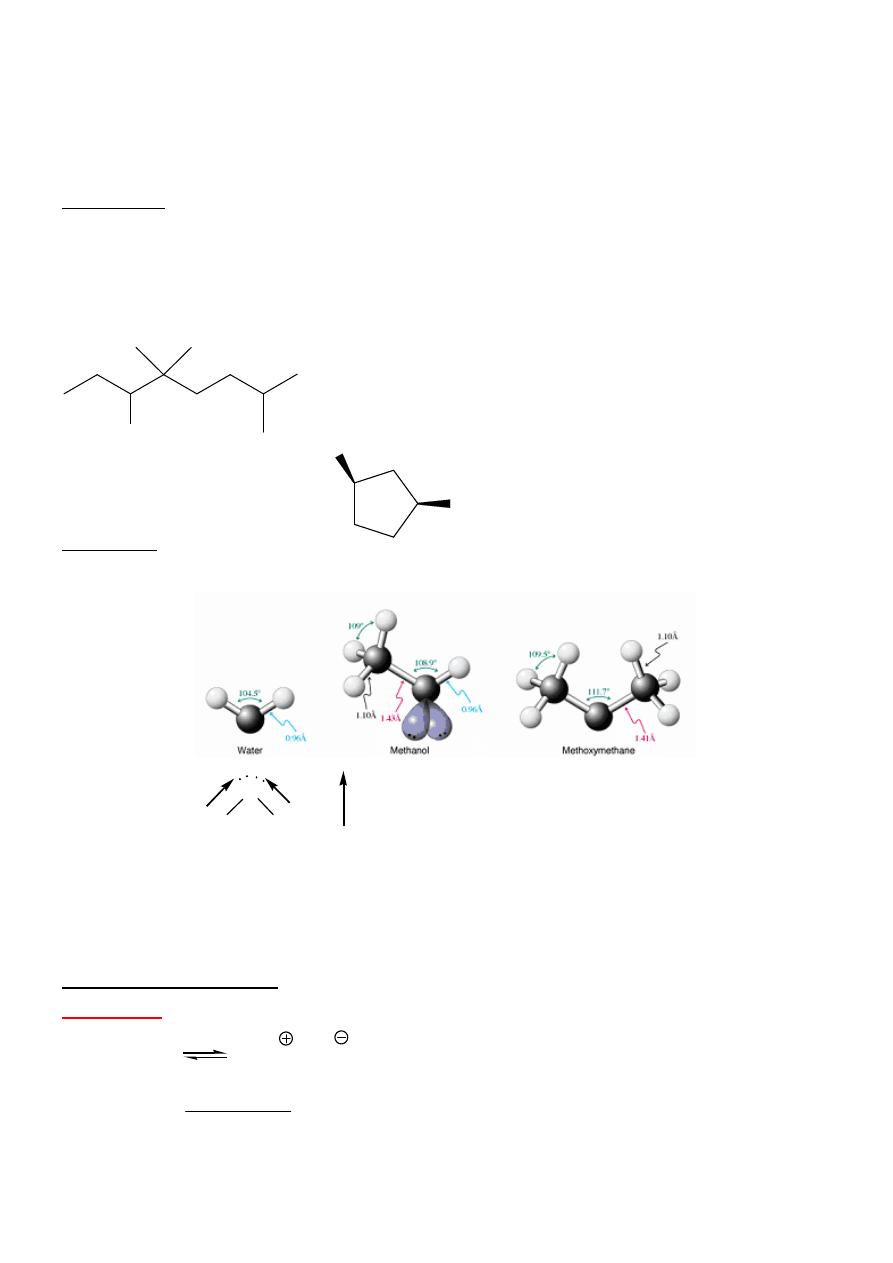
1
ALKOHOLE
R-OH: alkohol,- fenol, - enol
Nazewnictwo:
alkan
ol
- jak najni
ż
szy lokant podstawnika hydroksylowego
Alkohole 1
°
, 2
°
, 3
°
CH
3
CH
2
CH
2
OH propan-1-ol (1
°
)
CH
3
CH(OH)CH
2
CH
3
butan-2-ol = alkohol sec-butylowy (2
°
)
= 4,4,7-trimetylooktan-3-ol (2
°
)
cis-3-chlorocyklopentanol (2
°
)
Wła
ś
ciwo
ś
ci:
hybrydyzacja sp
3
O, O-H silniejsze, krótsze ni
ż
C-H
Zwi
ą
zki polarne
O
H
H
µ
Wi
ą
zania wodorowe
⇒
t.t., t.wrz., rozpuszczalno
ść
w wodzie - wy
ż
sze ni
ż
w alkanach i
halogenoalkanach. Woda - 100
°
C!
Cz
ęść
hydrofobowa i hydrofilowa
Alkohole s
ą
amfoteryczne
Kwasowo
ść
-
zbli
ż
ona do wody
ROH + H
2
O
H
3
O + RO
←
jon alkoksylowy
Ka=
[H
3
O
+
] [ RO
-
]
[ROH]
K[H
2
O] =
OH
Cl
HO
2
CH
3
OH + NaNH
2
NaOCH
3
+ NH
3
pK
a
= 15.5
pK
a
= 35
Kwasowo
ść
: 3
°
< 2
°
< 1
°
< CH
3
OH
Steryczne utrudnienie solwatacji jonu alkoksylowego:
Efekt indukcyjny
– przenoszenie ładunku przez wi
ą
zania
σ
(stabilizacja anionu)
pK
a
H
2
O
15.7
CH
3
CH
2
OH
15.9
(CH
3
)
3
COH
18.0
ClCH
2
CH
2
OH
14.3
CF
3
CH
2
OH
12.4
CF
3
CH
2
CH
2
OH
15.4
Zasadowo
ść
:
R-O-H
R-O-H
H
silny kwas
jon alkoksoniowy
[CH
3
OH
2
]
+
pK
a
= -2.2
R-O-H
R-O-H
H
silny kwas
zasada
silna zasada
kwas
RO
Metody otrzymywania alkoholi:
1. Przemysłowe
: katalityczna redukcja CO (gaz syntezowy), hydratacja etenu, 5% - fermentacja
2. Substytucja nukleofilowa halogenoalkanów:
Rzadko, cz
ęś
ciej odwrotnie; uboczna reakcja eliminacji (E2)
CH
3
CH
2
CH
2
CH
2
Br
NaOH
CH
3
CH
2
CH
2
CH
2
OH + NaBr
3.
Redukcja ketonów i aldehydów
a. Wodorowanie (hydrogenacja); kataliza heterogeniczna
1
°
CH
H
3
C
H
3
C
H
2
C
H
O
H
2
, Pd-C
CH
H
3
C
H
3
C
H
2
C
C
H
2
O
H
R
C
H
O
redukcja
utlenienie
R
C
H
OH
H
1
°
R
C
R'
O
redukcja
utlenienie
R
C
H
OH
R'
2
°
3
b. Redukcja wodorkami
C
O
+δ
−δ
NaBH
4
, LiAlH
4
; LiH, NaH – gorzej rozpuszczalne
H
C
O
+δ
−δ
+
H
OCH
2
CH
3
H
C
O
H
+
NaH
3
B(OCH
2
CH
3
)
NaH
3
B
4. Synteza z u
ż
yciem zwi
ą
zków organometalicznych
Zwi
ą
zki organometaliczne
:
CH
3
Br + Li
Et
2
O, 0
°
CH
3
Li + LiBr
(CH
3
)
2
CHI + Mg
(CH
3
)
2
CH
MgI
+ LiBr
THF
jodek 1-metyloetylomagnezowy
Victor Grignard – zw. magnezoorganiczne = zw. Grignarda
R
X + Mg
(CH
3
CH
2
)
2
O
R
Mg
X
CH
3
CH
2
OCH
2
CH
3
CH
3
CH
2
OCH
2
CH
3
R
M
+δ
−δ
+ H-OH
R-H + M-OH
M - metal
Halogenoalkan
→
alkan:
Mg
HOH
R-Br R-Mg-Br R-H
Aplikacja – otrzymywanie
deuteropochodnych:
Zwi
ą
zki metaloorganiczne w syntezie alkoholi:
O
C
δ−
+
M
R
δ−
δ
+δ
C
R
O
-
M
+
C
R
OH
aldehyd mrówkowy
→
1
°°°°
alkohole
(CH
3
)
3
C-Cl
1. Mg
2. D
2
O
(CH
3
)
3
C-D
4
CH
3
(CH
2
)
2
CH
2
MgBr + H
2
C O
Et
2
O
CH
3
(CH
2
)
2
CH
2
-C-OMgBr
H
H
H
+
, H
2
O
CH
3
(CH
2
)
2
CH
2
CH
2
OH
+ Mg(OH)Br
aldehydy
→
2
°°°°
alkohole
R-MgBr +
C
O
H
R'
R'
C
OMgBr
H
R
HOH
R'
C
OH
H
R
+ Mg(OH)Br
ketony
→
3
°°°°
alkohole
CH
3
(CH
2
)
2
CH
2
MgBr + CH
3
-C-CH
3
Et
2
O
H
+
, H
2
O
CH
3
(CH
2
)
2
CH
2
COH
CH
3
CH
3
O
+ Mg(OH)Br
Reakcje z estrami i chlorkami kwasowymi
→
3° alkohole
R
C
OCH
3
O
+ R'-MgBr
R C R'
OCH
3
OMgBr
C
O
R
R'
+ Mg(OCH
3
)Br
R
C
OCH
3
O
+ R'-MgBr
R C R'
OCH
3
OMgBr
C
O
R
R'
+ Mg(OCH
3
)Br
R'MgBr
R'
H
+
, H
2
O
R'MgBr
R'
H
+
, H
2
O
3°alkohol
C
R'
R
R'
C
R'
OH
R
OMgBr
mrówczany
→
2
°°°°
alkohole
5. Redukcja kwasów/estrów:
R
C
OR'
O
1. LiAlH
4,
eter
2. H
+
, H
2
O
R-CH
2
OH
R’ = H, alkil, aryl
NaBH
4
– tylko estry
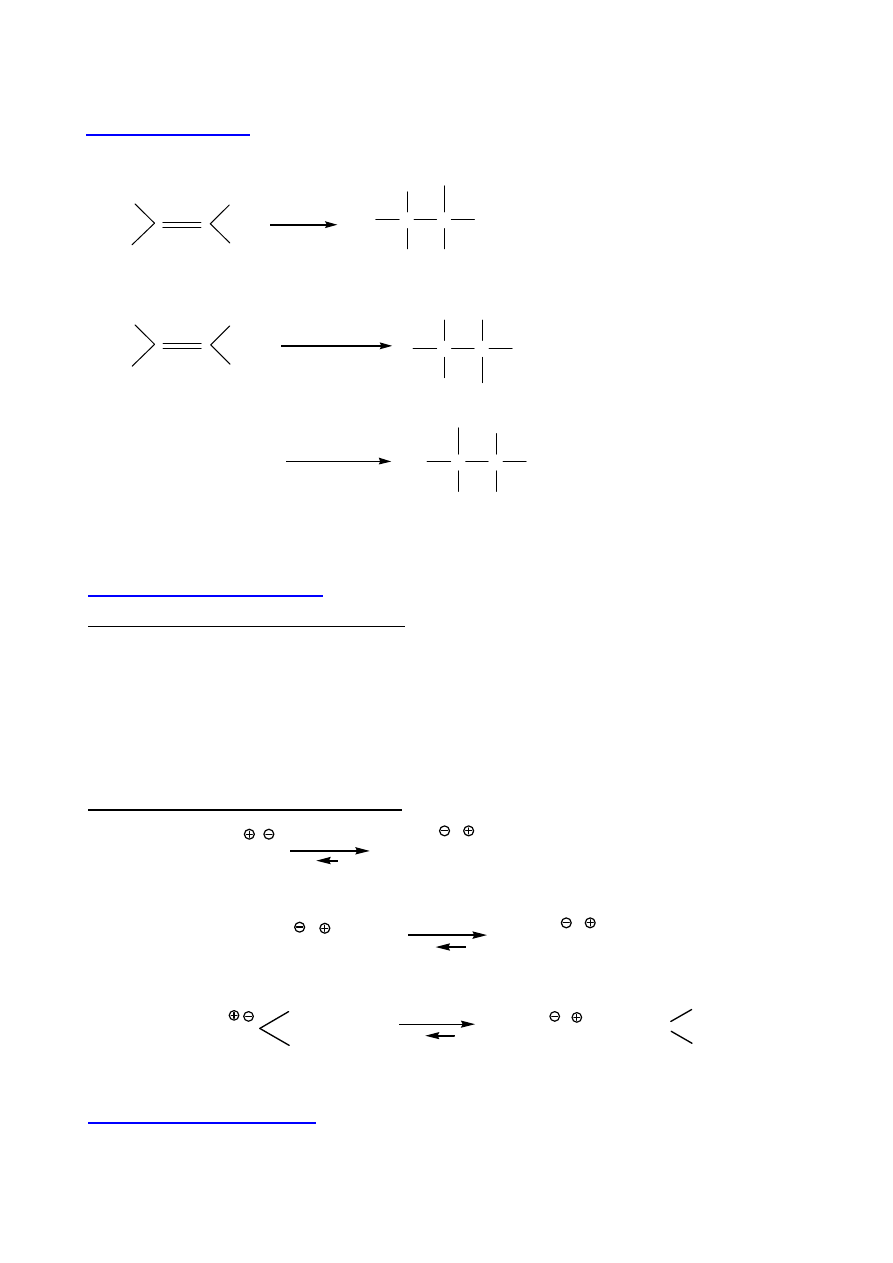
5
6. Hydratacja alkenów
a. bezpo
ś
rednia:
C C
R
H
R''
R'
H
+,
H
2
O
R
C
OH
R'
C
H
H
R''
b. po
ś
rednia
:
C C
R
H
R''
R'
R
C
OH
R'
C
R''
H
H
1. Hg(OCOCH
3
)
2
2. NaBH
4
1. BH
3
2. H
2
O
2,
OH
-
R
C
H
R'
C
OH
H
R''
REAKCJE ALKOHOLI
1.
Reakcje z silnymi zasadami:
A.
z metalami alkalicznymi (Na, Li, K, Cs)
2 H-OH + 2 Na
→
2 NaOH + H
2
2 R-OH + 2 Na
→
2 RO
-
Na
+
+ H
2
2 (CH
3
)
3
C-OH
→
2 (CH
3
)
3
CO
-
K
+
+ H
2
Reaktywno
ść
: CH
3
OH > 1
°
> 2
°
> 3
°
RO
-
M
+
- silne Nu/B ( np. + halogenki
→
etery)
B.
z silnymi zasadami (silniejsze od RO
-
): NaNH
2,
NaH, RMgX, RLi
CH
3
OH + K H
CH
3
O K + H-H
pKa = 15.5
pKa = 38
CH
3
OH + CH
3
(CH
2
)
3
Li
CH
3
O Li + CH
3
(CH
2
)
2
CH
3
pK
a
= 50
CH
3
OH + Li N
CH
3
O Li +
CH(CH
3
)
2
CH(CH
3
)
2
NH
CH(CH
3
)
2
CH(CH
3
)
2
pK
a
= 40
2. Reakcje z silnymi kwasami – podstawienie/dehydratacja (S
N
, E1):
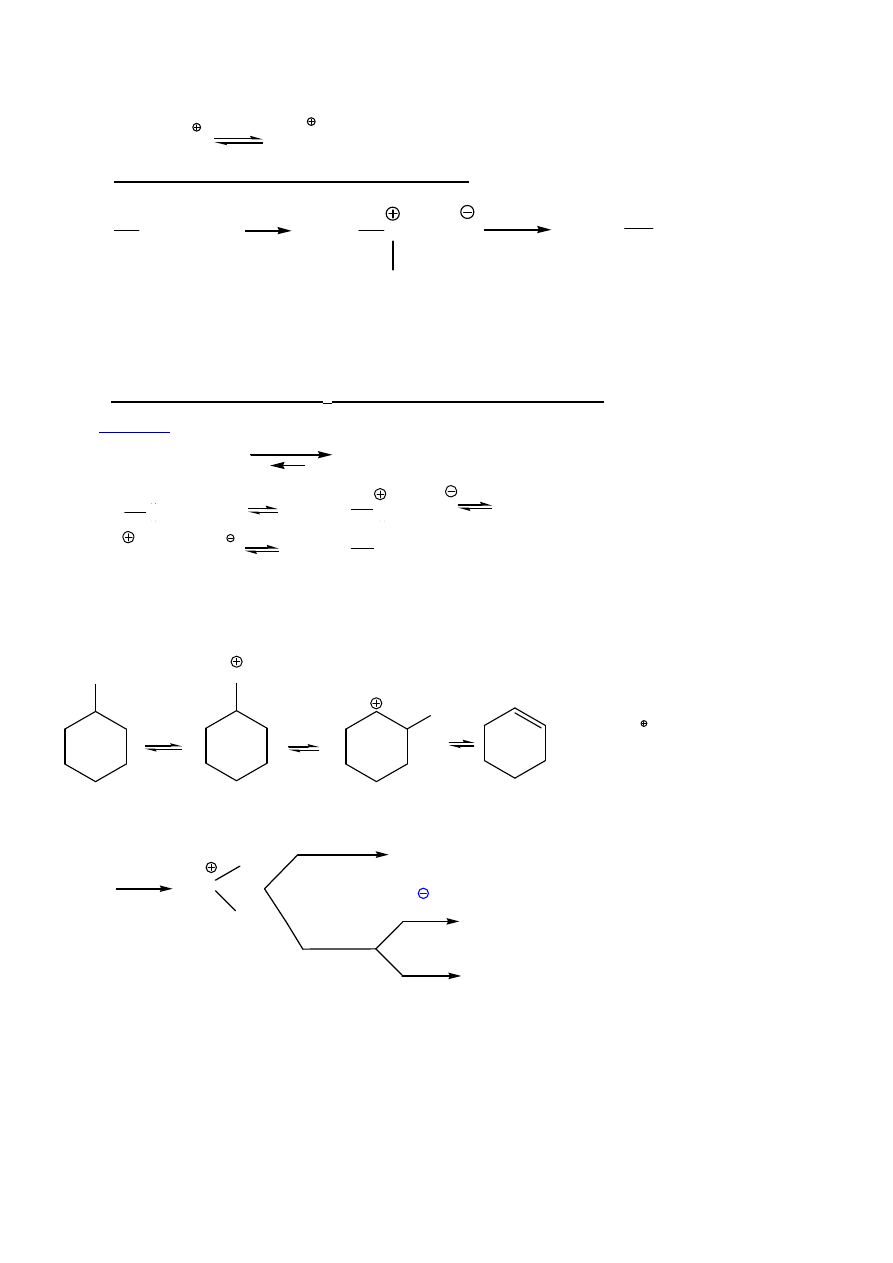
6
R-O-H + H
ROH
2
A.
Otrzymywanie halogenoalkanów z 1
°°°°
ROH:
RH
2
C
OH
+ HX
RH
2
C
OH
H
+ X
S
N
2
RH
2
C
X
+ H
2
O
X = Br, I
anion chloru za słabym Nu
SOCl
2
, PBr
3
B
. Reakcje z 2
°°°°
i 3
°°°°
ROH – S
N
1, E1; przegrupowanie kationów:
Gdy dobry Nu – S
N
1:
(CH
3
)
3
COH + HBr
(CH
3
)
3
CBr + H
2
O
(CH
3
)
3
C
OH
+ HBr
(CH
3
)
3
C
OH
2
+ Br
(CH
3
)
3
C + H
2
O + Br
(CH
3
)
3
C
Br + H
2
O
W wy
ż
szych temperaturach
→
E = dehydratacja alkoholi
POCl
3
– łagodny
ś
rodek odwadniaj
ą
cy dla 2º i 3º (0º, Py, E2)
H
2
SO
4,
T
OH
OH
2
H
+ H
2
O + H
- H
2
O
S
N
2
R - 1 rz.
R - 2, 3 rz.
ROH
R-O
H
H
X , S
N
1
E1
RX + H
2
O
RX
alken
Przegrupowania karbokationów/podstawienie:
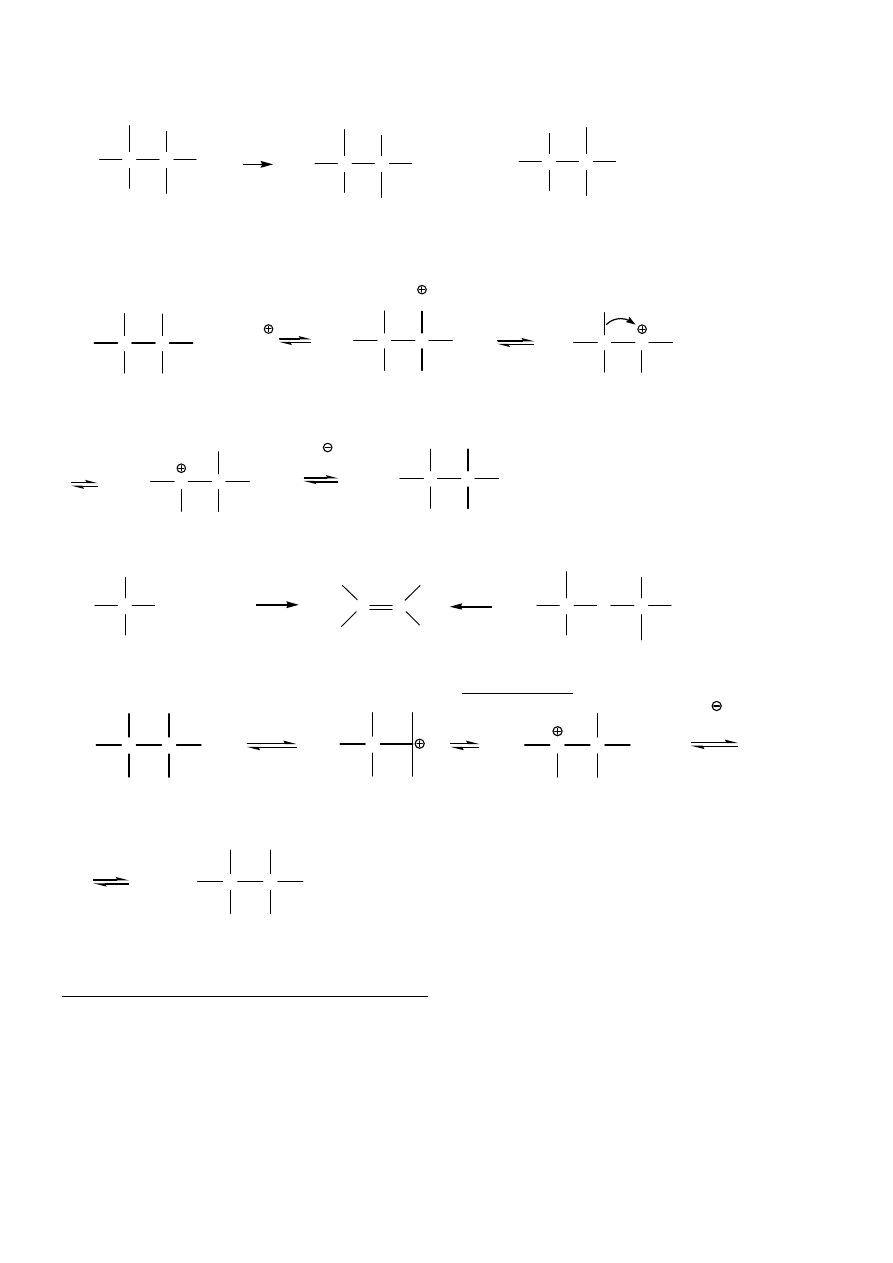
7
C
CH
3
H
3
C
H
C
OH
CH
3
H
C
CH
3
H
3
C
H
C
Br
CH
3
H
C
CH
3
H
3
C
Br
C
H
CH
3
H
+
HBr
0 C
°
°
+ H
2
O
główny produkt
E1 - przegrupowanie anionu wodorkowego/alkilu:
C
CH
3
H
3
C
H
C
OH
CH
3
H
+ H
C
CH
3
H
3
C
H
C
OH
2
CH
3
H
C
CH
3
H
3
C
H
C
CH
3
H
C
CH
3
H
3
C
C
H
CH
3
H
Br
C
CH
3
H
3
C
Br
C
H
CH
3
H
H
3
C
C
CH
3
OH
CH
2
CH
2
CH
3
H
2
SO
4
C
C
H
3
C
H
3
C
H
CH
2
CH
3
H
3
C
C
H
2
C
C
CH
3
CH
3
H
H
OH
T
H
2
SO
4
Gdy brak odpowiedniego 2
°
lub 3
°
atomu wodoru – migracja alkilu
C
CH
3
H
3
C
CH
3
C
CH
3
OH
H
HBr
C
CH
3
H
3
C
CH
3
CH
3
H
C
CH
3
H
3
C
C
CH
3
CH
3
H
Br
C
CH
3
H
3
C
Br
C
CH
3
CH
3
H
Szybko
ść
migracji wi
ę
ksza gdy prowadzi do 3
°
karbokationu.
Alkohole 1
°
te
ż
mog
ą
ulega
ć
przegrupowaniom - równoczesna z odej
ś
ciem wody migracja alkilu
(podstawienie utrudnia du
ż
e zatłoczenie):
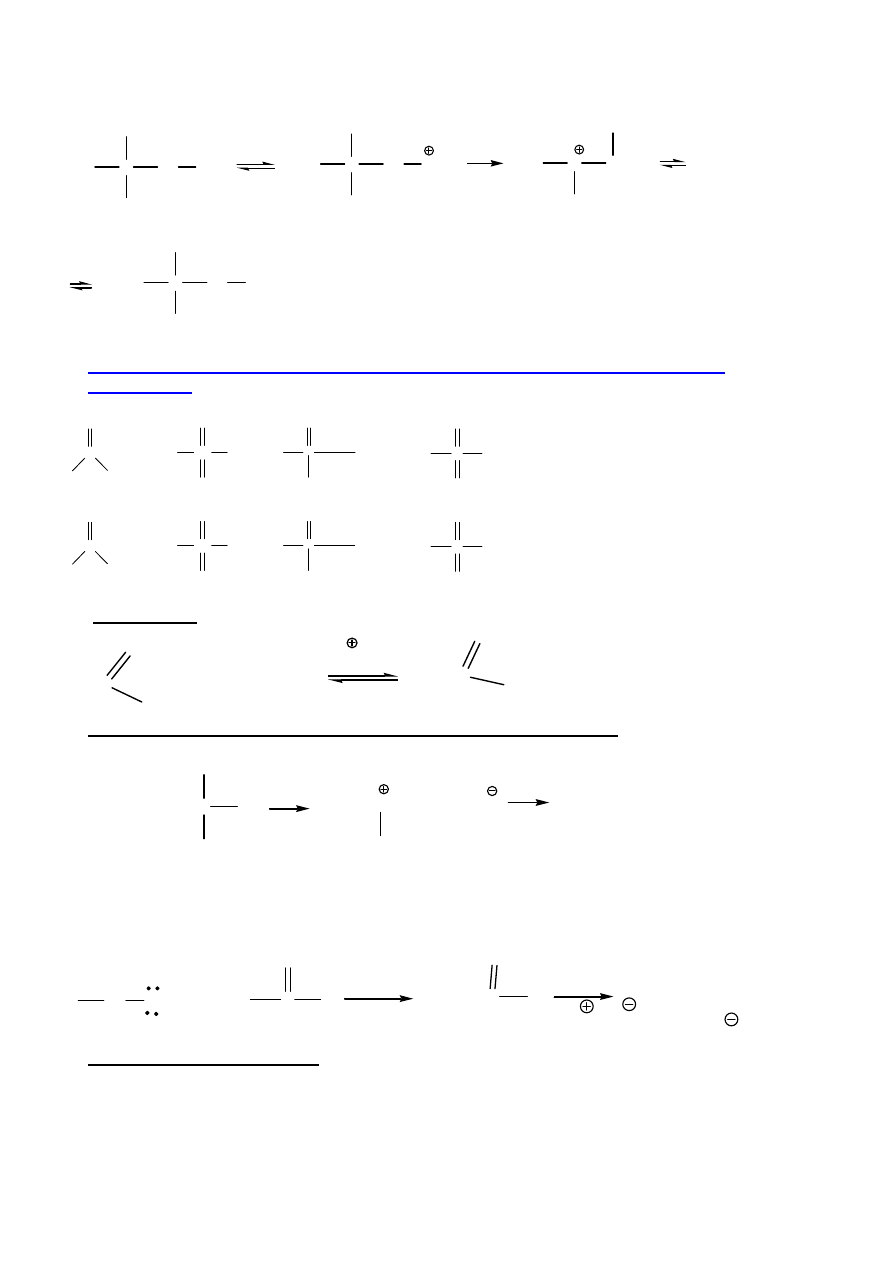
8
C
CH
3
H
3
C
CH
3
H
2
C
OH
C
CH
3
H
3
C
CH
3
H
2
C
OH
2
C
CH
3
H
3
C
CH
2
CH
3
C
CH
3
H
3
C
Br
H
2
C
CH
3
alkohol neopentylowy
3. Reakcje z kwasami organicznymi i tlenowymi kwasami nieorganicznymi i ich
pochodnymi
C
OR'
R
O
Cr
O
HO
OR
O
P
O
HO
OR
OH
S
O
R
OR'
O
C
OH
R
O
Cr
O
HO
OH
O
P
O
HO
OH
OH
S
O
R
OH
O
Estryfikacja:
CH
3
C
O
OH
+ CH
3
CH
2
OH
H
CH
3
C
O
OCH
2
CH
3
Estry kwasów nieorganicznych jako droga do halogenoalkanów
R-CH
2
-OH +
P
Br
Br
Br
RCH
2
-O-PBr
2
H
+ Br
S
N
2
RCH
2
Br + HOPBr
2
bromofosforan (III)
2 RCH
2
OH + HOPBr
2
→
2 RCH
2
Br + H
3
PO
3
chlorki – SOCl
2
, jodki – P + I
2
R
H
2
C
OH
+
S
O
Cl
Cl
OS
O
Cl
S
N
2
+ H + Cl
RCH
2
Cl
+ SO
2
+ Cl
RCH
2
Otrzymywanie sulfonianów (wszechstronnych substratów)
Sulfoniany – krystaliczne, łatwo oczyszcza
ć
.
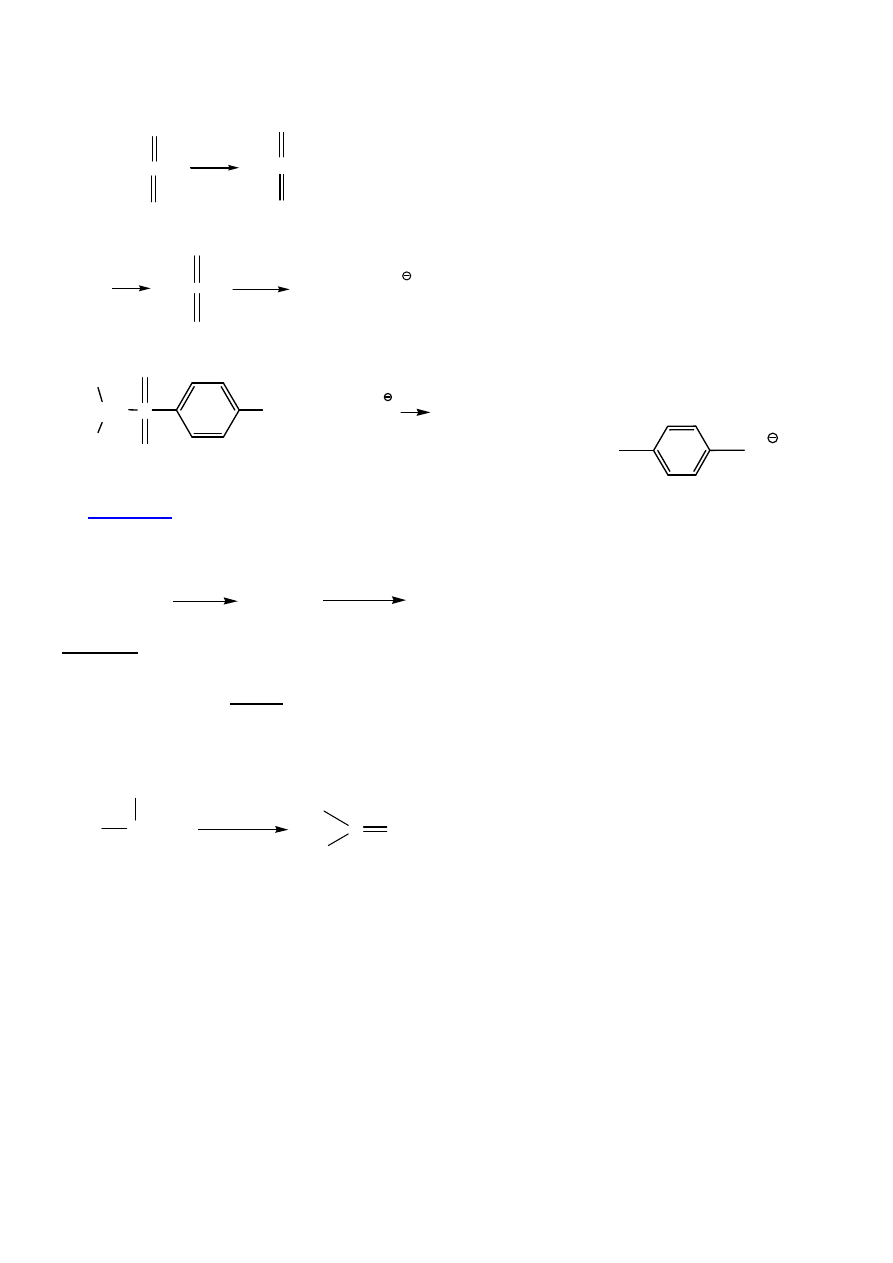
9
ROH + CH
3
-SCl
O
O
Py
O
O
+ Py x HCl
ROS-CH
3
ROH
RO-S-R'
O
O
Nu
R-Nu + R'SO
3
CH
3
S
CH
3
C-O
H
CH
3
+ CH
3
CH
2
S
(CH
3
)
2
CHSCH
2
CH
3
H
3
C
SO
3
+
+
O
O
4. Utlenianie
A. Alkohole 1
°
→
aldehydy/ kwasy
RCH
2
OH
RCHO
RCOOH
utl.
utl.
aldehydy – tylko wtedy gdy mo
ż
na je usuwa
ć
z roztworu lub selektywny utleniacz (CrO
3
w
acetonie, chlorochromian pirydyny (PCC) = CrO
3
+ HCl + Py)
KMnO
4
, K
2
Cr
2
O
7
→
kwasy
B.
Alkohole 2°
→
ketony
R
CHOH
R'
K
2
Cr
2
O
7
R'
C
R
O
Silne utleniacze - destrukcja
C.
Alkohole 3° - nie utleniaj
ą
si
ę
(brak H
α
)
FENOLE
ArOH, IUPAC – benzen
ole
Elektrony
π
nakładaj
ą
si
ę
z orb. p atomu tlenu. Delokalizacja – jak w anionie benzylowym.
Fenol – kwas karbolowy
→
dezinfektant, polimery …
Kopolimer fenolu i aldehydu mrówkowego –
ż
ywice fenolanowe (np. Bakelit)
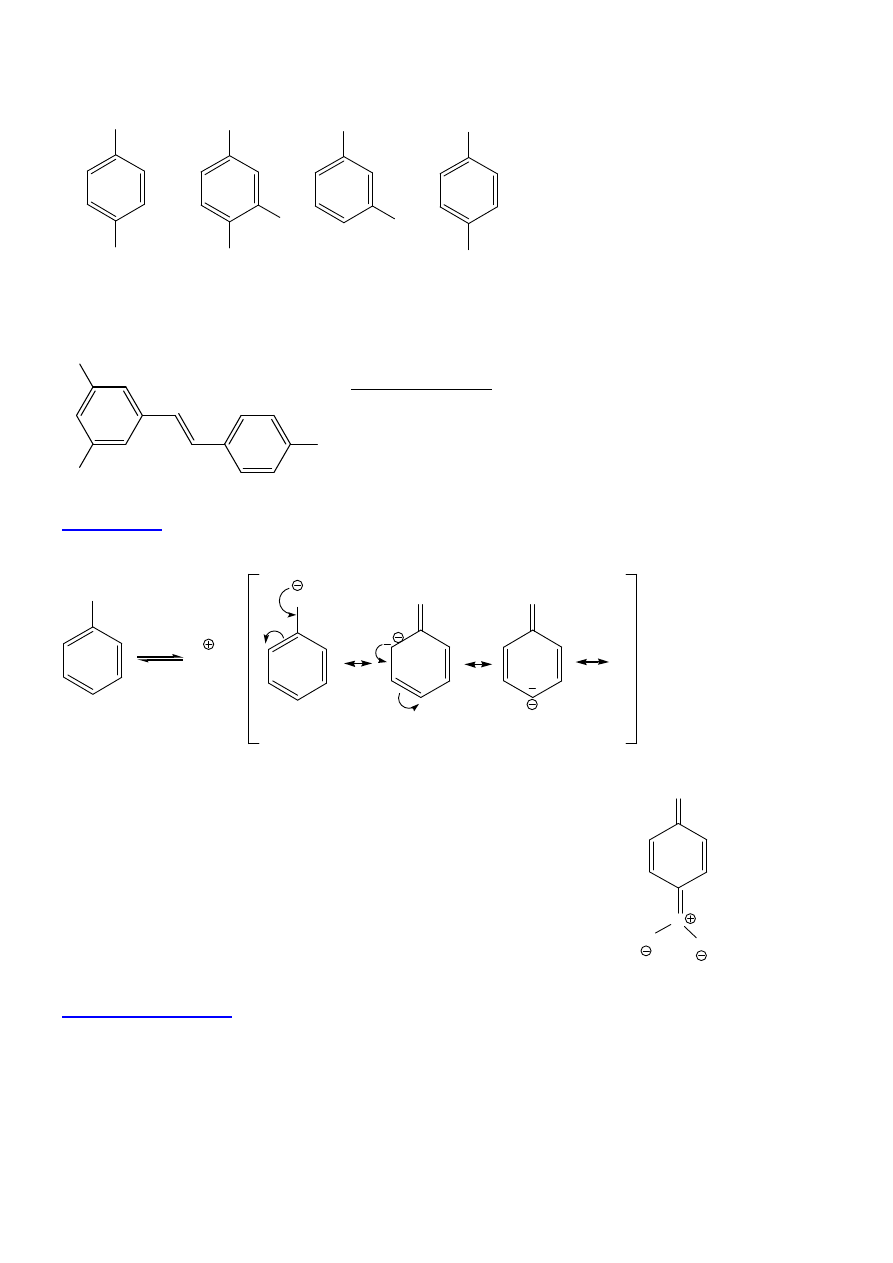
10
OH
CH
3
OH
NO
2
Cl
COOH
OH
OH
OH
4-metylofenol
(p-krezol)
4-chloro-3-nitrofenol
kwas
3-hydroksybenzoesowy
benzeno-1,4-diol
(hydrochinon)
trans - resweratrol
Kwasowo
ść
pK
a
= 8 ÷10 (kwasy – 3 ÷ 5, alkohole – 16 ÷ 18)
OH
O
O
O
H +
Sprz
ęż
ona zasada – stabilizowany rezonansowo anion fenolanowy
4-nitrofenol
pK
a
= 7.15
2,4,6-trinitrofenol
pK
a
= 0.25
C
6
H
5
OH + NaOH
→
C
6
H
5
O
-
Na
+
+ H
2
O
Otrzymywanie fenoli
Przez S
E
b.trudno
I. Przemysłowe otrzymywanie fenolu:
utlenianie lumenu; chlorobenzen + NaOH
- S
N
(Ar)
O
N
O
O
OH
HO
HO
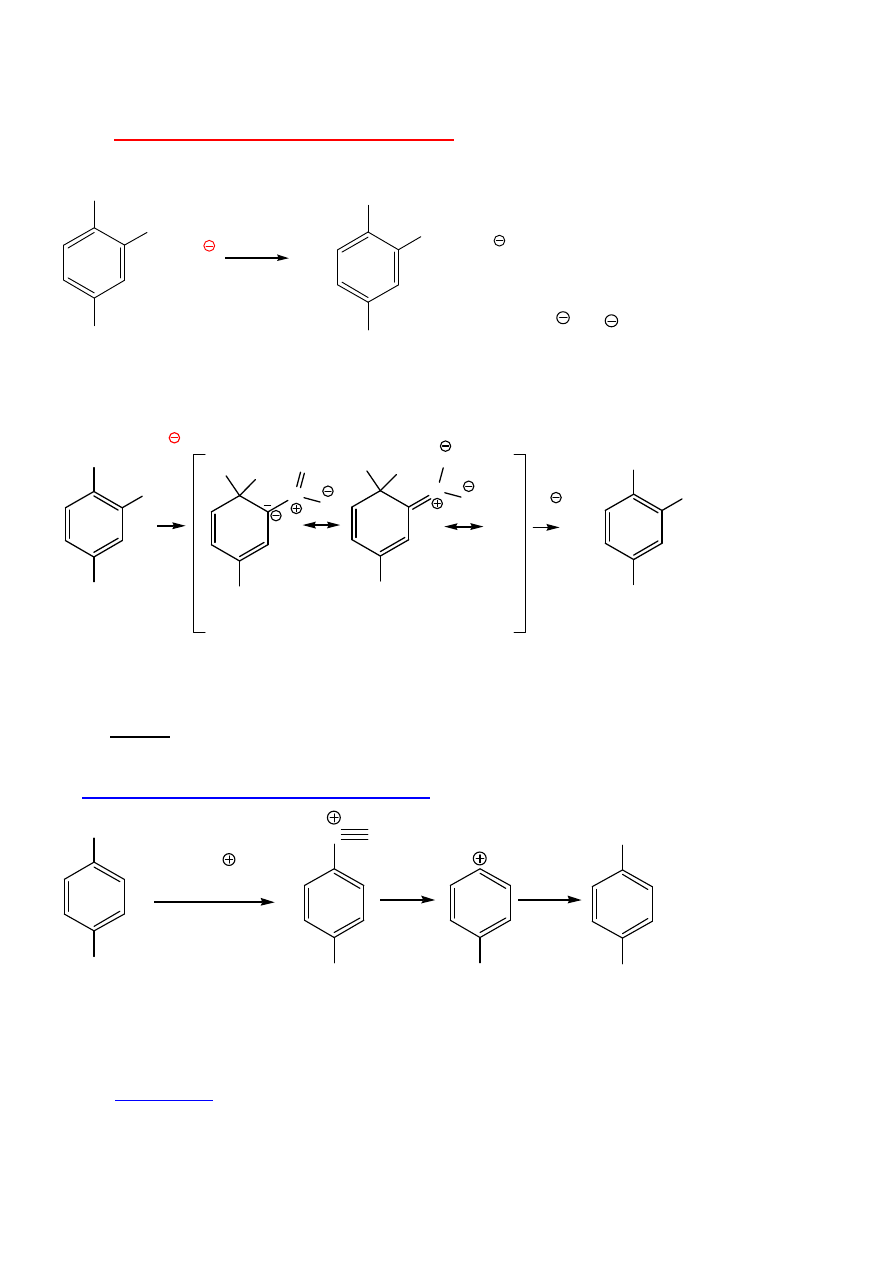
11
Nukleofilowe podstawienie aromatyczne -
S
N
(Ar)
Cl
NO
2
Nu
NO
2
+
Nu
+ Cl
NO
2
NO
2
Nu = OH , NH
2,
NH
3
Dwa etapy: addycja + eliminacja (jak pochodne kwasów)
A-E
Konieczne podst. akceptorowe (stabilizacja anionu); o-, p-
Cl
NO
2
NO
2
Nu
Cl
N
NO
2
Nu
O
O
Cl
N
NO
2
Nu
O
O
...
- Cl
Nu
NO
2
NO
2
Podstawniki meta nie wspomagaj
ą
rezonansowej stabilizacji.
E – A
Halogenoareny, które nie maj
ą
podstawników elektronoakceptorowych mog
ą
reagowa
ć
przez benzyn
II. Otrzymywanie fenoli z soli diazoniowych:
NH
2
R
NaNO
2,
H , H
2
O
N
R
R
OH
R
N
HOH
T
- N
2
REAKCJE FENOLI
Jak inne alkohole
:
1.
Estryfikacja
– z kwasami – endotermiczna; chlorki kwasowe:
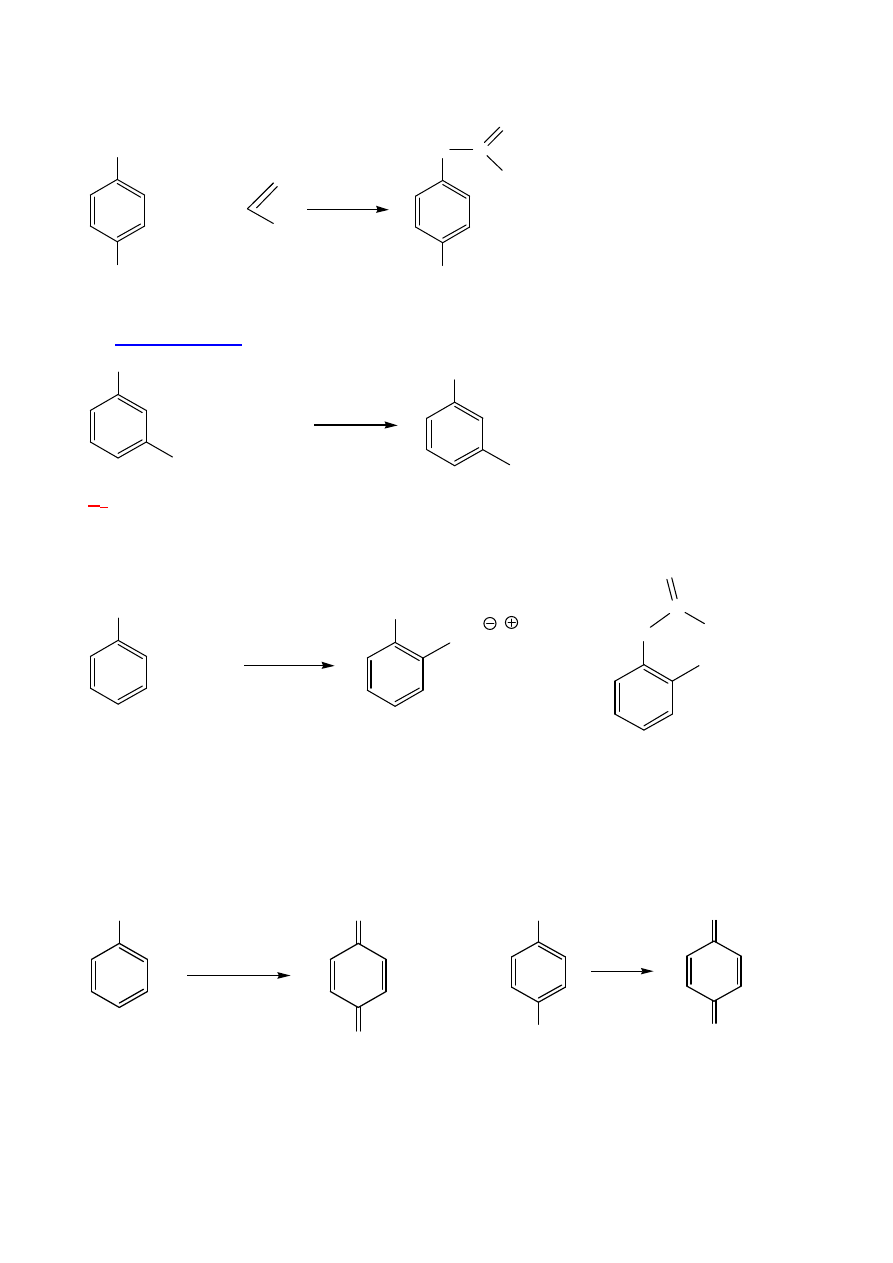
12
OH
CH
3
+ CH
3
CH
2
C
O
Cl
NaOH, H
2
O
O
CH
3
C
C
2
H
5
O
+ NaCl
2.
Synteza eterów
(Williamsona):
OH
+ CH
3
CH
2
CH
2
Br
NaOH, H
2
O
Cl
OCH
2
CH
2
CH
3
Cl
+ NaBr
S
E
Silna aktywacja o- i p-. Nawet rozc. HNO
3
powoduje nitrowanie.
Reakcj
ę
F.-C. komplikuje estryfikacja.
Reakcja Kolbego:
⇒
salicylan potasu
octan kwasu 2-hydroksybenzoesowego
kw. salicylowy – inhibicja tromboxanu A
2
(prostaglandyna)
1997 r. – 100-lecie aspiryny
3.
Utlenianie fenoli
Mechanizm transferu 1e
→
diony
OH
O
O
[O]
benzeno-1,4-diol
cykloheksa-2,5-dien-1,4-dion
( hydrochinon)
(chinon)
OH
+ CO
2
KHCO
3,
p
OH
COO K
O
COOH
C
O
CH
3
OH
OH
O
O
[O]
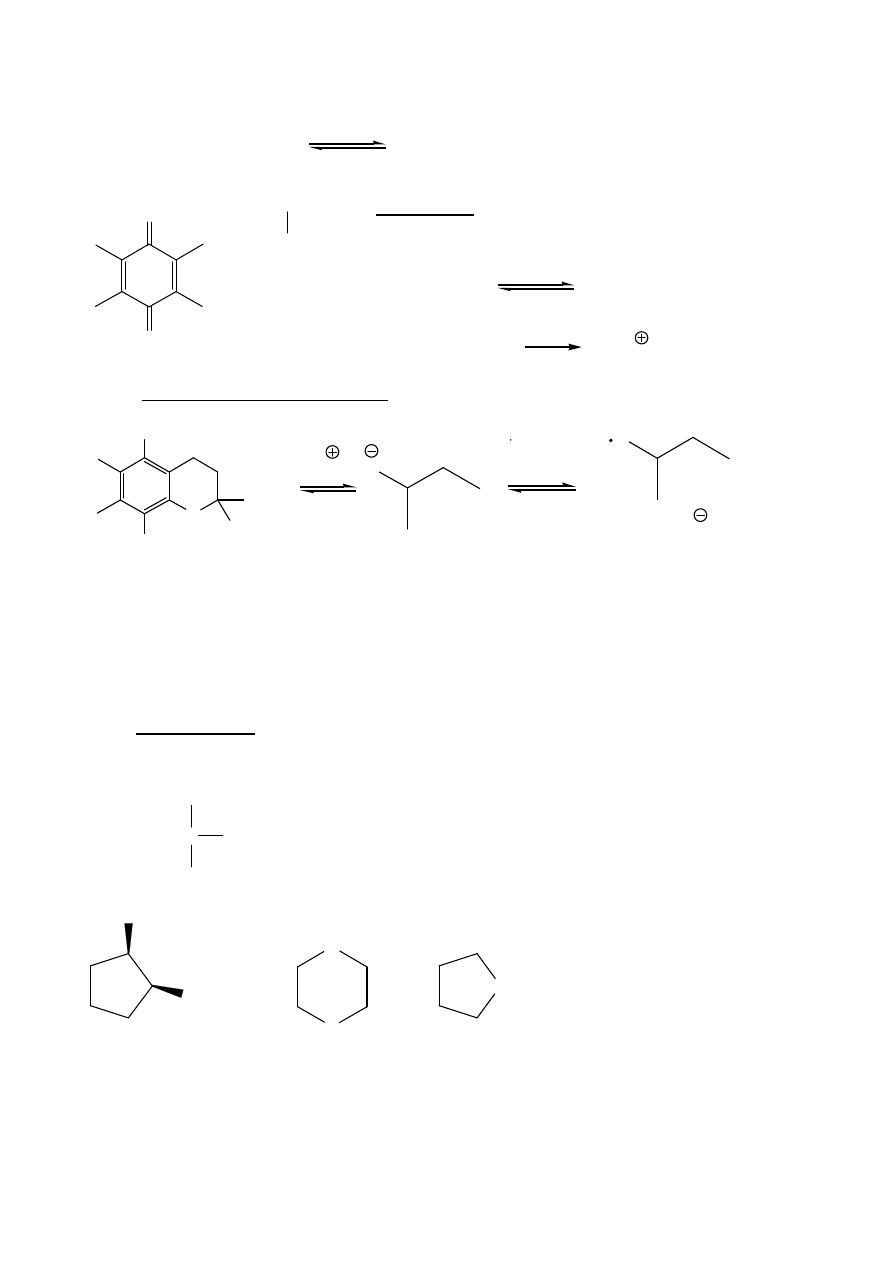
13
Para redoksowa benzochinon
hydrochinon w realizacji równowagowych procesów
utleniania w naturze.
ubichinony (koenzym Q) – reakcje redoksowe w
ła
ń
cuchu oddechowym aerobów (przeniesienie elektronów
z reduktazy NADH-Q do reduktazy cytochromowej)
ubichinon (Q)
ubichinol (QH
2
)
Sumarycznie:
NADH + 1/2 O
2
NAD + H
2
O
Naturalny antyoksydant (fenol) – witamina E – redukcja i protonowanie rodników
CH
3
CH
3
HO
H
3
C
O
R
CH
3
O
- H
O-lipid
O
+ O-lipid
Rodnik
α
-tokoferolu stosunkowo mało reaktywny (zatłoczenie, delokalizacja); redukowany np.
przez wit. C, metabolity – wydalane.
Syntetyczne analogi witaminy E – konserwanty:
ETERY
C
n
H
2n+2
O, R-O-R’
O sp
3
,
α
= 112º (eter dimetylowy)
IUPAC – alkoksyalkany; etery cykliczne
CH
3
OCH
2
CH
3
metoksyetan, eter etylowometylowy
2-
etoksy-2-metylopropan
CH
3
OCH
2
CH
2
OCH
3
1,2-dimetoksyetan
(eter dimetylowy glikolu)
oksolan
cis-1-etoksy-2-metoksycyklopentan
1,4-dioksan
(tetrahydrofuran, THF)
O
O
O
OCH
3
OCH
2
CH
3
CH
3
CH
2
OC
CH
3
CH
3
CH
3
O
O
H
3
CO
H
3
CO
(CH
2
CH=CCH
2
)
n
H
CH
3
CH
3
n = 6, 8, 10
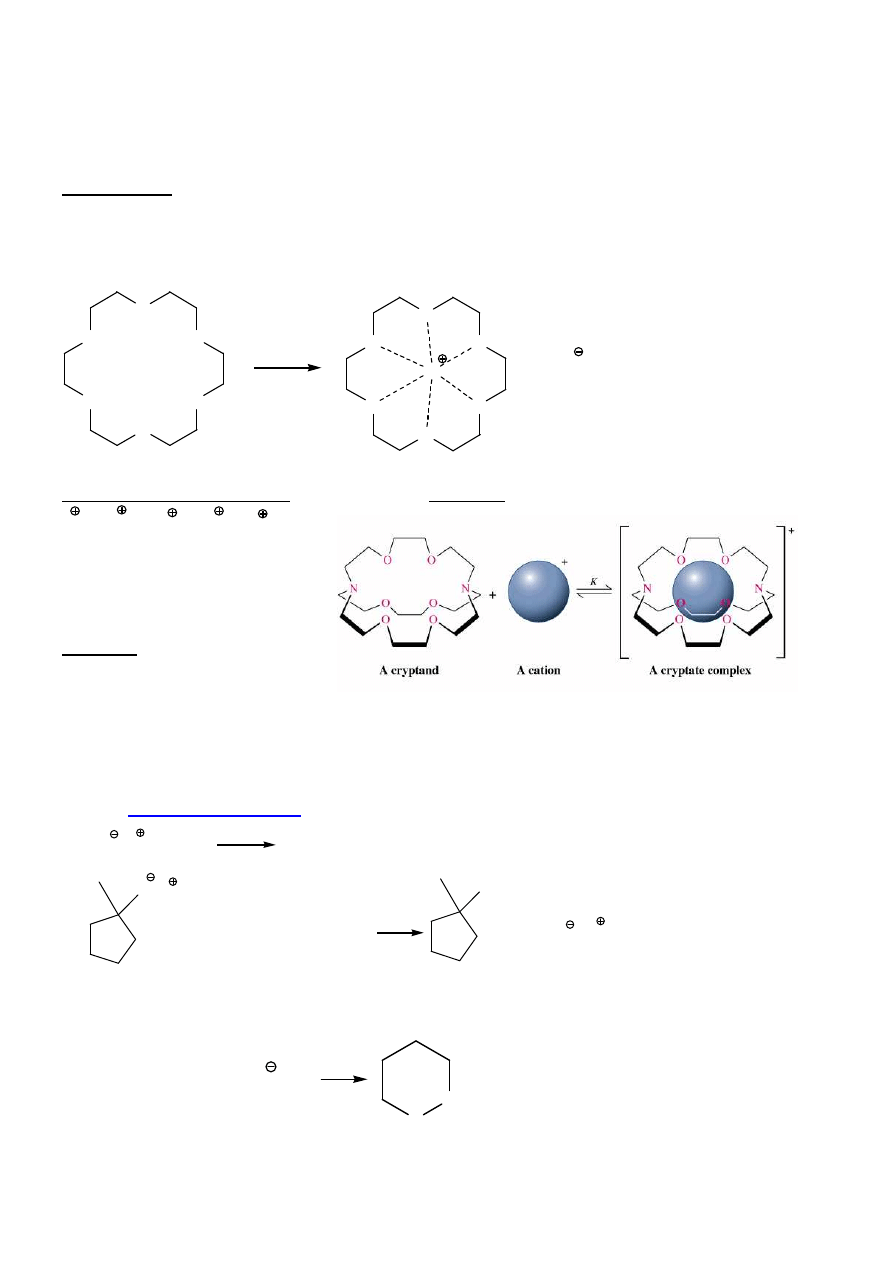
14
heterocykle
cykliczne polietery – etery koronowe
Wła
ś
ciwo
ś
ci:
Brak wi
ą
za
ń
wodorowych
→
niskie t.wrz., słaba rozpuszczalno
ść
w H
2
O
Solwatowanie kationów
→
rozpuszczanie soli nieorganicznych w rozp. niepolarnych
O
O
O
O
O
O
O
O
O
O
O
O
KMnO
4
C
6
H
6
K
MnO
4
Reagenty transportuj
ą
ce jony: etery koronowe, kryptandy (trójwymiarowe etery policykliczne)
K > Rb > Na > Cs > Li
K(K
+
) = 10
10
K(Li
+
) = 100
Jonofory – zwi
ą
zki otaczaj
ą
ce
przykoordynowany kation
Synteza eterów
1. Synteza Williamsona
RO Na
+ Cl-R'
R-O-R'
O Na
+ CH
3
(CH
2
)
15
CH
2
OSO
2
CH
3
DMSO
OCH
2
(CH
2
)
15
CH
3
+ CH
3
SO
3
Na
RO
-
- dobry Nu, silna B – tylko 1
°
czynniki alkiluj
ą
ce (konkurencja eliminacji)
Wewn
ą
trzcz
ą
steczkowa r. Willamsona:
H-O(CH
2
)
4
CH
2
Br
+ OH
O
CH
2
oksan (tetrahydropiran)
15
k
3
> k
5
> k
6
> k
4
> k
7
> k
8
←
czynniki entropowe + napr
ęż
enie pier
ś
cienia
(kompromis)
Halogenohydryna + NaOH
→
epoksyd/ oksiran
Wewn
ą
trzcz
ą
steczkowa reakcja Williamsona mo
ż
e by
ć
stereospecyficzna.
2. Działanie kwasów mineralnych na alkohole
– symetr. etery głównie 1
o
alkoholi
2 CH
3
CH
2
OH
H
2
SO
4
130
°
C
CH
3
CH
2
OCH
2
CH
3
Najsilniejszym Nu – niesprotonowany alkohol, S
N
2, konkurencja: E
CH
3
CHCH
2
OH
H
2
SO
4
180
°
C
CH
3
CH
H
E2,
CH
2
+ HOH
C
H
3
C
H
OH
CH
3
(CH
3
)
2
CH-O-CH(CH
3
)
2
+ H
2
O
H
2
SO
4
2-(1-metyloetoksy)propan
2
°
, 3
°
ROH – S
N
1
3. Alkoholiza halogenoalkanów/ sulfonianów - halogenki 2
°
, 3
°
H
3
C
Cl
H
3
C
OCH
2
CH
3
CH
3
CH
2
OH
H + Cl
4. Alkoksyrt
ę
ciowanie alkenów
1. Hg(OOCCF
3
)
2,
C
2
H
5
OH
2. NaBH
4
OC
2
H
5
REAKCJE ETERÓW
Tworzenie nadtlenków
- rodnikowe utlenienie na powietrzu
16
2 R-O-C-H +
O
2
2 R-O-C-
O-O
H
ROC-OO-C-OR
wodoronadtlenek eteru
nadtlenek eteru
Rozszczepienie przez silne kwasy (HBr, HI)
R-O-R'
R-Br + R'-OH
R-Br + R'-Br
HBr
HBr
H
Br
CH
3
CH
2
OH + PhCH
2
Br
CH
3
CH
2
-O-CH
2
Ph
CH
3
C-O-CH
2
Ph
H
Jony oksoniowe zawieraj
ą
ce 3
°
alkil
→
karbokationy (S
N
1, E1),
gdy 1
°
lub 2
°
- S
N
2
O
HI, H
2
O
OH
+
I
O
TFA
+
OH
REAKCJE EPOKSYDÓW
epoksyd = oksiran = 1,2-epoksyetan = tlenek glikolu
1. Nukleofilowe otwieranie pier
ś
cienia
⇒
pochodne alkoholi
O
H
2
C
CH
2
+ CH
3
S
HOH
HOCH
2
CH
2
SCH
3
S
N
2 – nietypowe. Nap
ę
d – likwidacja napr
ęż
enia.
O
CH
3
H
H
CH
3
CH
3
O, CH
3
OH
C
C
H
3
CO
OH
CH
3
H
H
CH
3
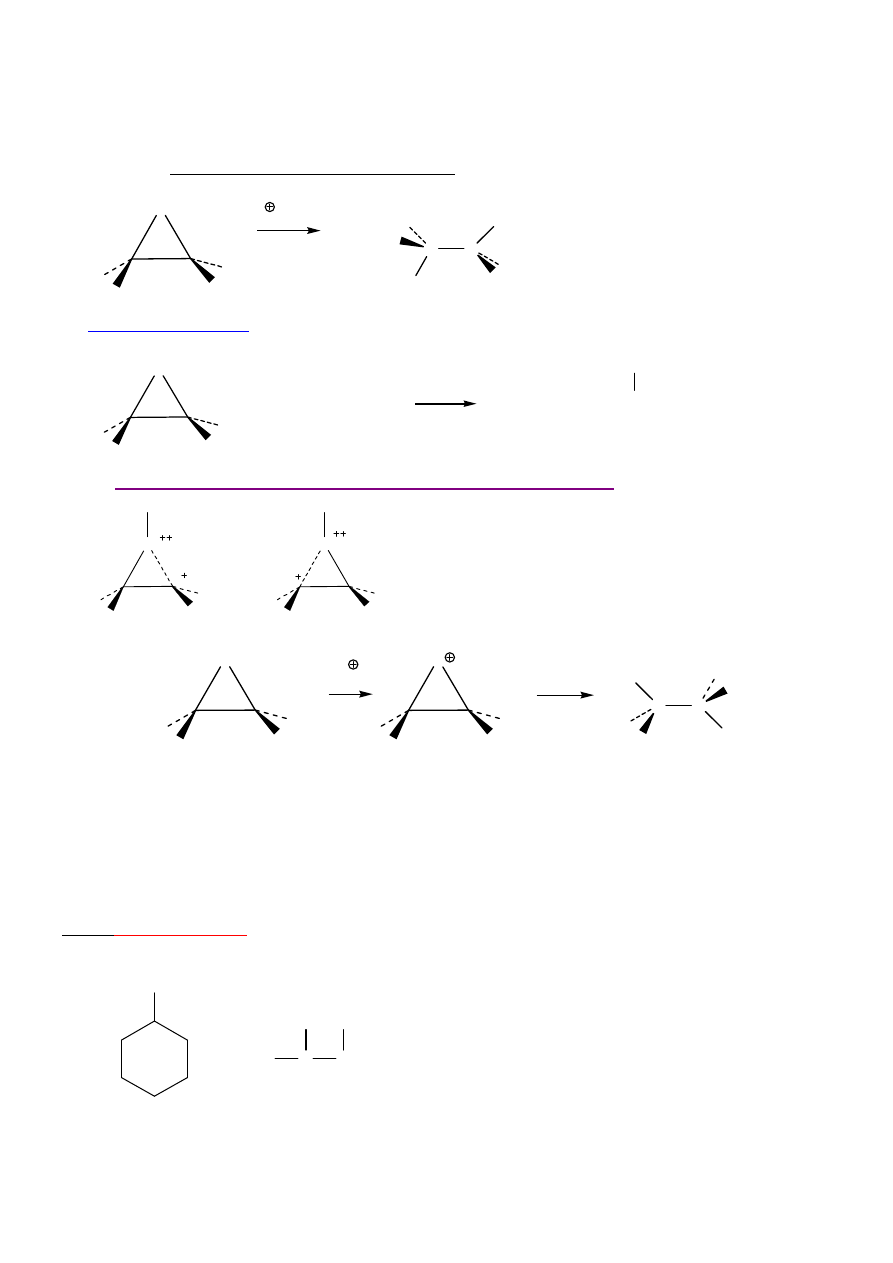
17
Regioselektywno
ść
←
zawada steryczna
, stereoselektywno
ść
-
anti
Reakcje z wodorkami i zw. organometalicznymi:
O
H
H
H
R
C
C
H
OH
H
H
H
R
1. LiAl
H
4
, Et
2
O
2. H , H
2
O
Hydroksyetylowanie:
O
H
H
H
R
+ CH
3
CH
2
CH
2
MgBr
CH
3
CH
2
CH
2
CH
2
CHR
O
H
2. Kwa
ś
no katalizowane otwieranie pier
ś
cienia epoksydowego
?
→
regiospecyficzno
ść
H
O
R'
H
H
R
H
O
R'
H
H
R
CH
3
OH
C
C
HO
OCH
3
R'
H
H
R
TIOLE i SULFIDY
siarkowe analogi alkoholi i eterów
Nazewnictwo:
alkano
tiole/merkaptany:
CH
3
SH
metanotiol
-SH gr. merkaptanowa
SH
H
3
C
C
H
CH
3
CHCH
2
CH
3
SH
cykloheksanotiol
2-metylopentano-3-tiol
O
R'
H
H
R
O
R'
H
H
R
δ
δ
δ
δ
H
H

18
HSCH
2
CH
2
OH 2-
merkapto
etanol (ni
ż
sza preferencja SH)
Tioetery = sulfidy
RS-
alkilotio-
CH
3
SCH
2
CH
3
etylometylosulfid
1,1-dimetyloetyloheptylosulfid
jon metanotiolanowy
sulfid dimetylowy = siarczek dimetylu
Wła
ś
ciwo
ś
ci:
Atom S –
wi
ę
kszy rozmiar
, słabo spolaryzowane S-H
⇒
słabe wi
ą
zania wodorowe. Lotno
ść
RSH
jak RX, a nie ROH. Zapach!
Słabe wi
ą
zanie S-H,
pK
a
= 9
÷
12
REAKCJE:
at. S
bardziej nukleofilowy
ni
ż
at. O
(CH
3
)
2
CH-Br + Na SH
EtOH
(CH
3
)
2
CH-SH + NaBr
Alkilowanie tioli
→
sulfidy
RSH + R'Br
NaOH
S
N
2
RSR' + NaBr + H
2
O
Nukleofilowo
ść
sulfidów
⇒
sole trialkilosulfoniowe
S
H
3
C
H
3
C
+ CH
3
I
S
H
3
C
H
3
C
CH
3
+ I
Sole sulfoniowe – S
N
na atom C (sulfid grup
ą
opuszczaj
ą
c
ą
)
S
H
3
C
H
3
C
CH
3
HO
+
HOCH
3
+ S(CH
3
)
2
Utlenienie
a/ łagodne
→
disulfidy
:
R-SH + I
2
R-S-S-R + 2 HI
b/ drastyczne
→
kwasy sulfonowe
:
H
3
C
C
CH
3
S(CH
2
)
6
CH
3
CH
3
CH
3
S
RSH + OH
RS + HOH
CH
3
SH
KMnO
4
(H
2
O
2
)
CH
3
S-OH
O
O
kwas metanosulfonowy
CH
3
-S-CH
3
H
2
O
2
CH
3
-S-CH
3
O
CH
3
-S-CH
3
O
O
sulfid dimetylowy
sulfotlenek dimetylowy
sulfon dimetylowy
DMSO
19
Zastosowanie, fizjologiczna rola alkoholi, eterów
CH
3
OH
– rozpuszczalnik, paliwo, toksyczny dodatek do EtOH, 30 ml – dawka
ś
miertelna (CH
2
=O
zaburza proces widzenia), ewent. prekursor benzyny
CH
3
CH
2
OH
– składnik napojów alkoholowych (depresant), rozpuszczalnik (perfumy, politury…)
dawki okołotoksyczne – przy zatruciach metanolem czy etano-1,2-diolem
spo
ż
ywczy:
(C
6
H
10
O
5
)
n
C
6
H
12
O
6
2 CH
3
CH
2
OH + 2CO
2
enzymy
enzymy
metabolizm 10ml/h (w
ą
troba
→
CO
2
)
Etano-1,2-diol (glikol etylenowy)
H
2
C
CH
2
[O]
O
H
2
O
HOCH
2
CH
2
OH
Niska t.t (-11,5
°
C), wysoka t.wrz. (198
°
C), pełna mieszalno
ść
z wod
ą
→
płyny przeciw
zamarzaniu.
Propano-1,2,3-triol (gliceryna, glicerol)
Ź
ródłem zasadowa hydroliza triglicerydów; nietoksyczna, lepka, rozpuszczalna w wodzie ciecz
(przemysł kosmetyczny, farmaceutyczny, skórzany)
Estry fosforowe – fosfoglicerydy (membrany komórkowe)
Triester kw. azotowego = nitrogliceryna
Naturalne alkohole wa
ż
ne fizjologicznie: dialkohol - morfina (diacetylowa pochodna – heroina),
tetrahydrokanabinol, cholesterol…
O
H
3
C
H
3
C
OH
C
5
H
11
CH
3
tetrahydrokanabinol
Niskocz
ą
steczkowe RSH, RSR’ –
ź
ródła nieprzyjemnych zapachów
CH
3
CH
2
SH – 50 ppm, (CH
3
)
2
CHCH
2
CH
2
SH (skunks)…
allicyna (zw. przeciwbakteryjny)
S
S
O
HO
CH
3
H
H
H
H
CH
3
CH
3
CH
3
CH
3
1
2
3
4
5
6
7
8
9
10
11
12
13
15
16
17
14
cholesterol
Wyszukiwarka
Podobne podstrony:
Prawo dewizowe 2010 09 id 38648 Nieznany
cwiczenia 09 id 124345 Nieznany
Alkohole i fenole 3 id 58102 Nieznany
Alkohole 13 id 58087 Nieznany (2)
gal08 09 id 185722 Nieznany
alkohole i fenole 2 id 58101 Nieznany (2)
B 09 x id 74805 Nieznany (2)
acad 09 id 50516 Nieznany (2)
E1 Teoria 2008 09 id 149145 Nieznany
I CSK 166 09 1 id 208206 Nieznany
Fizjologia Cwiczenia 09 id 1743 Nieznany
alkohol i kierowca id 58052 Nieznany
26429 09 id 31508 Nieznany (2)
IV CSK 297 09 1 id 220962 Nieznany
Alkohole monohydroksylowe id 58 Nieznany (2)
alkohole i fenole 2 id 58114 Nieznany (2)
lab 09 id 257545 Nieznany
I CSK 582 09 1 id 208220 Nieznany
Mikroekonomia I W 09 id 301257 Nieznany
więcej podobnych podstron