Główne szlaki biodegradacyjne związków aromatycznych
Na przestrzeni ostatnich kilkudziesięciu lat, w dobie burzliwego rozwoju różnych gałęzi przemysłu, w tym samochodowego i chemicznego oraz rozwoju miast i infrastruktury, dochodzi często do wzrostu zanieczyszczenia środowiska naturalnego różnorakimi szkodliwymi substancjami. Takie obce substancje, niespotykane naturalnie i zanieczyszczające przyrodę nazywa się ksenobiotykami. Do wykazu tych ekologicznych zagrożeń doszło ostatnio jeszcze jedna bardzo ważna pozycja: możliwość zatrucia środowiska naturalnego związkami aromatycznymi oraz ich pochodnymi. Na szczególną uwagę wśród tej grupy związków zasługują różnego rodzaju węglowodory aromatyczne, zarówno monopierścieniowe, jak i policykliczne.
Wzrost zanieczyszczeń środowiska związkami organicznymi skłania do poszukiwań jak najbardziej skutecznych metod ich utylizacji. W warunkach naturalnych jest to proces długotrwały i dlatego człowiek dąży do jego przyspieszenia. Do tych zabiegów wykorzystuje się osiągnięcia biotechnologii, gdzie drobnoustroje wykorzystują te związki jako źródło węgla i energii. Szlaki biodegradacji są nieustannie poznawane. Szczególną uwagę poświęca się mikrobiologicznej degradacji mieszanin związków sztucznie produkowanych przez człowieka, które naturalnie nie występują w naturze. Postępująca akumulacja tych toksycznych mieszanin w środowisku naturalnym jest szkodliwa dla biologicznych systemów. Badanie szlaków degradacji tych związków często prowadzi do rozpoznania nowych reakcji enzymatycznych.
Reakcją obronną natury na zanieczyszczenie w tym przypadku związkami aromatycznymi jest działalność wielu rodzajów drobnoustrojów zdolnych do biodegradacji tych zanieczyszczeń. Jednak procesy te są niekiedy zbyt powolne dlatego też działania biotechnologów są ukierunkowane na poznania mechanizmów działania tych organizmów, a następnie ulepszania i wykorzystania ich w procesach degradacji. Praktycznie dla każdego typu związku chemicznego można dobrać mikroorganizm zdolny do jego degradacji. Liczebność drobnoustrojów w środowisku naturalnym jest teoretycznie nieograniczona, jedynym praktycznie problemem jest odnalezienie odpowiedniego mikroorganizmu o uzdolnieniach do degradacji w kierunku konkretnego ksenobiotyku. W tym celu stosuje się także udoskonalanie szczepów na drodze inżynierii genetycznej.
W niektórych procesach biodegradacji konieczne jest współdziałanie dwóch lub większej liczby szczepów, ponieważ pojedynczo nie były by one w stanie doprowadzić tych procesów do końca.
W największym uproszczeniu proces biodegradacji kończy się, kiedy związek organiczny ulegnie rozpadowi do naturalnie występujących w środowisku związków, takich jak CO2 i H2O, czyli mineralizacji. Zanim jednak to nastąpi, w środowisku pojawiają się produkty pośrednie rozkładu tych substancji. Często takie pośrednie produkty rozkładu związków organicznych wykazują znacznie większą toksyczność w stosunku do mikroorganizmów, zwierząt i ludzi niż związki wyjściowe. Przykładem mogą być przemiany niektórych związków z grupy wielopierścieniowych węglowodorów aromatycznych (WWA). Część tych związków ma działanie prokancerogenne[5].
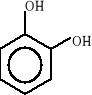
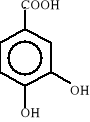
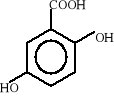
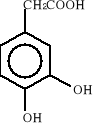
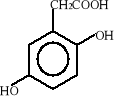
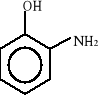
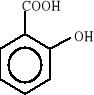
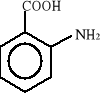
Związki aromatyczne ulegają biodegradacji do jednego z ośmiu podstawowych produktów pośrednich: 1) katecholu, 2) kwasu protokatechowego, 3) kwasu gentyzynowego, 4) kwasu homoprotokatechowego i 5) kwasu homogentyzynowego oraz do kilku niekonwencjonalnych związków, takich jak: 6) o-aminofenolu, 7) kwas salicylowego i 8) kwas antranilowego. Wzory strukturalne tych związków zostały przedstawione poniżej:
1) 
2) 
3) 
4) 
5) 
6) 
7) 
8) 
4.2. Drobnoustroje biorące udział w biodegradacji związków aromatycznych.
Reakcją środowiska na zanieczyszczenie związkami aromatycznymi jest działalność drobnoustrojów zdolnych te zanieczyszczenia biodegradować. Zdolność drobnoustrojów do biodegradacji węglowodorów uzależniona jest od [10]:
funkcjonowania odpowiednich systemów ich transportu do komórki
posiadania przez drobnoustroje potencjału genetycznego umożliwiającego wprowadzenie tlenu do cząsteczki węglowodoru;
specyficzności substratowej oksygenaz;
funkcjonowania mechanizmu indukowania enzymów takich jak: dehydrogenazy, hydrolazy czy dekarboksylazy.
Środowisko naturalne jest bogate w liczne gatunki drobnoustrojów zarówno tlenowych jak i beztlenowych, wykazujących uzdolnienia do wykorzystywania węglowodorów jako źródła węgla i energii a tym samym do ich degradacji. Wśród tych mikroorganizmów wyróżnić można zarówno bakterie, drożdże, glony i grzyby strzępkowe.
Drobnoustroje biorące udział w biodegradacji związków aromatycznych to:
bakterie: Aeromonas, Flavobakterium, Beneckea, Anabaena, Moraksella, Achromobacter, Mycobacterium, Nocardia, Pseudomonas, Chryzosporium, Micrococus, Alcaligenes, Vibrio, Acinetobakter, Bacillus, Arthrobacter.
grzyby nitkowate: Aspergillus, Penicylium, Mucor.
drożdże: Sacharomyces, Trichoderma.
promieniowce: Actinomyces, Nocardia, Streptomyces.
cyjanobakterie i glony z rodzajów: Oscillatoria, Anabaena, Nostoc, Chlorella, Chlamydomonas, Scenedesmus, Phormidium.
Spośród dużej grupy bakterii o uzdolnieniach do degradacji węglowodorów najlepiej zbadanymi i poznanymi są bakterie z rodzaju Pseudomonas, które utleniają katechol i pokrewne związki za pomocą dioksygenaz.
4.3. Biodegradacja związków aromatycznych w warunkach tlenowych - metabolizm tlenowy.
Jednym z czynników, który sprzyja szybkiej degradacji związków aromatycznych jest tlen. Metabolizm tlenowy charakteryzowany jest przez dwa podstawowe procesy związane z obecnością cząsteczki tlenu [11]:
biodegradacja substratu organicznego wykorzystująca system oksydoreduktaz z końcowym przeniesieniem elektronów poprzez łańcuch oddechowy na atom tlenu z wytworzeniem energii dla komórki w postaci ATP.
wprowadzeniem cząsteczki tlenu do substratu organicznego - aktywacja do dalszego metabolizmu.
Większość związków aromatycznych jest biodegradowalna według mechanizmu tlenowego poprzez rozszczepienie pierścienia aromatycznego. Etapem początkowym tego rozszczepienia jest włączenie atomu tlenu do cząsteczki związku aromatycznego. Wprowadzenie tlenu następuje przy wykorzystaniu systemu dioksygenaz. W następnym etapie odpowiednie dehydrogenazy odrywają dwa atomy wodoru, przez co przywracają aromatyczny charakter pierścienia. W wyniku tych reakcji węglowodory aromatyczne przekształcane są najczęściej do katecholu lub kwasu protokatechowego, które traktowane są jako metabolity pośrednie tych przemian. W dalszych etapach pierścień jest rozrywany między dwiema sąsiadującymi grupami hydroksylowymi, albo między hydroksylowanym i sąsiadującym niehydroksylowanym atomem węgla. Najważniejszymi typami otwarcia pierścienia aromatycznego są: rozszczepienie typu „orto”- rozszczepienie intradiolowe i typu „meta”- rozszczepienie ekstradiolowe [12].
Dzięki monooksygenazom można degradować takie związki jak: krezole, kwasy m- i p-hydroksybenzoesowy czy kwas salicylowy. Wykazują one bowiem wysoką specyficzność substratową.
Drugi mechanizm polega na jednoczesnym wprowadzeniu dwóch cząsteczek tlenu, w wyniku czego następuje zmiana konfiguracji na cis. Dotyczy to degradacji form orto-fenoli. W procesie tym wymagana jest obecność zarówno dioksygenazy, jak i dehydrogenazy. Enzymy te powodują degradację m.in.: kwasu benzoesowego, antranilowego, ftalowego i niektórych wielojądrowych węglowodorów. W wyniku metabolizmu bakterii powstawać mogą również formy trans, ale prawdopodobnie tylko jako związki pośrednie [11,13].
Inny mechanizm podwójnego hydroksylowania związków aromatycznych jest prowadzony przez grzyby w obecności drugiego źródła węgla. Reakcja prowadzi do powstania epoksydu, który może być uwodniony do trans-dihydrodiolu.
4.3.2. Rozszczepienie pierścienia aromatycznego typu „orto” i „para”.
Rozszczepienie typu „orto”-(intradiolowe). Rozszczepienie pierścienia między dwoma sąsiadującymi hydroksylowanymi atomami węgla prowadzi do powstania kwasów dikarboksylowych. Jest ono katalizowane przez dioksygenazy. Przypuszczalnie w reakcji tej zachodzi pierwotne przyłączenie cząsteczki O2 do atomów węgla obok grupy hydroksylowej z wytworzeniem cyklicznego nadtlenku. Z kolei, wewnątrzcząsteczkowe przekształcenia prowadzą do zerwania wiązania C-C z wytworzeniem kwasu cis,cis-mukonowego.
Rozszczepienie typu „para”-(ekstradiolowe). Rozszczepienie pierścienia między hydroksylowanym i niehydroksylowanym atomem węgla również jest katalizowane przez dioksygenazy. W wyniku reakcji powstają semialdehydy kwasu 2-hydroksymukonowego, które następnie wchodzą w szlaki metabolizmu pośredniego.
Jak wynika z badań szlaki kataboliczne związków aromatycznych znacznie różnią się sposobami rozszczepienia pierścienia, oraz reakcjami prowadzącymi do rozszczepienia pierścienia aromatycznego. U niektórych bakterii również faza wzrostu oraz warunki hodowli mogą decydować o tym, czy powstają enzymy katalizujące rozszczepienie typu orto, czy typu meta. U niektórych bakterii z rodzaju Pseudomonas związki aromatyczne katabolizowane poprzez katechol ulegają rozszczepieniu typu orto. Podczas gdy katabolizowane poprzez kwas protokatechanowy ulegają rozszczepieniu typu meta [14].
Dokładniej typy rozszczepienia pierścieni aromatycznych będą omówione w dalszych rozdziałach pracy przy omawianiu szlaków degradacji katecholu, kwasu protokatechowego i reszty związków pośrednich.
4.4. Biodegradacja związków aromatycznych w warunkach beztlenowych - metabolizm beztlenowy.
Związki aromatyczne mogą być degradowane beztlenowo. Sądzono, że benzen, kwas benzoesowy, kwas 4-hydroksybenzoesowy i wiele innych związków musi być opornych na beztlenową degradację. Okazało się to jednak nieprawdziwe. Fenol, a także kwas benzoesowy, czy nawet alkohol benzylowy są atakowane również w warunkach beztlenowych. Warunkiem jest obecność takich akceptorów wodoru, jak azotan, siarczan, CO2, które pozwalają na oddychanie beztlenowe, lub dostęp światła [14].
Procesy anaerobowej (beztlenowej) biodegradacji węglowodorów przebiegają znacznie wolniej niż procesy aerobowe (tlenowe). Są one istotne z uwagi na dalszą biotransformację metabolitów powstałych w tlenowych procesach biodegradacji węglowodorów, przede wszystkim alkoholi, fenoli, aldehydów oraz kwasów tłuszczowych. Związki te niejednokrotnie mogą wykazywać właściwości toksyczne dla organizmów żywych ze względu na zwiększoną polarność w porównaniu z wyjściowymi węglowodorami i mogą łatwiej rozpuszczać się w wodach gruntowych. Ograniczony dostęp tlenu faworyzować będzie przebieg procesów biodegradacji według mechanizmu beztlenowego [15].
Niektóre bakterie purpurowe, ale niesiarkowe np. Rhodospirillaceae wykorzystują proste aromatyczne kwasy karboksylowe, taki jak: kwas benzoesowy i hydroksybenzoesowy jako jedyne źródło węgla.
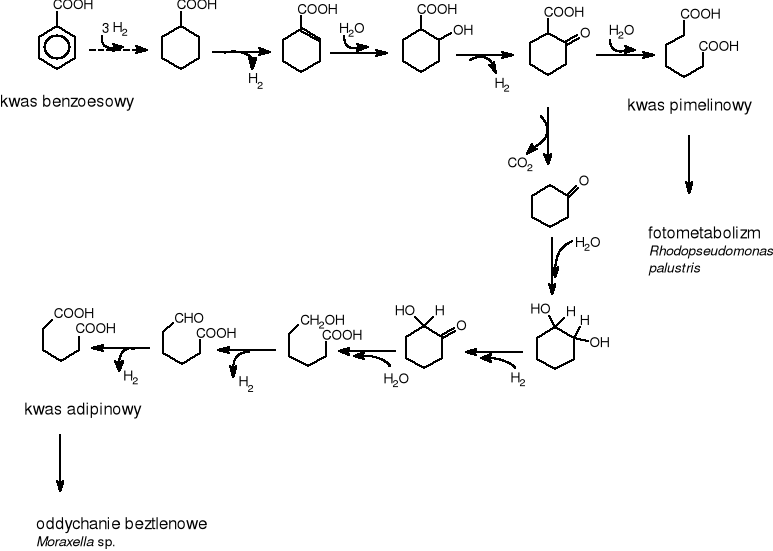
Rys.4 Beztlenowa biodegradacja kwasu benzoesowego przez Rhodopseudomonas palustris i Moraxella sp.
Pierwszy etap to redukcja pierścienia benzenowego przez przyłączenie 3 cząsteczek wodoru. Dalej zachodzi -oksydacja. Kwas benzoesowy degradowany jest do kwasu pimelinowego przez Rhodopseudomonas palustris, natomiast Moraxella sp. rozkłada ten sam substrat do kwasu adypinowego. We wczesnych etapach aktywacji benzoesanu uczestniczy koenzym-A i powstaje związek pośredni benzylo-CoA, który ulega hydratacji do pochodnej cyloheksanu, a następnie degradacji z uwolnieniem acetylo-CoA [11]. Beztlenowy rozkład podstawionego benzenu jest szlakiem reduktywnym. Możliwości beztlenowego rozkładu związków aromatycznych przez drobnoustroje nie zostały jeszcze w pełni poznane.
5. Biochemiczne szlaki biodegradacyjne głównych metabolitów pośrednich powstających podczas degradacji związków aromatycznych.
Jak już wspomniano we wcześniejszych rozdziałach, szlaki biodegradacji związków aromatycznych prowadzą zawsze przez któryś z następujących związków: katechol, kwas protokatechowy, kwas gentyzanowy, kwas antranilowy, kwas salicylowy, o-aminofenol, kwas homoprotokatechowy i kwas homogentyzanowy. Przy czym dwa ostatnie związki są rzadziej spotykane.
5.1. Katechol.
Katechol „posiada” dwa niezależne szlaki biodegradacyjne: szlak intradiolowy, zwany też szlakiem„orto katecholowym” i ekstradiolowy, czyli „para katecholowy”.
5.1.1. Szlak intradiolowy katecholowy („orto”)
Rozszczepienie pierścienia następuje między dwoma sąsiadującymi hydroksylowanymi atomami węgla. Rozszczepienie pierścienia katecholu następuje przy udziale 1,2-dioksygenazy katecholowej. Produktem tej enzymatycznej reakcji jest cis,cis-mukonian. Związek ten ulega dalszemu metabolizmowi poprzez kwas 3-ketoadypinowy, który jest aktywowany przez transferazę-CoA 3-ketoadypinianową i następnie zostaje rozszczepiony do bursztynylo-CoA i acetylo-CoA. Związki te są włączane do podstawowych szlaków katabolicznych [13,14]. (Rys.5).
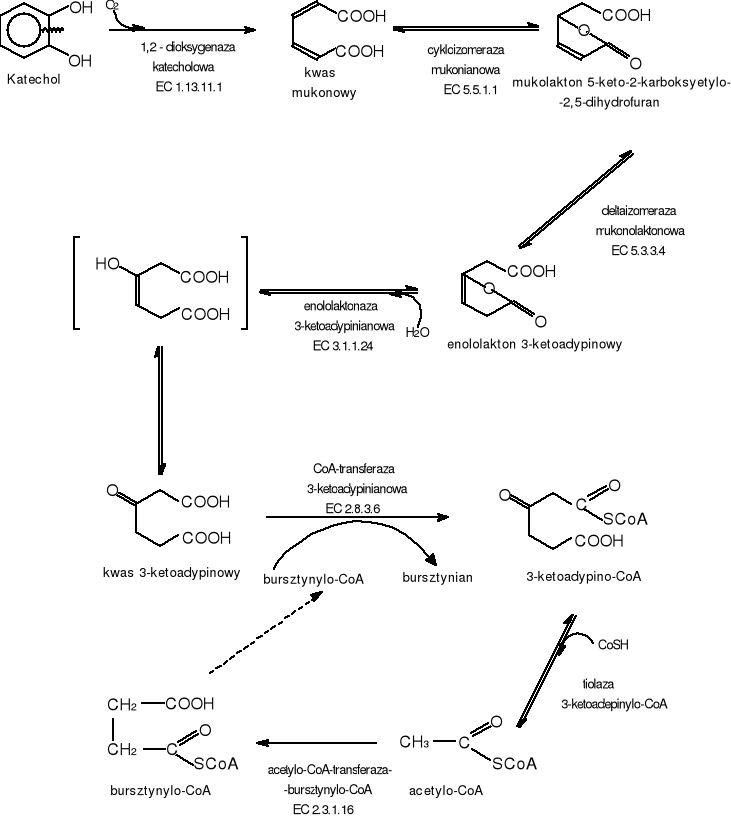
Rys.5 Szlak biodegradacji katecholu - intradiolowy (typu „orto”)
ENZYMY
1,2-dioksygenaza katecholowa EC 1.13.11.1 [55]
Nazwa systematyczna: 1,2-oksydoreduktaza katechol:tlen.
Komentarz: wymaga Fe3+; wciągana jest w metabolizm związków aromatycznych u Psedomonas putida. Włącza do substratu dwa atomy tlenu z cząsteczki.(rozszczepienie pierścienia aromatycznego)
cykloizomeraza mukonianowa EC 5.5.1.1 [56]
Nazwa systematyczna: liaza 2,5-dihdro-5-oksofurano-2-acetooctanowa.
Komentarz: wymaga Mg3+; działa także w reakcji odwrotnej.
delta-izomeraza mukonolaktonowa EC 5.3.3.4 [57]
enololaktonaza 3-ketoadypinianowa EC 3.1.1.24 [58]
Komentarz: należy do klasy hydrolaz, działa na produkty reakcji katalizowanych przez dekarboksylazę 4-karboksymukonolaktonową.
CoA-transferaza 3-ketoadypinianowa EC 2.8.3.6 [59]
Nazwa systematyczna: CoA-transferaza bursztynylo-CoA:3-ketoadypinian.
acetylo-CoA-transferaza-bursztynylo-CoA EC 2.3.1.16 [60]
Nazwa systematyczna: C-acylotransferaza acylo-CoA:acetylo-CoA.
5.1.2. Szlak ekstradiolowy katecholowy („meta”)
W tym przypadku rozszczepienie pierścienia aromatycznego następuje między hydroksylowanym i niehydroksylowanym atomem węgla. Proces ten jest katalizowany 2,3- dioksygenazę katecholową. W wyniku reakcji powstaje semialdehyd 2-hydroksymukonowy (rys.6), który następnie wchodzi w szlaki metabolizmu pośredniego poprzez pirogronian i aldehyd octowy. Semialdehyd 2-hydroksymukonowy może być przekształcony od razu do aliloszczawianu z pominięciem szlaku przez kwas 2-hydroksymukonowy i szczawiokrotonian. Reakcja ta jest katalizowana przez hydrolazę semialdehydu 2-hydroksymukonowego (EC 3.7.1.9).
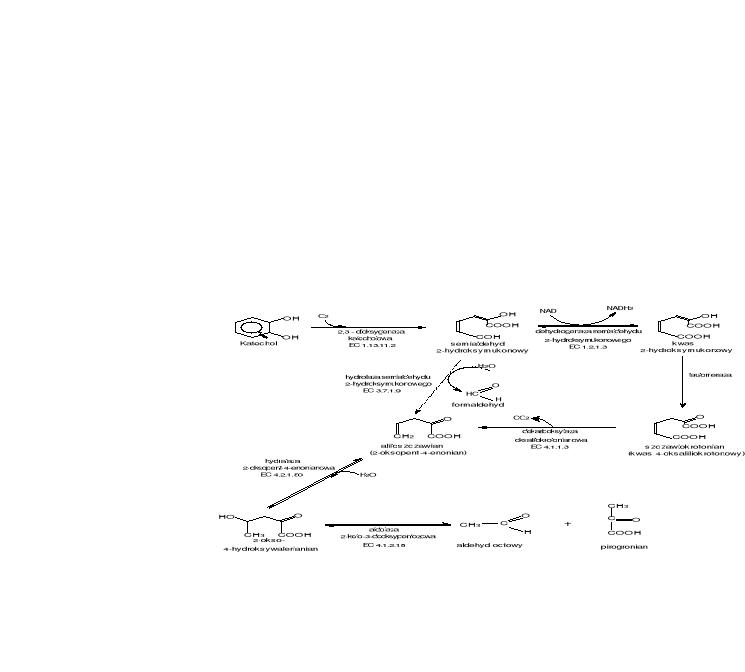
Rys.6. Szlak biodegradacji katecholu - ekstradiolowy (typu „meta”)
ENZYMY
2,3-dioksygenaza katecholowa EC 1.13.11.2 [61]
Nazwa systematyczna: 2,3-oksydoreduktaza katechol:tlen.
Komentarz: wymaga do działania dwuwartościowego żelaza Fe(II); występuje u Alcaligenes sp.; działa na katechol, włącza dwa atomy tlenu do substratu powodując rozszczepienie pierścienia.
dehydrogenaza semialdehydu 2-hydroksymukonowego EC 1.2.1.3 [62]
Nazwa systematyczna: oksydoreduktaza aldehyd:NAD+.
Komentarz: działa na aldehydy lub grupy ketonowe donorów; z NAD+ jako akceptor; szeroka specyficzność.
dekarboksylaza oksalilokrotonianowa EC 4.1.1.3 [63]
Nazwa systematyczna: karboksy-liaza oksaliloacetooctanowa.
Komentarz: pochodzi od Klebsiella aerogenes , jest biotynowa proteiną. Proces dekarboksylacji prowadzi przy udziale jonów sodu Na+. Niektóre enzymy pochodzenia zwierzęcego wymagają udziału jonów magnezu Mg2+.
hydrataza 2-oksopent-4-enonianowa EC 4.2.1.80 [64]
Nazwa systematyczna: hdro-liaza 4-hydroksy-2-oksopentanianowa.
Komentarz: działa także na cis-oksoheks-4-enonian ale o wiele wolniej, nie działa natomiast na trans-izomer.
aldolaza 2-keto-deoksypentozowa EC 4.1.2.18 [65]
Nazwa systematyczna: glikoaldehyd-liaza 2-dehydro-3-deoksy-L-pentanianianowa.
hydrolaza semialdehydu 2-hydroksymukonowego EC 3.7.1.9 [66]
Nazwa systematyczna: formylohydrolaza semialdehydu 2-hydroksymukonianowego.
5.2. Kwas protokatechowy.
Kwas protokatechowy podobnie jak katechol może być biodegradowany poprzez dwa rodzaje rozszczepienia pierścienia aromatycznego.
5.2.1. Szlak intradiolowy protokatchanowy („orto”)
Rozszczepienie pierścienia aromatycznego kwasu protokatechowego następuje pod wpływem 3,4-dioksygenazy protokatechanowej. Produktem tej reakcji jest kwas 3-karboksymukonowy, który ulega dalszemu metabolizmowi [14]. Związek ten jest przekształcany do 4-karboksymukonolaktonu. Reakcja ta jest katalizowana przez cykloizomerazę 3-karboksymukonianową. Następnie przy udziale dekarbosylazy 4-hydroksymukonolaktonowej powstaje enololakton 3-ketoadypinowy. Dalej droga biodegradacji przebiega jak w intradiolowym szlaku katecholowym aż do powstania bursztynylo-CoA i acetylo-CoA , włączanych następnie do metabolizmu pośredniego.
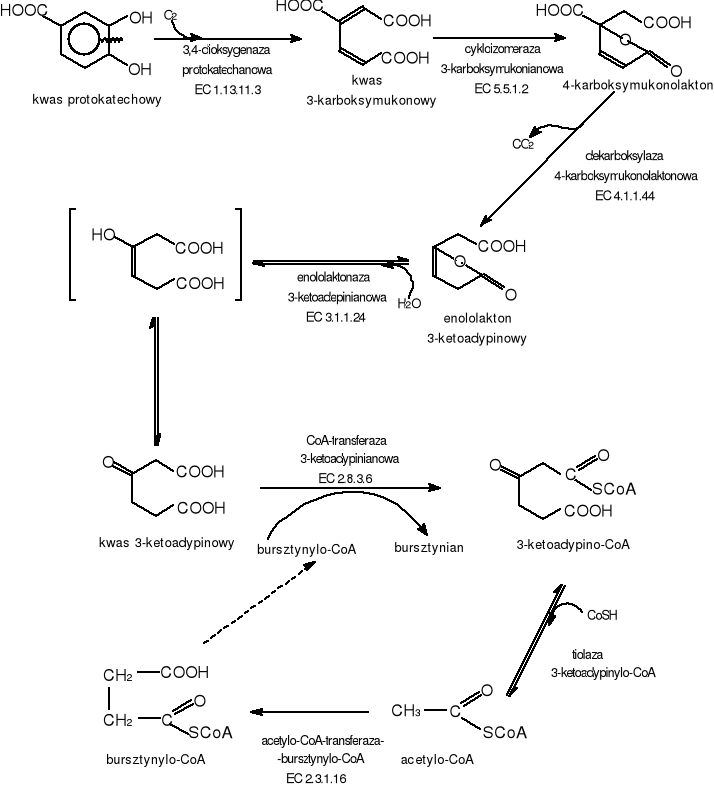
Rys.7 Intradiolowy szlak biodegradacji kwasu protokatechowego.
ENZYMY
3,4-dioksygenaza protokatechanowa EC 1.13.11.3 [67]
Nazwa systematyczna: 3,4-oksydoredukza protokatechan:tlen
Komentarz: działa na kwas protokatechowy włączając cząsteczkę do pierścienia aromatycznego powodując jego intradiolowe rozszczepienie. Wymaga do działania Fe3+.
cykloizomeraza 3-karboksymukonianowa EC 5.5.1.2 [68]
Nazwa systematyczna: liaza 2,5-dihydroksy-2-karboksy-5-oksofurano-2-octanowa.
dekarboksylaza 4-karboksymukonolaktonowa EC 4.1.1.44 [69]
Nazwa systematyczna: karboksy-liaza 4-karboksymukonolaktonowa
enololaktonaza 3-ketoadypinianowa EC 3.1.1.24 [58]
Komentarz: należy do klasy hydrolaz, działa na produkty reakcji katalizowanych przez dekarboksylazę 4-karboksymukonolaktonową.
CoA-transferaza 3-ketoadypinianowa EC 2.8.3.6 [59]
Nazwa systematyczna: CoA-transferaza bursztynylo-CoA:3-ketoadypinian
acetylo-CoA-transferaza-bursztynylo-CoA EC 2.3.1.16 [60]
Nazwa systematyczna: C-acylotransferaza acylo-CoA:acetylo-CoA.
5.2.2. Szlak ekstradiolowy protokatchanowy („meta”)
Ekstradiolowe rozszczepienie pierścienia aromatycznego kwasu protokatechowego zachodzi pod wpływem 4,5-dioksygenazy protokatechanowej. Enzym ten powoduje rozerwanie pierścienia między czwartym, a piątym atomem węgla. Produktem tej reakcji jest semialdehyd 2-hydroksy-4-karboksymukonowy. W kolejnym etapie w sposób spontaniczny powstaje semiacetal 2-hydroksy-4-karboksymukonowy. Przy udziale dehydrogenazy 6-semialdehydu-2hydroksy-4karboksymukonowego powstaje kwas 2-pirono-4,6-dikarboksylowy. Kolejnym enzymem katalizującym dalej reakcję jest laktonohydrolaza 2-pirono-4,6-dikarboksylanowa. Wynikiem tej reakcji jest powstanie kwasu 2-okso-4-karboksymukonowego, który następnie ulega hydratacji do 4-oksalilo-2-metylojabłczanu. Związek ten jest rozszczepiany przez aldolazę 4-hydroksy-4-metyloglutaranową, końcowe produkty biodegradacji kwasu protokatechowego czyli pirogronian i szczawiooctan, które są włączane do szlaków katabolicznych organizmów. (Rys.8)
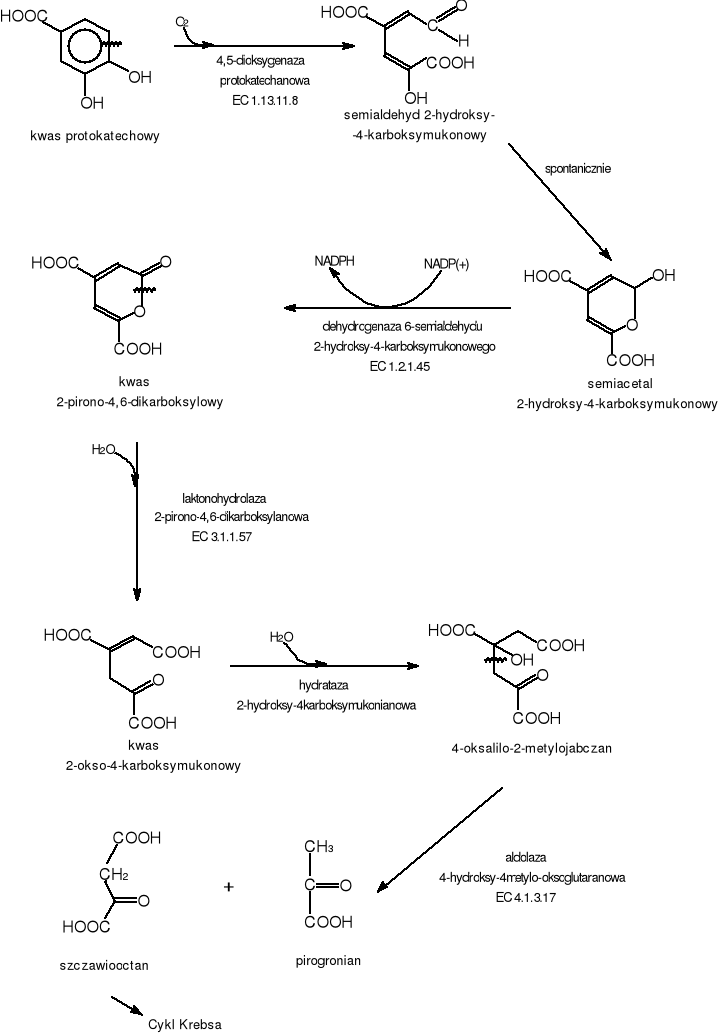
Rys.8 Ekstradiolowy szlak biodegradacji kwasu protokatechowgo
ENZYMY
4,5-dioksygenaza protokatechanowa EC 1.13.11.8 [70]
Nazwa systematyczna: 4,5-oksydoreduktaza protokatechan:tlen
Komentarz: działa na kwas protokatechowy, wbudowuje dwa atomy tlenu z cząsteczki przy 4 i 5 atomie węgla, powodując ekstradiolowe rozszczepienie pierścienia aromatycznego protokatechanu. Wymaga Fe2+.
dehydrogenaza 6-semialdehydu 2-hydroksy-4karboksymukonowego EC 1.2.1.45 [71]
Nazwa systematyczna: 6-oksydoreduktaza-2-hydroksy-4-karboksy-cis,cis-mukonianowa 6-semialdehyd:NADP+.
Komentarz: działa na aldehydy i grupy ketonowe donoru, przy udziale NAD+ jako akceptora, nie działa natomiast na nie podstawione aldehydy alifatyczne lub aromatyczne. NAD+ może być zastąpiony przez NADP+.
laktonohydrolaza 2-pirono-4,6-dikarboksylanowa EC 3.1.1.57 [72]
Nazwa systematyczna: laktonohydrolaza 2-pirono-4,6-dikarboksylanowa.
hydrataza 2-hydroksy-4-karboksymukonianowa.
aldolaza 4-hydroksy4-metylooksoglutaranowa EC 4.1.3.17 [73]
Nazwa systematyczna: pirogronian-liaza 4-hydroksy-4-metylo-2-oksoglutaranowa
Komentarz: działa również na 4-hydroksy-4-metylo-2-oksoadypinian oraz na 4-karboksy-4-hydroksy-2-oksoheksadionian.
Dodatkowo u drobnoustrojów możliwa jest degradacja kwasu protokatechowego poprzez przekształcenie go uprzednio do katecholu.(Rys.8a) Reakcja ta jest katalizowana przez odpowiedni enzym, którym jest dekarboksylaza protokatechanową EC 4.1.1.63 wytwarzany u Klebsiella aerogenes (Aerobacter aerogenes) [50].
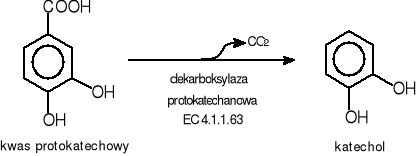
Rys. 8a. Biodegradacja kwasu protokatechowego do katecholu przez drobnoustroje.
5.3. Kwas gentyzynowy
Kwas gentyzynowy biodegradowany jest również poprzez rozszczepienie pierścienia, podobnie jak katechol czy kwas protokatechowy. Do rozszczepienie pierścienia niezbędne są również dioksygenazy. W tym przypadku jest to 1,2-dioksygenaza gentyzynianowa. Enzym ten powoduje rozerwanie wiązania miedzy atomem węgla połączonego z grupą karboksylowa a atomem węgla połączonym z grupą hydroksylową. Następnie poprzez 3-maleilopirogronian i 3-fumarylopirogronian kwas gentyzynowy przekształcany jest do końcowych produktów którymi są kwas fumarowy oraz pirogronian. (Rys.9) U niektórych organizmów występuje hydroliza z pominięciem izomeryzacji np. podczas degradacji ksylenu przez Pseudomonas sp. [11,16].
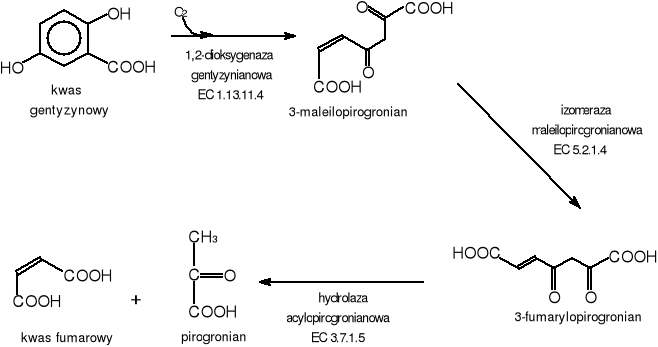
Rys.9 Szlak biodegradacji kwasu gentyzynowego.
ENZYMY
1,2-dioksygenaza gentyzynianowa EC 1.13.11.4 [74]
Nazwa systematyczna: 1,2-oksydoreduktaza gentyzynian:tlen
Komentarz: wbudowuje dwa atomy tlenu do pierścienia kwasu gentyzynowego powodując jego rozszczepienie; wymaga do działania Fe2+.
izomeraza maleilopirogronianowa EC 5.2.1.4 [75]
Nazwa systematyczna: cis-trans-izomeraza 3-maleilopirogronianowa.
hydrolaza acylopirogronianowa EC 3.7.1.5 [76]
Nazwa systematyczna: acylohydrolaza 3-acylopirogronianowa.
Komentarz: działa na formylopirogronian, 2,4-dioksopentanian, 2,4-dioksoheksanian i 2,4-dioksoheptanian.
5.4. Kwas homogentyzynowy
Degradacja kwasu homogentyzynowego jest wspólna zarówno dla mikroorganizmów jak i dla ssaków gdzie jest częścią szlaku metabolizmu tyrozyny. Przebieg szlaku biodegradacji kwasu homogentyzynowego jest analogiczny do szlaku degradacji kwasu gentyzynowego.
Pod wpływem działania 1,2-dioksygenazy homogentyzynianowej, pierścień aromatyczny ulega rozszczepieniu. Produktem tej reakcji jest 4-maleiloacetooctan, który jest następnie przekształcany przez izomeraze maleiloacetooctanową do 4-fumaryloacetooctanu. Końcowymi produktami biodegradacji kwasu homogentyzynowego są kwas fumarowy i acetylooctan, które powstają w reakcji katalizowanej przez hydrolazę fumaryloacetooctanową (fumaryloacetoacetaze).
Kwas homogentyzynowy może być również biodegradowany poprzez aldehyd homogentyzynowy i następnie kwas gentyzynowy którego biodegradacja została opisana w poprzednim punkcie.(Rys 10)
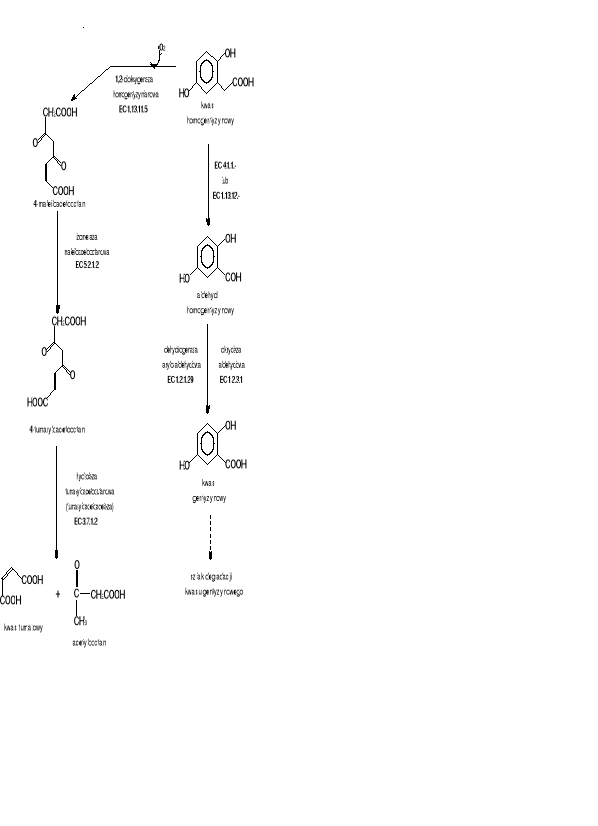
Rys.10 Szlak biodegradacji kwasu homogentyzynowego.
ENZYMY
1,2-dioksygenaza homogentyzynianowa EC 1.13.11.5 [77]
Nazwa systematyczna: 1,2-oksydoreduktaza homogentyzynian:tlen
Komentarz: Komentarz: wbudowuje dwa atomy tlenu do pierścienia kwasu homogentyzynowego powodując jego rozszczepienie; wymaga do działania Fe2+.
izomeraza maleiloacetooctanowa EC 5.2.1.2 [78]
Nazwa systematyczna: cis-trans-izomeraza 4-maleiloacetooctanowa.
Komentarz: działa również na maleilopirogronian.
hydrolaza fumaryloacetooctanowa (fumaryloacetoacetaza) EC 3.7.1.2 [79]
Nazwa systematyczna: fumarylohydrolaza 4-fumaryloacetooctanowa.
Komentarz: działa również na inne 3,5- i 2,4-diokso kwasy.
oksydaza aldehydowa EC 1.2.3.1 [80]
Nazwa systematyczna: oksydoreduktaza aldehyd:tlen
Komentarz: zawiera molibden, centra [2Fe-2S] oraz FAD.
dehydrogenaza arylo-aldehydowa EC 1.2.1.29 [81]
Nazwa systematyczna: oksydoreduktaza arylo-aldehyd:NAD+.
Komentarz: działa na aldehydowe lub ketonowe grupy donoru przy udziale NAD+ jako akceptor. Działa na aldehydy aromatyczne, nie działa na alifatyczne.
5.5. o-Aminofenol
Związek ten jest podstawowym metabolitem pośrednim przy biodegradacji aryloamin (głównie aniliny i jej pochodnych). Pierścień aromatyczny o-aminofenolu rozszczepiany jest przy udziale 1,6-dioksygenazy 2-aminofeolowej (EC 1.13.11.?). Enzym ten jest jeszcze nie do końca poznany, badania nad nim są w toku. W wyniku reakcji rozszczepienia powstaje semialdehyd 2-aminomukonowy (rys.11) [17-21].
Semialdehyd 2-aminomukonowy utleniany jest w obecności NAD do kwasu 2-aminomukonowego. Proces ten zachodzi u Pseudomonas pseudoalcaligenes JS45 [17,19-20] oraz u Comamonas sp. JS765 [21] 2-aminomukonian transformowany jest natomiast do kwasu 2-hydroksymukonowego w procesie hydrolitycznej deaminacji katalizowanym przez deaminazę 2-aminomukonową. Dalsze przemiany kwasu 2-hydroksymukonowego są typowe dla ekstradiolowego szlaku katecholowego (rozdz.5.5.1) [24-25]. Końcowymi metabolitami tego szlaku są pirogronian i aldehyd octowy. Wyniki badań sugerują [17,23], że powyższa droga biodegradacji 2-aminofenolu funkcjonuje także w przypadku drobnoustrojów wyizolowanych z zanieczyszczonych wód gruntowych.
W hodowlach Pseudomonas arvilla [22] oraz w warunkach beztlenowych lub w przypadku braku NAD [17,20] z powstałego w pierwszym etapie procesu semialdehydu 2-aminomukonowego, spontanicznie powstaje kwas pikolinowy i jest to prawdopodobnie ślepa uliczka procesu biodegradacji.
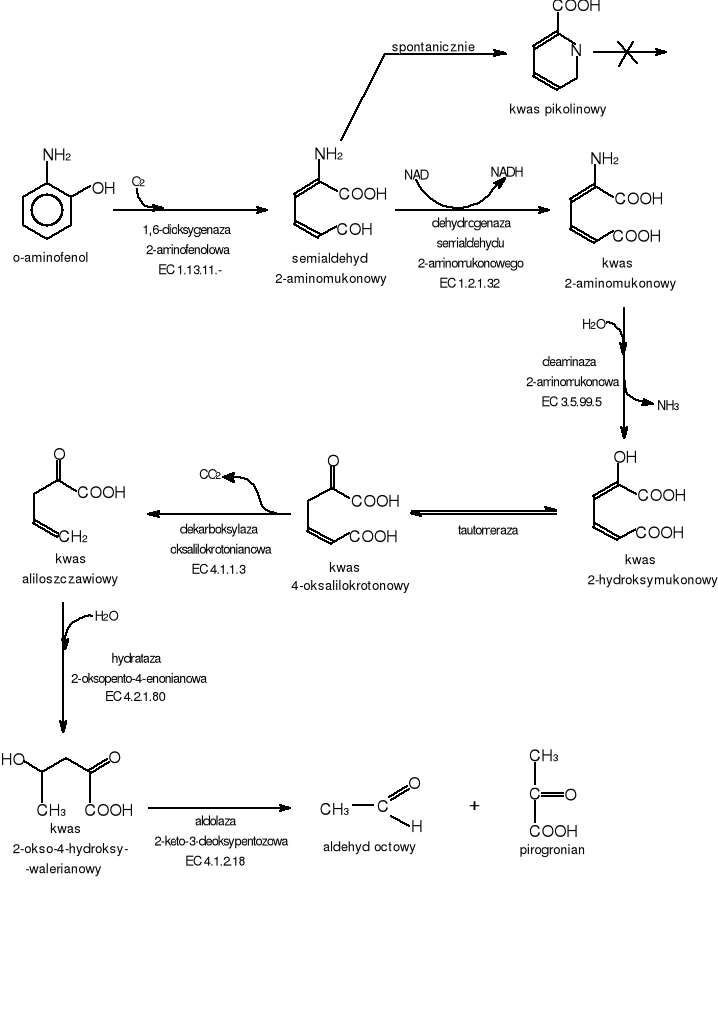
Rys.11 Szlak biodegradacji o-aminofenolu
ENZYMY
1,6-dioksygenaza 2-aminofenolowa EC 1.13.11.-
dehydrogenaza semialdehydu 2-aminomukonowego EC 1.2.1.32 [82]
Nazwa systematyczna: 6-oksydoreduktaza 2-aminomukono-6-semialdehydu:NAD+. (ang. 2-aminomuconate-6-semialdehyde:NAD+ 6-oxidoreductase)
Komentarz: działa na aldehydowa lub ketonowa grupę donoru z NAD+ jako akceptor. Działa również na aldehyd 2-hydroksymukonowy.
deaminaza 2-aminomukonowa EC 3.5.99.5 [83]
Nazwa systematyczna: aminohydrolaza 2-aminomukonianowa.
Komentarz: pośredniczy w biodegradacji nitrobenzenu przez Pseudomonas alcaligenes JS45. Reakcja zachodzi spontanicznie w środowisku kwaśnym.
dekarboksylaza oksalilokrotonianowa EC 4.1.1.3 [63]
Nazwa systematyczna: karboksy-liaza oksaliloacetooctanowa.
Komentarz: pochodzi od Klebsiella aerogenes , jest biotynowa proteiną. Proces dekarboksylacji prowadzi przy udziale jonów sodu Na+. Niektóre enzymy pochodzenia zwierzęcego wymagają udziału jonów magnezu Mg2+.
hydrataza 2-oksopent-4-enonianowa EC 4.2.1.80 [64]
Nazwa systematyczna: hdro-liaza 4-hydroksy-2-oksopentanianowa.
Komentarz: działa także na cis-oksoheks-4-enonian ale o wiele wolniej, nie działa natomiast na trans-izomer.
aldolaza 2-keto-3-deoksypentozowa EC 4.1.2.18 [65]
Nazwa systematyczna: glikoaldehyd-liaza 2-dehydro-3-deoksy-L-pentanianianowa.
5.6. Kwas antranilowy
Kwas antranilowy jest jednym z ważniejszych aromatycznych związków azotowych w metabolizmie zwierząt i mikroorganizmów [26]. Jego estry (metylowy, etylowy), o zapachu pomarańczy, stosowane są w przemyśle kosmetycznym i spożywczym [27].
Kwas antranilowy może bezpośrednio ulec przemianie do katecholu pod wpływem 1,2-dioksygenazy antranilanowej (dekarboksylujaco-deaminującej)[28-29]. Enzym ten powoduje wprowadzenie dwóch atomów tlenu do substratu z jednoczesną jego dekarboksylacją i deaminacjią [30]. Mechanizm ten występuje u bakterii Pseudomonas fluorescens.
Inaczej biodegradacja antranilanu przebiega u grzybów. Inicjacja polega tu na jego transformacji do kwasu 2,3-dihydroksybenzoesowego przy udziale 3-monooksygenazy antranilanowej (deaminującej) występującej np. u Aspergillus niger [31,33,34]. Trichosporon cutaneum [32] i Claviseps paspali [35]. Następnie kwas 2,3-dihydroksybenzoesowy ulega przekształceniu do katecholu na drodze dekarboksylacji. Enzymem katalizującym tą reakcję jest dekarboksylaza o-pirokatechanowa[36,37].
Kwas 2,3-dihydroksybenzoesowy może być również dalej degradowany poprzez bezpośrednie rozszczepienie pierścienia. U Pseudomonas fluorescens 23D-1 (w komórkach którego nie stwierdzono obecności dekarboksylazy o-pirokatechanowej) reakcję katalizuje 3,4-dioksygenaza 2,3-dihydroksybenzoesanowa (EC 1.13.11.14) [38].
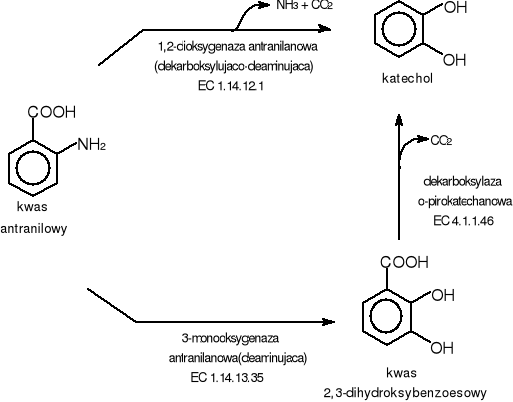
Rys.12 Szlak biodegradacji kwasu antranilowego
ENZYMY
1,2-dioksygenaza antranilanowa(dekarboksylująco-deaimnująca) EC 1.14.12.1 [84]
Nazaw systematyczna: oksydoreduktaza antranilanowa, NAD(P)H:tlen (deaminująca, dekarboksylująca).
Komentarz: dzaiała na rozmieszczone parami donory z włączeniem molekularnego tlenu. NADPH jako donor; wymaga Fe2+ .
3-monooksygenaza antranilanowa(deaminująca) EC 1.14.13.35 [85]
Nazwa systematyczna: oksydoreduktaza antranilanowa NADPH: tlen (deaminująca). Enzym ten pochodzący z Aspergillus niger jest żelazaoproteiną, natomiast pozyskany od Trichosporon cutaneum jest flawoproteiną (FAD).
dekarboksylaza o-pirokatechanowa EC 4.1.1.46 [86]
Nazwa systematyczna: karboksy-liaza 2,3-dihydroksybenzoesanowa.
Kwas antranilowy może być również biodegradowany w warunkach beztlenowych. Produktem jest w tym przypadku benzoil-CoA (Rys.12a), który ostatecznie na drodze wielu reakcji enzymatycznych jest przekształcany do acetylo-CoA [51].
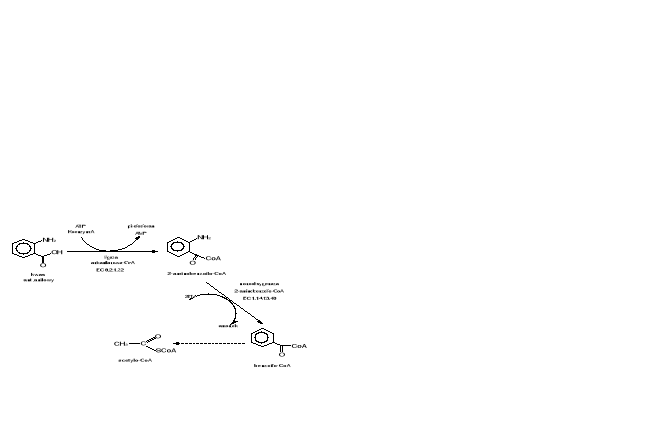
Rys.12a. Schemat biodegradacji kwasu antranilowego w warunkach beztlenowych
ENZYMY
ligaza antranilanowa-CoA EC 6.2.1.32 [87]
Nazwa systematyczna: ligaza antranilan:CoA
monooksygenaza 2-aminobenzoilo-CoA EC 1.14.13.40 [88]
Nazwa systematyczna: oksydoreduktaza 2-aminobenzoilo-CoA, NAD(P)H: tlen.
Komentarz: jest flawoproteiną (FAD).
5.7. Kwas salicylowy
Kwas salicylowy jest pośrednim związkiem biodegradacji takich substancji jak np.: naftalen, fluoren czy dibenzofuran. Poniżej przedstawiono schemat biodegradacji naftalenu przez bakterie z grupy Pseudomonas (Rys.13) [39]. Związek ten jest rozkładany poprzez 1,2-dihydroksynaftalen i aldehyd salicylowy do kwasu salicylowego.
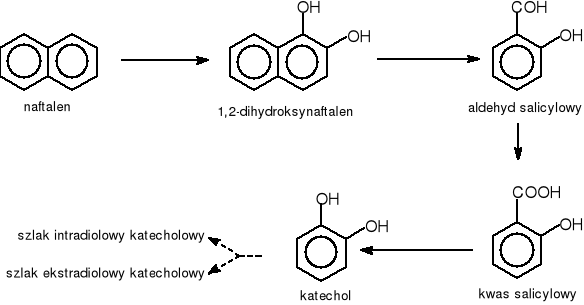
Rys.13. Schemat procesu degradacji naftalenu w warunkach tlenowych przez bakterie z grupy Pseudomonas [39].
Kwas salicylowy może być biodegradowany na różne sposoby w zależności od drobnoustrojów prowadzących proces biodegradacji. Klasycznym przykładem jest proces biodegradacji prowadzony przez Pseudomonas putida. Salicylan jest tu przekształcany bezpośrednio do katecholu przy udziale 1-monooksygenazy salicylanowej. W reakcji uwalniany jest również ditlenek węgla.(Rys.14).
Natomiast u Ralstonia sp. Kwas salicylowy jest przekształcany do kwasu gentyzynowego przy udziale 5-hydroksylazy salicylanowej, która jest wytwarzana przez te drobnoustroje [52].
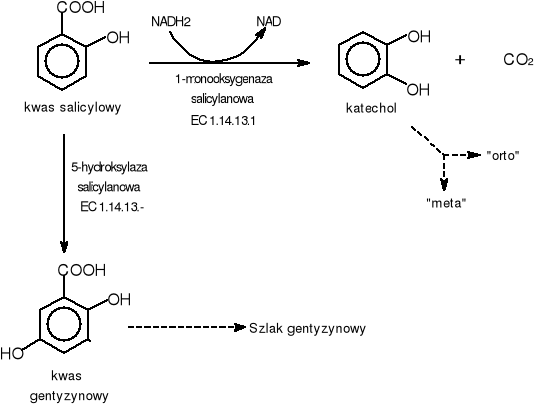
Rys.14. Szlak biodegradacji kwasu salicylowego u drobnoustrojów.
ENZYMY
1-monooksygenaza salicylanowa EC 1.14.13.1 [89]
Nazwa systematyczna: oksydoreduktaza salicylanowa NADH:tlen (dekarboksylująca)
Komentarz: jest flawoproteiną, wymaga koenzymu (FAD).
5-hydroksylaza salicylanowa EC 1.14.13.-
5.8. Kwas homoprotokatechowy
Gatunki Acinetobacter oraz bakterie Pseudomonas putida, które wzrastają w obecności kwasu 4-hydroksyfenylooctowego dają ekstrakt komórkowy, który konwertuje kwas homoprotokatechowy (3,4-dihydroksyfenylooctowy) do ditlenku węgla, pirogronianu i bursztynianu [53].
Biodegradacja homoprotokatechanu obejmuje nastepujące etapy: rozszczepienie pierścienia aromatycznego przez 2,3-dioksygenazę 3,4-dihydroksyfenylooctanową. Następnie powstały semialdehyd 2-hydroksy-5-karboksymetylomukonowy ulega dehydrogenacji do kwasu 2-hydroksy-5-karboksymetylomukonowego. Reakcja ta katalizowana jest przez odpowiednią dehydrogenazę przy udziale NAD. Kolejnym etapem jest dekarboksylacja przy udziale dekarboksylazy 5-oksopent-3-eno-1,2,5-trikarboksylanowej, w wyniku której powstają nowe metabolity: kwas 2-hydroksyhepta-2,4-dieno-1,7-dionowy i kwas 2-oksohept-3-eno-1,7-dionowy. Związek ten ulega następnie hydratacji i dalej następuje rozszczepienie aldolowe powstałego kwasu 2,4-dihydroksyhept-2-eno-1,7-dionowego do pirogronianu i semialdehydu bursztynowego. Końcowym produktem biodegradacji kwasu homoprotokatechowego jest kwas bursztynowy powstały z semialdehydu przy udziale dehydrogenazy semialdehydu bursztynianowego [53].
ENZYMY
2,3-dioksygenaza-3,4-dihydroksyfenylooctanowa. EC 1.13.11.15 [90]
Nazwa systematyczna: 2,3-oksydoreduktaza 3,4-dihydroksyfenylooctan: tlen.
Komentarz: jest żelazoproteiną.
dehydrogenaza semialdehydu 2-hydroksy-5-karboksymetylomukonowego. EC 1.2.1.60 [91]
Nazwa systematyczna: oksydoreduktaza semialdehyd 5-karboksymetylo-2-hydroksymukonowy : NAD+.
Komentarz: jest włączany w szlaku degradacji tyrozyny u Aspergillus sp.
delta-izomeraza 2-hydroksy-5-karboksymetylomukonianowa EC 5.3.3.10 [92]
dekarboksylaza 5-oksopent-3-eno-1,2,5-trikarboksylanowa EC 4.1.1.68 [93]
Nazwa systematyczna: karboksy-liaza 5-oksopent3-eno-1,2,5-trikarboksylanowa.
hydrataza 2-oksopent-3-eno-1,7-dionianowa EC 4.2.1.-
aldolaza 2,4-dihydroksyhept-3-eno-1,7-dionianowa EC 4.2.1.-
dehydrogenaza semialdehydu bursztynianowego EC 1.2.1.16 [94]
Nazwa systematyczna: oksydoreduktaza semialdehyd bursztynianowy: NAD(P)+.
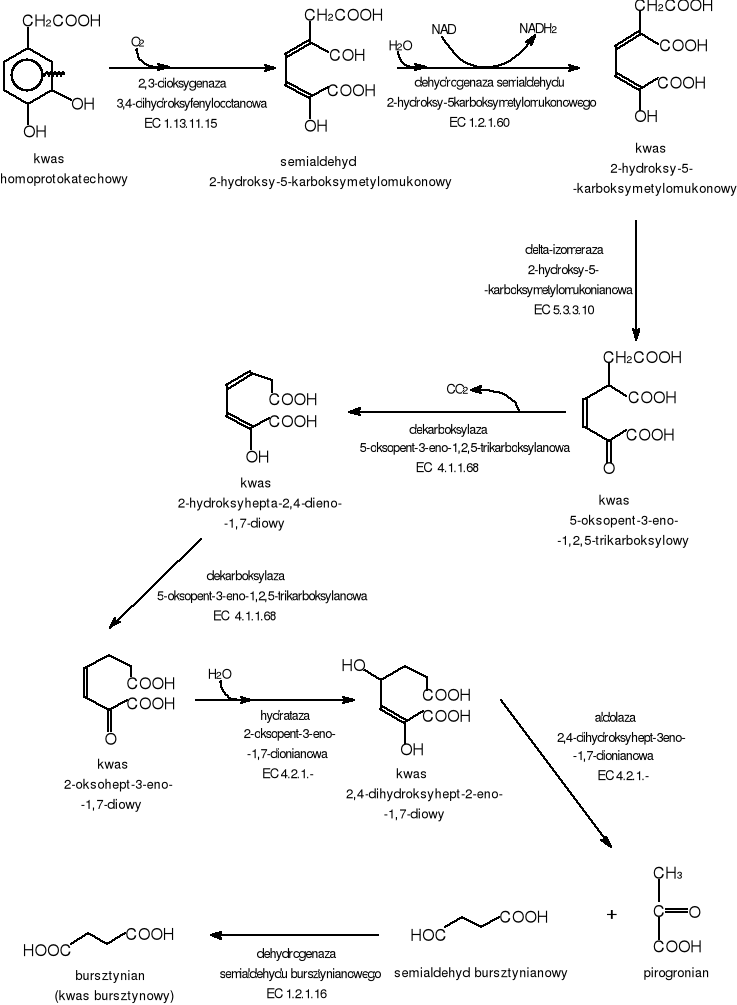
Rys.15 Szlak biodegradacji kwasu homoprotokatechowgo.
6. Enzymy biorące udział w biodegradacji związków aromatycznych
Przy biodegradacji związków aromatycznych największy udział mają następujące klasy enzymów: monooksygenazy, dioksygenazy, dehydrogenazy, hydrolazy i liazy.
Monooksygenazy
Monooksygenazy są to enzymy, które wbudowują jeden atom z cząsteczki tlenu. Drugi natomiast ulega redukcji do wody. Rolę dodatkowego donora wodoru pełni w przypadku tych enzymów NADH lub NADPH. Rozpoczęcie procesu degradacji jest możliwe dzięki działaniu monooksygenaz. Mikroorganizmy wykorzystują te enzymy w celu przekształcenia węglowodorów do odpowiednich alkoholi [101].
Dużą rolę przy biodegradacji związków aromatycznych mają monooksygenazy flawoproteinowe. Katalizują one reakcje, w których jeden atom z cząsteczki tlenu jest wprowadzany do cząsteczki substratu, dając pochodną hydroksylową, a drugi do cząsteczki wody [41].
Dioksygenazy
Dioksygenazy natomiast są enzymami, które katalizują reakcje wprowadzania obu atomów tlenu z cząsteczki substratu. Przypuszczalnie enzymy te zawierają w swej cząsteczce jon metalu powodujący aktywację cząsteczki tlenu i substratu, dzięki czemu możliwe jest ich połączenie [102].
Większość dioksygenaz wymaga obecności jonów żelaza związanych w układzie niehemowym, ale nie tworzących też standardowych kompleksów Fe/S. Zaledwie kilka enzymów z tej grupy zawiera hem jako grupę prostetyczną, dwa są flawoproteinami, a jeden miedzioenzymem [41]. Dioksygenazy odgrywają dużą rolę przy biodegradacji związków aromatycznych gdyż katalizują reakcje rozszczepienia pierścienia aromatycznego co umożliwia dalszą biodegradacje tych związków. Mechanizm reakcji katalizowanych przez diooksygenazy są jednak jeszcze mało poznane i mają charakter wysoce spekulatywny.
Hydrolazy
Są to enzymy rozkładające substrat hydrolitycznie, z jednoczesnym przyłączeniem cząsteczki wody. Zazwyczaj są to białka proste przeprowadzające reakcje rozpadu z udziałem wody. Enzymy te rozkładają wiązania w cząsteczkach używając wody - (hydroliza wiązań peptydowych, glikozydowych, estrowych) [54].
Reakcje hydrolizy katalizowane przez hydrolazy są specyficznym przypadkiem przenoszenia grup funkcyjnych z cząsteczki donora do cząsteczki akceptora, którym jest woda. Reakcje katalizowane przez te enzymy a w szczególności reakcje hydrolizy wiązania peptydowego, należą do najlepiej zbadanych reakcji enzymatycznych [41].
Do tej grupy należą endopeptydazy, czyli enzymy proteolityczne rozszczepiające białka, lipazy czyli enzymy lipolityczne rozkładające tłuszcze, ureazy rozkładające mocznik na amoniak i dwutlenek węgla. Spośród hydrolaz można także wyróżnić następujące grupy: esterazy, glikozylohydrolazy, peptydazy. W biodegradacji węglowodorów największe znaczenie mają esterazy, które rozszczepiają wiązanie estrowe substratu, a jako produkty uzyskujemy alkohole i kwasy [103]. Dzięki hydrolazom zostają skrócone łańcuchy cząsteczek węglowodorów długołańcuchowych, które po dalszych przemianach mogą być włączane w szlaki metabolizmu centralnego.
Liazy
Liazy katalizują inne niż hydrolityczne i utleniające, rozerwanie wiązań pojedynczych w związkach organicznych. W wyniku tych reakcji powstają dwa produkty. Jednym z nich jest najczęściej dwutlenek węgla, amoniak, woda lub cząsteczka aldehydu, które odrywają się z cząsteczki substratu, powodując powstanie w nim wiązania podwójnego lub układu cyklicznego. Tak więc drugim produktem końcowym reakcji jest związek nienasycony lub cykliczny. Produkt nienasycony nie musi być produktem końcowym reakcji, a może być produktem pośrednim jak na przykład ma to miejsce w przypadku dekarboksylaz [41].
Dehydrogenazy
Mianem dehydrogenaz określa się grupę enzymów z klasy oksydoreduktaz. Enzymy te odszczepiają od utlenianego substratu 2 atomy wodoru i przyłączają do różnych przenośników. Rozróżnia się: dehydrogenazy współdziałające z NAD lub NADP (dostarczają zredukowanego NAD do łańcucha oddechowego), dehydrogenazy związane z flawinami (flawoproteiny) i współdziałające z kwasem liponowym. Dehydrogenazy charakteryzuje wysoka specyficzność substratowa. Dzięki możliwości przenoszenia atomów wodoru między dwoma substratami, jeden z nich ulega utlenieniu drugi redukcji. Ten typ reakcji wykorzystywany jest w warunkach beztlenowych [40].
Obecność dehydrogenaz jest niezbędna przy degradacji węglowodorów. Pod wpływem tych enzymów I-rzędowe alkohole utleniane są do aldehydów i kwasów. Alkohole II-rzędowe do ketonów i estrów. Dehydrogenazy degradujące węglowodory zależne są od NAD.
6. Podsumowanie
Badania nad biodegradacją ksenobiotyków mają w ostatnim czasie duże znaczenie, ponieważ zanieczyszczenie środowiska naturalnego nieustannie postępuje. Istnieje pilna konieczność eliminacji skażeń środowiskowych, gdyż stanowią one ogromne zagrożenie dla roślin, zwierząt i ludzi, wywołując niejednokrotnie niebezpieczne choroby i zakłócenie równowagi biologicznej.
Duże znaczenie w usuwaniu ksenobiotyków ze środowiska naturalnego uzyskały w ostatnich latach metody biologiczno-biochemiczne, które likwidują zagrożenia w sposób ostateczny, a jednocześnie nie wymagają dużych nakładów finansowych. Rozwój nauki i głębsze poznanie biochemicznych mechanizmów związanych z unieszkodliwianiem organicznych odpadów przez różne drobnoustroje umożliwiają zastosowanie wyspecjalizowanych bakterii, grzybów i roślin do walki z odpadami. W przyrodzie występuje ogromna ilość mikroorganizmów, które są przystosowane do bytowania w różnorodnych warunkach. Dlatego też uważa się, że praktycznie dla każdego ksenobiotyku można znaleźć, odpowiednie drobnoustroje zdolne do jego biodegradacji lub detoksyfikacji. Przy dobieraniu mikroorganizmu należy zwrócić uwagę przede wszystkim na zasób enzymów oraz ich specyficzność oraz odporność na mogące pojawić się w procesie degradacji substancje mające w stosunku do tego enzymu działanie inhibitujące.
Szerokie wykorzystanie metod biologicznych w walce z zanieczyszczeniami w środowisku naturalnym (w wodach, glebach i powietrzu) wiąże się z wieloma korzystnymi czynnikami, z których najistotniejsze są:
bezpośrednia degradacja zanieczyszczenia (ksenobiotyku), a nie jego transfer pomiędzy różnymi mediami, jak to często dzieje się w przypadku metod fizykochemicznych.
wykorzystanie szlaków i cykli metabolicznych mikroorganizmów prowadzących do końcowych produktów przemian ksenobiotyku tj. do H2O i CO2.
źródłem energii potrzebnej do unieszkodliwienia zanieczyszczeń są one same.
możliwe jest zastosowanie bioremediacji zanieczyszczeń in situ, czyli w miejscu zanieczyszczenia, co nie prowadzi do dewastacji krajobrazu.
Stwierdzić jednak należy, iż niektóre biochemiczne mechanizmy biodegradacji związków aromatycznych przez drobnoustroje nie zostały jeszcze do końca poznane. Działanie i rola niektórych enzymów również stanowi w wielu przypadkach niewiadomą i nie zostały one jeszcze sklasyfikowane. Mechanizmem jeszcze nie do końca wyjaśnionym jest na przykład biodegradacja związków aromatycznych w warunkach beztlenowych.
Badania nad procesami biodegradacji związków aromatycznych są nadal w toku. Cały czas poznawane są nowe biochemiczne mechanizmy, odnajdywane są nowe szczepy drobnoustrojów mających zdolność wytwarzania wielu enzymów niezbędnych prowadzenia tych przemian. Dzięki tym badaniom dziedzina ta nieustannie się rozwija dając coraz to szersze możliwości wykorzystywane w ochronie środowiska i nie tylko.
7. Literatura.
Kot - Wasik A., Namieśnik J.: Chem. Inż. Ekol. 8, 867 (2001).
Walton B.T., Anderson T.A.: Chemosphere, 17,1501 (1988)
Pehkonen S.O., Zhang Q.: Crit. Rev. Environ. Sci. Technol., 32, 17 (2002)
MeeKyung K, O'Keefe P.W., Chemosphere, 41, 793 (2000)
Coats J.R.: Chemtech., 25 (1993)
Mastalerz P.: Chemia organiczna, 209, Warszawa (1986)
Prince R.C., Drake E.N.,: Transformation and fate of polycyclic aromatic hydrocarbons in soil, in: Bioremediation of Contaminated Soils, Argonomy Monograph vol. 37, 677, S. Segoe Rd., Madison USA 1999.
Gotvajn A.Z., Zagorec-Koncan J.: Wat. Sci. Tech., 39, 375 (1999).
Stryer L.: Biochemia, Wydawnictwo Naukowe PWN, Warszawa 1997.
Kwapisz E.: 1999. Problemy biodegradcji węglowodorów ropy naftowej. VI Ogółnopolskie Sympozjum Naukowo-Techniczne „Biotechnologia Środowiska”.
Rehm J., Reed G.: Biotechnology vol. 6,8. (1996)
Schlegel H.G.: Mikrobiologia ogólna (2003).
Evans W.C.: J. Gen. Microbiol. 32, 177-184 (1963).
Schlegel H.: Mikrobiologia ogólna 528-535 (1993)
Remediacja i bioremedjacja zanieczyszczonych wód i gruntów oraz wykorzystanie modelowania i technik informatycznych w inżynierii środowiska. Pod redakcją A. Olszanowskiego, M. M. Sozańskiego, A. Urbaniaka, A. Voelkela, Wydawnictwo Politechniki Poznańskiej, Poznań 2001. 85-99
Chapman P.J., Dagley S. J. Gen. Microbiol.(1962) 28 ,251-256
Nishino S.F., Spain J.C., (1993), Appl. Environ. Microbiol. 59, 2520-2525
Lendenmann U., Spain J.C., (1996), J. Bacteriol. 178, 6227-6232
He Z., Spain J.C., (1998), J. Bacteriol. 180, 2502-2506
Katsivela E., Wray V., Pieper D., Wittich R-M., (1999), Appl. Environ. Microbiol. 65, 1405-1412
Nishino S.F., Spain J.C., (1995), Appl. Environ. Microbiol. 61, 2308-2313
Que L., (1978), Biochem. Biophys. Res. Commun. 84, 123-129
He Z., Spain J.C., (1997), Appl. Environ. Microbiol. 63, 4839-4843
Sala-Trepat J.M., Evans W.Ch., (1971), Eur. J. Biochem. 20, 400-413
Harayama S., Rekik M., Ngai K-L., Ornstor L.N., (1989), J. Bacteriol. 171, 6251-6258
Hayaishi O., Stanier R.Y., (1952) , J. Biol. Chem. 195, 735-740
Nowa Encyklopedia Powszechna PWN, tom 1,Wyd.Naukowe PWN, W-wa 1995, s.181
Ichihara A., Adachi K., Hosokawa K., Takeda Y., (1962), J. Biol. Chem. 237, 2296-2302
Kobayashi S., Hayaishi O., (1970), Methods Enzymol. 17A, 505-510
Kobayashi S., Kuno s., Itasa S., Hayaishi O., (1964), Biochem. Biophys. Res. Commun. 16, 556-561
Subba Rao P.V., Moore K., Towers G.H.N., (1967), Biochem. Biophys. Res. Commun. 28, 1008-1012
Anderson J.J., Dagley S., (1981), J. Bacteriol. 146 291-297
Subramanian V., Vaidyanathan C.S., (1984), J. Bacteriol. 160, 651-655
Premkumar R., Sreeleela N.S., Subba Rao P.V., Vaidyanathan C.S., (1973), J. Bacteriol. 113, 1213-1216
Floss H.G., Guenther H., Groeger D., Erge D., (1969), Arch. Biochem. Biophys. 131, 319-324
Subba Rao P.V., Moore K., Towers G.H.N., (1967), Arch. Biochem. Biophys. 122, 466-470
Ramachandran A., Subramanian V., Sugumaran M., Vaidyanathan C.S., (1979), FEMS Microbiol. Lett. 5, 421-42
Ribbons D.W., (1966) , Biochem J. 99, 30P
Ellis L.B.M., Hershberger C.D., Bryan E.M., Wackett L.P.: Nucleic Acids Res., 29, 340 (2001)
Kłyszejko-Stefanowicz L. 1982. Ćwiczenia z biochemii.
Kafarski P., Lejczak B.: Chemia bioorganiczna.,79,140,149 Wydawnictwo Naukowe PWN Warszawa 1994.
http://www.mlyniec.gda.pl/~chemia/organiczna/areny.htm
http://www.psse-krakow.internetdsl.pl/pdf/weglowodory-aromatyczne-2005-01-31.pdf
http://www.wsse.krakow.pl/Files/Attachments/phpObN8Xl_WWA.doc
http://www.dioksyny.pl/files/Informacje_o_dioksynach.pdf
http://www.ochronaprzyrody.republika.pl/zanie_powietrza.html
http://levis.sggw.waw.pl/~ozw1/zintegrowgospwod/ZintergrowanagospwodOZW20/jakoscwod/rodz_zan_fenole.html
htttp://levis.sggw.waw.pl/~ozw1/zintegrowgospwod/ZintergrowanagospwodOZW20/jakoscwod/rodz_zan_aromatyczne.html
http://levis.sggw.waw.pl/~ozw1/zintegrowgospwod/ZintergrowanagospwodOZW20/jakoscwod/rodz_zan_pestycydy.html
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.1.1.63
http://biocyc.org/META/NEW-MAGE?type=PATHWAY&object=2AMINOBENZDEG-PWY
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=134886
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=245745
http://www.pmp.p-net.pl/biologia/enzymy.htm
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.13.11.1
http://www.genome.ad.jp/dbget-bin/www_bget?ec:5.5.1.1
http://www.genome.ad.jp/dbget-bin/www_bget?ec:5.3.3.4
http://www.genome.ad.jp/dbget-bin/www_bget?ec:3.1.1.24
http://www.genome.ad.jp/dbget-bin/www_bget?ec:2.8.3.6
http://www.genome.ad.jp/dbget-bin/www_bget?ec:2.3.1.16
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.13.11.2
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.2.1.3
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.1.1.3
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.2.1.80
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.1.2.18
http://www.genome.ad.jp/dbget-bin/www_bget?ec:3.7.1.9
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.13.11.3
http://www.genome.ad.jp/dbget-bin/www_bget?ec:5.5.1.2
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.1.1.44
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.13.11.8
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.2.1.45
http://www.genome.ad.jp/dbget-bin/www_bget?ec:3.1.1.57
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.1.3.17
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.13.11.4
http://www.genome.ad.jp/dbget-bin/www_bget?ec:5.2.1.4
http://www.genome.ad.jp/dbget-bin/www_bget?ec:3.7.1.5
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.13.11.5
http://www.genome.ad.jp/dbget-bin/www_bget?ec:5.2.1.2
http://www.genome.ad.jp/dbget-bin/www_bget?ec:3.7.1.2
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.2.3.1
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.2.1.29
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.2.1.32
http://www.genome.ad.jp/dbget-bin/www_bget?ec:3.5.99.5
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.14.12.1
http://www.chem.qmul.ac.uk/iubmb/enzyme/EC1/14/13/35.html
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.1.1.46
http://www.genome.ad.jp/dbget-bin/www_bget?ec:6.2.1.32
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.14.13.40
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.14.13.1
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.13.11.15
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.2.1.60
http://www.genome.ad.jp/dbget-bin/www_bget?ec:5.3.3.10
http://www.genome.ad.jp/dbget-bin/www_bget?ec:4.1.1.68
http://www.genome.ad.jp/dbget-bin/www_bget?ec:1.2.1.16
http://biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=P183-PWY
http://biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=CATECHOL-ORTHO-CLEAVAGE-PWY
http://biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=P184-PWY
http://biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=PROTOCATECHUATE-ORTHO-CLEAVAGE-PWY
http://www.genome.ad.jp/dbget-bin/show_pathway?MAP00350+C00628
http://umbbd.ahc.umn.edu/
Murray R.K.: Biochemia Harpera, Wydawnictwo Lekarskie PZWL, Warszawa 1995.
Schlegel H.G.: Mikrobiologia ogólna, pod red. nauk. Zdzisłąwa Markiewicza, Wydawnictwo Naukowe PWN, 1996.
Filipowicz B.: Biochemia dziś i jutro, Państwowe Wydawnictwo Naukowe, Warszawa 1973.
30
Wyszukiwarka
Podobne podstrony:
Główne szlaki biodegradacji związków aromatycznych, Naukowe PL, Biotechnologia, Enzymologia, Genetyk
Biodegradacja węglowodorów aromatycznych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Biodegradacja nitrowych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Bakteriocyny i L-antybiityki, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Rośliny transgeniczne, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Lipazy, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Biodegradacja heterocyklicznych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
Biodegradacja halogenopochodnych, Naukowe PL, Biotechnologia, Enzymologia, Genetyka
ZWIAZKI AROMATYCZNE
notatek pl , Biotechnologie, BAKTERIE LAB
Witaminy, Naukowe PL, Witaminy
Biotechnologia i inżynieria genetyczna
Zwiazki aromatyczne 09
IZOL.DNA Z ŻELI, Biotechnologia notatki, Genetyka - biologia molekularna
fermentacja beztlenowa 2014-2015, Biotechnologia PŁ, Biotechnologia środowiska
Cw 5 Zwiazki aromatyczne
więcej podobnych podstron