Nitrowanie węglowodorów alifatycznych (alkanów)
WSTĘP - reakcje rodnikowe:
W odróżnieniu od nitrowania węglowodorów aromatycznych reakcja kwasu azotowego z alkanami ma charakter procesu rodnikowego. Jedną z najważniejszych różnic między reakcjami rodnikowymi a jonowymi jest to, że rodniki istnieją z reguły tylko w bardzo małych stężeniach. Aniony i kationy mogą koegzystować w dużych stężeniach dzięki stabilizacji solwatacyjnej. Rodniki są solwatowane w dużo mniejszym stopniu i dlatego dążą do rekombinacji z utworzeniem wiązania kowalencyjnego. W konsekwencji większość reakcji rodnikowych posiada etap indukcji oraz wymaga inicjacji, czyli wstępnego wytworzenia rodników. Inicjacja zachodzi najczęściej przez termiczną lub fotochemiczną homolizę słabego wiązania. Najpopularniejszymi źródłami wolnych rodników są nadtlenki (np. nadtlenek wodoru, nadtlenki alkilowe, acylowe) lub chlorowce, których cząsteczki rozpadają się pod wpływem światła.
Drugi etap polega na zaniku wolnych rodników. Zachodzi on w reakcji odwrotnej do etapu pierwszego, tzn. przez rekombinację dwóch jednakowych lub różnych rodników z utworzeniem wiązania kowalencyjnego.
Zakończenie reakcji nie następuje bezpośrednio po jej zainicjowaniu. Dzieje się tak, ponieważ większość rodników jest bardzo reaktywna i dlatego mogą one reagować z pierwszym dostępnym indywiduum, z którym wejdą w kontakt. W zwykłej sytuacji, gdy stężenie wolnych rodników jest małe, bardziej prawdopodobne jest, że indywiduum tym będzie cząsteczka, a nie inny wolny rodnik. W reakcji wolnego rodnika (nieparzysta liczba elektronów) i cząsteczki (parzysta liczba elektronów) musi powstać produkt zawierający nieparzysta liczbę elektronów czyli nowy rodnik.
Powstały wolny rodnik może reagować z następną cząsteczką wytwarzając kolejny rodnik itd., aż do chwili, gdy dwa rodniki spotkają się ze sobą i zakończą proces. Taka reakcja nosi nazwę reakcji łańcuchowej. Pomiędzy zainicjowaniem, a zakończeniem łańcucha mogą zachodzić setki lub tysiące etapów wzrostu. Gdy wolne rodniki są bardzo reaktywne (np. rodniki alkilowe), to reakcje mogą przebiegać z wieloma cząsteczkami i wtedy łańcuchy są długie. W każdej reakcji łańcuchowej zachodzi zwykle wiele różnych etapów wzrostu i różnych zakończeń. Dlatego powstaje w nich wiele różnorodnych produktów.
NITROWANIE ALKANÓW:
Węglowodory alifatyczne łatwo ulegają nitrowaniu w fazie ciekłej lub gazowej przy działaniu na nie kwasem azotowym w podwyższonej temperaturze. Mechanizm tej reakcji był przedmiotem wielu badań, jednak dotychczas uzgodniono tylko pogląd, że ma ona charakter wolnorodnikowej reakcji łańcuchowej, której szybkość limitowana jest etapem zainicjowania łańcucha. W związku z tym zachodzi przez cały szereg stadiów, komplikowanych dodatkowo utlenianiem węglowodorów. Opracowano kilkanaście schematów przebiegu reakcji, które są zgodne z danymi kinetycznymi. Jeden z tych schematów (uwzględniających tylko zasadnicze etapy) zakłada, że czynnikiem nitrującym jest w tych warunkach cząsteczka dwutlenku azotu posiadająca niesparowany elektron i będąca wolnym rodnikiem. Oddziaływanie *NO2 z alkanem prowadzi do wytworzenia rodnika alkilowego, który może bardzo szybko rekombinować z następną cząsteczką dwutlenku azotu z utworzeniem pochodnej nitrowej.
Jednocześnie zachodzi regeneracja dwutlenku azotu:
Na podkreślenie zasługuje fakt, że łatwość podstawienia wodoru grupą nitrową rośnie w szeregu:
Potwierdza to wolnorodnikowy mechanizm nitrowania alkanów, bowiem w tym samym kierunku rośnie stabilność rodników alkilowych (dzięki delokalizacji niesparowanego elektronu).
Reakcji nitrowania towarzyszy reakcja uboczna prowadząca do wytworzenia azotynu alkanu na skutek przeniesienia aktywnego centrum w *NO2 z atomu azotu na atom tlenu.
Powstały ester kwasu azotawego jest mało stabilny i wstępuje w szereg reakcji prowadzących do złożonych produktów:
Ostatecznie działanie kwasu azotowego na alkany prowadzi jednocześnie do tworzenia znacznych ilości produktów utlenienia (złożone etery, alkeny, aldehydy, ketony, kwasy karboksylowe, alkohole). Ich wydajność nierzadko jest wyższa niż wydajność pochodnej nitrowej. Sytuacja taka ma miejsce zwłaszcza wtedy gdy proces prowadzony jest w wysokiej temperaturze, ponieważ współczynnik temperaturowy szybkości reakcji utlenienia jest większy niż reakcji nitrowania.
Prowadzenie procesu nitrowania w fazie gazowej (temperatura procesu 350÷500 oC) sprzyja intensywnemu rozkładowi kwasu azotowego, a także i destrukcji alkanów. W tym przypadku, obok pochodnej nitrowej wyjściowego alkanu tworzą się również pochodne nitrowe wszystkich produktów jego destrukcji. Jeden z możliwych schematów przebiegu reakcji nitrowania alkanów w fazie gazowej jest następujący:
a) nitrowanie;
b) nitrowanie destrukcyjne
W ten sposób przy nitrowaniu wyższych alkanów w fazie gazowej uzyskuje się pochodne nitrowe produktów jego destrukcji. Skład produktów nitrowania zależy od stosunku reagentów, czasu trwania reakcji i temperatury procesu. Wyizolowanie z mieszaniny poreakcyjnej poszczególnych nitroalkanów przeprowadza się na drodze rektyfikacji pod obniżonym ciśnieniem.
Reasumując, można stwierdzić, że reakcja bezpośredniego nitrowania alkanów zachodzi według mechanizmu substytucji rodnikowej. Etapem decydującym o szybkości reakcji jest etap zainicjowania łańcucha. W związku z tym reakcja ta jest inicjowana i przyśpieszana przez typowe źródła wolnych rodników. Przebieg reakcji w fazie ciekłej i gazowej jest bardzo podobny. Podwyższenie temperatury procesu prowadzi do pojawienia się pochodnych nitrowych niższych alkanów będących produktami destrukcji (krakingu) wyjściowego alkanu. Bezpośrednie nitrowanie alkanów prowadzi tylko do związków mononitrowych. Polinitrowe pochodne szeregu alifatycznego otrzymuje się metodami pośrednimi, wychodząc zwykle ze związków mononitrowych.
Wygodnym, laboratoryjnym sposobem otrzymywania pierwszo- i drugorzędowych mononitroalkanów oraz polinitroalkanów z izolowanymi grupami nitrowymi jest reakcja odpowiednich halogenopochodnych alkilowych z azotynem srebra lub azotynem sodu (reakcja Meyera). Reakcja ta zachodzi według mechanizmu substytucji nukleofilowej z przejściowym utworzeniem anionu karboniowego.
Produktami ubocznymi są azotyny alkilowe, powstające w zmiennych ilościach, zależnie od budowy substratów i warunków reakcji. Możliwość dwukierunkowego przebiegu reakcji wyjaśnia budowa elektronowa anionu azotynowego, w którym charakter nukleofilowy posiadają zarówno atomy tlenu jak i atom azotu. Reakcję halogenopochodnych alkilowych z AgNO2 przeprowadza się najczęściej w środowisku bezwodnego eteru dietylowego, natomiast w przypadku wykorzystywania NaNO2 optymalnym rozpuszczalnikiem jest dimetyloformamid. Wydajność pierwszorzędowych nitroalkanów przy zastosowaniu azotynu srebra jako reagenta dochodzi do 80 % wydajności teoretycznej. W przypadku syntezy drugorzędowych nitroalkanów korzystniejsze jest stosowanie azotynu sodu.
Alifatyczne polintrozwiązki można otrzymać poprzez addycję tlenków azotu do olefin. Reakcje te mają jednak ograniczone znaczenie preparatywne, ponieważ prowadzą w większości przypadków do złożonych mieszanin, zawierających poza pochodnymi nitrowymi także produkty utlenienia olefin.
Dogodną metodą syntezy gem-dinitroalkanów jest reakcja 1-nitro-1-halogenoalkanów z azotynem sodu w środowisku zasadowym (reakcja Ter Meera).
Reakcja ta może być także wykorzystana do otrzymywania tetranitroalkanów.
Reakcja Ter Meera zachodzi przez stadium izomeryzacji chloronitroalkanu do jego formy aci, po którym następuje nukleofilowe podstawienie chloru jonem azotynowym.
Mechanizm ten tłumaczy nieaktywność drugorzędowych nitrochlorowcoalkanów w reakcji Ter Meera (brak wodoru w pozycji α względem grupy NO2).
Nitrowanie amin aromatycznych i alifatycznych
WSTĘP:
Nitrowe pochodne amin aromatycznych otrzymuje się na drodze nitrowania odpowiednich amin, w których grupa nitrowa może podstawiać zarówno wodór związany z węglem ≡≡CH _→ C_NO2, jak i wodór związany z azotem grupy aminowej ==NH _→ N_NO2. W pierwszym przypadku powstaje związek C-nitrowy, a w drugim N-nitrowy. Przy bezpośrednim nitrowaniu produktem reakcji jest mieszanina związków C-nitro i N-nitro, ponieważ warunki ich tworzenia są bardzo podobne.
MECHANIZM:
Czynnikiem nitrującym w procesie bezpośredniego N-nitrowania amin aromatycznych za pomocą stężonego kwasu azotowego lub klasycznej mieszaniny nitrującej (HNO3/H2SO4), jest kation nitroniowy. Podobnie jak przy nitrowaniu arenów w pierścieniu aromatycznym, również w tym przypadku reakcja zachodzi według mechanizmu substytucji elektrofilowej, zawierającego dwa zasadnicze etapy. Najpierw na skutek elektrofilowego ataku kationu nitroniowego na wolną parę elektronową azotu grupy aminowej, następuje jego przyłączenie i utworzenie przejściowego kationu amoniowego (ładunek dodatni zlokalizowany na azocie grupy aminowej).
Następnie, produkt przyłączenia stabilizuje się w reakcji deprotonowania zachodzącej z udziałem obecnej w środowisku reakcji cząsteczki zasady B-.
Równolegle z N-nitrowaniem zachodzi intensywne nitrowanie pierścienia aromatycznego,
ponieważ grupa aminowa jako jeden z najsilniejszych podstawników elektrodonorowych, aktywuje pierścień na substytucję elektrofilową. Wprowadzenie grupy nitrowej do pierścienia aromatycznego następuje zarówno wskutek bezpośredniego C-nitrowania, jak i w rezultacie przegrupowania nitroaminowego, którego mechanizm omówiłem na poprzednim wykładzie. Według schematu zapisanego prezentowanymi reakcjami reagują również trzeciorzędowe aminy alifatyczno-aromatyczne. Wówczas odszczepieniu i utlenieniu ulega jedna z grup alkilowych związanych z azotem grupy aminowej.
Aktywujący wpływ grupy aminowej na konkurencyjne procesy utlenienia substratu w warunkach nitrowania może być ograniczony przez wstępne zablokowanie jej grupą acylową _COCH3 lub przez przeprowadzenie jej do postaci znacznie stabilniejszej soli kwasu siarkowego. Uzyskiwanie w tym ostatnim przypadku produktów C-nitrowania pierścienia w położeniu orto i para wykazuje, że nitrowaniu ulega wolna postać aminy znajdująca się w równowadze z jej formą protonowaną.
Gdyby C-nitrowaniu ulegał wodorosiarczan aminy wówczas w produktach powinien występować w znacznych ilościach izomer meta, ponieważ grupa amoniowa kieruje atak elektrofilowy właśnie w tę pozycję.
W niektórych przypadkach N-nitrowanie amin aromatycznych można przeprowadzić przez stadium tworzenia azotanu aminy zachodzące w wyniku oddziaływania na aminę rozcieńczonym kwasem azotowym.
Powstały azotan aminy, w wyniku działania stężonego kwasu azotowego lub bezwodnika octowego ulega dehydratacji z utworzeniem pochodnej N-nitrowej.
NITROWANIE AMIN ALIFATYCZNYCH:
Bezpośrednie N-nitrowanie amin alifatycznych za pomocą stężonego kwasu azotowego lub jego mieszanin z kwasem siarkowym jest możliwe tylko w przypadku amin o słabo zaznaczonych właściwościach zasadowych. W tym przypadku nitrowanie zachodzi według znanego już mechanizmu, zawierającego elektrofilowy atak kationu nitroniowego na wolną parę elektronową atomu azotu z przejściowym utworzeniem kationu nitroamoniowego.
Im wyższa jest zasadowość aminy, tym większa jej część występuje w mieszaninie reakcyjnej w postaci protonowanej [R2NH2]+, która z oczywistych względów nie jest podatna na elektrofilowy atak kationu nitroniowego. Pierwszorzędowe aminy nitruje się w tych warunkach bardzo trudno, również z powodu małej stabilności monopodstawionego jonu nitroamoniowego.
oraz z powodu niestabilności tautomerycznej formy nitroaminy (izonitroaminy) w środowisku silnego kwasu.
W takich przypadkach dogodnym sposobem wprowadzania grupy nitrowej jest metoda pośrednia, polegająca na wstępnym acylowaniu aminy i następnym nitrowaniu powstałego amidu.
Powstały N-nitroamid poddaje się zasadowej hydrolizie prowadzącej do otrzymania odpowiedniej soli formy kwasowej nitroaminy. Uwolnienie nitroaminy następuje po zakwaszeniu środowiska reakcji.
Silnie zasadowe aminy ulegają nitrowaniu z wysokimi wydajnościami za pomocą mieszaniny kwasu azotowego z bezwodnikiem octowym w obecności katalizatorów takich jak HCl czy ZnCl2. Katalizowane anionami chlorkowymi nitrowanie drugorzędowych amin jest reakcją łańcuchową, którą można przedstawić schematem.
Powstająca w tej reakcji chloroamina R2NCl jest znacznie słabszą zasadą niż odpowiadająca jej amina i w związku z tym stężenie nieprotonowanej formy podatnej na elektrofilowy atak kationu NO2+, jest większe niż dla wyjściowej aminy.
Ponadto tworzenie wiązania N_N, zgodnie z powyższym schematem jest łatwiejsze jeżeli od przejściowego kationu nitroamoniowego odszczepiany jest kation Cl+ a nie H+, ponieważ wiązanie N_Cl jest słabsze od wiązania N_H.
Mocznik i jego pochodne alkilowe oraz guanidyna i jej pochodne alkilowe mogą zostać znitrowane poprzez przekształcenie ich w azotany i następną dehydratację za pomocą stężonego kwasu siarkowego.
W monoalkilomocznikach grupa nitrowa jest przyłączana do drugorzędowej grupy amidowej, natomiast pochodne nitrowe guanidyn mają budowę nitroimidową.
NITROLIZA (aminy trzeciorzędowe):
Nitrowanie amin trzeciorzędowych (w tym cyklicznych) za pomocą stężonego kwasu azotowego lub jego mieszanin z bezwodnikiem octowym prowadzi do rozerwania wiązania węgiel - azot. W rezultacie tego, obok N-nitroaminy tworzy się alkohol, ulegający następnie estryfikacji w reakcji z kwasem azotowym. Proces ten może przebiegać również z udziałem cząstek jonowych. Tworzący się wówczas kation alkilowy reaguje z anionem azotanowym dając wykrywany w produktach reakcji azotan alkilowy.
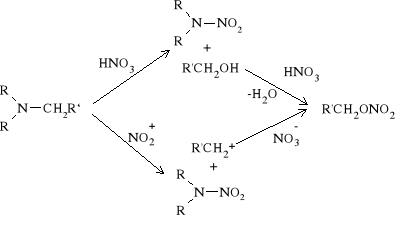
Nitrowanie przebiegające z rozerwaniem wiązania C_N nazywane jest nitrolizą. Typowym przykładem nitrolizy jest proces otrzymywania heterocyklicznych polinitroamin (heksogenu i oktogenu) poprzez oddziaływanie na heksametylenotetraaminę (urotropinę) stężonym kwasem azotowym lub jego mieszaninami z bezwodnikiem octowym. Obecnie uważa się, że nitroliza urotropiny kwasem azotowym przebiega z udziałem kationu nitroniowego. Zgodnie z tym mechanizmem rozerwanie wiązania C_N zachodzi z wytworzeniem kationu karboamoniowego;
powstającego w środowisku reakcyjnym z kationu nitroniowego solwatowanego cząsteczką aminy.
Jeżeli kation karboamoniowy przechwyci anion azotanowy NO3-, to ulega stabilizacji w postaci azotanu.
Natomiast po przechwyceniu anionu hydroksylowego tworzy hydroksymetyloaminę zdolną do reakcji z kationem nitroniowym. Powstały złożony jon:
rozpada się do nitroaminy i kationu karboksoniowego [HO_CH2]+, który po stracie protonu przekształca się do formaldehydu - jednego z produktów reakcji.
W przypadku nitrolizy prowadzonej w warunkach bezwodnych (np. w obecności bezwodnika octowego), założenie o występowaniu w środowisku reakcji anionu OH- jest trudne do przyjęcia. Dla tych reakcji zaproponowano mechanizm nitrolizy z udziałem hydratowanego kationu nitroniowego H2O⋅NO2+ .
Mechanizm ten zgadza się z rzeczywistymi danymi dotyczącymi reakcji prowadzących bezpośrednio do syntezy heksogenu i oktogenu. Jednakże nie można na jego podstawie wyjaśnić faktu tworzenia się produktów ubocznych w postaci liniowych polimetylenopolinitroamin z dwoma końcowymi grupami azotanowymi. Tworzenie się tych związków jest natomiast uzasadnione, jeżeli przyjmie się mechanizm zakładający udział w reakcji kationu nitroniowego.
Ostatecznie można stwierdzić, że istnieją dane eksperymentalne świadczące zarówno na korzyść molekularnych cząstek nitrujących jak i wolnego kationu nitroniowego. Nie można wykluczyć, że obydwa te mechanizmy występują w różnych stadiach całego procesu syntezy.
Estryfikacja alkoholi kwasem azotowym
MECHANIZM:
Estry kwasu azotowego otrzymuje się najczęściej przez estryfikację alkoholi kwasem azotowym lub jego mieszaniną z kwasem siarkowym. Reakcja estryfikacji zachodzi w tych warunkach według mechanizmu substytucji elektrofilowej, przy czym podstawieniu ulega wodór grupy hydroksylowej. W związku z tym estryfikacja może być traktowana jako O-nitrowanie. Analogicznie jak przy C- i N-nitrowaniu, cząstką nitrującą jest kation nitroniowy, a proces zachodzi przez dwa stadia. Najpierw, wskutek elektrofilowego ataku kationu nitroniowego na atom tlenu grupy hydroksylowej następuje jego przyłączenie i utworzenie przejściowego kompleksu - kationu oksoniowego.
W następnym etapie, produkt przyłączenia odszczepia kation H+ i stabilizuje się w postaci związku O-nitrowego, czyli azotanu alkilowego.
Kation nitroniowy stanowiący w myśl tego mechanizmu aktywną cząsteczkę estryfikującą tworzy się w reakcjach:
Etap tworzenia przejściowego kompleksu (kationu oksoniowego) jest etapem najwolniejszym, limitującym szybkość całego procesu. Poza tym ma on charakter reakcji odwracalnej, a zatem kation oksoniowy może się rozpadać zarówno na produkty końcowe jak i na substraty. W związku z tym, aby reakcja przebiegała w pożądanym kierunku, niezbędna jest obecność w środowisku reakcji aktywnych akceptorów protonu. Rolę tę pełnią tworzące się w tych reakcjach aniony azotanowe NO3- i wodorosiarczanowe HSO4-.
Reakcja estryfikacji zachodzi tym szybciej im większe jest stężenie aktywnych cząstek estryfikujących (obdarzonych możliwie dużym ładunkiem dodatnim) oraz im większe jest stężenie akceptorów protonu. Z tych względów mieszanina stężonych kwasów azotowego i siarkowego jest znacznie silniejszym czynnikiem estryfikującym niż czysty kwas azotowy. Należy jednak pamiętać, że kwas siarkowy nie tylko przyśpiesza estryfikację. Przy określonym jego nadmiarze powoduje także rozkład azotanów i ich reestryfikację.
Reaktywność alkoholi w reakcji O-nitrowania przebiegającej według mechanizmu elektrofilowego podstawienia atomu wodoru grupą nitrową, jest tym większa im większy jest ładunek ujemny zlokalizowany na atomie tlenu grupy hydroksylowej. Wielkość tego ładunku jest zdeterminowana efektami indukcyjnymi oddziaływującymi na rozkład gęstości elektronowej wzdłuż łańcucha atomów stanowiących cząsteczkę alkoholu. Obecność przy sąsiednim atomie węgla grupy elektronoakceptorowej, wywierającej ujemny efekt indukcyjny, obniża reaktywność atakowanej grupy hydroksylowej. Z tego powodu w polialkoholach, grupy hydroksylowe związane z pierwszorzędowymi atomami węgla są bardziej podatne na estryfikację niż grupy związane z węglem drugorzędowym. Na przykład mechanizm estryfikacji gliceryny może być zapisany schematem.
Otrzymanie triazotanu gliceryny (nitrogliceryny) wymaga użycia bezwodnej mieszaniny nitrującej (HNO3/H2SO4), natomiast diazotan powstaje w warunkach nitrowania gliceryny czystym kwasem azotowym.
Bezpośrednie O-nitrowanie alkoholi za pomocą mieszaniny nitrującej jest jedyną przemysłową metodą otrzymywania estrów kwasu azotowego. Reakcję prowadzi się w niskiej temperaturze, bowiem jej podwyższenie zwiększa nie tylko szybkość estryfikacji, ale również szybkość reakcji odwrotnej oraz przyśpiesza konkurencyjne procesy utleniania.
11
Wyszukiwarka
Podobne podstrony:
WYKLAD15.DOC, C - nitrowe pochodne amin aromatycznych - TNAnilina, Heksyl, TATB
WYKLAD13.DOC, Chemia i technologia nitrowych pochodnych chlorobenzenu. 2,4-chlorodinitrobenzen, trin
WYKLAD12.DOC, Temat: Nitrowe pochodne ksylenu i naftalenu.
Ćwiczenia – węglowodory alifatyczne, Studia, Biotechnologia, Chemia, Chemia organiczna, Wykłady II
WYKLAD3.DOC, Mechanizm reakcji nitrowania. Nitrowanie jako elektrofilowa substytucja w pierścieniu a
WYKLAD11.DOC, Temat: Nitrowe pochodne toluenu.
WYKLAD4.DOC, Mechanizm reakcji nitrowania.
Porównanie węglowodorów alifatycznych
Biochemia wykład 13 Metabolizm węglowodanów
ginmaterialy, gin Krzysiek1, Ginekologia - wykład, doc
Fizyka wykłady doc
SPG wyklady doc, Wrokflow WFMC OMC, Wprowadzenie
Węglowodory alifatyczne
weglowodory charakterystyka alkanow
Fizyka1 wykłady doc
Węglowodory alifatyczne 2
SPG wyklady doc, Wstęp pojecia, Systemy pracy grupowej
ginmaterialy, GIN KRZ2, Ginekologia - wykład, doc
GO, notatek pl wyklad 7 odpady torfowe weglowe wyklad
więcej podobnych podstron