
YWNOŚĆ
to mieszanina o złożonym składzie:
substancje pożądane:
budulcowe (białka, związki mineralne)
energetyczne (sacharydy i lipidy)
regulacyjne (witaminy)
nadające pożądane cechy sensoryczne (aromaty, barwniki, alkohole)
kształtujące cechy użytkowe – w tym reologiczne (regulatory kwasowości, substancje zagęszczające)
substancje niepożądane:
zanieczyszczenia
związki szkodliwe
w celu nadania produktom spożywczym oczekiwanych właściwości stosowane są substancje dodatkowe:
barwniki
substancje konserwujące
przeciwutleniacze
emulgatory
substancje zagęszczające i żelujące
stabilizatory
regulatory kwasowości
substancje słodzące
substancje wzmacniające smak i zapach
substancje spulchniające
substancje utrzymujące wilgotność
substancje wypełniające
substancje pianotwórcze
substancje aromatyczne
oprócz składników o charakterze budulcowym i energetycznym, duży nacisk kładzie się na wrażenie sensoryczne –
smakowe i zapachowe (wrażenia sensoryczne są jednym z najważniejszych czynników decydujących o ponownym
zakupie produktu)
kwasy i hydroksykwasy organiczne (np. kwasy: jabłkowy, cytrynowy, winowy, szczawiowy i bursztynowy)
występujące w owocach w postaci wolnej lub w warzywach związane w solach wapnia, potasu i sodu → nadają
smak i obniżają pH ograniczając jednocześnie rozwój drobnoustrojów
metyloketony powstające podczas jełczenia ketonowego tłuszczów ( -oksydacja) w bardzo małych ilościach
(1 ppm) nadają korzystne cechy smakowo-zapachowe serom dojrzewającym, ale w większych ilościach (60 ppm)
dyskwalifikują tłuszcz przed wykorzystaniem do celów spożywczych
FAŁSZOWANIE PRODUKTÓW SPO YWCZYCH
może pociągnąć za sobą, oprócz obniżenia jakości, zagrożenie dla zdrowia konsumentów – w przypadkach, gdy
prowadzi do ukrywania wad i szkodliwości produktów.
Najbardziej znanymi substancjami
służącymi do fałszowania żywności są:
woda rozcieńczająca mleko, soki i piwo,
kawa zbożowa i prażone korzenie cykorii dodawane do naturalnej kawy,
olej rzepakowy dodawany do słonecznikowego i sojowego,
glikol etylenowy osładzający wino,
smalec i przetworzone tłuszcze roślinne dodawane do masła,
mąka ryżowa i pasza kukurydziana fałszujące droższe gatunku mąki,
galaretka jabłkowa używana zamiast droższych przetworów owocowych,
skrobia dodawana do rozcieńczonej śmietany,
dodatek sody oczyszczonej maskuje rozwój drobnoustrojów chorobotwórczych w mleku.
mogą zostać wprowadzone do żywności na etapie wzrostu roślin i zwierząt (np. metale ciężkie),
podczas wytwarzania żywności (konserwanty, katalizatory technologiczne) i podczas
bezpośredniego przygotowywania do spożycia (N-nitrozoaminy)
Substancje dodatkowe mogą pełnić jednocześnie 2- lub więcej
różnych funkcji.
Podczas produkcji żywności stosuje się dodatek kilku substancji
konserwujących (np. benzoesanów – hamujących rozwój drożdży,
pleśni oraz bakterii z pochodnymi tlenku siarki(IV) –
inhibitujących bakterie mlekowe i octowe). Ponad to substancje
te działają na siebie synergistycznie – pozwalają zmniejszyć
dodawaną porcję środka konserwującego.

BIAŁKA
podstawowe składniki odżywcze i główne źródło związków azotowych w artykułach żywnościowych → zawartość
białka w produktach spożywczych stanowi jeden z czynników określających ich wartość odżywczą
białka wykazują funkcje budulcowe, regulacyjne (enzymy, hormony), ale także kształtują cechy sensoryczne
białka to liniowe produkty kondensacji ok. 20 -L-aminokwasów połączonych wiązaniami trans-peptydowymi,
których wzajemna konformacja nadaje charakterystyczne właściwości biologiczne i żywieniowe białek
struktura molekularna białek:
I-
rzędowa: kolejność (sekwencja) ułożenia aminokwasów stabilizowana przez wiąz. peptydowe i determinująca
właściwości cząsteczki
II-
rzędowa: przestrzenna konformacja: -helisa lub -harmonijka stabilizowana przez wiąz. wodorowe
III-
rzędowa: struktura II-rzędowa zwinięta w przestrzenną bryłę i stabilizowana przez wiele rodzajów wiązań (m.in.
wodorowe, hydrofobowe, jonowe, mostki siarczkowe i oddziaływania Van der Waalsa) determinująca
aktywność biologiczną białek
IV-
rzędowa: białka o dużej masie cząsteczkowej złożone z mniejszych podjednostek
punkt izoelektryczny – wartość pH przy której cząsteczki białka są obdarzone jednakową liczbą ładunków
elektrycznych dodatnich i ujemnych
– są elektrycznie obojętne
cząsteczka białka staje się dipolem z powodu nierównomiernego rozmieszczenia grup anionowych i kationowych
w punkcie izoelektrycznym białko wykazuje najmniejszą rozpuszczalność i najłatwiej można je wytrącić z
roztworu
powyżej pkt. izoelektrycznego (po stronie zasadowej) białko występuje w formie anionu, a poniżej (po stronie
kwasowej) w formie kationu
OZNśCZśNIE ZśWśRTO CI BIśŁKś
metoda pośrednia związana z oznaczeniem zawartości azotu (średnia zawartość azotu w białku produktów
spożywczych wynosi ok. 16%)
aby uwolnić azot z aminokwasów stosuje się mineralizację w stęż. kwasach (H
2
SO
4
) lub w przypadku niektórych
produktów mlecznych w stężonych roztworach wodorotlenków (NaOH, KOH)
azot wydziela się w postaci amoniaku
REAKCJA BIURETOWA
:
reakcja charakterystyczna białek i związków zawierających wiązanie peptydowe (-CO-NH-), np. biuret
w środowisku alkalicznym (Np. NaOH) w obecności jonów miedzi Cu
2+
(np. CuSO
4
)
tworzą się barwne kompleksy
barwa kompleksów zależy od długości łańcucha polipeptydowego (liczby wiązań peptydowych):
dipeptydy
→ niebieskie
tripeptydy
→ fioletowe
p
olipeptydy → czerwone
PRÓBś KSśNTOPROTEINOWś
białka poddane działaniu stężonego kwasu azotowego(V) (HNO
3
) barwią się na kolor żółtopomarańczowy
reakcja ta polega na nitrowaniu aminokwasów zawierających pierścień aromatyczny (tyrozyna, fenyloalanina,
tryptofan)
białko ogrzewane w środowisku zasadowym ulega przemianom powodującym zmianę jego struktury oraz zawartości
niektórych aminokwasów (w przypadku cysteiny i cystyny produktem ubocznym jest siarkowodór)
DENATURACJA BIAŁKA
nieodwracalne zmiany w strukturze cząsteczek białka, w wyniku których białko traci swoje charakterystyczne
właściwości biologiczne i fizykochemiczne
trwałość białek wiąże się z ich przestrzenną konfiguracją → podczas denaturacji pod wpływem czynników
fizycznych lub chemicznych następuje deformacja lub zniszczenie struktur, na skutek zmian w wiązaniach
stabilizujących
wywoływana przez:
wysoką temperaturę
promieniowanie ultrafioletowe lub jonizujące
stężone kwasy i zasady
sole metali ciężkich (jony)
alkohole i etery
większość białek traci stabilność swojej natywnej struktury w temp. ok. 60°C
w trakcie procesu denaturacji cieplnej białka dochodzi do osłabienia lub rozpadu wiązań stabilizujących
strukturę II-, III- lub IV-rzędową
zmiany te powodują rozrywanie, rozwinięcie lub rozfałdowanie łańcuchów polipeptydowych i nowe, często
przypadkowe zwinięcie łańcucha denaturowanego białka
w czasie takich przekształceń w cząsteczce białka następuje odsłanianie nowych wiązań i grup funkcyjnych
(np. grupy niepolarne = hydrofobowe są odsłaniane, a grupy polarne – hydrofilowe chowane do wewnątrz →
denaturujące białko uwalnia znaczne ilości związanej wody)
w niektórych przypadkach poprzez denaturację cieplną białka uzyskuje się inaktywację enzymów niekorzystnie
wpływających na właściwości produktu (np. blanszowanie kostki jabłkowej)

KOAGUŻACJA BIAŁKA
denaturacja cieplna białek w odpowiednich warunkach prowadzi do łączenia się układu koloidalnego białek
w większe zespoły → rozwinięcie łańcuchów polipeptydowych i ich połączenie w duże agregaty
przy wysokim stężeniu białek rozpuszczalnych ich denaturacja i koagulacja prowadzi do utraty płynności –
powstaje stała masa (żel), w której połączone łańcuchy białka tworzą przestrzenną sieć, w której „oczkach”
zostaje zatrzymana cała woda
zdolność białek do tworzenia żelu zależy od długości łańcucha białka i sposobu jego pofałdowania w wyniku
denaturacji
(im dłuższy i bardziej wyprostowany, tym mniej łańcuchów potrzeba do wytworzenia żelu)
odporność żelu na ogrzewanie zależy od ilości reszt hydrofobowych w łańcuchach bocznych aminokwasów
> 30% =
żele nie ulegające upłynnieniu w wysokiej temperaturze (np. białko jaja kurzego)
< 30% = żele ulegające upłynnieniu po ogrzaniu, a po ochłodzeniu ponownie żelujące (np. kolagen)
tworzenie żelu jest ważnym zjawiskiem nadającym odpowiednie cechy strukturalne wielu produktom spożywczym
i potrawom (m.in. wędlinom drobnozmielonym, galaretom mięsnym i rybnym, deserom zestalanym żelatyną, serom
i potrawom z sera)
stopień i jakość usieciowania kolagenu oraz jego współdziałanie z innymi składnikami tkanek łącznych wpływa na
kruchość mięsa po obróbce cieplnej
WYSAŻANIE BIAŁKA
odwracalne przemiany w przestrzennej konfiguracji białek
rozpuszczalność białek w wodzie zależy od ich powinowactwa do wody (zdolności do hydratacji) → w roztworach
wodnych cząsteczki wody tworzą otoczkę wokół obdarzonych ładunkiem białek, zmniejszając ich wzajemne
oddziaływanie i stabilizując układ koloidalny
zobojętnienie ładunków elektrostatycznych makrocząsteczek białek powoduje ich agregację i wydzielenie się z
roztworu
→ wzrost stężenia soli zmniejsza rozpuszczalność białek i następuje ich wytrącanie z roztworu
dokonuje się tego przez odpowiednio dobrane pH (punkt izoelektryczny) lub przez dodatek niektórych soli (Na
2
SO
4
,
NaCl, MgSO
4
, (NH
4
)
2
SO
4
) które adsorbują się na powierzchni białek i zobojętniają ładunek
FERMENTACJA MLEKA
przemiany mikrobiologiczne zachodzące w mleku podczas przechowywania wywołane działaniem bakterii kwasu
mlekowego (m.in. bacillus Delbrucki)
podczas tego procesu mono- i disacharydy ulegają przemianie do kwasu mlekowego, który dysocjując wywołuje
kwaśny odczyn pH mleka
w środowisku kwaśnym następują zmiany konformacji białek (wywołane zmianami ładunku i jego rozkładu na
łańcuchach aminokwasowych), co powoduje zmiany ich rozpuszczalności → białka zostają wytrącone, a mleko
ulega „zakwaszeniu”
sztuczne alkalizowanie mleka jest niedozwoloną metodą jego fałszowania – substancja alkalizująca wiąże wolne
protony powstającego kwasu mlekowego, przez co białka pozostają w stabilnej konformacji
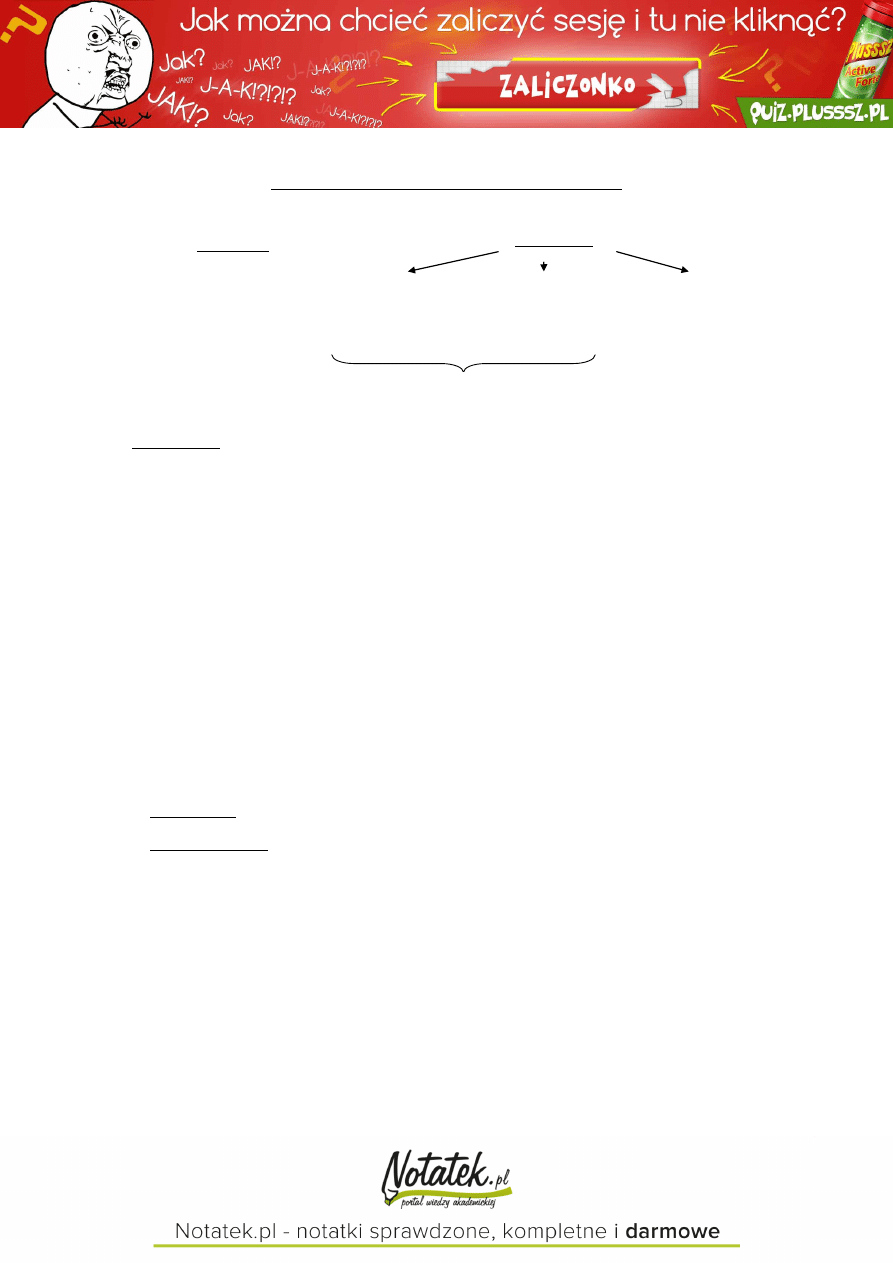
CUKRY PROSTE i OLIGOSACHARYDY
hydroksyaldehydy (aldozy) / hydroksyketony (ketozy)
GLIKOZYDY
są szczególnym przykładem acetali zbudowanych z jednostki cukrowej (glukozy) i aglikonu (np. reszty alkoholu,
fenolu, aminy, kwasu organicznego)
glukoza z metanolem
– tworzy 2 anomeryczne glikozydy:
-O-metylo-D-glukozyd i -O-metylo-D-glukozyd
aglikon może być połączony za pomocą tlenu (O-glikozydy), azotu (N-glikozydy), siarki (S-glikozydy)
anomery glikozydów są dużo trwalsze od anomerycznych postaci cukrów i nie redukują odczynnika Fehlinga ani
Benedicta
glikozydami są m.in. nukleozydy (składniki DNA i RNA), antocyjany (barwniki), streptomycyna (antybiotyk)
szczególnym przypadkiem glikozydów są HOLOZYDY, w których aglikonem jest reszta cukrowa – np. cukry złożone
CZYNNO Ć OPTYCZNś CUKRÓW:
cukry są związkami optycznie czynnymi, tzn. mają zdolność skręcania płaszczyzny światła spolaryzowanego
czynność optyczna jest konsekwencją asymetrycznej budowy cząsteczek (występowanie asymetrycznych atomów
węgla → atomów węgla, którego 4 wiązania wysycone są różnymi podstawnikami)
w związku z asymetrycznością cząsteczki cukru mogą występować w 2 odmiennych formach przestrzennych, które
są swoimi wzajemnymi, nienakładanymi odbiciami lustrzanymi –
CHIRśLNO Ć
izomery optyczne mają zbliżone właściwości chemiczne i fizyczne, ale różnią się kątem skręcania płaszczyzny
światła spolaryzowanego
odmiana prawoskrętna (+)
od
miana lewoskrętna (-)
STEREOIZOMERIA = izomeria optyczna
:
ENANCJOMERY: pary cukrów będących swoimi wzajemnymi odbiciami lustrzanymi
(np. D-(+)-glukoza i L-(-)-glukoza)
DIASTEREOIZOMERY
:
izomery różniące się przestrzenną konfiguracją przy asymetrycznych atomach węgla, ale nie
są dla siebie enancjomerami
(np. D-(+)-
glukoza ma 14 innych izomerycznych D i L aldoheksoz z pominięciem L-(-)-glukozy)
EPIMERY – różnią się ułożeniem podstawników przy jednym centrum asymetrii
ANOMERY – różnią się ułożeniem podstawników przy węglu anomerycznym (nowym, asymetrycznym atomie
węgla, wytworzonym przy zamknięciu pierścienia) → w przypadku aldoz C
1
/ ketoz C
2
cukry proste
(monosacharydy)
mannoza
galaktoza
glukoza
fruktoza
ryboza
deoksyryboza
cukry zło one
dwucukry
(disacharydy)
2 monozy
maltoza (2x glukoza)
sacharoza (glukoza + fruktoza)
laktoza (glukoza + galaktoza)
kilkocukry
(oligosacharydy)
3-6 monoz
wielocukry
(polisacharydy)
> 6 monoz
skrobia
glikogen
celuloza
krystaliczne, dobrze rozpuszczalne w H
2
O,
słodki smak
analogicznie: O-fruktozydy i O-mannozydy
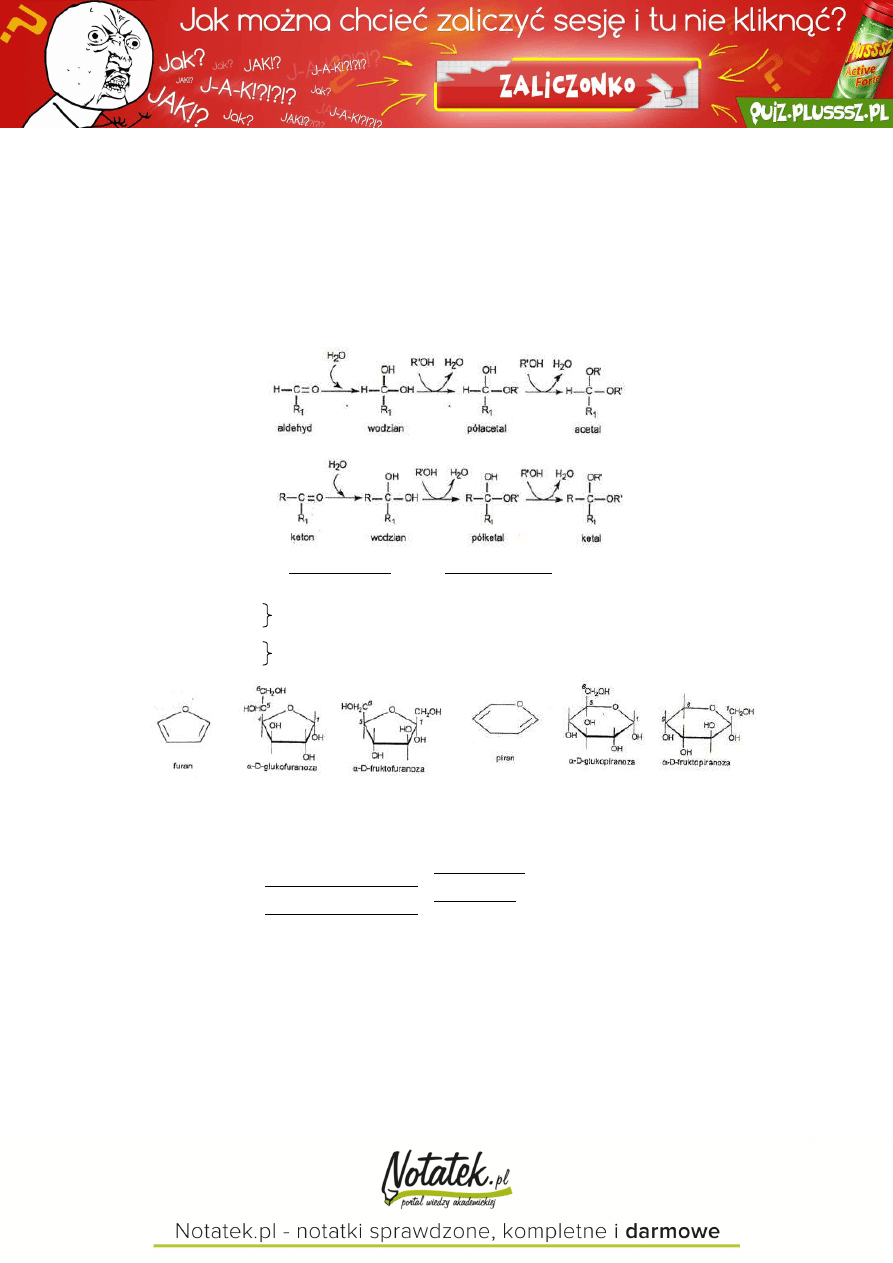
BUDOWś PIER CIENIOWś
oprócz formy łańcuchowej cukry mogą tworzyć pierścienie heterocykliczne
forma
łańcuchowa
↔
forma pierścieniowa
TAUTOMERIA
= zjawisko występowania związków chemicznych w dwóch postaciach izomerycznych mogących
swobodnie przechodzić jedna w drugą
cukry pierścieniowe zalicza się do cyklicznych półacetali/półketali, które tworzą się w wyniku reakcji
aldehydu/ketonu z alkoholem
półacetale (półketale) tworzą się gdy wodzian aldehydu (ketonu) reaguje z jedną cząsteczką alkoholu, z kolei
acetale (ketale)
w przypadku reakcji z dwiema cząsteczkami alkoholu
podczas cyklizacji cukrów grupa karbonylowa reaguje z grupą hydroksylową tworząc półacetalową/półketalową
formę cykliczną
atom tlenu grupy karbonylowej wiąże się z atomem tlenu grupy hydroksylowej
ALDOZY C
1
z C
4
KETOZY C
2
z C
5
ALDOZY C
1
z C
5
KETOZY C
2
z C
6
utworzenie formy cyklicznej powoduje powstanie w cząsteczkach nowego asymetrycznego atomu węgla i
zwiększenie liczby izomerów
każdy monocukier cykliczny może występować w dwóch odmianach: i
dla szeregu D:
forma = grupa hydroksylowa przy at. C
1
występuje po prawej stronie we wzorze rzutowym Fishera
występuje pod powierzchnią pierścienia w perspektywie Hawortha
forma = grupa hydroksylowa przy at. C
1
występuje po lewej stronie we wzorze rzutowym Fishera
występuje nad powierzchnią pierścienia w perspektywie Hawortha
dla szeregu L zależności są odwrotne
powstaje 5-
członowy pierścień tetrahydrofuranu
FURśN → furanoza
powstaje 6-
członowy pierścień tetrahydropiranu
PIRśN → piranoza
MUTAROTACJA
przechodzenie form anomerycznych jedna w drugą ( ↔ )
zachodzi w wodnych roztworach mono- i oligosacharydów
formy i różnią się przede wszystkim skręcalnością właściwą
dla -D-
glukozy +112,2°
dla -D-glukozy +19°
wzajemne stopniowe przechodzenie form zachodzi aż do ustalenia się stanu równowagi dynamicznej
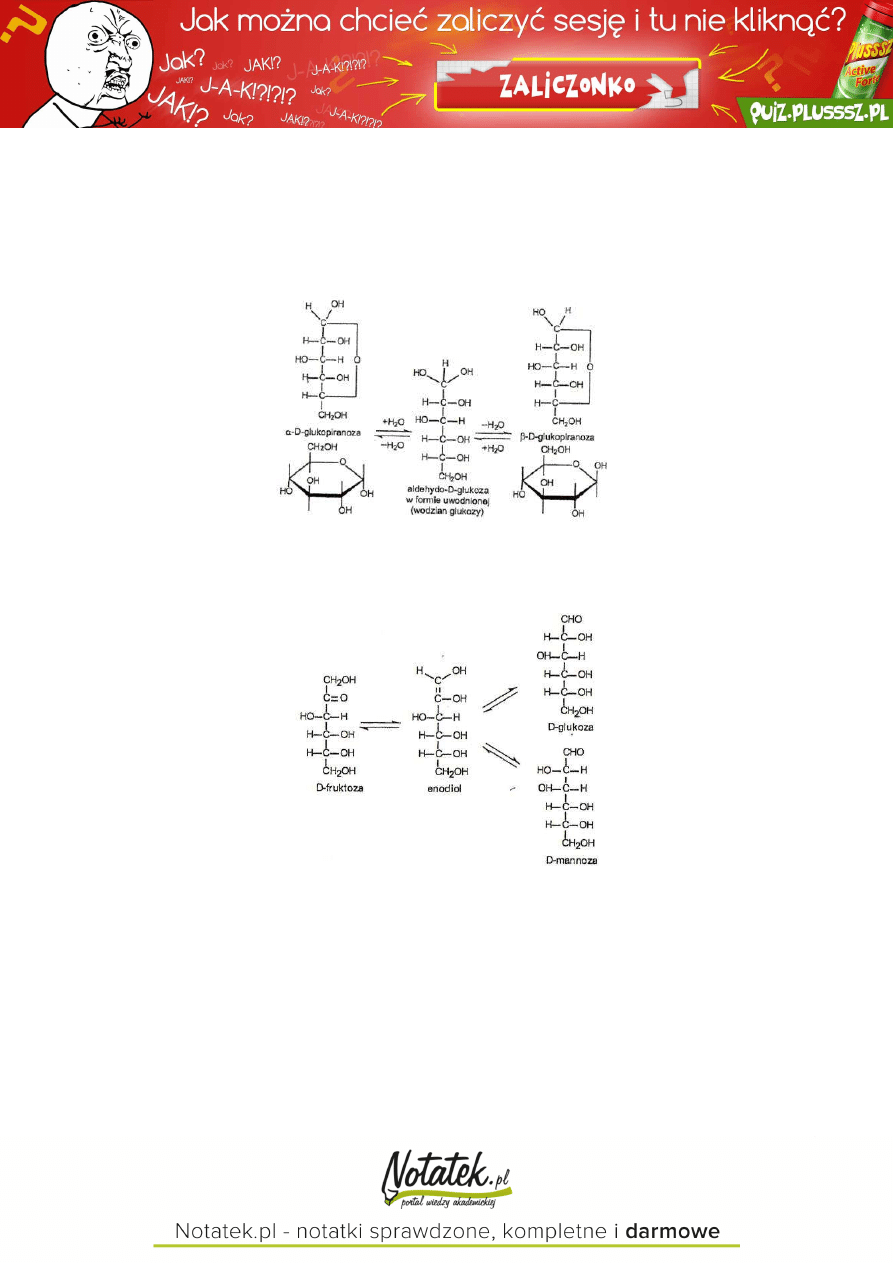
EPIMERYZACJA
specyficzna izomeryzacja heksoz, zachodząca w r-r rozcieńczonych wodorotlenków
podczas tej przemiany następuje przegrupowanie podstawników przy atomach C
1
i C
2
→ tworzy się enodiol, który
jest w równowadze z ketozą i dwoma aldozami
aldozy są dla siebie epimerami – izomerami o różnej konfiguracji przy tylko jednym chiralnym atomie węgla (ale
różnym od C
1
), z kolei ketoza jest dla nich izomerem konsytucyjnym
ogrzewanie cukrów w stężonych r-r zasad powoduje przesunięcie wiązania enolowego w inne pozycje szkieletu
węglowego, wraz z przemianami polimeryzacyjnymi i degradacyjnymi → zmiana zabarwienia na żółte, czerwone
aż do czarnego oraz wydzielanie się zapachu karmelu (próba Mohra)
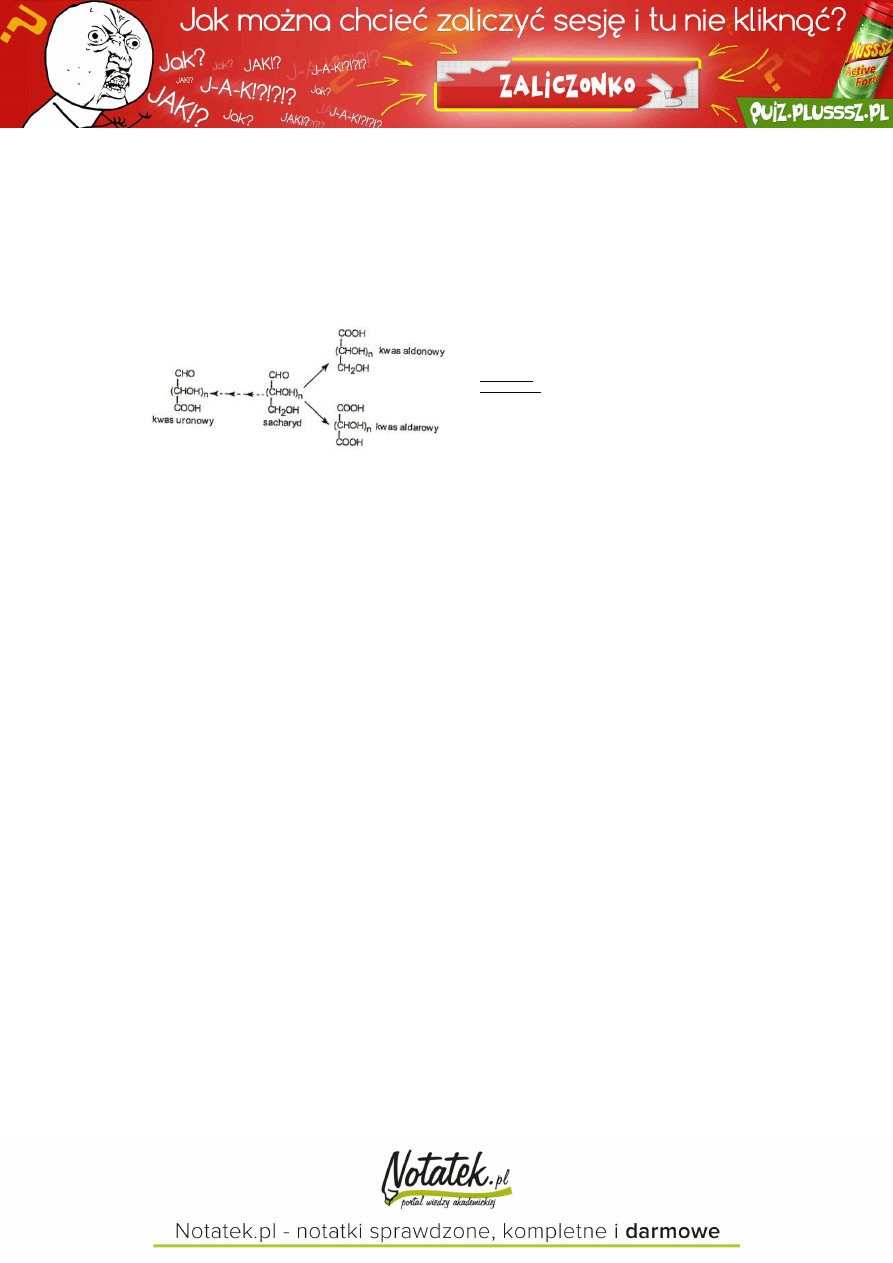
UTLENIANIE
przebieg utleniania jest uzależniony od rodzaju utleniacza
w zależności od warunków reakcji produktami utleniania są:
kwasy glikonowe (aldonowe, onowe) → utlenienie grupy aldehydowej/ketonowej (woda bromowa, odczynnik
Tollensa, odczynnik Trommera)
kwasy glikarowe (aldarowe, cukrowe) → utlenienie krańcowych atomów węgla (HNO
3
w obecności tlenu
i katalizatora platynowego)
kwasy glikuronowe (uronowe) → utlenienie atomu C
6
i wcześniejsze zablokowanie grupy
aldehydowej/ketonowej
kwas i aldehyd mrówkowy → rozerwanie wiązań C-C w pierścieniu (meta jodan(VII) HIO
4
)
śNśLIZś JśKO CIOWś I ILO CIOWś CUKRÓW
reakcje utleniania, redukcji, kondensacji oraz odwodnienia sacharydów wykorzystywane są do ich analizy
jakościowej i ilościowej
PRÓBA FEHLINGA → cukry redukujące
utleniane cukrów w środowisku zasadowym (NaOH) do hydroksykwasów, a jony Cu
2+
(pochodzące z kompleksu
z winianem sodowo-potasowym) redukowane do Cu
2
O
redukcja kationów miedzi powoduje wytworzenie nierozpuszczalnego tlenku miedzi(I) o barwie ceglastoczerwonej
P
RÓBA BENEDICTA → cukry redukujące
utlenianie sacharydów w środowisku zasadowym (Na
2
CO
3
), połączone z redukcją jonów Cu
2+
(z kompleksu
z cytrynianem) do Cu
2
O
osad tlenku miedzi(I) w miarę zwiększania agregacji cząsteczek zmienia barwę od zielonej, przez żółtą do
ceglastoczerwonej
PRÓBA BARFOEDA → rozróżnienie redukujących cukrów prostych od dwucukrów
utlenianie sacharydów w środowisku lekko kwaśnym (kwas mlekowy), połączone z redukcją jonów Cu
2+
(z
kompleksu winianem)
odpowiednio dobrany czas reakcji pozwala na rozróżnienie cukrów prostych i dwucukrów (glukoza – szybko;
laktoza
– wolno)
PRÓBA SELIWANOWA → rozróżnienie aldoz i ketoz
odczynnik Seliwanowa (H
2
O + HCl + rezorcyna)
rezorcyna daje barwne kompleksy z pochodnymi cukrów, powstającymi podczas odwodnienia z użyciem
stężonego kwasu mineralnego
KETOZY → barwa łososiowa → szybko ulegają reakcji (fruktoza, sacharoza)
ALDOZY
→ wolno ulegają reakcji (maltoza, glukoza)
PRÓBA MOLISCHA → rozróżnienie cukrów od innych zw. organicznych
reakcja z -naftolem w środowisku kwaśnym H
2
SO
4
furfuralowe produkty odwodnienia cukrów dają kompleksy o czerwonowiśniowej barwie
bardzo czuła reakcja dająca pozytywny wynik nawet w śladowej obecności cukrów (pozytywny wynik reakcji
dają niektóre związki nie będące cukrami: aceton, aldehydy)
praktyczną wartość ma ujemny wynik reakcji, jako wykluczający obecność cukru
PRÓBA BIALA → rozróżnienie pentoz i heksoz
reakcja z orcyną w środowisku kwaśnym HCl
furfuralowe produkty odwodnienia cukrów dają barwne kompleksy z orcyną
PENTOZY: furfural + orcyna → ciemnozielony
HEKSOZY: hydroksymetylofurfural + orcyna → żółtobrązowy
PRÓBA TAUBERA → rozróżnienie pentoz i heksoz
barwna reakcja z benzydyną
PENTOZY: czerwonowiśniowe
HEKSOZY:
żółte/brunatne
dla glukozy: kwas glukonowy, cukrowy, glukuronowy
dla galaktozy
: kwas galaktonowy, śluzowy, galakturonowy

PRÓBA TOLLENSA → rozróżnienie pentoz i heksoz
reakcja z fluoroglucyną w środowisku kwaśnym HCl
wynik analogiczny jak w przypadku próby Traubera
REAKCJA Z ANTRONEM
→ rozróżnienie cukrów od innych zw. organicznych
reakcja z antronem
odwodnione cukry (furfurale) tworzą niebieskozielony związek
POLISACHARYDY
podstawowa forma występowania węglowodanów w komórkach
są związkami optycznie czynnymi, ale nie wykazują właściwości redukujących i nie mają smaku słodkiego
podział ze wzg. na pełnione funkcje biologiczne:
polisacharydy szkieletowe = budulcowe (np. celuloza, chityna, pektyny)
zazwyczaj nierozpuszczalne w wodzie i pęczniejące w bardzo małym stopniu
polisacharydy zapasowe (np. skrobia, glikogen)
duża zdolność pęcznienia i łatwość tworzenia roztworów koloidalnych
podział ze wzg. na strukturę chemiczną:
homopolisacharydy
cząsteczki zbudowane z reszt tych samych monosacharydów
glukany (złożone z glukozy → skrobia, glikogen, celuloza)
fruktany
(złożone z fruktozy → inulina)
mannany
(złożone z mannozy → mannan)
galaktazy
(złożone z galaktozy → galaktogen)
heteropolisacharydy
cząsteczki zbudowane z reszt różnych monosacharydów, niekiedy dodatkowo połączonych związkami
niecukrowymi
np. glikolipidy, polisacharydy kwaśne, mukopolisacharydy
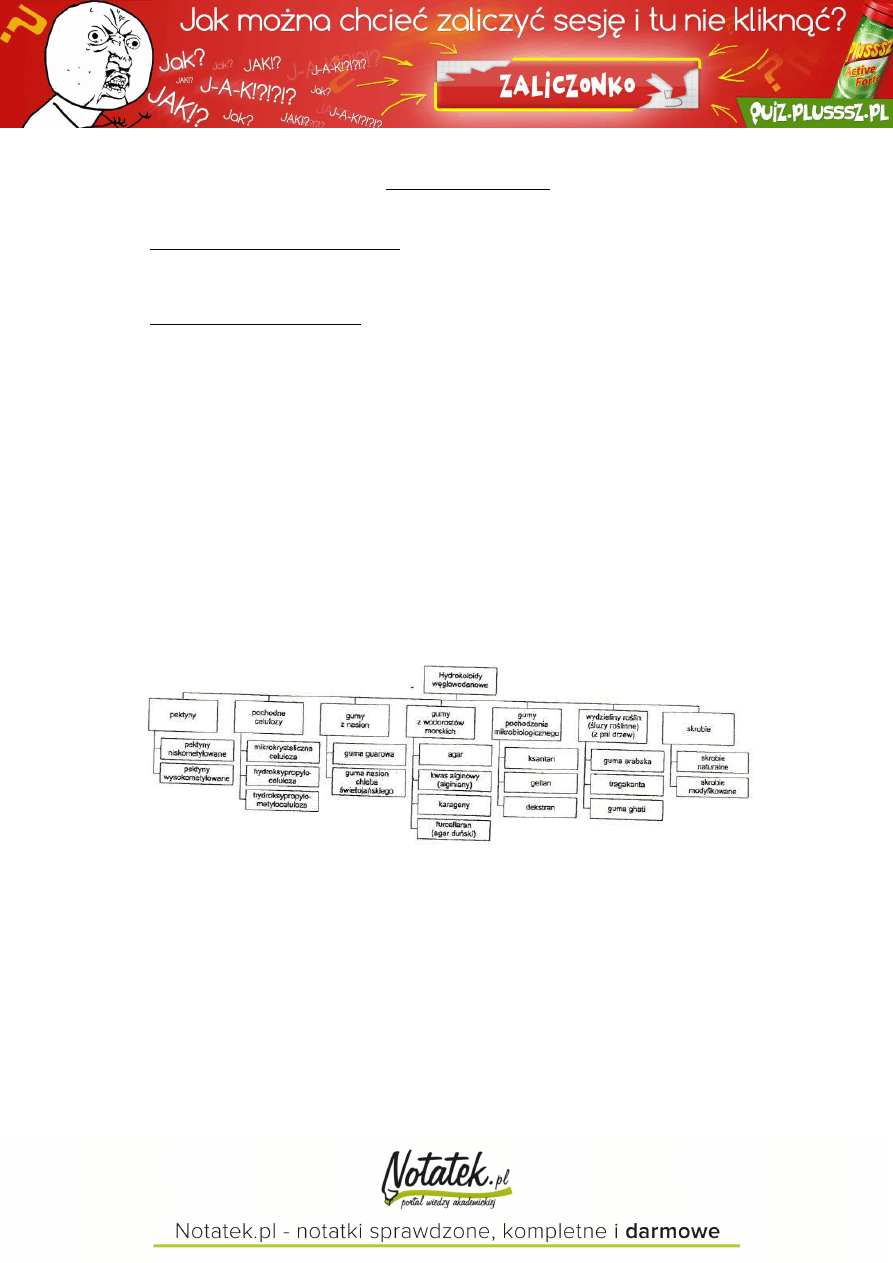
HYDROKOLOIDY
wielkocząsteczkowe związki hydrofilowe wykazujące duże powinowactwo do wody
są głównie produktami pochodzenia roślinnego – naturalnymi lub modyfikowanymi (większość polisacharydów
zaliczana jest do hydrokoloidów)
w wodzie hydrokoloidy dobrze się rozpuszczają, rozpraszają lub silnie pęcznieją
wykazują właściwości emulgujące, stabilizujące, wypełniające i teksturo twórcze
hydrokoloidy zwiększają lepkość roztworów lub tworzą żele (lepkość r-r hydrokoloidów rośnie w miarę wzrostu
ich stężenia)
ogrzewanie r-r hydrokoloidów do temperatur wyższych niż temperatura, w której osiągają maksymalną lepkość,
przyczynia się do obniżenia ich lepkości na skutek degradacji polimeru
ochładzanie gorących r-r hydrokoloidów prowadzi do wzrostu ich lepkości, co spowodowane jest rozpoczęciem
procesu żelowania
NIESKROBIOWE HYDROKOLOIDY POLISACHARYDOWE
ich cechą funkcjonalną jest zdolność do tworzenia żeli już przy niskim ich stężeniu (0,1-0,5%)
siła żelowania polisacharydu zwiększa się wraz ze wzrostem ciężaru cząsteczkowego, a maleje wraz ze wzrostem
stopnia jego rozgałęzienia
żelowanie hydrokoloidów może zachodzić pod wpływem różnych czynników:
temperatury (np. guma ksantanowa)
związków chemicznych (np. alginiany tworzą wiązania chemiczne z metalami, tj Ca, Mg)
działania synergistycznego dwóch różnych koloidów (np. guma ksantanowa i galaktomannany)
hydrokoloidy nieskrobiowe pełnią funkcję zagęstników, stabilizatorów, emulgatorów oraz środków żelujących
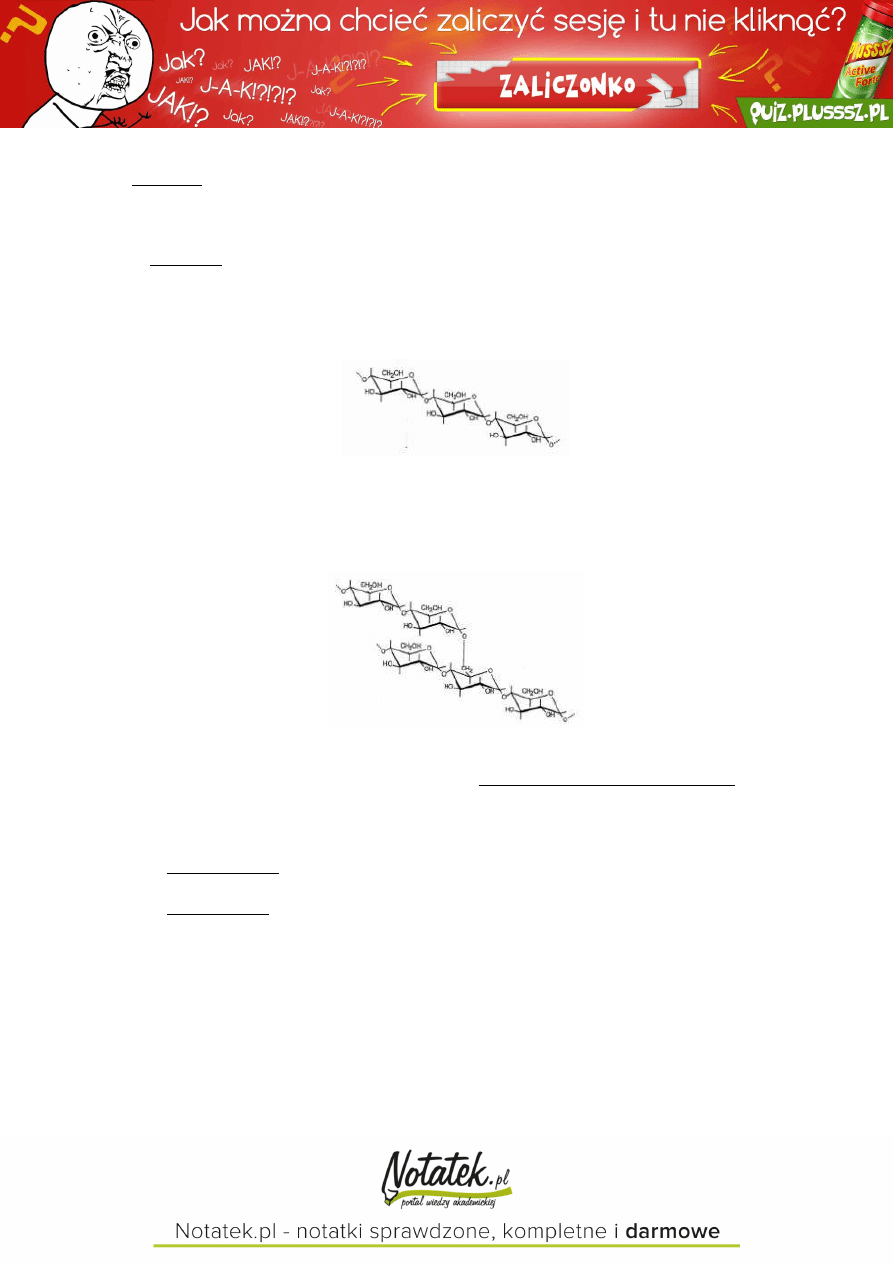
SKROBIA
zapasowy węglowodan roślinny, gromadzony głównie w nasionach (ziarnach zbóż), owocach i bulwach ziemniaka
występuje w postaci ziaren o zróżnicowanym kształcie i wielkości
nie jest substancją jednorodną – oprócz składnika węglowodanowego zawiera minimalne ilości substancji
mineralnych oraz nieznaczne ilości wyższych kwasów tłuszczowych
frakcje skrobi:
AMYLOZA
→ frakcja liniowa
→ długie, nierozgałęzione łańcuchy utworzone z reszt D-glukopiranozy połączonych wiąz. -1,4-glikozydowymi
→ stopień polimeryzacji: 250 – 1000 reszt glukozowych
→ 15-30% udziału w skrobi
→ dobrze rozpuszczalna w wodzie i podatna na hydrolizę (w r-r wodnych łańcuch zwija się w spiralę)
AMYLOPEKTYNA
→ frakcja rozgałęziona
→ od liniowych łańcuchów (analogicznych do amylozy) co 24-30 reszt glukozowych poprzez wiązania rozgałęziające
-1,6-
glikozydowe odchodzą łańcuchy boczne
→ stopień polimeryzacji: ok. 1000 reszt glukozowych
→ 40-85% udziału w skrobi
→ nierozpuszczalna w wodzie i trudno ulega hydrolizie (w r-r wodnych tylko zewnętrzne łańcuchy wykazują
skłonność do przyjmowania struktury spiralnej)
w roztworach zawierających wolny jod i jony jodkowe (płyn Lugola) ziarna skrobi barwią się na kolor
ciemnoniebieski:
obie frakcje skrobi wykazują różne powinowactwo do jodu: amyloza wiąże 40x więcej jodu niż amylopektyna
amyloza
– jod: na jeden skręt spirali przypada jedna cząsteczka jodu → barwa granatowa
amylopektyna
– jod: jod układa się wewnątrz zwiniętych w spiralę bocznych łańcuchów → fioletowo czerwona
ze wzrostem masy cząsteczkowej frakcji amylozy wzrasta intensywność niebieskiej barwy tworzonego kompleksu
różnice w zachowaniu się obu frakcji skrobi wobec cząsteczkowego jodu pozwalają na ilościowe oznaczanie
proporcji amyloza
– amylopektyna, tj. wyznaczanie wartości niebieskiej
środowisko zasadowe = charakterystyczne zabarwienie kompleksów jodowo-skrobiowych nie powstaje, ponieważ
jod reaguje z zasadą:
I
2
+ 2NaOH → NaIO + NaI + H
2
O
środowisko kwaśne = reakcja przebiega w kierunku odwrotnym
NaIO + NaI + 2HCl → 2NaCl + I
2
+ H
2
O
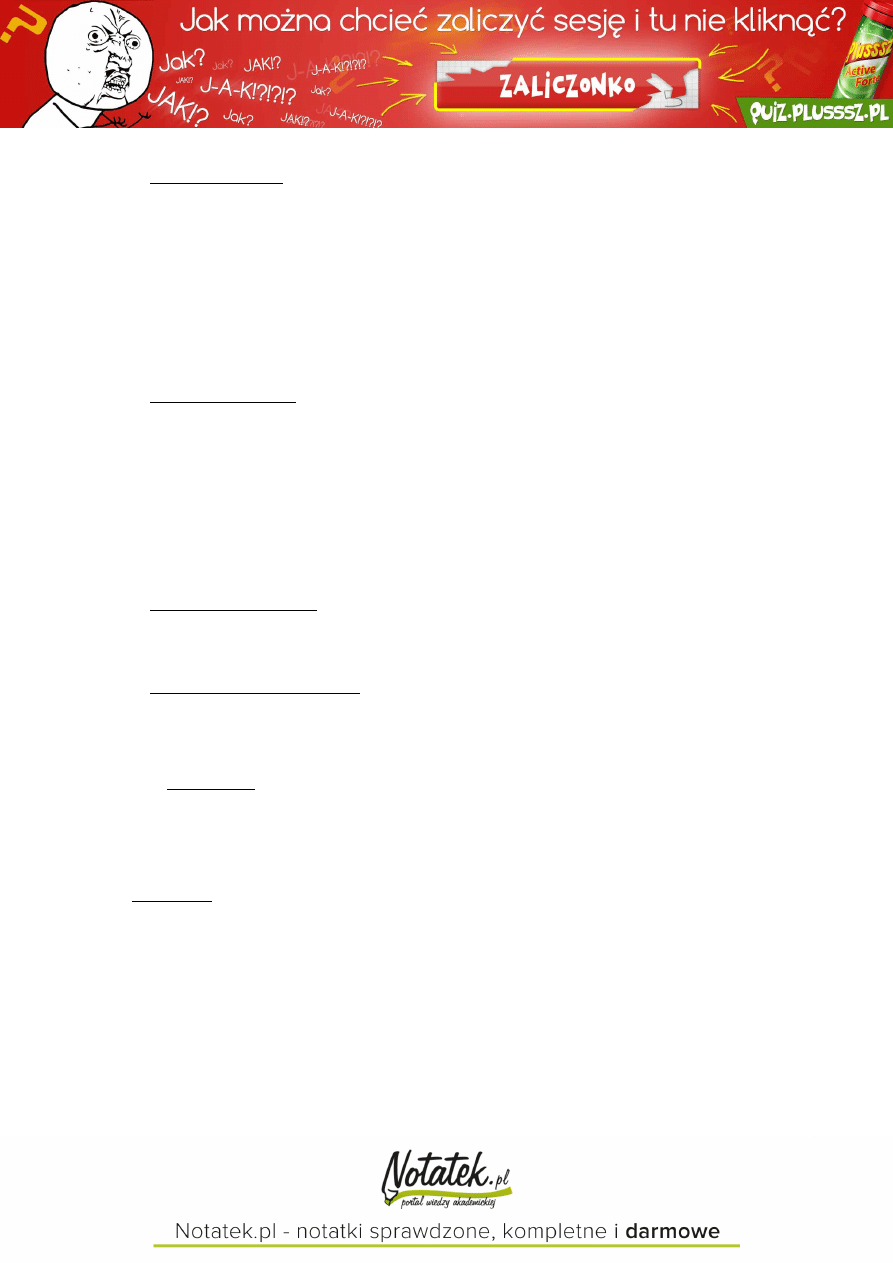
HYDROLIZA SKROBI:
skrobia ulega częściowej lub całkowitej hydrolizie pod wpływem kwasów mineralnych: HCl lub H
2
SO
4
reakcja przebiega przez etapy pośrednie:
1) tworzenie dekstryn, barwiących się w obecności jodu:
amylodekstryny → barwa niebieskofiołkowa
erytrodekstryny
→ barwa czerwona
achrodekstryny → nie dające zabarwienia
2) powstawanie maltozy
3) powstawanie glukozy
hydroliza skrobi może być przeprowadzona również z udziałem enzymów:
-
amylaza, -amylaza, glukoamylaza → rozpad wiąz. -1,4-glikozydowych
pululanaza
→ rozpad wiąz. -1,6-glikozydowych
transferaza glukozylowa cyklodekstryn → powstanie cyklodekstryn (cyklicznych oligosacharydów)
zróżnicowane warunki w jakich może zachodzić hydroliza skrobi prowadzą do otrzymania różnych produktów,
np. syropów skrobiowych, maltodekstryn i glukozy
KLEIKOWANIE SKROBI:
skrobia w wodzie jest prawie nierozpuszczalna – w zimnej wodzie ulega pęcznieniu, a podczas ogrzewania
kleikowaniu i wytworzeniu zoli, które podczas ponownego schładzania przekształcają się w żele
kleiki skrobi naturalnej są mało stabilne, a ich lepkość zmienia się pod wpływem zmian temperatury,
kwasowości oraz mieszania
dodanie butanolu do skleikowanej skrobi powoduje wytrącenie się amylozy – jako kompleksu z butanolem
skrobia ulega rozpuszczeniu m.in. w dimetylosulfotlenku (DMSO), kwasie chlorowym(VII) oraz stężonym r-r
chlorku wapnia zakwaszonym kwasem octowym
w czasie przechowywania zolu lub żelu skrobiowego następuje retrogradacja skrobi → tworzenie się wiąz.
wodorowych
pomiędzy sąsiadującymi cząsteczkami amylozy → wytrącenie osadu poprzez powstanie struktury
krystalicznej skrobi, mętnienie kleiku, twardnienie i wydzielanie wody (synereza)
retrogradacja i synereza powodują obniżenie jakości produktów spożywczych o dużej zawartości skrobi (np.
pieczywa)
czynnikiem sprzyjającym retrogradacji jest niska temperatura ok. 0°C
MODYFIKOWANIE SKROBI:
w celu uniknięcia niekorzystnych procesów zachodzących podczas przechowywania zoli i żeli skrobiowych
skrobię naturalną poddaje się modyfikacji
skrobią modyfikowaną jest skrobia, w której wywołano niewielkie zmiany struktury lub składu metodami
fizycznymi, chemicznymi, enzymatycznymi lub ich kombinacjami
PRZEMIANY TERMICZNE SKROBI
przemiany skrobi pod wpływem wysokich temperatur wpływają na strukturę i konsystencję oraz w mniejszym
stopniu na smak i barwę produktów
przemiany termiczne skrobi można podzielić na suche oraz takie, które zachodzą w obecności wody
w obecności wody: przemiany związane z rozpuszczalnością, zdolnością wiązania wody, kleikowaniem
i retrogradacją
w warunkach bezwodnych: dekstrynizacja (105-210°C) i piroliza (>210°C)
dekstrynizacja:
polega na rozrywaniu łańcucha polisacharydowego na mniejsze fragmenty – dekstryny (białe i żółte)
stopień dekstrynizacji = stopień zniszczenia łańcucha skrobiowego (im wyższy, tym mniejszą masę
cząsteczkową mają powstające produkty rozkładu)
wyższe temperatury obróbki powodują większy stopień dekstrynizacji
proces dekstrynizacji i pirolizy zachodzi m.in. w skórce w pieczonych wyrobach z ciasta, w warstwie panierki
w produktach smażonych, w czasie prażenia płatków śniadaniowych
GLIKOGEN
węglowodan zapasowy występujący w organizmach zwierzęcych, gromadzony w formie ziaren o średnicy 10-40nm
w wątrobie: 2-10%
w mięśniach: 0,5-1,5%
zbudowany podobnie jak amylopektyna, ale posiadający bardziej rozgałęzioną strukturę:
liczniejsze odgałęzienia boczne (co 8 – 10 reszt glukozowych)
krótsze odgałęzienia boczne (zawierające 10 – 18 reszt glukozowych)
sporadycznie występujące wiązania -1,3-glikozydowe
zróżnicowany stopień polimeryzacji: kilka tysięcy (mięśnie) – kilkadziesiąt tysięcy (wątroba)
glikogen lepiej niż amylopektyna rozpuszcza się w zimnej wodzie tworząc opalizujący koloidalny roztwór
(roz
puszczalność w wodzie jest możliwa dzięki obecności licznych, krótkich odgałęzień bocznych łańcucha)
kompleks glikogen – jod: ma barwę czerwonobrunatną
stosunkowo łatwo ulega hydrolizie kwasowej i enzymatycznej do glukozy

łatwo ulega wytrąceniu z roztworu w obecności dobrze zdysocjowanych soli nieorganicznych (np. siarczanu(VI)
amonu, chlorku sodu)
następuje dehydratacja glikogenu oraz agregacja cząsteczek na skutek samorzutnie tworzących się wiązań
wewnątrz- i międzycząsteczkowych
wytrącenie glikogenu z roztworu następuje również pod wpływem rozpuszczalników organicznych (np. aceton,
alkohol etylowy)
glikogen zmagazynowany w org. zwierzęcia bezpośrednio po uboju ulega rozkładowi z wytworzeniem kwasu
mlekowego, zakwaszającego środowisko i powodującego hydrolizę białek (kruszenie mięsa)
CELULOZA
polisacharyd strukturalny najbardziej rozpowszechniony w przyrodzie
stanowi główny składnik ścian komórkowych w komórkach roślinnych (najwięcej celulozy zawiera włókno bawełny
ok. 87
– 98%)
celuloza jest polimerem -D-glukopiranozy, w którym reszty glukozowe połączone są wiąz. -1,4-glikozydowymi
stopień polimeryzacji: 14 000 reszt glukozowych
zwarta, nitkowata (fibrylarna) budowa
charakteryzuje się wysoką odpornością na działanie czynników chemicznych
nierozpuszczalna w wodzie, ani w innych znanych rozpuszczalnikach - jedynymi rozpuszczalnikami celulozy są:
odczynnik Schweitzera = wodorotlenek tetraaminamiedzi(II) [Cu(NH
3
)
4
](OH)
2
odczynnik Cross-Bewana
= chlorek cynku(II) w stężonym kwasie solnym
kwasy i zasady działają na celulozę w znikomym stopniu (długotrwałe gotowanie ze stężonymi kwasami
mineralnymi pod ciśnieniem powoduje jej hydrolityczny rozkład do glukozy)
nie jest trawiona przez enzymy człowieka → zaliczana jest do błonnika pokarmowego
enzym -celulaza (wytwarzany przez niektóre drobnoustroje i grzyby) hydrolizuje celulozę do celobiozy i glukozy
w przemyśle spożywczym ma znaczenie celuloza modyfikowana – głównie etery, które są albo rozpuszczalne
w wodzie, albo ulegają w niej pęcznieniu i są stosowane jako emulgatory i subst. zapobiegające pienieniu
celuloza mikrokrystaliczna jest cennym dodatkiem do żywności, pełniącym rolę wypełniacza w produktach
o obniżonej zawartości tłuszczu oraz jako subst. zapobiegająca krystalizacji w żywności mrożonej
DEKSTRAN
polisacharyd syntetyzowany z sacharozy przez bakterie Leuconostoc mesenteroides
zbudowany z reszt D-glukopiranozy
reszty glukozowe połączone są w łańcuchu głównym wiąz. -1,6-glikozydowymi, a w miejscach rozgałęzień wiąz.
-1,4- , -1,3- i -1,2-glikozydowymi
od których odchodzą krótkie łańcuchy boczne (ok. 5 reszt glukozy)
stopień polimeryzacji: 100 – 200 000 reszt glukozy
duża lepkość dekstranu decyduje o jego zastosowaniu jako zamiennika gum i śluzów roślinnych
PEKTYNY
polisacharydy kwaśne, których podstawowymi składnikami są kwasy uronowe
szeroko rozpowszechnione – tworzą składnik strukturalny roślin
nie są polisacharydami jednorodnymi: protopektyna, pektyna właściwa, kwas pektynowy
protopektyna
nierozpuszczalna w wodzie
stanowi łańcuch ramnozogalakturonianu zawierający łańcuchy boczne galaktanu i arabinianu (dodatkowo
zawiera atomy wapnia tworzące mostki między cząsteczkami pektyn)
pod wpływem obróbki termicznej owoców i warzyw oraz pod wpływem enzymów hydrolizuje do pektyny
właściwej
pektyna wła ciwa
zbudowana z kwasu pektynowego połączonego wiąz. kowalencyjnymi z łańcuchami galaktanu i arabinianu
kwas pektynowy
długi, nierozgałęziony łańcuch kwasu -poligalakturonowego, w którym część grup karboksylowych w pozycji
C-6 zestryfikowana jest alkoholem metylowym
rozpuszczają się w zimnej wodzie, ale nie rozpuszczają się w etanolu o stężeniu wyższym niż 20%
wykazują zdolność wiązania znacznych ilości wody - wodochłonno ć (dwukrotnie więcej niż ich masa
cząsteczkowa)
zdolność żelowania i warunki tworzenia żelu zależą od stopnia zmetylowania pektyn:
pektyny wysokozmetylowane
(zawierają >50% zestryfikowanych grup karboksylowych) → tworzą żele w bardzo
kwaśnym środowisku (pH 2,0-3,5) i przy wysokim stężeniu cukru (ponad 50%)
pektyny niskozmetylowane
(zawierają <50% zestryfikowanych grup karboksylowych) → żelują w szerokim zakresie
(pH 2,5-
5,5) i przy niższym stężeniu cukru (od 20%), ale wymagają obecności jonó
wapnia
znajdują zastosowanie jako substancje żelujące w produkcji dżemów, konfitur i galaretek owocowych oraz jako
stabilizatory do kremów, budyniów i deserów
Wyszukiwarka
Podobne podstrony:
Notatki na I kolokwium, Patofizjologia
Notatki na kolokwium 3
rekreacja notatki na kolokwium, pedagogika czasu wolnego, rekreacja, metodyka rekreacji
Biologiczne mechanizmy zachowania - notatki na kolokwium 16.04.2010, KN, rok I, Biologiczne mechaniz
notatki na kolokwium z filmu, Studia, Film, media i audiowizualna
Notatki na 1 kolokwium- wykłady + prelekcje, Studia, Stomatologia Łódź, Rok III, Immunologia, Immuno
Finanse notatki na kolokwium z zeszytu wykład, Finanse publiczne i prawo podatkowe, Wykład
Notatka na kolokwium
Notatki na kolokwium - zimna wojna, Studia, Stosunki Międzynarodowe
TK.NERWOWA-notatki na kolokwium, Biotechnologia, Histologia
Biologiczne mechanizmy zachowania - notatki na kolokwium 07.06.2010, KN, rok I, Biologiczne mechaniz
Notatki na kolokwium z ćwiczeń
Notatki na kolokwium z upodobnień
gospodarka wodna notatki na kolokwium, Gospodarka wodna i zaopatrzenie ludności w wodę
Notatki na kolokwium Barwnik,związki?nolowe
SOCJOLOGIA (ćwiczenia) Notatki na kolokwium zaliczajace
Pedagogika KF notatki na kolokwium
logika notatki na kolokwium
więcej podobnych podstron