stapleton stwierdził, że konsorcjum grzybów i bakterii mogło by mineralizować naftalen w kwasowych warunkach (pH≠3). Niestety tylko pojedyńcze przykłady takiego konsorcjum wykazały niskie poziomy mineralizacji naftalenu.
FENANTREN
Znane są również wiadomości dotyczące metabolizmu fenantrenu przez mikroflorę [Ortega-Calvo J.J. 1998]. Jest to trzypierścieniowy węglowodór aromatyczny. Wraz z antracenem jest szeroko rozpowszechniony w środowisku jako rezultat procesów pirolitycznych oraz gazyfikacji węgla i procesów skraplania (Blumer w 1976 [Carnigilia C.E. 1984]). Chociaz fenantren nie posiada cech mutagennych i rakotwórczych to jest toksyczny dla małżów, skorupiaków i ryb. Jest uważany za zwiazek modelowy w badaniach nad degradacją policyklicznych węglowodorów aromatycznych w środowisku. Fenantren jest najprostszym węglowodorem posiadającym niszę (region zatokowy) oraz obszar „K”. Nisza to zawada przestrzenna powstająca w wyniku połączenia pierścieni benzenowych w sposób kątowy. W fenantrenie obszar ten powstaje między 4 a 5 atomem węgla rys. 15. Natomiast obszar „K” to wiązanie podwójne o wysokiej gęstości elektronowej.
Fenantren jest metabolizowany przez system monooksygenazy działającej z cytochromem P-450 do trans 1,2-, 3.4-, 9,10-dihydroli fenantrenowych. Degradacja fenantrenu jest zapoczątkowana przez podwójne hydroksylowanie prowadzące do powstania cis-3,4-dihydroksy-3,4-dihydrofenantrenu, który dalej jest przekształcany enzymatycznie do 3,4-dihydroksyfenantrenu i następnie do kwasu 1-hydroksy-2-naftalenowego. To jest ogólny szlak degradacji fenantrenu. W dalszym etapie kwas 1-hydroksy-2-naftalenowy może zostać zdegradowany na dwa sposoby. Pierwszy sposób to utlenienie do 1,2-dihydroksynaftalenu, który ulega rozszczepieniu do kwasu salicylowego [Evans, Gibson, Subramanian 1984]. Kwas salicylowy może dalej zostać zdegradowany do katecholu. W innym szlaku kwas 1-hydroksy-2-naftalenowy ulega rozszczepieniu do kwasu o-ftalowego i kwasu protokatechowego, który ostatecznie prowadzi do cyklu TCA [Ghosh i Mishara 1983, Houghton i Shanley 1994, Kiyohara i Nagao 1978, Kiyohara 1976].
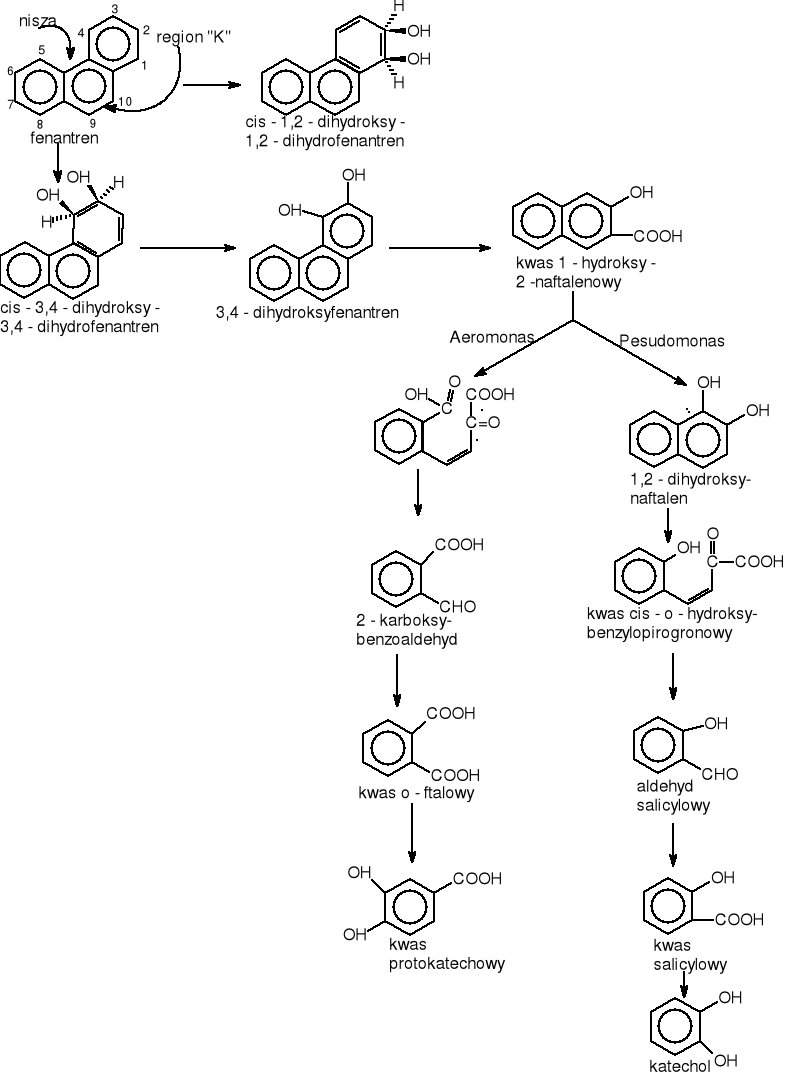
Rysunek 15 Różne drogi utleniania fenantrenu przez bakterie
Prowadzone doświadczenia nad utlenianiem fenantrenu przez grzyby wykazały, iż inicjacja metaboliczna przebiega inaczej niż w przypadku bakterii rys.16.Odbywa się ona przy pomocy monooksygenazy i hydrolazy epoksydowej.
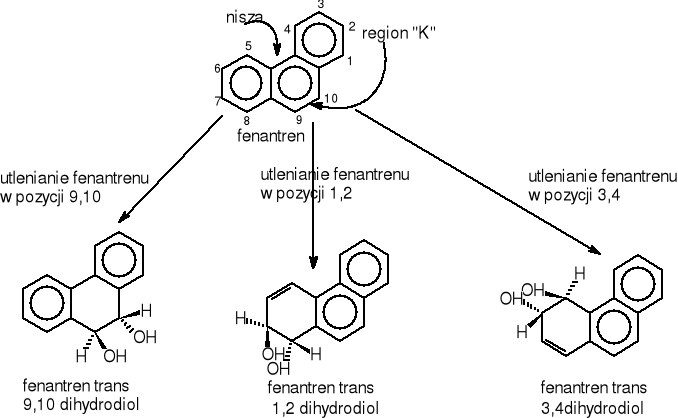
Rysunek 16 Reakcje inicjujące proces biodegradacji fenantrenu przez grzyby
ANTRACEN
Antracen jest węglowodorem aromatycznym o trzech pierścieniach skondensowanych. Jest to substancja krystaliczna, nierozpuszczalna w wodzie, dobrze rozpuszczalna w benzenie na gorąco. Pod względem własności chemicznych jest podobny do naftalenu. Występuje w oleju antracenowym, który stanowi 5% frakcję smoły węglowej. Wykorzystywany jest do produkcji barwników antrachinonowych oraz jako luminofor w licznikach scyntylacyjnych.
Antracen może być degradowany przez bakterie z grupy Pseudomonas rys.17
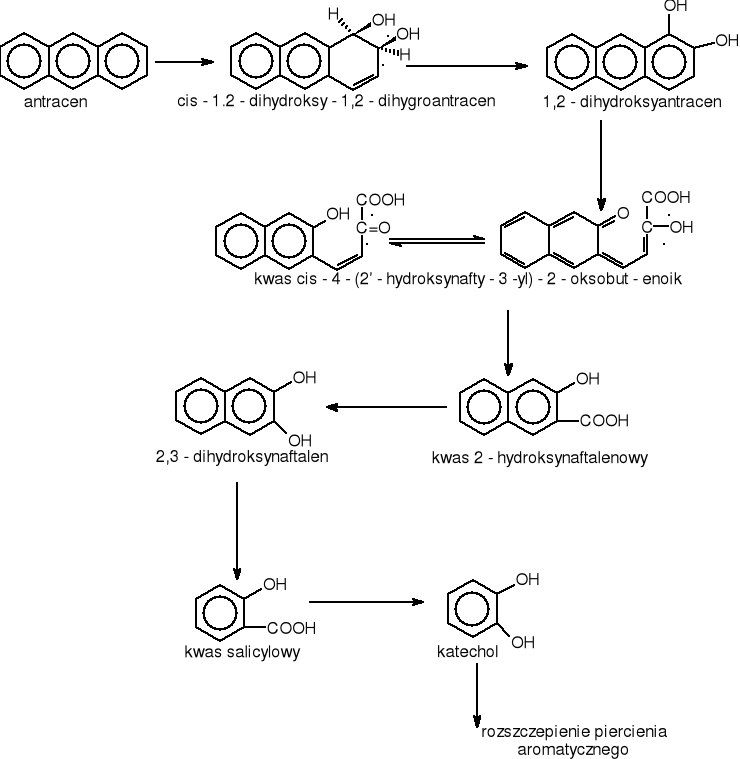
Rysunek 17 Drogi utleniania antracenu przez bakterie
Prowadzono również wiele badań nad degradacją antracenu przez grzyby. Otrzymane wyniki pozwoliły stwierdzić fakt, iż wstępne przekształcenie antracenu przebiega zgoła odmiennie niż to miało miejsce u bakterii rys.18
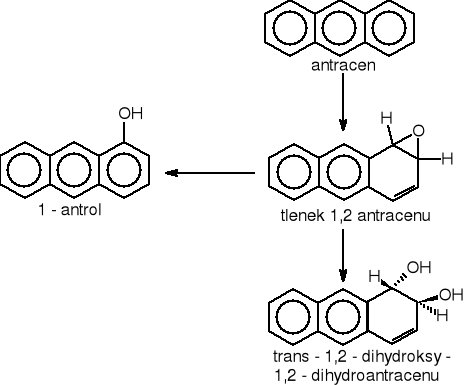
Rysunek 18 Droga wstępnej degradacji antracenu przez grzyby
PIREN
Jest to węglowodór czteropierścieniowy, odkryty przez Greabego w 1871 roku, w smole węglowej, skąd otrzymuje się go w ograniczonych ilościach. Otrzymywany jest podczas produkcji benzyny i innych procesów spalania. Piren nie jest związkiem rakotwórczym ale został włączony w bakteryjny kometabolizm benzo[a]pienu, który ma silne właściwości kancerogenne.
Piren jest jednym z najczęściej występujących wielopierścieniowych węglowodorów aromatycznych w środowisku. Znajduje się jako jeden z głównych związków zanieczyszczających na liście Agencji Ochrony Środowiska Stanów Zjednoczonych [Keith i Telliard 1979].
Początkowe produkty degradacji pirenu i jego droga kataboliczna nie zostały jak dotąd do końca określone. Bakterie z grupy Mycobacterium posiadają zdolność do utlizacji pirenu jako jedynego źródła węgla i energii. Mycobacteium flavescenes wyhodowany na pirenie atakuje go w pozycji 4,5, przypuszczalnie przy udziale dioksygenazy i tworzy cis-4,5-dihydroksy-4,5-dihydropiren. Nastepnie powstały 4,5-dihydroksypiren jest degradowany do kwasu fenantreno-4,5-dikarboksylowego, który ulega dekarboksylacji do kwasu fenantreno-4-karboksylowego. Końcowym produktem degradacji pirenu jest 3,4-dihydroksyfenantren.
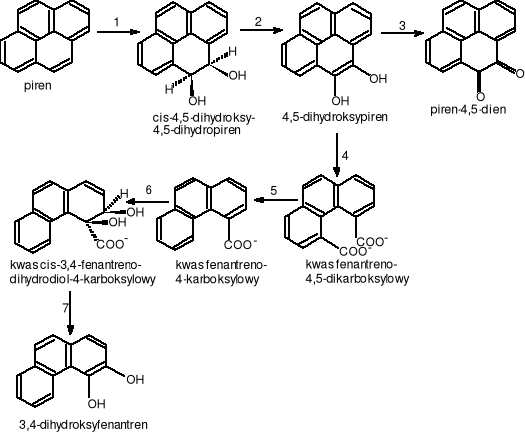
gdzie:
1.EC.1.13.11.?. - dioksygenaza pirenu; 2.EC.1.3.?.?. - dehydrogenaza cis-4,5-dihydroksy-4,5-dihydropirenu; 3. autooksydacja (nie enzymatyczna); 4.EC.1.13.11.?. -dioksygenaza 4,5-dihydroksypirenu; 5.EC.4.1.1.?. - dekarboksylaza kwasu fenantreno-4,5-dikarboksylowego; 6.EC.1.13.11.?. - dioksygenaza kwasu fenantreno-4-karboksylowego; 7.EC.1.3.?.?. - dehydrogenaza kwasu cis-3,4-fenantrnodihydrodiol-4-karrboksylowego
Rysunek 19 Droga biodegradacji pirenu przez bakterie
Piren może być również degradowany przez grzyby, a dokładnie grzyby białej zgnilizny, gdzie proces ten jest zainicjowany przez pozakomórkowe enzymy ligniny [Hammel 1986]. W konsekwencji , częściowo utleniony piren, staje się dostępny w pozakomórkowych przestrzeniach i dalej degradowany jest przez mikroorganizmy glebowe.
7.3. BiodegradacjA węglowodorów Ropopochodnych
1. Drobnoustroje degradujące węglowodory
W świecie mikroorganizmów istnieje duża grupa drobnoustrojów posiadająca zdolności do rozkładu węglowodorów. W grupie tej występują zarówno bakterie, drożdże, jak i grzyby strzępkowe. Tabela 3 zawiera przegląd drobnoustrojów degradujących węglowodory [13].
Tabela 3. Drobnoustroje utylizujące węglowodory alifatyczne
Bakterie |
Drożdże |
Grzyby nitkowate |
Acetobacter |
Candida |
Aspergillus |
Acinetobacter |
Cryptococcus |
Cladosporium |
Actinomyces |
Debaryomyces |
Corollaspora |
Alcaligenes |
Hansenula |
Dendryphiella |
Bacillus |
Pichia |
Glicladium |
Beneckea |
Rhodotorula |
Lulworthia |
Corynebacterium |
Sporobolomyces |
Penicillium |
Flavobacterium |
Torulopsis |
Varicospora |
Mycobacterium |
Trichosporon |
|
Nocardia |
|
|
Pseudomonas |
|
|
Xentomonas |
|
|
Spośród dużej grupy mikroorganizmów posiadających zdolności do degradacji węglowodorów, najlepiej zbadanymi są bakterie z rodzaju Pseudomonas, Acinetobacter i Bacillus. Przeprowadzone badania utylizacji różnego typu węglowodorów pozwoliły na wykrycie w ich komórkach enzymów odpowiedzialnych za prowadzenie tego procesu.
Wśród wszystkich drobnoustrojów asymilujących węglowodory wyróżnia się grupę organizmów utleniających metan. Są to tak zwane metylotrofy. Drobnoustroje te mogą wykorzystywać jako jedyne lub dodatkowe źródło węgla metanol, formaldehyd, kwas mrówkowy oraz metyloaminę, względnie związki wielowęglowe zawierające grupę metylową, lecz pozbawione wiązań C-C, takie jak: di- i trimetyloaminy czy aceton.
Metylotrofy dzielimy na dwie grupy:
metylotrofy bezwzględne - należą do nich rodzaje: Methylobacter, Methylococcus, Methylocystis, Methylomonas, Methylophilus, Methylosinus;
metylotrofy względne - jest to duża grupa drobnoustrojów, wykorzystujących do wzrostu różne związki organiczne, w tym również metanol. Właściwości te wykazują między innymi: bakterie z rodzaju Corynebacterium, Flavobacterium, Achromobacter, Arthrobacter, Bacillus, Brevibacterium, Hyphomicrobium, Micrococcus, Protominobacter, Vibrio i Pseudomonas; grzyby Gliocladium deliguescens, Paecilomyces varioti i Trichoderma lignorum; drożdże z rodzajów Candida, Hansenula, Kloeckera, Pichia i Torulosporis.
2. Asymilacja węglowodorów ze środowiska
Pobieranie węglowodorów ze środowiska przez mikroorganizmy może zachodzić w drodze:
bezpośredniego przyswajania węglowodorów rozpuszczonych w fazie wodnej;
utworzenia węglowodorowych makrokropli w środowisku wodnym;
utworzenia węglowodorowych mikrokropli (o średnicy mniejszej od średnicy komórki drobnoustroju)[13].
Przyswajanie węglowodorów o niskiej masie cząsteczkowej, takich jak etan czy propan, z fizycznego punktu widzenia nie stanowi większego problemu, ze względu na ich stosunkowo dużą rozpuszczalność w wodzie. Asymilacja węglowodorów cięższych, trudno rozpuszczalnych w wodzie, jest bardzo utrudniona. Przechodzenie tych węglowodorów z fazy hydrofobowej do fazy wodnej jest możliwe dzięki procesowi rozproszenia ich w formie kropli. Proces ten odbywa się dzięki produkcji biosurfaktantów, których synteza zwykle poprzedza proces biodegradacji węglowodorów. Związki o charakterze substancji powierzchniowo czynnych powodują zwiększenie rozpuszczalności węglowodorów w fazie wodnej oraz utworzenie emulsji węglowodorowej, a tym samym zwiększenie powierzchni między fazami. Przykładem biosurfaktanta produkowanego podczas asymilacji węglowodorów są 6,6-dimykolowe estry trehalozy wytwarzane przez bakterie z rodzaju: Corynebacterium, Nocardia, Mycobacterium, Rhodococcus.
Szybkość przyswajania węglowodorów zależy od [14]:
długości łańcucha węglowego,
stopnia jego nasycenia,
budowy cząsteczki.
Węglowodory alifatyczne są łatwiej degradowane niż aromatyczne; długołańcuchowe parafiny są chętniej rozkładane, niż krótkołańcuchowe; nasycone związki są łatwiej utylizowane niż nienasycone; związki o łańcuchach rozgałęzionych są trudniej degradowane od nierozgałęzionych. Do trudno przyswajalnych należą wielopierścieniowe węglowodory aromatyczne, chloropochodne bifenyli i inne.
Zdolność drobnoustrojów do biodegradacji węglowodorów uzależniona jest od funkcjonowania odpowiednich systemów ich transportu do komórki oraz od obecności takich enzymów, jak: oksygenazy, dehydrogenazy, hydrolazy, dekarboksylazy.
3. Warunki biodegradacji węglowodorów
Efektywność procesu biodegradacji związków ropopochodnych uzależniona jest od następujących parametrów:
Składniki pokarmowe
Podstawowym źródłem węgla i energii dla dużej grupy drobnoustrojów są związki organiczne, w tym także węglowodory. Są one częściowo wykorzystywane przez mikroorganizmy do budowy masy komórkowej. Pozostała część metabolizowana jest w niezbędną do wzrostu i rozmnażania energię. Oprócz źródła węgla drobnoustroje wymagają do wzrostu soli mineralnymi. Są to związki azotu, fosforu, siarki, wapnia i magnezu. Nieskażone ekosystemy zawierają wszystkie te składniki w określonych proporcjach, przez co warunkują harmonijny wzrost bytującej w nim mikroflory. W wyniku skażenia środowiska naturalnego substancjami ropopochodnymi zostaje naruszona równowaga między ilością węgla organicznego, azotu i fosforu. Mimo obfitości źródła węgla drobnoustroje nie są w stanie go wykorzystać, ponieważ w podłożu występuje niedobór azotu i fosforu uniemożliwiający prawidłowy rozwój drobnoustrojów. Dlatego niezbędne jest wzbogacenie środowiska w wymienione wcześniej pierwiastki.
Z badań przeprowadzonych przez Rasiaha [15] wynika, że najkorzystniejszy stosunek węgla do azotu wynosi 18:1. Bardzo istotna jest także postać, w jakiej dostarczane są do podłoża pierwiastki. Azot może być dodawany w postaci jonów NH4+ lub NO3-. Za najbardziej odpowiednie źródło azotu Rasiah uważa azotan (V) wapnia[15].
W celu przyspieszenia procesu utylizacji zanieczyszczeń zalecane jest dodawanie do skażonego środowiska fosforu. Fosfor i siarka są najłatwiej przyswajane przez mikroorganizmy w postaci jonów PO43- i SO42-.
Temperatura
Temperatura jest czynnikiem silnie wpływającym na procesy życiowe drobnoustrojów, na ich aktywność, a tym samym na intensywność biodegradacji węglowodorów. Optimum temperaturowe dla flory bakteryjnej degradującej węglowodory wynosi zwykle 22-30°C. W większości procesów mikrobiologicznych wykorzystywane są przede wszystkim drobnoustroje mezofilne.
pH środowiska
pH należy do podstawowych parametrów fizykochemicznych decydujących o rozwoju drobnoustrojów. Jony H+ i OH- odgrywają istotną rolę w regulacji metabolizmu. Wpływają na szybkość reakcji enzymatycznych, na stopień przepuszczalności błony komórkowej. Jony wodorotlenowe mogą hamować syntezę białek i aktywność oddechową komórek. Dlatego też znalezienie optymalnego pH jest niezwykle istotne.
Drożdże i grzyby degradujące węglowodory wymagają środowiska lekko kwaśnego, o pH 3,5-6,0. Natomiast bakterie posiadają swoje optimum pH w zakresie 6,0-7,5.
Natlenienie środowiska
Tlen należy do kluczowych czynników kształtujących warunki rozwoju drobnoustrojów. Wpływa na szybkość ich wzrostu, plon biomasy oraz na fizjologię komórek. W krańcowych przypadkach jego stężenie może decydować o całkowitym zahamowaniu wzrostu mikroorganizmów U bezwzględnych i względnych tlenowców tlen jest końcowym akceptorem elektronów w przemianach katabolicznych [16].
Utlenianie węglowodorów wymaga włączenia w kolejnych etapach metabolizmu dużych ilości tlenu[15][17]. Najczęściej drobnoustroje włączają tlen cząsteczkowy bezpośrednio, wiążąc go przy udziale oksygenaz. Tlen może być również pobierany z substratów nieorganicznych, tj. niektórych jonów (np. SO42-, NO3-) lub CO2 a nawet H2O.
Uwodnienie środowiska
Wszystkie drobnoustroje wymagają do swojego rozwoju środowiska zawierającego wodę. Różnią się jednak pomiędzy sobą pod względem wymagań co do jej zawartości w środowisku, a ściślej do jej dostępności. Aby możliwe było porównanie substancji stałych oraz roztworów wodnych pod względem dostępności zawartej w nich wody, stosuje się parametr zwany aktywnością wodną. Aktywność wodna charakteryzuje dostępność wody dla mikroflory i zależy ona od oddziaływań pomiędzy cząsteczkami wody a substancjami w niej rozpuszczonymi.
Większość drobnoustrojów wymaga do swojego rozwoju aktywności wodnej w zakresie 0,95-0,99. Generalnie drożdże i pleśnie wykazują znacznie większą tolerancję na odwodnienie środowiska w porównaniu do bakterii [16].
Obecność surfaktantów
Niektóre węglowodory (długołańcuchowe i policykliczne) są trudno rozpuszczalne w wodzie i w związku z tym ich szybkość biodegradacji jest znacznie ograniczona[18]. Drobnoustroje lepiej przyswajają węglowodory, jeśli zostanie zwiększona ich rozpuszczalność i rozproszenie w fazie wodnej.
Surfaktanty są związkami o charakterze hydrofilowo-hydrofobowym [19][20]. Działają na granicy faz i powodują zwiększenie stężenia związków hydrofobowych w fazie wodnej przez ich emulgację lub rozpuszczenie. Znane są surfaktanty wytwarzane syntetycznie lub na drodze biologicznej (biosurfaktanty). Biosurfaktanty mają dużą przewagę nad swoimi odpowiednikami syntetycznymi z uwagi na ich niską toksyczność, możliwość regulacji poziomu ich syntezy. Mają szeroki zakres działania jeśli chodzi o temperaturę, pH i zasolenie. Biosurfaktanty są zazwyczaj związkami o charakterze glikolipidów (trehalozolipid, ramnolipid, glukolipid, celobiozolipid), rzadziej lipopeptydów, glioprotein, fosfolipidów, kwasów tłuszczowych lub obojętnych lipidów. Wśród mikroorganizmów wykazujących zdolność do produkcji biosurfaktantów wymienia się bakterie z rodzaju: Pseudomonas, Acinetobacter, Bacillus, Rhodococcus, Torulopsis.
8. Enzymy biorące udział w procesie biodegradacji
węglowodorów
Wśród enzymów biorących udział w procesie biodegradacji węglowodorów na uwagę zasługują następujące klasy enzymów:
monooksygenazy;
dioksygenazy;
dehydrogenazy;
hydrolazy;
liazy.
monooksygenazy
Monooksygenazy wbudowują do substratu tylko jeden atom tlenu, drugi zaś ulega redukcji do wody. Niezbędna jest więc obecność dodatkowego donora elektronów lub kosubstratu. Rolę tę pełni najczęściej NADH lub NADPH. Działanie systemu monooksygenaz jest niezbędne do rozpoczęcia procesu degradacji, ich działanie jest pierwszym etapem utylizacji węglowodorów przez większość mikroorganizmów w celu przekształcenie ich w odpowiednie alkohole [Murray R.K. 1994].
Woods i Murrell [Watkinson R.J. 1990] w 1989 roku wykazali, że bakterie Rhodococcus rhodochrous utleniają alkany o łańcuchach od C2 do C8 przy wykorzystaniu oksygenaz współdziałających z cytochromem P-450. Występowanie tych systemów określono na podstawie badań spektrofotometrycznych, wykorzystując charakterystyczne widma odpowiednio formy utlenionej i zredukowanej. System monooksygenaz zależnych od cytochromu P-450 znaleziono ponadto u następujących mikroorganizmów:
Candida tropicalis obecny w mikrosomach system, składający się z: cytochromu P-450, reduktazy NADPH2-cytochromowej oraz fosfolipidów stabilnych w wysokich temperaturach;
Pseudomonas aeruginosa odpowiedzialny za oksydację n-heksadekanu do n-heksadekanolu. Powyższy system składa się z: niehemowych żelazo-protein, hemowych protein i flawoprotein o niskocząsteczkowych;
Cunninghamella elegans odpowiedzialny za degradację policyklicznych węglowodorów aromatycznych;
Agmenellum guadruplicatum odpowiedzialny również za degradację policyklicznych związków aromatycznych. [Carnigilia C.E. 1984]
Systemy monooksygenaz niezależnych od cytochromy P-540 są jeszcze mało zbadane. U szczepów Pseudomonas olevarus var putidia [Burry S.J. 1993] stwierdzono występowanie monooksygenaz zależnej od rubrydoksyn. Układ ten jest szeroko specyficzny, katalizuje nie tylko oksydację kwasów tłuszczowych o długich łańcuchach, ale także uczestniczy w epoksydowaniu 1-alkenów.
dioksygenazy
Dioksygenazy przyłączają całą cząsteczkę tlenu do węglowodorowego substratu. Mechanizm ich działania nie jest do końca wyjaśniony. Sądzi się, że w swej cząsteczce zawierają jon metalu, który powoduje aktywację cząsteczki tlenu i substratu, co umożliwia połączenie obu substancji. Dioksygenazy mogą zawierać kilka podjednostek, np.: system dioksygenazy naftalenowej, odpowiedzialny za wstępny etap degradacji naftalenu przez bakterie, zawiera trzy składniki białkowe. Są to: flawoproteina, żelazo-siarkowa proteina oraz dwużelazowo-dwusiarkowa ferrodoksyna. Składniki te są podobne do tych występujących w systemach dioksygenaz benzenowej i toluenowej [Schlegel H.G. 1996]. Według hipotez Leadbettera i Fostera przyłączenie tlenu może prowadzić do powstania pierwszo- lub drugorzędowych związków przejściowych, które dalej są redukowane do odpowiednich alkoholi [Ng T.K 1989].
dehydrogenazy
Dehydrogenazy katalizują odrywanie atomów wodoru od utlenianego substratu i przenoszą je na inne enzymy czy związki pośrednie, a nie mają zdolności przenoszenia elektronów bezpośrednio na tlen. Różnią się one rodzajem koenzymów, które są właściwymi chwytnikami atomów wodorów. Mogą nimi być: NAD+, NADP+, FMN, FAD lub liponian. Dehydrogenazy te wykazują swoistość zarówno do substratów, jak i do koenzymów. Dzięki możliwości przenoszenia atomów wodoru między dwoma substratami, jeden z nich ulega utlenieniu drugi redukcji. Ten typ reakcji wykorzystywany jest w warunkach beztlenowych [Kłyszejko-Stefanowicz L. 1982]. Dla degradacji węglowodorów obecność dehydrogenaz jest niezbędna. Pod wpływem tych enzymów I-rzędowe alkohole utleniane są do aldehydów i kwasów. Alkohole II-rzędowe do ketonów i estrów. Dehydrogenazy degradujące węglowodory zależne są od NAD. W niektórych gatunkach drożdży Candida wykryto zamiast dehydrogenazy zależnej od NAD oksydazę alkoholową. Fakt ten potwierdził: Blasig [Watkinson R.J. 1990] w 1988 roku prowadząc proces z udziałem drożdży Candida maltosa, Kemp [Watkinson R.J. 1990] w 1988 dla Candida tropicalis, Hommel[Watkinson R.J. 1990] i Ratledge[Watkinson R.J. 1990] w 1990 dla Candida bombicola.
hydrolazy
Do klasy hydrolaz zaliczamy enzymy katalizujące proces rozpadu substratu z udziałem cząsteczek wody. Hydrolazy stanowią bardzo dużą i szeroko rozpowszechnioną grupę enzymów. Znaczną ich część stanowią enzymy trawienne, występujące w dużym stężeniu w ślinie, w soku żołądkowym, trzustkowym i jelitowym. Spośród enzymów z klasy hydrolaz można wyróżnić następujące grupy: esterazy, glikozylohydrolazy, peptydazy. W biodegradacji węglowodorów największe znaczenie mają esterazy, które rozszczepiają wiązanie estrowe substratu, a jako produkty uzyskujemy alkohole i kwasy [Filipowicz B. 1973]. Dzięki hydrolazom zostają skrócone łańcuchy cząsteczek węglowodorów długołańcuchowych, które po dalszych przemianach mogą być włączane w szlaki metabolizmu centralnego.
liazy
Do klasy liaz zaliczamy enzymy, które niehydrolitycznie odrywają od substratów pewne grupy uwalniając podwójne wiązania bądź przyłączają pewne grupy do tych wiązań. Liazy jest to dość duża grupa enzymów katalizujących głównie procesy degradacji (procesy kataboliczne). Bardzo rozpowszechnionymi liazami mającymi duże znaczenie w procesach biodegradacji węglowodorów są karbo-liazy zwane dekarboksylazami. Łańcuchy aminokwasów, ketokwasów i innych działaniem dekarboksylaz skracane są o jeden atom węgla. Z grupy karboksylowej wyzwala się wówczas CO2. W roślinach i w komórkach drożdżowych stwierdzono obecność takich dekarboksylaz, dzięki którym w procesie biodegradacji węglowodorów przekształceniu ulegają ketokwasy do kwasów tłuszczowych [Filipowicz B. 1973].
9.PODSUMOWANIE
5.6.3.1. wpływ temperatury na wzrost, stopień degradacji i uzdolnienia emulgacyjne
Właściwości fizyczne węglowodorów, a tym samym i proces ich degradacji zależą od temperatury w jakiej się znajdują. Wysoka temperatura powoduje: wzrost parowania alkanów krótkołańcuchowych i węglowodorów aromatycznych o małej masie cząsteczkowej oraz zwiększa rozpuszczalność węglowodorów w wodzie, dzięki czemu stają się one bardziej dostępne dla bakterii. Skład gatunkowy drobnoustrojów i ich metabolizm zależne są również od temperatury. Z jej wzrostem zwiększa się aktywność działania enzymów również tych odpowiedzialnych za biodegradację węglowodorów. Niska temperatura z kolei zmniejsza rozpuszczalność węglowodorów, wydłuża czas aklimatyzacji drobnoustrojów i obniża szybkość działania enzymów [Boszczyk-Maleszka H. 2000]. Optymalna temperatura dla bakterii i grzybów zdolnych do rozkładu produktów ropopochodnych mieści się w granicach 22-34oC.
Temperatura wywiera również wpływ na proces degradacji węglowodorów. Przykładem tego mogą być wyniki uzyskane przez Liu i wsp. [Liu i wsp 1991] , którzy stwierdzili, że najlepszy efekt degradacji 2,4,6-trichlorofenolu przez szczep Azotobacter sp. Ponieważ zwiększenie temperatury z 30 do 370C powoduje prawie 2-krotnie większe odparowanie obu grup węglowodorów, a ich stopień zużycia wzrasta zaledwie o 9-11j% (węglowodory ogółem) i 17-19j% (węglowodory aromatyczne)nie ma większego sensu prowadzić proces degradacji w tej temperaturze. Pamiętać należy, iż większe odparowanie przyczynia się do przedostania się węglowodorów z podłoża do atmosfery.
5.8. zależności między składnikami stanowiącymi mieszaninę węglowodorową
W skażonym środowisku węglowodory aromatyczne rzadko występują jako pojedyncza grupa związków, spotkamy je obok innych węglowodorów cyklicznych i alifatycznych. Do tej pory mało wiadomo na temat degradacji węglowodorów w mieszaninie obok innych związków, a w szczególności o wpływie obecności określonych węglowodorów na zdolność do biodegradacji wybranego węglowodoru aromatycznego. Wiadomym jest, że niektóre związki podlegają degradacji przy udziale drobnoustrojów jedynie wówczas, gdy w środowisku obecne są inne wybrane substancje. Badania degradacji różnych węglowodorów wykazały, że istnieje wzajemny wpływ między węglowodorami w mieszaninie powodujący zmianę szybkości ich biodegradacji. W niniejszej pracy do zbadania zależności między omawianymi związkami użyto jednopierścieniowe węglowodory aromatyczne: benzen, toluen i ksylen.
Węglowodory aromatyczne są trudniej degradowane niż węglowodory alifatyczne, aczkolwiek występowanie węglowodorów alifatycznych wywiera znaczący wpływ na proces ich degradacji [Razak C.N.A. 1999]. Znanych jest wiele przykładów z literatury [Bayly R.C. 1988, Stapletion R.D. 1998, Buhler M. 1985, Oh Y.S. 1994], w których nie tylko węglowodory alifatyczne przyczyniają się do poprawy procesu degradacji węglowodorów aromatycznych. W literaturze [Buhler M. 1985, Chang M.K. 1993, Stapletion R.D. 1998] można znaleźć również wzmianki o oddziaływaniu na siebie prostych węglowodorów aromatycznych jakimi są: benzen, toluen i ksylenu.
6 PODSUMOWANIE WYNIKÓW
W wielu krajach świata poważnym problem jest wysoki poziom skażenia środowiska substancjami ropopochodnymi. Taki stan rzeczy wynika z intensywnego wydobycia surowca naturalnego jakim jest ropa naftowa oraz wykorzystania znacznych jej ilości, co związane jest z szybkim rozwojem przemysłowym wielu krajów na świecie. Skażeniu związkami ropopochodnymi ulegają woda a także gleba. Bardzo ważne jest opracowanie metod likwidacji tych zanieczyszczeń. Prowadzone są poszukiwania efektywnych metod oczyszczania skażonych terenów metodami zarówno fizyko - chemicznymi jak i biotechnologicznymi. Dotychczasowe doświadczenia wskazują, że zastosowanie metod biotechnologicznych jako ostatniego etapu oczyszczania tych środowisk stanowi najbardziej racjonalny sposób ich remediacji. Przedsięwzięcie to jest nie łatwe, wymaga ono bowiem ustalenia wielu parametrów procesu, na które składają się czynniki: biologiczne, chemiczne i techniczne. Często koniecznym jest użycie biopreparatów (zestawów drobnoustrojów charakteryzujących się specyficzną aktywnością do występującego skażenia). Mikroorganizmy wchodzące w jego skład współdziałają z autochtoniczną mikroflorą danego środowiska, zachowując swoją aktywność mimo, że warunki procesu często odbiegają od optymalnych (zbyt niskie temperatury, za wysokie stężenia ksenobiotyków, nieodpowiedni odczyn środowiska).
Według danych Światowej Organizacji Zdrowia spośród związków ropopochodnych najbardziej toksyczna i rakotwórcza jest grupa węglowodorów aromatycznych. Wśród węglowodorów jednopierścieniowych szczególnie BTEX (benzen, toluen, etylobenzen, o,m,p-ksylen) a wśród wielopierścieniowych szczególnie (benzo[a]piren).
W skażonym środowisku węglowodory aromatyczne rzadko występują jako pojedyncza grupa związków, spotkamy je obok innych węglowodorów cyklicznych i alifatycznych. Do tej pory mało wiadomo na temat degradacji węglowodorów w mieszaninie obok innych związków, a w szczególności o wpływie obecności określonych węglowodorów na zdolność do biodegradacji wybranego węglowodoru aromatycznego. Wiadomym jest, że niektóre związki podlegają degradacji przy udziale drobnoustrojów jedynie wówczas, gdy w środowisku obecne są inne wybrane substancje. Zjawisko to określane jest terminem kometabolizmu. Badania degradacji różnych węglowodorów wykazały, że istnieje wzajemny wpływ między węglowodorami w mieszaninie powodujący zmianę szybkości ich biodegradacji [Hubert C. 1999, Haigler B.E. 1993, Soli G. 1972, Bastianes L. 1997, Bauer J.E. 1988, Millette D. 1995]
Z uwagi na ogromną kancerogenność ksenobiotyków jakimi są węglowodory aromatyczne, a szczególnie BTX celem pracy było wyselekcjonowanie drobnoustrojów degradujących te związki w najwyższym stopniu. W trakcie prowadzonej selekcji napotkano na problem związany z dużym odparowaniem substratu aromatycznego (benzenu, toluenu, ksylenu). Z uwagi na powyższy fakt zakres pracy należało dodatkowo rozszerzyć o dobór sposobu hodowli mikroorganizmów na podłożu zawierającym lotne związki. W ramach pracy zbadano również zjawisko kometabolizmu występujące w mieszaninie BTX i podjęto nieśmiało próbę ustalenia drogi degradacji toluenu przez wytypowane szczepy bakteryjne. Poświecono również uwagę doborowi optymalnego składu ilościowego i jakościowego pożywki hodowlanej (źródło azotu, fosforu, łatwo przyswajalne źródła węgla, obecność makro i mikroelementów) oraz warunków procesu degradacji (pH, temperatura, natlenienie), dzięki którym uzyskuje się wysoki stopień degradacji węglowodorów aromatycznych. Nieco uwagi poświecone zostało również bioremediacji gleby skażonej węglowodorami oleju napędowego (w tym aromatycznymi) i podjęto próbę ustalenia formy biopreparatu, który najefektywniej usunie skażenie węglowodorowe z badanej gleby.
Do dalszych badań wykorzystano dwa szczepy: NS2 zidentyfikowany jako Bacillus sp. i R51 zidentyfikowany jako Ochrobactrum anthropi.
Z przeprowadzonych badań można wyciągnąć następujące wnioski:
Wiele z węglowodorów aromatycznych w naturalnych warunkach jest lotnych. Stwarza to poważne komplikacje podczas badań, zwłaszcza na etapie hodowli drobnoustrojów w podłożach z ich dodatkiem. Na podstawie przeprowadzonych badań (pkt. 5.2.) stwierdzono, iż problem ten nie jest łatwy do realizacji, wymaga zapewnienia jednakowych warunków podczas hodowli (zarówno sprzętowych jak i jednorodności samego podłoża). Z zastosowanych w badaniach układów dających pożądane efekty i dużą powtarzalność wyników godny uwagi pozostaje wariant z dodatkiem oleju napędowego jako związku rozpuszczającego substancję lotną. W tym przypadku hodowli zapewnione jest „zatrzymanie” rozpuszczalnika w podłożu. Odparowanie benzenu po 3 dniach jest na poziomie 4%, przy 12%-ym odparowaniu oleju napędowego w ciągu 10 dni hodowli.
W wyniku kontroli hodowli obejmującej pomiar stężenia biomasy, ubytku węglowodorów aromatycznych (BTX), oznaczenia aktywności emulgacyjnej ustalono, że przyjęte do badań szczepy wykazują zróżnicowane preferencje substratowe. Szczep R51 w największym stopniu wykorzystuje toluen (88%), następnie benzen (84%), a w najmniejszej ilości ksylen (40%). Niewielka różnica w efektywności procesu degradacji toluenu i benzenu przez badany szczep może sugerować, iż węglowodory te wykorzystywane są zamiennie. Szczep NS2 z kolei w najwyższym stopniu wykorzystuje benzen (88%), następnie toluen (34%) i najsłabiej ksylen (28%). Wyraźnie widać, iż badany szczep ma silnie sprecyzowane preferencje substratowe.
Zjawisko kometabolizmu jest dość szeroko rozpowszechnione lecz mechanizm jego działania mało zbadany. Wyniki dotychczasowych badań wykazują, że wykorzystanie tego zjawiska może przynieść duże korzyści np. w oczyszczalni ścieków przemysłowych. W tej samej oczyszczalni bowiem można by umieścić syntetyczne związki oporne na degradację razem ze ściekami komunalnymi. Przeprowadzenie doświadczeń mających na celu poznanie zjawiska kometabolizmu (ptk. 5.9.1 i 5.9.2) pozwoliło jednoznacznie stwierdzić, iż dla szczepów Bacillus sp. (NS2) i Ochrobactrum anthropi (R51) benzen i toluen są kosubstratami w procesie degradacji ksylenu przez te drobnoustroje. Proces jego degradacji przez badane szczepy bakteryjne zachodzi na drodze kometabolizmu.
Wyniki dotychczasowych badań wykazują, że forma i stężenie źródła azotu to jedne z najważniejszych parametrów decydujących o efektywności procesu degradacji węglowodorów. Problem doboru właściwej formy i stężenia pożywki azotowej jest jednak skomplikowany. Wynika to z faktu, że w procesie degradacji udział bierze nie jeden lecz grupa mikroorganizmów, często o odmiennych preferencjach substratowych. Generalnym wnioskiem z prowadzonych prób dla badanych w ramach pracy szczepów jest stwierdzenie, że obecność w podłożu jonów amonowych sprzyja zwykle szybkiemu i obfitemu wzrostowi populacji bakteryjnych. Aktywność degradacyjna biomasy (liczona jako zużycie węglowodorów ogółem na jednostkę biomasy) badanych szczepów jest jednak wyższa w obecności azotanów.
Forma źródła azotu wywiera wpływ na zdolności do biodegradacji węglowodorów przez badane szczepy. Każdy z nich wykazuje zróżnicowane wymagania odnośnie tej pożywki. I tak: szczep R51 efektywnie degraduje węglowodorów bez względu na formę użytego źródła azotu. Obserwacji te dotyczą zarówno biodegradacji węglowodorów ogółem jak i aromatycznych. Zużycie węglowodorów ogółem jest w granicy 92% - 94%, a węglowodorów aromatycznych w granicy 94% - 95%. Dla szczepu NS2 jony azotu w postaci NaNO3 są zdecydowanie odpowiedniejszym źródłem azotu dla biodegradacji węglowodorów zarówno ogółem jak i aromatycznych w porównaniu z NH4Cl. Różnice w stopniu biodegradacji węglowodorów ogółem dochodzą do 30 jednostek procentowych. Wobec tego źródła azotu biodegradacja węglowodorów aromatycznych wynosi 91%, a przy zastosowaniu pozostałych źródeł azotu 89% - 90%. Wyniki uzyskane w obecności NH4NO3 są na zbliżonym poziomie w porównaniu do rezultatów uzyskanych w obecności NH4Cl
W wyniku doświadczeń mających na celu ustalenie właściwej dawki soli azotowych podczas procesu degradacji ustalono, że optymalna ilość źródła azotu użyta w stosowanych warunkach doświadczenia jest 1[gN/l]. Zmiana stężenia źródła azotu powoduje zmiany w plonie biomasy, lecz nie towarzyszy jej wzrost zużycia węglowodorów.
W ramach pracy badano wpływ dodatku naturalnego źródła azotu (w postaci otrąb pszennych i wytłoków sojowych) na proces biodegradacji węglowodorów aromatycznych zawartych w oleju napędowym. Jak wynika z przeprowadzonych doświadczeń badane surowce w stężeniu 1[gN/l] mogą korzystnie wpływać na proces degradacji wyżej wymienionej grupy węglowodorów. Na podstawie przeprowadzonej analizy chromatograficznej produktów procesu degradacji w obecności naturalnych źródeł azotu stwierdzono gromadzenie się podczas hodowli wielu pośredników metabolizmu (o czym świadczy duża ilość pików). Sytuacji takiej nie obserwowano podczas hodowli badanych szczepów na pożywce z nieorganicznymi źródłami azotu.
Ogromny wpływ na wzrost populacji bakteryjnej i jej zdolności do biodegradacji węglowodorów wywiera natlenienie środowiska. Stosując zmienne wypełnienie kolb podłożem hodowlanym w zakresie: 50 ml, 100 ml i 150 ml co odpowiada zastosowaniu warunków napowietrzenia 0,6, 0,47 i 0,31 [mMO2/l*min] stwierdzono, że szczególnie ważne jest ograniczenie napowietrzenia w zakresie 0,6 do 0,47 [mMO2/l*min] tj. zmiana wypełnienia kolby podłożem z 50 ml do 100 ml. Przy tej zmianie wypełnienia kolby zaobserwowano dla szczepów R51 i NS2 znaczne obniżenie wzrostu odpowiednio o 70% i 62.5%.
Prawidłowe funkcjonowanie każdego drobnoustroju jest ściśle związane z odczynem środowiska w jakim dany mikroorganizm się znajduje. Dla szczepu R51 przy pH o wartości 7,5 obserwujemy najlepsze wykorzystanie węglowodorów aromatycznych. Dla szczepu NS2 efektywność procesu degradacji węglowodorów aromatycznych uzyskujemy dla pH=6,0.
Funkcje życiowe mikroflory, właściwości fizyczne węglowodorów, a tym samym i proces ich degradacji zależą od temperatury w jakiej się znajdują. Jak widać z przeprowadzonych doświadczeń podwyższanie temperatury owocuje polepszeniem warunków degradacji węglowodorów aromatycznych dla obu badanych szczepów. Ponieważ zwiększenie temperatury z 30 do 370C powoduje prawie 2-krotnie większe odparowanie węglowodorów, a ich stopień zużycia wzrasta zaledwie o 17-19% nie ma większego sensu prowadzić proces degradacji w tej temperaturze.
Obecność makro i mikroelementów w odpowiednim stężeniu w środowisku hodowlanym jest niezbędna dla prawidłowego rozwoju i funkcjonowania drobnoustrojów. Mikroelementy (np.: Fe2+, Fe3+, Mn2+, Zn2+) często odpowiedzialne są za prawidłowy metabolizm komórkowy, ponieważ pełnią funkcje aktywatorów enzymów. Wiadomo bowiem, iż część jonów metali dwuwartościowy jest jednostką centralną w enzymie. W razie nieobecności określonego jonu metalu lub zbyt niskiego jego stężenia enzym staje się nieaktywny. Makroelementy takie jak np.: Mg2+, Ca2+, K+ mogą również być aktywatorami enzymów ponad to wpływają na strukturę, budowę i przepuszczalność ściany komórkowej. Wymienione jony niezbędne są dla komórki w większym stężeniu i pełnią wielorakie funkcje w komórce drobnoustrojowej. Z przeprowadzonych doświadczeń wynika, że obecność pojedynczych makro czy mikroelementów w podłożu hodowlanym nie przynosi znaczącej poprawy degradacji szczególnie węglowodorów aromatycznych. Występowanie obok siebie dwu makro czy mikro elementów lub mieszaniny tych pierwiastków poprawia wykorzystanie węglowodorów aromatycznych nawet o 8-13% w porównaniu do próby wyjściowej bez żadnego ze związków pomocniczych.
Zastosowanie w podłożu hodowlanym wszystkich omawianych makro i mikroelementów mimo obniżenia plonu biomasy badanych szczepów pozwala na osiągnięcie 30% poprawy stopnia degradacji węglowodorów aromatycznych.
Proces degradacji ksenobiotyków jest procesem ogromnie złożonym. Na jego efektywność wpływa wiele różnorodnych czynników (pkt. 2.3.). Jednym z nich jest „garnitur” enzymatyczny w jaki wyposażony jest dany drobnoustrój. W procesie degradacji węglowodorów aromatycznych udział biorą skomplikowane i wyspecjalizowane systemy enzymatyczne. Szczególną rolę odgrywają tu oksygenazy. Wśród tej grupy enzymów można wyróżnić oksygenazy właściwe (dioksygenazy) i oksygenazy mieszane (monooksygenazy). Dioksygenazy włączają w strukturę pierścienia aromatycznego oba atomy tlenu. Monooksygenazy z kolei wprowadzają do ksenobiotyku jeden atom tlenu, drugi zaś wiązany jest w cząsteczkę wody z udziałem czynnika redukującego. Enzymy te mogą lecz nie musza współdziałać z przekaźnikami elektronów. Do najczęściej spotykanych czynników redukujących należą: NADH, NADPH, cytochrom P-450, rubredoksyny.
Z uwagi na złożoną budowę oksygenaz uczestniczących w procesie degradacji ksenobiotyków oznaczenie ich aktywności jest trudne i wymagające skomplikowanej procedury oczyszczania poszczególnych podjednostek [Lindstrom M.E. 1990, Subramanian V. 11981, 1985]. Wiadomo także, iż proces ustalenia szczegółowej drogi biodegradacji interesującego nas związku jest długi, pracochłonny i trudny.
Otrzymane wyniki sugerują, iż szczep Ochrobactrum anthropi (R51) degraduje toluen z wykorzystaniem monoksygenazy metanowej. Zastanawiające jest natomiast pojawienie się w fazie wodnej w 16-tej godzinie hodowli kwasu protokatechowego. Znana jest z literatury (rys 20) droga degradacji toluenu przez kwas protokatecholowy (rys 63).
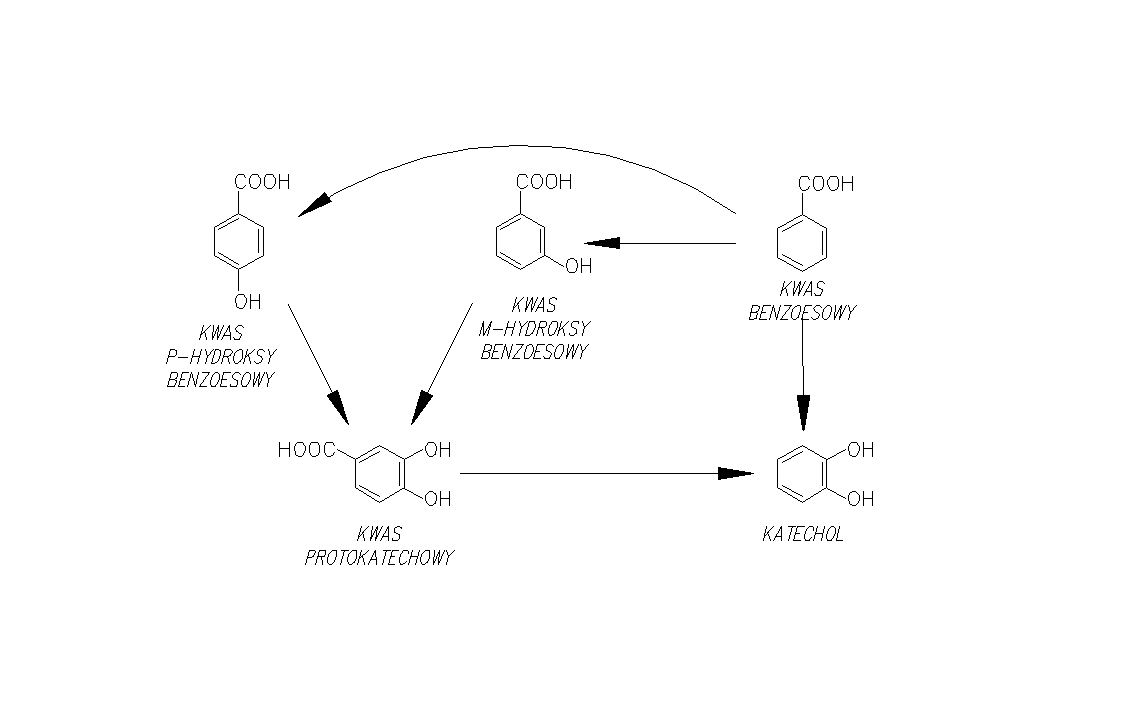
Rysunek 4 Przejście kwasu benzoesowego do katecholu i kwasu protokatechanowego
Trudne do racjonalnego wytłumaczenia jest dlaczego badana populacja bakteryjna wykorzystuje dłuższą drogę dojścia do katecholu (na chromatogramie większy pik kwasu protokatechowego niż katecholu). Z uwagi na oznaczenie kwasu mukonowego w dalszym czasie hodowli przypuszcza się działanie na katechol 1,2 dioksygenaz czyli szlak „orto” katecholowy.
W literaturze [Shields M.S. 1989] opisywano zazwyczaj trzy drogi degradacji toluenu (rys.9): pierwsza-monoutlenienie grupy metylowej przez Pseudomonas putidia mt-2 posiadającą plazmid TOL, druga - p-hydroksylację pierścienia aromatycznego przez monooksygenazę toluenową obecną w komórkach Pseudomonas mendocina dającą p-krezol, trzecia - podwójne utlenienie pierscienia aromatycznego przez dioksygenazę toluenową obecną w komórkach Pseudomonas putidia F1 co w rezultacie prowadziło do powstania cis-2,3-toluene dihydrodiolu, który następnie utleniany jest do 3-metylokarecholu przez dehydrogenazę. Pomijano natomiast drogę, zaobserwowaną u szczepu G4, w której pierścień aromatyczny toluenu ulega podwójnej hydroksylacji przez dwa niezależne sekwencyjnie monoutlenienia: pierwsze w pozycji „orto”, drugie w pozycji „meta”. W tym przypadku również powstaje 3-metylokatechol ale jako wynik dwustopniowej hydroksylacji. Prawdopodobnie u szczepu G4 transformacja toluenu do 3-metrylokatecholu jest efektem nie dwu enzymów ale jednego katalizującego obie reakcje.
W przypadku szczepu Bacillus sp. NS2 można domniemywać, iż występuje podobna zależność jak u szczepu G4 [Shields M.S. 1989]. Co prawda podczas ustalania metabolitów pośrednich występuje dużo więcej niejasności niż w przypadku szczepu Ochrobactrum anthropi R51 czy literaturowego G4. Wynikają one z braku niektórych wzorców, co pozwoliłoby na jednoznaczne określenie oznaczonego w hodowli metabolitu. Dotyczy to głównie 3-metylokatecholu i m-krezolu. Z literatury [Shields M.S. 1989] wiadomo, że piki chromatograficzne 3-metylokatechol i 4-metylokatechol czy m- i o-krezolu są położone bardzo blisko siebie (różnice w czasie retencji są znikome). Dlatego też nie mając obu metabolitów trudno jednoznacznie określić, który ze związków wykryto w czasie hodowli
W trakcie hodowli szczepu Bacillus sp. NS2 udało się natomiast zidentyfikować kwas ketopropionowy i pośrednio alliloszczawian, co sugerowało by przejście jednego z krezoli (m- lub o-) w 3-metylokatechol i dalszą drogę 2,3-dioksygenaz (czyli szlak „meta” katecholowy). Z kolei fakt istnienia u tego drobnoustroju dioksygenazy toluenowej (tabela 31) oraz obecność w cieczy pohodowlanej kwasu ketopropionowego i alliloszczawianu uniemożliwia jednoznaczne wskazanie drogi degradacji toluenu przez ten szczep.
Przeprowadzone przeze mnie badania miały jedynie wstępnie wskazać kierunek przemian biochemicznych toluenu przez badane szczepy bakteryjne: Ochrobactrum anthropi (R51) i Bacillus sp. (NS2). Aby rezultaty otrzymanych badań były bardziej wiarygodne należałoby poświęcić im jeszcze więcej czasu, i przeprowadzić żmudne badania ze związkami znaczonymi węglem C14 z wykorzystaniem wysokoselektywnych technik chromatograficznych zawierających bardzo czułe detektory promieniowania UV. Z uwagi na duże niejasności we wskazaniu jednoznacznie szlaku biochemicznych przemian toluenu przez badane populacje zrezygnowałam z prób wskazania szlaku degradacji benzenu przez te drobnoustroje.
Jednym z najtrudniejszych problemów dotyczących likwidacji skażeń substancjami ropopochodnymi jest bioremedjacja gruntów. Poważnym problemem jest tutaj silna sorbcja węglowodorów w pory gleby. Stwierdzono, iż zastosowanie biomasy bakterii wysuszonej przez 72 godziny daje nieco lepsze rezultaty biodegradacji węglowodorów, niż użycie biomasy tuż po jej odwirowaniu z podłoża po hodowli wstrząsanej. Jak już wspomniano w pkt. 5.12 wyniki otrzymane w tej części pracy są trudne do racjonalnego wytłumaczenia. Zostały one już w znacznej mierze przedyskutowane przy okazji ich omawiania. Wypada uzupełnić, że teoretycznie aktywność drobnoustrojów jest tym wyższa im środowisko ich bytowania jest bogatsze w wodę. Większość mikroorganizmów potrzebuje do życia aktywności wodnej z zakresu 0,95-0,99. Nie da się jednoznacznie stwierdzić, jak wygląda sytuacja pod tym względem podczas działania biopreparatu w złożu skażonym olejem napędowym. Jeżeli uznać, że podczas przeprowadzania tego eksperymentu nie popełniono żadnego błędu i uzyskane wyniki są wiarygodne, to należy stwierdzić, że suszenie przez 72 godziny biomasy odwirowanej z podłoża po hodowli testowanych bakterii wpływa aktywująco na ich uzdolnienia w kierunku biodegradacji węglowodorów oleju napędowego. Wysunięto hipotezę, że być może na etapie suszenia biomasy w suszarce dochodzi do zakażenia obcą mikroflorą, która uaktywnia się w czasie testowania biopreparatów na złożu stałym, nieoczekiwanie korzystnie zwiększając skuteczność ich działania. Dłuższy czas suszenia biomasy może zwiększać ilość zakażenia i stąd lepsze rezultaty. Potwierdzenie tej hipotezy wymaga dalszych szczegółowych badań.
Zastosowanie mieszaniny suszonej przez 72 godziny biomasy bakterii Ochrobactrum anthropi R51 i Bacillus sp. NS2 nieoczekiwanie nie przyniosło poprawy rezultatów degradacji węglowodorów oleju napędowego skażającego złoże stałe. Teoretycznie współdziałanie dwóch szczepów degradujących węglowodory oleju napędowego powinno dać lepsze rezultaty, niż działanie pojedynczego szczepu. Możliwe jest jednak wystąpienie pewnych antagonistycznych oddziaływań pomiędzy tymi dwoma gatunkami mikroorganizmów. Jeden ze szczepów może produkować związki, które szkodzą drugiemu i odwrotnie. Tak więc można stwierdzić, że stosowanie mieszaniny Ochrobactrum anthropi R51 i Bacillus sp. NS2 w warunkach eksperymentu do usuwania oleju napędowego skażającego grunt nie jest celowe, gdyż nie przynosi poprawy uzyskiwanych efektów, a będzie zwiększało koszty procesu.
W pracy zbadano także wpływ rodzaju stosowanego złoża stałego na stopień utylizacji oleju napędowego. Porównano oddziaływania drobnoziarnistego piasku, gleby z ogródka warzywnego i ziemi do kwiatów. Zużycie węglowodorów we wszystkich tych złożach było zbliżone i wahało się od 78% w piasku do 84% w przypadku ziemi do kwiatów. Zauważono różnicę w ilości oleju, który odparowuje ze złoża w ciągu 25 dni testu. Najwięcej węglowodorów odparowało z ziemi do kwiatów. Spowodowane to jest prawdopodobnie jej luźniejszą strukturą, większymi porami, a tym samym lepszą wentylacją w porównaniu do piasku i gleby ogrodowej. Ziemie kwiatowa była najlepiej napowietrzona i znajdowały się w niej wszystkie niezbędne do prawidłowego rozwoju drobnoustrojów mikroelementy, w odpowiednich względem siebie stosunkach ilościowych. Te czynniki spowodowały, że mikroorganizmy najlepiej „czuły się” właśnie w tych warunkach i stąd stopień biodegradacji węglowodorowych był najwyższy. Najsłabsze efekty utylizacji węglowodorów otrzymano w piasku. Złoże to jest najbardziej ubogie w składniki mineralne i najgorzej napowietrzone w stosunku do dwóch pozostałych.
9.PODSUMOWANIE
Zanieczyszczenie środowiska stanowi coraz poważniejszy problem w wielu krajach świata, nie wyłączając Polski. Skala problemu zależy od powierzchni zanieczyszczonego obszaru, głębokości penetracji zanieczyszczeń oraz ich składu chemicznego.
Bardzo ważne jest opracowanie metod likwidacji tych zanieczyszczeń. Prowadzone są poszukiwania efektywnych metod oczyszczania skażonych terenów metodami zarówno fizyko-chemicznymi jak i biotechnologicznymi. Dotychczasowe doświadczenia wskazują, że zastosowanie metod biotechnologicznych jako ostatniego etapu oczyszczania tych środowisk stanowi najbardziej racjonalny sposób ich remediacji. Przedsięwzięcie to nie jest jednak łatwe, wymaga ono bowiem ustalenia wielu parametrów procesu, na które składają się czynniki: biologiczne, chemiczne i techniczne. Często koniecznym jest użycie biopreparatów, czyli zestawów drobnoustrojów charakteryzujących się specyficzną aktywnością do występującego skażenia. Mikroorganizmy wchodzące w jego skład współdziałają z autochtoniczną mikroflorą danego środowiska, zachowując swoją aktywność mimo, że warunki procesu często odbiegają od optymalnych, np. zbyt niskie temperatury lub nieodpowiedni odczyn środowiska.
W skażonym środowisku węglowodory aromatyczne rzadko występują jako pojedyncza grupa związków, spotykamy je obok innych węglowodorów cyklicznych i alifatycznych. Jednak wśród tej grupy są one najtrudniej degradowalne. Do tej pory mało wiadomo o wpływie obecności określonych węglowodorów na zdolność do biodegradacji wybranego węglowodoru aromatycznego. W 1999 roku Razak stwierdził, że wystepowanie węglowodorów alifatycznych wywiera znaczący wpływ na proces ich degradacji. Wiadomym jest również, że niektóre związki podlegają degradacji przy udziale drobnoustrojów jedynie wówczas, gdy w środowisku obecne są inne wybrane substancje. Zjawisko to nosi nazwę kometabolizmu.
Biodegradacja węglowodorów aromatycznych jest istotnym problemem pozbawiającym środowisko naturalne groźnych zanieczyszczeń. Główną rolę odgrywają tu drobnoustroje naturalnie bytujące w przyrodzie, które prowadzą w warunkach naturalnych rozkład wielu zwiazków aromatycznych wchodzących w skład struktury roślin. W oparciu o badania istnieje przekonanie, że bakterie prowadzą proces biodegradacji węglowodorów bardziej kompletnie, natomiast grzyby atakują te związki w dużej mierze w celu ich detoksykacji i nie prowadzą ich rozkładu do końca. Grzyby atkują często substancje należące do grupy szcególnie trudnodegradowalnych i jednocześnie bardzo toksycznych, przekształcając je w substancje o niższej tokyczności i łatwiej degradowlne. Pośredniki te mogą być potem degradowane z udziałem bakterii. Bakterie z rodzaju Pseudomonas utleniają np. katechol, który może być pośrednim metabolitem rozkładu ksylenu, toluenu i innych związków aromatycznych.
Dużą rolę przy biodegradacji związków aromatycznych odgrywają enzymy produkowane przez mikroorganizmy. Rozróżniamy tutaj dioksygenazy, które włączają dwa atomy tlenu przy jednoczesnym rozszczepieniu wiązania pomiędzy dwoma węglami oraz monooksygenazy, włączające tylko jeden atom tlenu, zaś drugi zamieniany jest w wodę. Przy udziale oksygenaz przebiegają zwykle pierwsze etapy utleniania pierścieni aromatycznych.
Wiele gatunków drobnoustrojów, zarówno bakterii jak i grzybów wykazuje zdolność do degradacji węglowodorów aromatycznych w wrunkach tlenowych, jak również, choć gorzej w warunkach beztlenowych.
Z obecnych w środowisku mikroorganizmów, w reakcjach biodegradacji związków aromatycznych najczęściej uczestniczą bakterie z rodzaju Pseudomonas, które utylizują np. benzen, toluen, etylobenzen, ksylen, fenantren jako źródło węgla i energii.
Monopierścieniowe węglowodory aromatyczne są dobrze rozpuszczalne w wodzie i przez to są bardzo toksyczne i mutagenne, a wśród nich najbardziej niebezpieczny jest benzen, ponieważ jest rakotwórczy. Mimo że te węglowodory (BTEX - benzen, toluen, etylobenzen, ksylen) są strukturalnie podobne, to jednak o-ksylen jest najbardziej opornym węglowodorem do degradacji wśród tych związków.
WWA reprezentują ważną klasę polutantów, które również wykazują cechy mutagenne i rakotwórcze. Jednak z powodu swej niskiej rozpszczalności w wodzie często występują w osadach.
Warto zauważyć, że w kilku przypadkach biochemiczny mechanizm biodegradacji węglowodorów aromatycznych nie jest do końca znany. Podobnie jest z wieloma enzymami uczestniączacymi w tych przemianach, które nie zostały dotychczas sklasyfikowane lub nawet bliżej określone.
10. LITERATURA
Białecka-Floriańczyk E., Włostowska E. 2003. Chemia organiczna.
Chen C.J., Taylor R.T. 1997. Thermoppilic biodegradation of BTEX by two
vonsortia of anaerobic bacteria. Appl. Micribiol. Biotechnol. vol. 48: 121-128.
Chmiel A. 1998. Biotechnologia: podstawy mikrobiologiczne i biochemiczne.
Cho M.C., Kang D.O., Yoon B.D., Lee K. 2000. Toluene degradation pathway from Psudomonas putida F1: substrate specificity and gene induction by 1-substituted benzenes. Journal of Industrial Microbiology & Biotechnology vol. 25: 163-170.
Collin L.D., Daugulis A.J. 1999. Benzene / toluene / p-xylene degradation. Appl.Microbiol. Biotechnol. vol. 52: 360-365.
Dean- Ross D., Cernigilia C.E. 1996. degradation of pyrene by Mycobacterium flavescens. Appl. Microbiolo. Biotechnol. vol.46: 307-312.
Dong Jun Oh. 2002. Uniwersytet Minnesota. o-xylene graphical patway map -
strony www.
Dore S.Y., Clancy Q.E., Rylec S.M., Kulpa Jr. C.F. 2003. Naphthalene - utilizing and mercury - resistant bacteria isolated from an acidic enviroment. Appl. Microbiol.Biotechnol.
Encyklopedia PWN. 1995.
Enzyme Nomenklature. 2003 - strony www.
Evans P.J., Mang D.T., Young L.Y. 1991. Degradation of toluene and m-xylene and transformation of o-xylene by denitritying enrichment cultures. Appl. Environ. Microbiol. vol. 57: 450-454.
Faber R,W., Schonewille A.B.J.A., R.F.M. von Gorcom, Duine J.A. 2001.
Constitutive and inducible hydroxylase acitivities involved in the degradation of naphthalene by Cuninghamella eleans. Appl. Microbiol. Biotechnol. vol. 55: 486-491.
Garbić-Galić D., Vogel T.M. 1987. Transformation of toluene and benzene by mixed metanogenic cultures. Appl. Environ. Microbiol. vol. 53: 254-260.
Grzywa E., Molenda J. Technologia podstawowych syntez organicznych.
Hanumanthanaik P. Doddamani, Harichandra Z. Ninnekar. 2000. Biodegradation of phenanthrene by a Bacillus species. Current Microbiology vol. 41: 11-14.
Hyat B.A., Oh D.J. 2002. Uniersytet Minnesota. m-xylene graphical pathway map - strony www.
Juhasz A.L., Stanley G.A., Britz M.L. 2002. Metabolite repression inhibites degradation of benzo[a]pyrene and dibenzo[a,h]anthracene by Stenotrophomonas maltophilia VUN 10.003. Journal of Industrial Microbiol. & Biotechnol. vol. 28: 88-96.
Keith N. 2003. Uniwersytet Minnesota. Pyrene graphical pathway map - strony www.
Kim E., Zylstra G.J. 1999. Functional analysis of genes involved in biphenyl, naphthalene, phenanthrene, and m-xylene degradation by Sphingomonas yanoikuyae B1. Journal of Industrial Microbiol. & Biotechnol. vol. 23: 294-302.
Klimiuk E., Łebkowska M. 2003. Biotechnologia w ochronie środowiska.
Kłyszejko-Stefanowicz L. 1982. Ćwiczenia z biochemii.
Kunicki-Goldfinger. Życie bakterii.
Kwapisz E. 1999. Problemy biodegradcji węglowodorów ropy naftowej. VI Ogółnopolskie Sympozjum Naukowo-Techniczne „Biotechnologia Środowiska”.
Lee S.K., Lee S.B. 2001. Isolation and characterization of a thermotolerant bacterium Ralstonia sp. strain PHS1 that degrades benzene, toluene, ethylobenzene, and o-xylene. Appl. Microbiol. Biotechnol. vol. 56: 270-275.
Lim T.G.H., Gan K.D., Hughes T.A., Hayasaka S.S. 2001. Toluene mineralization and growth potential of Pseudomonas putida PaW164 under toluene - limiting conditions. Arch. Environ. Contam. Toxical. vol. 41: 117-122.
Manohar S., Karegaudar T.B. 1998. Degradation of naphthalene by cells of Pseudomonas sp. strain NGK 1 immobilized in alginate, agar and polyacrylamide. Appl. Microbiol. Biotechnol. vol. 49: 785-792.
Manohar S., Karegaudar T.B., Kim C.K. 2001. Enhanced degradation of naphthalene by immobilization of Pseudomonas sp. strain NGK 1 in polyurethane foam. Appl. Microbiol. Biotechnol. vol. 55: 311-316.
Martens R., C. in der Wiesche, Zadrazil F. 1996. Two-step degradation of pyrene by white-rot fungi and soil microorganisms. Appl. Microbiol. Biotechnol. vol. 46: 653-659.
Mastalerz P. 1996. Podręcznik chemii organicznej.
Morrison R.T., Boyd R.N. 1985. Chemia organiczna, tom 1.
Nenitescu C.D. 1969. Chemia organiczna, tom 2.
Onyang J., Fitzgerald M. 2003. Uniwersytet Minnesota. Phenenthrene graphical pathway map - strony www.
Ortega-Calvo J.J., Saitz-Jimenez C. 1998. Effect of humi fractions and clay on biodegradation of phenanthren by a Pseudomonas fluorescens strain isolated from soil. Appl. Environ. Microbiol. vol. 64: 3123-3126.
Parales R.E., Ditty J.L., Harwood C.S. 2000. Toluene - degrading bacteria are chemotactic towards the enviromental pollutans benzene, toluene and trichloroethylene. Appl. Environ. Microbiol. vol. 66: 4098-4104.
Pedro J., Alvarez J., Vogel T.M. 1991. Substrate interactions of benzene, toluene and p-xylene during microbial degradation by pure cultures and mixed culture aquifer slurries. Appl. Environ. Microbiol. vol. 57: 2981-2985.
Preuss R., Angerer j., Drexler H. 2003. Naphtalene - an enviromental and
occupational toxicant. Int. Arch. Occup. Environ. Health. vol. 76: 556-576.
Ravelet C., Crossel C., Krivoboks S., Montuelle B., Alary J. 2001. Pyrene
degradation by two fungi in a freshwater sediment and evaluation of fungal biomass by ergosterol content. Appl. Microbiol. Biotechnol. vol. 56: 803-808.
Rehm J., Reed G. 1996. Biotechnology vol. 6,8.
Russel S. 1990. Biotechnologia.
Samanta S.K., Chakraborti A.K., Jain R.K. 1999. Degradation of phenanthrene by different bacteria: evidence for novel transformation sequences involving the formation of 1-naphtol. Appl. Environ. Microbiol. vol. 53: 98-107.
Schlegel H.G. 2003. Mikrobiologia ogólna.
Shuttleworth K.L., Cernigilia C.E. 1995. Enviromental aspects of PAH biodegradation. Appl. Bioch. Biotechnol. vol . 54: 291-302.
Stapletion R.D., Savage D.S., Sayler D.S., Stacey G. 1998. Biodegradation of aromatic hydrocarbons in an extremly acidic enviroment. Appl. Environ. Microbiol. vol. 64: 4180-4184.
Stepheus S., Joen M. 2002. Uniwersytet Minnesota. p-xylene graphical pathway map - strony www.
Strinfellow W.T., Aitken M.D. 1995. Competitve metabolism of naphthalene, methylnaphthalene and fluorene by phenanthrene - degrading pseudomonads. Appl. Environ. Microbiol. vol. 61: 357-362.
Trzmiel T. PŁ Wydział BiNŻ, Instytut Biochemii technicznej.
Tsao C.W., Song H.G., Bartha R. 1998. Metabolism of benzene, toluene and xylene hydrocarbons in soil. Appl. Environ. Microbiol. vol. 64: 4924-4929.
Wackett L. 2002. Uniwersytet Minnesota. Naphthalene graphical pathway map - strony www.
Wackett L. 2002. Uniwersytet Minnesota. Toluene graphical pathway map - strony www.
Wilson L.P., Bouwer E.J. 1997. Biodegradation of aromatic compounds under mixed oxygen/denitryfitying conditions: a raview. Journal of Industrial Microbiol. & Biotechnol. vol. 18: 116-130.
Yerushalmi L., Guiot S.R. 1998. Kinetics of biodegradation of gasoline and its hydrocarbon constitutents. Appl. Microbiol. Biotechnol. vol. 49: 475-481.
Zheng Z., Breedveld G., Aagaard P. 2001. Biodegradation of soluble compounds of jet fuel under anaerobic conditions: laboratory batch experiments. Appl. Microbiol. Biotechnol. vol. 57: 572-578.
Zhuang W.Q., Tay J.H., Maszenan A.M. 2002. Bacillus naphthovorans sp. from oil - contaminated tropical marine sediments and its role in naphthalene biodegradation. Appl. Microbiol. Biotechnol. vol. 58: 547- 553.
Żiriakow W.G. 1976. Chemia organiczna.
7
2. Część teoretyczna 2.2. Warunki biodegradacji
3-13
1. Wstęp
1-78
6. Podsumowanie wyników
Wyszukiwarka
Podobne podstrony:
Główne szlaki biodegradacji związków aromatycznych, Naukowe PL, Biotechnologia, Enzymologia, Genetyk
związki aromatycznych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Biodegradacja nitrowych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Biodegradacja heterocyklicznych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Biodegradacja halogenopochodnych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Bakteriocyny i L-antybiityki, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Rośliny transgeniczne, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Lipazy, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
wyk 4 węglow aromat
notatek pl , Biotechnologie, BAKTERIE LAB
WĘGLOWODORY AROMATYCZNE
Witaminy, Naukowe PL, Witaminy
Biotechnologia i inżynieria genetyczna
BT węglowodory aromatyczne (wykład III)
kl 2, kartkowka-benzen, Benzen - C6H6 Wzór Elektronowy - 6¬¬C KL 1H K Węglowodory Aromatyc
Oznaczanie wybranych węglowodorów aromatycznych przy zastosowaniu chromatografii gazowej(1)
więcej podobnych podstron