
Neurologia Praktyczna • 6/2011
15
Copyright wydania polskiego © 2011 by Wydawnictwo Czelej Sp. z o.o.
WSTĘP
Kryteria diagnostyczne stwardnienia rozsiane-
go (multiple sclerosis – MS) ewoluowały w ciągu
ostatnich 50 lat. Chociaż kolejne ich wersje kła-
dły nacisk na różne aspekty choroby, wszyst-
kie wymagały udokumentowanego kryteria-
mi klinicznymi, paraklinicznymi lub laborato-
ryjnymi potwierdzenia rozproszenia objawów
w przestrzeni i czasie. Ponadto kryteria diagno-
styczne MS kładły nacisk na konieczność roz-
ważenia i wykluczenia innych alternatywnych
schorzeń mogących wyjaśnić objawy kliniczne
[1-4].
py g
y
p
g
y y
j p
Diagnostyka różnicowa w przypadkach
podejrzenia stwardnienia
rozsianego: konsensus
Tł u m a c z e n i e a r t y ku ł u :
Diff erential diagnosis of suspected multiple sclerosis: a consensus approach
D.H. Miller
1
, B.G. Weinshenker
2
,
M. Filippi
3
, B.L. Banwell
4
, J.A. Cohen
5
,
M.S. Freedman
6
, S.L. Galetta
7
,
M. Hutchinson
8
, R.T. Johnson
9
,
L. Kappos
10
, J. Kira
11
, F.D. Lublin
12
,
H.F. McFarland
13
, X. Montalban
14
, H.
Panitch
15
, J.R. Richert
16
, S.C. Reingold
16
,
17
i C.H. Polman
18
1
Department of Infl ammation, Institute of Neurology, NMR
Research Unit, University College London, Wielka Brytania
2
Department of Neurology, Mayo Clinic College
of Medicine, Rochester, Minnesota, USA
3
Neuroimaging Research Unit, Department of
Neurology, Ospedale San Rafaele, Mediolan, Włochy
4
Th
e Hospital for Sick Children, Department of Paediatrics,
Division of Neurology, Toronto, California, USA
5
Th
e Mellen Center, Cleveland Clinic, Cleveland, Ohio, USA
6
MS Research Unit, Department of Medicine
(Neurology), University of Ottawa, Th
e Ottawa Hospital
– General Campus, Ottawa, Kalifornia, USA
7
Department of Neurology, University of Pennsylvania
Hospital, Philadelphia, Pennsylvania, USA
8
St Vincent’s University Hospital, Department
of Neurology, Dublin, Irlandia
9
Th
e Johns Hopkins Hospital, Department
of Neurology, Baltimore, Maryland, USA
10
Department of Neurology, University
Hospitals, Basel, Szwajcaria
11
Department of Neurology, Kyushu University, Kyushu, Japonia
12
Corrine Goldsmith Dickinson Center for Multiple Sclerosis,
Mt. Sinai School of Medicine, New York City, New York, USA
13
Neuroimmunology Branch, NINDS, National
Institutes of Health, Bethesda, Maryland, USA
14
Unitat de Neuroimmunologia Clinica, Hospital
Universitari Vall d’Hebron, Barcelona, Hiszpania
15
Neurology Service, University of Vermont College
of Medicine, Burlington, Vermont, USA
16
Research and Clinical Programs Department, National
Multiple Sclerosis Society, New York City, New York, USA
17
Scientifi c and Clinical Review Associates,
LLC, New York City, New York, USA
18
Department of Neuroinfl ammation, Institute of Neurology,
University College London, Wielka Brytania
Słowa kluczowe:
rozpoznanie, diagnostyka różnicowa, stwardnienie rozsiane.
S t re s z c z e n i e
Założenia i cele. Rozpoznanie stwardnienia rozsianego (multiple sclerosis – MS)
wymaga wykluczenia chorób, które mogą stanowić lepsze wyjaśnienie objawów
klinicznych i paraklinicznych. Dotychczas nie opisano uporządkowanego procesu
wykluczania alternatywnych rozpoznań. Międzynarodowy Panel Ekspertów zaj-
mujący się Stwardnieniem Rozsianym (International Panel of MS experts) opracował
ujednolicony algorytm diagnostyki różnicowej MS.
Metodyka. W oparciu o dostępne piśmiennictwo opracowano ujednolicone wy-
tyczne diagnostyki różnicowej MS. Zwrócono szczególną uwagę na wykluczenie
stanów mogących potencjalnie przypominać MS, rozpoznanie najczęstszych izolo-
wanych zespołów klinicznych stanowiących manifestację MS oraz na różnicowanie
MS i innych idiopatycznych zapalnych chorób demielinizacyjnych.
Wyniki. Prezentujemy zalecenia odnoszące się do: 1) klinicznych i paraklinicznych
„czerwonych fl ag” sugerujących schorzenie inne niż MS; 2) bardziej precyzyjnej
defi nicji „izolowanych zespołów klinicznych” (clinically isolated syndrome – CIS),
będących często pierwszą prezentacją MS lub innych schorzeń alternatywnych;
3) algorytmów diagnostycznych trzech częstych izolowanych zespołów klinicznych
związanych z MS, obejmujących objawy z nerwów wzrokowych, pnia mózgu i rdze-
nia kręgowego; 4) schematu klasyfi kacji i kryteriów diagnostycznych idiopatycznych
demielinizacyjnych chorób zapalnych ośrodkowego układu nerwowego.
Wnioski. Diagnostyka różnicowa prowadząca do rozpoznania MS lub innych scho-
rzeń alternatywnych jest złożona, a ponadto brakuje w tym aspekcie silnych danych
opartych na faktach. Ujednolicone wytyczne stanowią praktyczną ścieżkę diagno-
styczną i będą użyteczne dla neurologów niebędących specjalistami w dziedzinie
MS. Zaleca się, aby wytyczne poddano walidacji i ocenie w perspektywie przyszłych
badań. Wytyczne dotyczące procesu diagnostycznego w przypadkach podejrzenia
MS zwiększą dokładność i precyzję rozpoznania.
Reprinted from
Multiple Sclerosis
2008; 14: 115 7-11 74,
DH Miller, BG Weinshenker, M Filippi, BL Banwell, JA Cohen, MS Freedman, SL Galetta, M Hutchinson,
RT Johnson, L Kappos, J Kira FD Lublin, HF McFarland, X Montalban, H Panitch, JR Richert, SC
Reingold, CH Polman, Differential diagnosis of suspected multiple sclerosis: a consensus approach,
© SAGE Publications 2008, with permission from Elsevier
Licencjodawca nie odpowiada za kompletność i dokładność tłumaczenia
Neurologia Praktyczna • 6/2011
16
Najnowsze Kryteria McDonalda formalnie
włączają dane z rezonansu magnetycznego (ma-
gnetic resonance imaging – MRI) i koncentrują się
na wczesnym rozpoznaniu w przypadkach wy-
stąpienia izolowanych zespołów klinicznych (cli-
nically isolated syndrome – CIS) sugerujących MS
(np. jednostronne zapalenie nerwu wzrokowe-
go, porażenie międzyjądrowe, częściowa mielo-
patia) [3, 4]. Ponieważ u większości spośród tych
pacjentów drugi epizod objawów klinicznych
pojawi się w ciągu miesięcy lub lat, kryteria dia-
gnostyczne postrzegane są bardziej jako pro-
gnostyczne w odniesieniu do przyszłej aktywno-
ści klinicznej (czy kolejny rzut wystąpi?) niż dia-
gnostyczne (instrument różnicujący MS od in-
nych chorób).
U pacjentów, u których podejrzewa się MS,
w pierwszym badaniu fi zykalnym można stwier-
dzić obecność zespołów neurologicznych jedno-
ogniskowych klinicznie (brak rozsiania w prze-
strzeni, dla którego wytłumaczeniem jest poje-
dyncze uszkodzenie OUN), wieloogniskowych
klinicznie (rozsianie w przestrzeni, dla którego
wytłumaczeniem jest obecność przynajmniej
dwóch zmian ogniskowych w różnych częściach
OUN), które mają charakter jednofazowy (poje-
dynczy epizod), wielofazowy (nawracający) lub
postępujący. Podobne objawy kliniczne mogą
wystąpić u pacjentów z chorobami infekcyjnymi,
nowotworowymi, wrodzonymi, metaboliczny-
mi, naczyniowymi lub innymi niż MS idiopatycz-
nymi demielinizacyjnymi chorobami zapalnymi
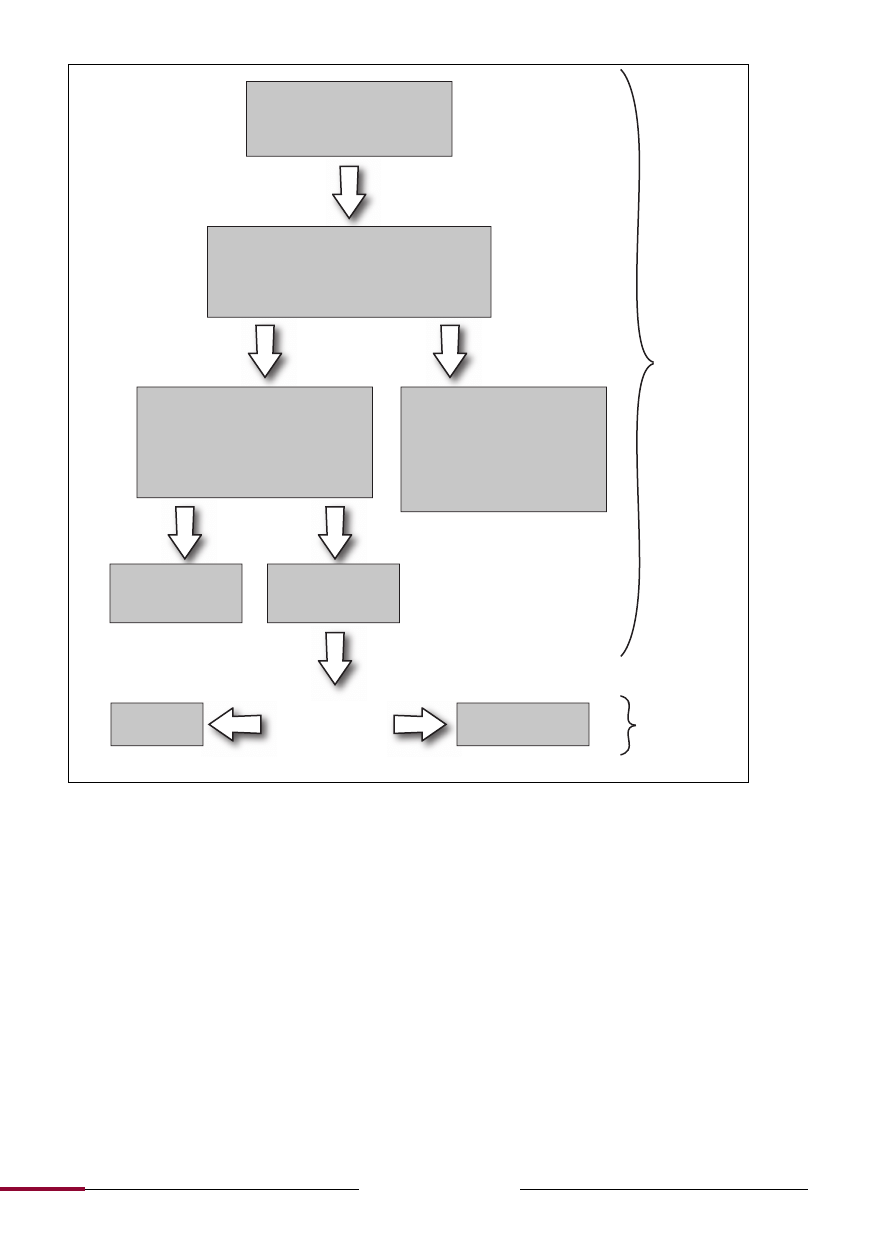
Ryc. 1.
Etapy diagnostyki różnicowej MS.
Objawy odpowiadające zapalnej
chorobie demielinizacyjnej (ze-
społy jednoogniskowe lub wielo-
ogniskowe)
Wykluczenie niedemielinizacyjnego zespołu
objawów klinicznych (w oparciu o dane demo-
graficzne, specyficzne dolegliwości i objawy
kliniczne, przebieg kliniczny, wyniki badań ra-
diologicznych i testów laboratoryjnych)
Zaklasyfikowanie idiopatycznej zapal-
nej choroby demielinizacyjnej (w opar-
ciu o dane demograficzne, przebieg
kliniczny, specyficzne dolegliwości
i objawy kliniczne, wyniki badań radio-
logicznych i testów laboratoryjnych)
Nie MS (MNO,
ADEM, niesklasyfi-
kowana)
Ustalenie roz-
poznania MS
Rozpoznanie MS nie
jest jeszcze ustalone
Objawy zgodne
z prototypowym MS
(włączając CIS)
Rozproszenie w cza-
sie i przestrzeni (kry-
teria McDonalda)
Określenie rozpoznania niezapal-
nej choroby demielinizacyjnej
(rozpoznanie „czerwonych flag”
sugerujących specyficzne rozpo-
znanie lub wyczerpująca ocean
kliniczna, jeśli rozpoznanie nie
jest oczywiste)
No
wa ścieżk
a diag
nost
y
czna – dok
ładniejsza oc
ena warunku
„braku
lepsz
ego w
y
tłumacz
enia
” w istniejąc
y
ch k
ryt
eriach diag
nost
y
c
zn
y
c
h
H
ist
or
y
czna
ścieżk
a
diag
nost
y
k
i MS

Neurologia Praktyczna • 6/2011
17
(idiopathic infl ammatory demyelinating disease –
IIDD). Inne IIDD mogą mieć objawy przypomina-
jące MS [np. neuromyelitis optica (NMO), wzroko-
wo-rdzeniowa postać MS w populacji azjatyckiej
(opticospinal MS – OSMS), ostre rozsiane zapale-
nie mózgu i rdzenia kręgowego (acute dissemi-
nated encephalomyelitis – ADEM)], jednak różnią
się przebiegiem klinicznym, patofi zjologią, le-
czeniem i rokowaniem (patrz ryc. 1). Możliwość
właściwego rozpoznania w jak najwcześniejszym
momencie ma istotne znaczenie w kontekście
postępowania, doradztwa i optymalizacji terapii.
Nie istnieje pojęciowa struktura diagnostyki
różnicowej MS. Europejska grupa MAGNIMS zde-
fi niowała „czerwone fl agi” MRI w kontekście po-
dejrzenia MS, sugerujące możliwość alternatyw-
nego rozpoznania [5], nieznajdujące jednak za-
stosowania w kontekście innych zgodnych ob-
jawów klinicznych czy badań laboratoryjnych.
Poniższy artykuł opisuje międzynarodową ujed-
noliconą próbę pokierowania klinicznej, labora-
toryjnej i obrazowej oceny pacjentów, u których
możliwe jest rozpoznanie MS, co ma na celu speł-
nienie wymagania „braku lepszego wytłumacze-
nia”, stanowiącego integralny element wszyst-
kich kryteriów diagnostycznych MS.
METODYKA
Skład i misja Międzynarodowej Grupy
Roboczej
W 2006 roku Międzynarodowa Komisja Doradcza
dla Prób Klinicznych w MS wchodząca w skład
Narodowego Stowarzyszenia MS w Stanach
Zjednoczonych zleciła Grupie Roboczej Diagno-
styki Różnicowej MS opracowanie zagadnienia
praktycznego podejścia klinicznego do zasady
„braku lepszego wytłumaczenia” w przypadkach
podejrzenia MS. Grupa Robocza składała się z 18
międzynarodowych (USA, Kanada, Europa, Japo-
nia) ekspertów w dziedzinie chorób demieliniza-
cyjnych, z różnym doświadczeniem klinicznym
i badawczym (neurologia, okulistyka, choroby
infekcyjne, MRI).
Początkowo misją grupy było: opracowanie
ujednoliconego i wypływającego z dostępnych
danych postępowania diagnostycznego u pa-
cjentów, u których stwierdza się objawy i obiek-
tywne dowody kliniczne sugerujące chorobę
istoty białej OUN; włączenie do wytycznych wła-
ściwego zestawu badań klinicznych, radiologicz-
nych i/lub laboratoryjnych, które należy wykonać
w celu wykluczenia alternatywnych rozpoznań,
szczególnie tych poddających się właściwemu
leczeniu; opracowanie praktycznych narzędzi
ułatwiających neurologom stawianie trafnych
rozpoznań i kierowanie procesem diagnostycz-
nym, stanowiących uzupełnienie Kryteriów Dia-
gnostycznych McDonalda. Intencją nie było za-
prezentowanie obszernego piśmiennictwa lub
przeglądu koncepcji, gdyż spektrum możliwości
diagnostycznych jest olbrzymie. Skoncentrowa-
no się na pacjentach z obiektywnymi objawami
sugerującymi chorobę istoty białej OUN, a po-
nadto uwzględniono osoby pozornie bezobja-
wowe lub pacjentów z innymi powszechnymi
odmiennymi klinicznie jednostkami chorobowy-
mi (np. migreną), u których w MRI stwierdza się
zmiany sugerujące chorobę istoty białej.
Plan pracy Grupy Roboczej
W celu odniesienia się do wszystkich problemów
Grupa Robocza podzieliła się na podgrupy kon-
centrujące się na: wykluczeniu potencjalnych
rozpoznań alternatywnych dla MS, diagnosty-
ce częstych izolowanych zespołów klinicznych
w kontekście MS, a także różnicowaniu MS od in-
nych niż MS IIDD. W przebiegu serii spotkań kon-
ferencyjnych i spotkań podgrup roboczych na
przestrzeni roku oraz spotkania całej Grupy Ro-
boczej w lutym 2007 roku osiągnięto konsensus
i opracowano wytyczne. Opracowanie nie obej-
mowało formalnego przeglądu piśmiennictwa,
chociaż publikowane dane wpływały na prace
członków Grupy Roboczej. Konsensus oparty na
poglądach powołanych ekspertów dotyczył po-
stępowania diagnostycznego i klasyfi kacji, dla
których brakowało dowodów naukowych.
Wykluczenie rozpoznań alternatywnych
Jedna z podgrup koncentrowała się na wyklu-
czeniu rozpoznań alternatywnych w stosunku do
MS. Opracowano liczne kliniczne i paraklinicz-
ne „czerwone fl agi”, które powinny spowodo-
wać odejście od rozpoznania MS. W czasie pracy
grupy przeglądowi poddano wybrane piśmien-
nictwo odnoszące się do cech demografi cznych,
objawów ogólnych i neurologicznych, danych
paraklinicznych i laboratoryjnych (obejmujących
różnorodne techniki obrazowe i testy laborato-
ryjne, takie jak analiza osocza i płynu mózgowo-
rdzeniowego oraz wzrokowe potencjały wywo-
łane) dotyczących całej gamy chorób uwzględ-
nianych w diagnostyce różnicowej MS.
Po uzmysłowieniu sobie, że pierwszym kro-
kiem w ocenie pacjenta z podejrzeniem choroby
istoty białej OUN jest wykonanie badania klinicz-
nego i zaplanowanie badań obrazowych oraz in-
nych testów laboratoryjnych, opracowano tabe-
lę zawierającą 79 cech demografi cznych, klinicz-
nych, laboratoryjnych i obrazowych. Tabela oce-

Neurologia Praktyczna • 6/2011
18
niana była niezależnie przez 6 członków podgrup
roboczych z zastosowaniem skali 1-5, klasyfi kują-
cej te cechy jako główne „czerwone fl agi” (4 lub
5 punktów) defi nitywnie wskazujące na inne niż
MS specyfi czne rozpoznanie alternatywne lub
jako drugorzędowe „czerwone fl agi”, wskazujące
na konieczność rozważenia rozpoznania innego
niż MS. Pośredni wynik (3 punkty) wskazywał na
brak pewności. Wyniki dla każdej cechy, uzyska-
ne od poszczególnych badaczy zsumowano i na
tej podstawie obliczono odchylenie standardo-
we (SD) (wysokie SD odzwierciedla niski stopień
zgodności pomiędzy badaczami). Zbiór 79 „czer-
wonych fl ag” podzielono następnie na trzy gru-
py zgodnie z poniższymi kryteriami:
Główne „czerwone fl agi”: całkowity wynik ≥ 24
punkty lub 23 i nie więcej niż jedna 3-punkto-
wa ocena indywidualna (SD ≤ 0,41).
Pośrednie „czerwone fl agi”, wskazujące na
brak zgodności pomiędzy badaczami co do
ich znaczenia: całkowity wynik ≥ 13 i ≤ 23
punkty, przy więcej niż jednej 3-punktowej
ocenie indywidualnej (SD ≥ 4,1).
Drugorzędowe „czerwone fl agi”: całkowi-
ty wynik ≤ 12 lub 13 punktów przy nie więcej
niż jednej 3-punktowej ocenie indywidualnej
(SD ≤ 0,41).
Algorytmy diagnostyczne częstych izolowanych
zespołów klinicznych sugerujących MS
Druga podgrupa skoncentrowała się na CIS sta-
nowiących często pierwszą manifestację cho-
roby, diagnozowanej ostatecznie jako MS. Pod-
sumowano, że termin CIS jest mylący w kontek-
ście diagnostycznym, ponieważ nie jest jasne,
czy odnosi się do zespołu izolowanego w cza-
sie, przestrzeni lub obu. Ponadto terminowi bra-
kuje specyfi czności patologicznej [6-9]. Opraco-
wano dokładniejszą defi nicję CIS. Ponadto pod-
grupa opracowała algorytmy diagnostyczne
trzech najbardziej typowych CIS (neuropatii ner-
wu wzrokowego, zespołów pnia mózgu i rdzenia
kręgowego) i wprowadziła rozróżnienie pomię-
dzy CIS poprzedzającymi zwykle MS a rzadszymi
i atypowymi objawami wymagającymi rozważe-
nia alternatywnych rozpoznań i rozszerzenia dia-
gnostyki.
Różnicowanie MS i innych IIDD
Trzecia podgrupa oceniała czynniki kliniczne,
demografi czne i parakliniczne różnicujące pro-
totypowy MS od „wariantów” takich jak NMO
i ADEM. W kontekście najnowszych danych do-
tyczących zmian obrazowych i biomarkerów za-
proponowano ujednolicone kryteria ich rozpo-
znania. Opracowano również roboczą klasyfi ka-
cję IIDD, jednocześnie uznając niedostateczność
danych, na których taka klasyfi kacja powinna się
opierać.
Perspektywy konsensusu
W ramach Panelu zgodzono się, że diagnostyka
różnicowa pacjentów z objawami sugerującymi
MS powinna być prowadzona zgodnie z ustalo-
ną strategią:
Pierwszy krok pozwala wykluczyć choroby,
które nie sugerują MS czy innych niż MS IIDD
(na przykład infekcyjne, nowotworowe, wro-
dzone, metaboliczne, naczyniowe i inne) oraz
dostarcza specyfi cznych wskazówek odnoszą-
cych się do diagnostyki różnicowej częstych
objawów początkowych, w kontekście pato-
logii nerwu wzrokowego, pnia mózgu i rdze-
nia kręgowego.
Drugi krok pozwala zróżnicować prototypo-
wy MS od innych niż MS IIDD, a ponadto su-
geruje schemat klasyfi kacji i kryteria diagno-
styczne IIDD innych niż MS.
Eliminacja możliwych rozpoznań
alternatywnych dla MS
W schemacie diagnostyki różnicowej poświęca
się niewiele uwagi pacjentom z klinicznymi ob-
jawami chorób OUN podobnymi do MS, u któ-
rych jednak nie rozwinie się MS. W tej grupie pa-
cjentów mogą być chorzy, u których ostatecz-
nie ustalone zostanie rozpoznanie na przykład
schorzeń naczyniowych lub infekcyjnych. Strate-
gie oceny diagnostycznej znajdują zastosowanie
w stosunku do osób z:
Klinicznymi, laboratoryjnymi i obrazowymi ce-
chami „typowymi” dla MS, podczas gdy żad-
ne dane nie sugerują rozpoznania alternatyw-
nego. MS jest schorzeniem prawdopodob-
nym. Najprawdopodobniej nie są potrzebne
dodatkowe badania i testy wykraczające poza
schemat diagnostyczny wyczerpujący kryte-
ria McDonalda.
Objawami zgodnymi z MS, jednak pojawiają-
cymi się wraz z innymi cechami („czerwone
fl agi”) sugerującymi możliwość rozpoznania
alternatywnego. MS może być rozpoznane
wyłącznie po wykonaniu badań wykluczają-
cych rozpoznania alternatywne. W sytuacjach
niejednoznacznych, przed ustaleniem osta-
tecznego rozpoznania zaleca się obserwa-
cję pacjenta oraz powtarzanie badań obrazo-
wych i laboratoryjnych.
Klinicznymi i/lub paraklinicznymi „czerwo-
nymi fl agami” wskazującymi na rozpoznanie
inne niż MS. Rozpoznanie MS nie jest praw-

Neurologia Praktyczna • 6/2011
19
Tabela I.
„Czerwone fl agi”
„Czerwona fl aga”
Typ
Wynik
całkowity
SD
„Czerwona
fl aga”
Przykłady rozpoznań alternatywnych
Uszkodzenia kości
Kliniczna
30
0,00
Główna
Histiocytoza; choroba Erdheima Chestera
Zajęcie płuc
Kliniczna
30
0,00
Główna
Sarkoidoza; ziarniniakowatość limfoidalna
Neuropatie wielu ner-
wów czaszkowych lub
poliradikuloneuropatia
Kliniczna
30
0,00
Główna
Przewlekłe zapalenie opon mózgowo-rdzeniowych obejmu-
jące sarkoidozę i gruźlicę; borelioza
Neuropatia obwodowa
Kliniczna
30
0,00
Główna
Niedobór witaminy B12; adrenoleukodystrofi a; leukodystro-
fi a metachromatyczna; borelioza
Żółtaki ścięgien
Kliniczna
30
0,00
Główna
Ksantomatoza mózgowo-ścięgnista
Zakrzepica zatok żylnych
mózgowia
MRI
30
0,00
Główna
Choroba Behçeta; zapalenie naczyń; przewlekłe zapalenie
opon mózgowo-rdzeniowych; zespoły antyfosfolipidowe
lub antykardiolipinowe
Choroba serca
Kliniczna
29
0,41
Główna
Mnogie zawały mózgowia; ropień mózgu w przypadku
zapalenia wsierdzia lub przecieku prawo-lewo na pozio-
mie serca
Miopatia
Kliniczna
29
0,41
Główna
Encefalomiopatia mitochondrialna (np. MELAS); zespół
Sjögrena
Zajęcie nerek
Kliniczna
29
0,41
Główna
Zapalenie naczyń; choroba Fabry’ego; toczeń rumieniowaty
Zawały korowe
MRI
29
0,41
Główna
Choroba zatorowa; zakrzepowa plamica małopłytkowa;
zapalenie naczyń
Krwotoki/mikrokrwotoki
MRI
29
0,41
Główna
Angiopatia amyloidowa; choroba Moya-Moya; CADASIL;
zapalenie naczyń
Wzmocnienie kontrastowe opon
mózgowo-rdzeniowych
MRI
29
0,41
Główna
Przewlekłe zapalenie opon mózgowo-rdzeniowych; sarko-
idoza; chłoniakowatość; zapalenie naczyń OUN
Objawy pozapiramidowe
Kliniczna
28
0,52
Główna
Choroba Whipple’a; zanik wieloukładowy; choroba Wilsona
Siność siatkowata
(livedo reticularis)
Kliniczna
28
0,52
Główna
Zespół antyfosfolipidowy; układowy toczeń rumieniowaty;
zespół Sneddona
Retinopatia
Kliniczna
28
0,52
Główna
Encefalomiopatia mitochondrialna; zespół Susaca i inne
zapalenia naczyń (zawał nerki); neuronalna lipofuscynoza
ceroidowa (NCL)
Zwapnienia w obrazach TK
MRI
28
0,52
Główna
Cysticerkoza; toksoplazmoza; choroby mitochondrialne
Moczówka prosta
Kliniczna
28
0,82
Główna
Sarkoidoza; histiocytoza; neuromyelitis optica
Zwiększone stężenie kwasu
mlekowego w surowicy
Kliniczna
27
0,55
Główna
Choroba mitochondrialna
Wybiórcze zajęcie przedniej części
płata skroniowego i podstawy
płata czołowego
MRI
27
0,55
Główna
CADASIL
Objawy hematologiczne
Kliniczna
27
0,84
Główna
Zakrzepowa plamica małopłytkowa; niedobór witaminy B12;
choroba Wilsona (anemia hemolityczna); niedobór miedzi
Zawały lakunarne
MRI
27
0,84
Główna
Choroba nadciśnieniowa; CADASIL; zespół Susaca
Trwałe wzmocnienie gadolinowe
i ciągłe powiększanie się zmian
MRI
27
0,84
Główna
Chłoniak; glejak; zapalenie naczyń; sarkoidoza
Owrzodzenia błon śluzowych
Kliniczne
27
1,22
Główna
Choroba Behçeta
Miorytmia
Kliniczna
27
1,22
Główna
Choroba Whipple’a
Zaburzenia funkcji podwzgórza
Kliniczna
26
0,52
Główna
Sarkoidoza; neuromyelitis optica; histiocytoza
Powtarzające się spontaniczne
poronienia lub epizody
zakrzepowe
Kliniczna
26
0,52
Główna
Zespół antyfosfolipidowy; zakrzepowa plamica małopłyt-
kowa; stan nadkrzepliwości w przebiegu nowotworu
rozsianego
Jednoczesne wzmocnienie kon-
trastowe wszystkich zmian
obrazowych
MRI
26
0,52
Główna
Zapalenie naczyń; chłoniak; sarkoidoza
(ciąg dalszy na następnej stronie)

Neurologia Praktyczna • 6/2011
20
„Czerwona fl aga”
Typ
Wynik
całkowity
SD
„Czerwona
fl aga”
Przykłady rozpoznań alternatywnych
Wysypka
Kliniczna
26
0,82
Główna
Układowy toczeń rumieniowaty; chłoniak T-komórkowy;
borelioza; choroba Fabry’ego
Hiperintensywność jąder zębatych
w sekwencjach T2-zależnych
MRI
MRI
26
0,82
Główna
Ksantomatoza mózgowo-ścięgnista
Zapalenie stawów, bóle wielosta-
wowe; bóle mięśni
Kliniczna
26
1,63
Główna
Układowy toczeń rumieniowaty; borelioza; fi bromialgia
Zaniki mięśni
Kliniczna
25
0,75
Główna
Stwardnienie zanikowe boczne; jamistość rdzenia;
poliradikulopatia
Ból głowy lub zespół oponowy
Kliniczna
25
0,98
Główna
Zakrzepica zatok żylnych mózgowia; przewlekłe zapalenie
opon mózgowo-rdzeniowych; chłoniak lub glejak; zapale-
nie naczyń; układowy toczeń rumieniowaty
Hiperintensywność poduszek
wzgórza w sekwencjach
T1-zależnych MRI
MRI
25
0,98
Główna
Choroba Fabry’ego; encefalopatia wątrobowa; zatrucie
manganem
Stałe objawy jednoogniskowe
Kliniczna
24
0,63
Główna
Uszkodzenia strukturalne (np. malformacja Arnolda-
-Chiariego); guz mózgu
Duże naciekające zmiany w obrę-
bie pnia mózgu
MRI
24
1,10
Główna
Choroba Behçeta; glejak mostu
Dominująca lokalizacja zmian
w okolicy korowo-podkorowej
MRI
23
0,41
Główna
Zawały zatorowe; zapalenie naczyń; postępująca wieloogni-
skowa leukoencefalopatia (PML)
Wodogłowie
MRI
23
0,98
Pośrednia
Sarkoidoza lub inne przewlekłe zapalenia opon mózgowo-
rdzeniowych; chłoniak lub inne nowotwory OUN
Punktowate wzmocnienie kontra-
stowe parenchymy mózgowej
MRI
23
0,98
Pośrednia
Sarkoidoza; zapalenie naczyń
Zespół suchości
Kliniczna
23
1,33
Pośrednia
Zespół Sjögrena
Hiperintensywność w sekwen-
cjach T2-zależnych włókien
U na sklepistości, torebce
zewnętrznej i w rejonie wyspy
MRI
22
1,37
Pośrednia
CADASIL
Objawy żołądkowo-jelitowe
Kliniczna
22
1,51
Pośrednia
Choroba Whipple’a; celiakia i inne stany zaburzeń wchłania-
nia jelitowego, prowadzące do niedoboru witaminy B12
i miedzi
Regionalna atrofi a pnia mózgu
MRI
21
0,55
Pośrednia
Choroba Behçeta; choroba Aleksandra rozpoczynająca się
w wieku dorosłym
Rozlany wzrost stężenia mlecza-
nów w MRS mózgowia
MRI
21
0,84
Pośrednia
Choroba mitochondrialna
Wyraźny zanik hipokampów i ciał
migdałowatych
MRI
21
0,84
Pośrednia
Hiperhomocysteinemia
Utrata słuchu
Kliniczna
21
0,38
Pośrednia
Zespół Susaca; glejak; zawał w obszarze kręgowo-
-podstawnym
Przebieg piorunujący
Kliniczna
20
0,82
Pośrednia
Zakrzepowa plamica małopłytkowa; chłoniak wewnątrz-
naczyniowy; ostre rozsiane zapalenie mózgu i rdzenia
(ADEM)
Symetryczna lokalizacja zmian
MRI
20
0,82
Pośrednia
Leukodystrofi a
Zmiany hiperintensywne w se-
kwencjach T2-zależnych
w zwojach podstawy, wzgórzu
i podwzgórzu
MRI
20
1,03
Pośrednia
Choroba Behçeta; encefalomiopatie mitochondrialne; zespół
Susaca; ostre zapalenie mózgu i rdzenia
Rozlane zmiany sygnału sznurów
tylnych rdzenia kręgowego
MRI
20
1,37
Pośrednia
Niedobór witaminy B12; niedobór miedzi; schorzenie
paranowotworowe
Wzrost stężenia ACE w osoczu
Kliniczna
20
1,86
Pośrednia
Sarkoidoza; histiocytoza
Wyraźny wywiad rodzinny
Kliniczna
19
0,41
Pośrednia
W zależności od wzorca dziedziczenia sugerowanego przez
wywiad rodzinny: dziedziczna parapareza spastyczna;
leukodystrofi a; choroba Wilsona; choroba mitochondrial-
na; CADASIL
Tabela I.
„Czerwone fl agi”(ciąg dalszy)

Neurologia Praktyczna • 6/2011
21
„Czerwona fl aga”
Typ
Wynik
całkowity
SD
„Czerwona
fl aga”
Przykłady rozpoznań alternatywnych
Objawy ogólnoustrojowe
Kliniczna
19
1,17
Pośrednia
Sarkoidoza; choroba Whipple’a; zapalenie naczyń
Zmiany zlokalizowane na grani-
cach istoty szarej i białej
MRI
19
1,17
Pośrednia
Uszkodzenie hipoksyczno-ischemiczne; zapalenie naczyń;
układowy toczeń rumieniowaty
Hiperintensywne zmiany w se-
kwencjach T2-zależnych
zlokalizowane w biegunie
płata skroniowego
MRI
19
1,17
Pośrednia
CADASIL
Pełne obrączkowate wzmocnienie
kontrastowe
MRI
18
0,63
Pośrednia
Ropień mózgu; glioblastoma; przerzuty nowotworowe
Izolowana postępująca ataksja
Kliniczna
18
1,10
Pośrednia
Zanik wieloukładowy; dziedziczna ataksja rdzeniowo-
-móżdżkowa; paranowotworowy zespół móżdżkowy
Zmiany zlokalizowane w central-
nej części pnia mózgu
MRI
17
0,75
Pośrednia
Środkowa mielinoliza mostu; uszkodzenia hipoksyczno-
-ischemiczne; zawał
Zmiany zlokalizowane głównie
w pniu mózgu i móżdżku
MRI
17
0,75
Pośrednia
Choroba Behçeta; glejak mostu
Zespół zaburzeń
neuropsychiatrycznych
Kliniczna
17
1,33
Pośrednia
Zespół Susaca; układowy toczeń rumieniowaty; choroba
Wilsona; gangliozydoza GM2
Zmiany zlokalizowane w środko-
wej części ciała modzelowa-
tego, z oszczędzeniem części
obwodowej
MRI
17
1,33
Pośrednia
Zespół Susaca
Napady padaczkowe
Kliniczna
16
1,63
Pośrednia
Choroba Whipple’a; zapalenie naczyń; przerzuty do OUN
Poszerzenie przestrzeni Virchova-
Robina
MRI
15
0,55
Pośrednia
Hiperhomocysteinemia; pierwotne zapalenie naczyń OUN
Zapalenie błony naczyniowej oka
(uveitis)
Kliniczna
15
0,84
Pośrednia
Sarkoidoza; chłoniak; choroba Behçeta
Zmiany korowo-podkorowe
przekraczające granice
unaczynienia
MRI
14
1,21
Pośrednia
Leukoencefalopatia niedokrwienna; CADASIL; zapalenie
naczyń
Izolowane zajęcie układu
piramidowego
Kliniczna
13
0,75
Pośrednia
Pierwotne stwardnienie boczne jako wariant ALS; dziedzicz-
na parapareza spastyczna
Duże zmiany bez obecności lub
z rzadko występującym efek-
tem masy i wzmocnieniem
kontrastowym
MRI
13
0,98
Pośrednia
Postępująca wieloogniskowa leukoencefalopatia
Stopniowo postępujący prze-
bieg kliniczny od początku
zachorowania
Kliniczna
13
1,17
Pośrednia
Mielopatia związana z infekcją wirusem HTLV-1; adreno-
mieloneuropatia; adrenoleukodystrofi a; leukodystrofi a
metachromatyczna; niedobór witaminy B12
Brak „ukrytych” zmian w NAWM
MRI
13
1,33
Pośrednia
Borelioza; izolowane zapalenie rdzenia kręgowego; CADASIL
Zespół pnia mózgu
Kliniczna
7
0,41
Drugorzędowa Glejak mostu; naczyniak jamisty; niedokrwienie obszaru
kręgowo-podstawnego
Brak wzmocnienia kontrastowego MRI
8
0,52
Drugorzędowa Postępująca wieloogniskowa leukoencefalopatia; zmiany
niedokrwienne; leukodystrofi a metachormatyczna
Izolowana mielopatia
Kliniczna
9
0,55
Drugorzędowa Malformacja Arnolda-Chiariego typu 1; ucisk rdzenia kręgo-
wego obejmujący spondylozę szyjną; niedobór witaminy
B12 lub miedzi; HTLV1
Brak zmian w obrębie nerwów
wzrokowych
MRI
9
0,55
Drugorzędowa Przerzuty nowotworowe; glejakowatość mózgowa;
toksoplazmoza
Zachorowanie przed 20. rokiem
życia
Kliniczna
10
0,52
Drugorzędowa Encefalomiopatia mitochondrialna; leukodystrofi a; ataksja
Friedricha
Brak zmian w obrębie rdzenia
kręgowego
MRI
10
0,52
Drugorzędowa Mnogie zawały; zapalenie naczyń; postępująca wieloogni-
skowa leukoencefalopatia
Nagły początek
Kliniczna
11
1,17
Drugorzędowa Zawał mózgu; krwotok śródmózgowy; zakrzepica zatok
żylnych mózgowia
Duże zmiany
MRI
11
0,75
Drugorzędowa Glioblastoma; chłoniak; postępująca wieloogniskowa
leukoencefalopatia
(ciąg dalszy na następnej stronie)
Tabela I.
„Czerwone fl agi”(ciąg dalszy)

Neurologia Praktyczna • 6/2011
22
dopodobne. Należy dołożyć starań, aby roz-
poznać schorzenie alternatywne, szczególnie
kiedy poddaje się ono leczeniu.
Klinicznymi i/lub paraklinicznymi cechami su-
gerującymi obecność MS z innym nakładają-
cym się schorzeniem. Potwierdzenie współ-
istnienia dwóch chorób wymaga wykonania
właściwych badań obrazowych i laboratoryj-
nych.
W tabeli I zaprezentowano 79 klinicznych i para-
klinicznych cech stanowiących „czerwone fl agi”,
dotyczące pacjentów z objawami choroby OUN,
u których rozważa się rozpoznanie MS. Na drodze
głosowania przeprowadzonego wśród członków
podgrupy, jak opisano w sekcji „Metodyka”, zi-
dentyfi kowano 36 głównych „czerwonych fl ag”,
wskazujących jednoznacznie na rozpoznanie
inne niż MS. Większość z nich to objawy klinicz-
ne. Zidentyfi kowano 11 drugorzędowych „czer-
wonych fl ag”, sugerujących rozważenie innych
schorzeń alternatywnych przy możliwości roz-
poznania MS. Decyzja nie może zostać podję-
ta wyłącznie na podstawie samej oceny klinicz-
nej. Dodatkowe 32 wskaźniki kliniczne, parakli-
niczne i laboratoryjne, spośród których wiele ma
charakter obrazowy, okazały się mieć pośrednie
znaczenie. Otrzymały one stosunkowo wysoki
średni wynik w procesie oceny, jednak przy du-
żym SD. Wskazuje to na brak zgodności co do ich
znaczenia. Wartość tych wskaźników w diagno-
styce różnicowej zależy od ogólnego kontekstu,
w jakim się pojawiają (tzn. dodatkowe dane kli-
niczne, laboratoryjne i parakliniczne).
Diagnostyka różnicowa pierwszych izolowanych
prezentacji klinicznych
W chwili wystąpienia pierwszych objawów su-
gerujących MS można doszukać się w wywiadzie
więcej niż jednego rzutu choroby lub objawów
o przebiegu postępującym. Jednakże u większo-
ści pacjentów, u których ostatecznie rozpoznano
MS, pierwszym objawem choroby jest ostry epi-
zod kliniczny odzwierciedlający patologię isto-
ty białej, określany ogólnym terminem CIS. Ze
względu na dużą częstość tego typu objawów
oraz brak danych potwierdzających rozprosze-
nie objawów w czasie, co uniemożliwia natych-
miastowe rozpoznanie MS, koncentrujemy się na
diagnostyce różnicowej tego typu izolowanych
zespołów klinicznych.
Defi nicja i klasyfi kacja CIS
Objawy CIS najczęściej obejmują jeden nerw
wzrokowy, rdzeń kręgowy lub pień mózgu, cho-
ciaż możliwe jest występowanie również innych
izolowanych zespołów, na przykład dotyczących
półkul mózgowych (np. niedowidzenie połowi-
cze). Pomimo częstego stosowania w znacze-
niu pierwszej manifestacji choroby demielini-
zacyjnej, termin CIS nie jest dokładny: używany
był w różnych kontekstach do grupowania pa-
cjentów z: 1) jednym epizodem klinicznym i ob-
jawami wskazującymi na pojedyncze ognisko
uszkodzenia (zatem chorobą izolowaną w czasie
i przestrzeni); 2) nawracającymi epizodami w po-
jedynczej lokalizacji (zatem chorobą izolowaną
w przestrzeni, ale nie w czasie, np. nawracające
zapalenie nerwu wzrokowego) [10]; 3) pojedyn-
czym epizodem klinicznym, podczas gdy obja-
wy i/lub badanie neurologiczne sugeruje obec-
ność dwóch lub większej liczby ognisk uszko-
dzenia, w różnych lokalizacjach (zatem chorobą
izolowaną w czasie, ale nie w przestrzeni).
Celem większości badań klinicznych obej-
mujących pacjentów z CIS było określenie roli
interferonu-β w opóźnianiu czasu do klinicznie
defi nitywnego MS. Włączano pacjentów z jed-
nofazowym epizodem klinicznym, ale z wielo-
ma objawami klinicznymi, u których badania
wykazywały mnogie objawy OUN (np. pacjent
z ostrym zapaleniem nerwu wzrokowego i obja-
wem Lhermitte’a lub pacjent z zapaleniem nerwu
wzrokowego i dodatnim objawem Babińskiego)
„Czerwona fl aga”
Typ
Wynik
całkowity
SD
„Czerwona
fl aga”
Przykłady rozpoznań alternatywnych
Brak zmian hipointensywnych
w sekwencjach T1-zależnych
(black holes)
MRI
11
0,75
Drugorzędowa Niedokrwienna leukoencefalopatia zwyrodnieniowa; postę-
pująca wieloogniskowa leukoencefalopatia
Zachorowanie po 50. roku życia
Kliniczna
12
0,89
Drugorzędowa Zawał mózgu; angiopatia amyloidowa; chłoniak
Znaczna asymetria zmian w obrę-
bie istoty białej
MRI
12
0,89
Drugorzędowa Glioblastoma; chłoniak; zawał mózgu
a
„Czerwone fl agi” uporządkowano od najbardziej „głównych” do najbardziej „drugorzędowych” zgodnie z klasyfi kacją ustaloną w podgrupach, opisaną w tekście. Główne
„czerwone fl agi” wskazują defi nitywnie na rozpoznanie inne niż MS; drugorzędowe „czerwone fl agi” mogą potwierdzać rozpoznanie MS lub rozpoznania alternatywne. W od-
niesieniu do pośrednich „czerwonych fl ag” brak było zgodności między osobami oceniającymi ich znaczenie, a ponadto nie było pewności co do ich wagi w diagnostyce różnico-
wej MS, szczególnie przy braku innych objawów, oznak i wyników badań. Drugorzędowe „czerwone fl agi” sugerują, że należy wziąć pod uwagę i wyczerpująco zbadać choroby
inne niż MS, jednak nie wyklucza to rozpoznania MS.
Tabela I.
„Czerwone fl agi”(ciąg dalszy)

Neurologia Praktyczna • 6/2011
23
[7, 8]. W jednej z prób klinicznych dotyczących
CIS u 48% pacjentów z CIS występowały dowo-
dy wskazujące na chorobę wieloogniskową [11].
Zaproponowano zatem strategię postępowania
mającą na celu zredukowanie różnic w interpre-
tacji wyników prób klinicznych, polegającą na
rozróżnieniu wstępnej manifestacji jednoogni-
skowej od wieloogniskowej i na tej podstawie
stratyfi kację rekrutacji [8]. Ponadto termin CIS
ignoruje początkowe manifestacje choroby, mo-
gące nie mieć charakteru klinicznego, natomiast
mogące mieć istotę objawów paraklinicznych
i laboratoryjnych [12]; wśród pacjentów z poje-
dynczą prezentacją kliniczną nie różnicuje tych,
którzy mają lub nie objawowe zmiany w MRI, co
wiąże się z różnym rokowaniem [13-16].
Ze względu na powyższe niejasności interpre-
tacji oraz znaczenie CIS w procesie diagnostyki
różnicowej członkowie Panelu czuli obowiązek
dokładniejszego zdefi niowania tego terminu.
Uzgodniono, że CIS należy defi niować jako mani-
festację jednofazową (monophasic presentation),
u której podłoża leży zapalna choroba demieli-
nizacyjna. „Jednofazowa manifestacja” sugeruje
pojedynczy epizod kliniczny będący pierwszym
objawem choroby, o względnie nagłym począt-
ku. Istnieje wiele możliwości jednoczesnych ma-
nifestacji klinicznych/paraklinicznych (odzwier-
ciedlających rozproszenie objawów w przestrze-
ni), chociaż rozproszenie objawów w czasie nie
musi być oczywiste. Zatem w zależności od jed-
no- lub wieloogniskowej charakterystyki jedno-
fazowych objawów klinicznych lub obrazowych
(MRI) można zdefi niować cztery klasy CIS (tab. II;
typy CIS 1-4). Ostatecznie muszą istnieć rozsąd-
ne przesłanki, aby podejrzewać zapalną chorobę
demielinizacyjną.
MRI może mieć wpływ na decyzję, czy obja-
wy kliniczne związane są z uszkodzeniem jed-
no- czy wieloogniskowym oraz na prawdo-
podobieństwo ostatecznego rozpoznania MS
(tab. II). Pacjenci, u których stwierdza się przynaj-
mniej jedną typowo demielinizacyjną, bezobja-
wową zmianę w MRI, z wysokim prawdopodo-
bieństwem spełnią kryteria MS w późniejszym
okresie (typ 1 i 2 CIS); rokowanie jest zróżnico-
wane i nie jest silnie skorelowane z liczbą i loka-
lizacją zmian [13, 14]. U pacjentów z jednoogni-
skowymi objawami klinicznymi przy braku bez-
objawowych, typowo demielinizacyjnych zmian
w MRI prawdopodobieństwo późniejszego speł-
nienia kryteriów MS jest względnie niskie (typ 3
CIS) [16]. Mało prawdopodobna jest manifesta-
cja kliniczna z objawami wieloogniskowymi, bez
wykrywanych w MRI bezobjawowych zmian de-
mielinizacyjnych (typ 4 CIS), zatem tacy pacjen-
ci wymagają dalszej obserwacji w celu rozpozna-
nia MS lub innego schorzenia.
Chociaż objawy i oznaki choroby jednofazo-
wej stanowią niezbędny warunek do rozpozna-
nia CIS, istnieje wyjątkowy scenariusz tłumaczą-
cy włączenie typu 5 CIS (tab. II): pacjenci bez ob-
jawów lub jedynie z objawami niespecyfi cznymi
(np. ból głowy, zaburzenia równowagi), u któ-
rych MRI wykazuje wieloogniskowe zmiany ty-
powe dla choroby demielinizacyjnej. Tego ro-
dzaju pacjenci są coraz częściej identyfi kowani
za pomocą MRI, zlecanego z innych powodów
(np. bólu głowy), szczególnie przy silnym polu
magnetycznym, wykazującym dużą czułość dla
tego typu zmian [17]. Aktualne kryteria wyklu-
czają rozpoznanie MS bez obiektywnych klinicz-
nych dowodów uszkodzenia OUN, zatem możli-
wość ustalenia pewnego rozpoznania MS u tych
osób i naturalny przebieg schorzenia powinny
być tematem badań prospektywnych [18].
Diagnostyka różnicowa CIS zajmujących nerwy
wzrokowe, pień mózgu i rdzeń kręgowy
Defi nicja CIS obejmuje szeroki zakres izolowa-
nych zespołów klinicznego uszkodzenia OUN,
spotykanych w kontekście procesu diagnostycz-
nego przy możliwości rozpoznania MS. Jak wspo-
mniano, niektóre z nich silniej sugerują ostatecz-
ne rozpoznanie MS niż inne, w zależności od cha-
rakterystyki obrazowej. Jednakże ich kliniczne
objawy w chwili zachorowania mogą również do-
starczyć pewnych wskazówek co do prawdopo-
dobieństwa późniejszego rozpoznania MS. W ta-
beli III objawy CIS u pacjentów, u których osta-
tecznie rozpoznano MS skategoryzowano jako:
1) typowe dla pacjentów z późniejszym rozpo-
znaniem MS; 2) rzadsze, niemniej jednak mogące
być wstępną manifestacją choroby u pacjentów
z późniejszym rozpoznaniem MS, lub też mogą
wskazywać na inna chorobę; 3) nietypowe, suge-
rujące alternatywne rozpoznanie.
Tabela II.
Izolowane zespoły kliniczne (CIS) w diagnostyce różni-
cowej MS
Typ 1 CIS: klinicznie jednoogniskowy, przynajmniej jedno bezobjawowe ogni-
sko uszkodzenia w MRI
Typ 2 CIS: klinicznie wieloogniskowy; przynajmniej jedno bezobjawowe ognisko
uszkodzenia w MRI
Typ 3 CIS: kliniczne jednoogniskowy, MRI może wyglądać prawidłowo; brak
bezobjawowych ognisk uszkodzenia w MRI
Typ 4 CIS: klinicznie wieloogniskowy, MRI może wyglądać prawidłowo; brak
bezobjawowych ognisk uszkodzenia w MRI
Typ 5 CIS: brak objawów klinicznych sugerujących chorobę demielinizacyjną,
ale MRI sugeruje obecność tej choroby
Uwaga: objawowe ogniska uszkodzenia muszą mieć typowy wygląd zmian demielinizacyj-
nych; mogą być zlokalizowane w mózgowiu lub rdzeniu kręgowym; chociaż częściej poja-
wiają się w mózgowiu; aktualne dane na temat wartości prognostycznej bezobjawowych
ognisk uszkodzenia pochodzą głównie z badań obrazowych.

Neurologia Praktyczna • 6/2011
24
Prawdopodobnie do trzech najczęstszych ze-
społów CIS spotykanych w procesie diagnostycz-
nym MS należą zespoły zajmujące nerw wzroko-
wy, pień mózgu i rdzeń kręgowy. Schematy pre-
zentujące diagnostykę różnicową tych zespołów
przedstawiono na rycinach 2-4. Schematy te ilu-
strują niektóre główne elementy oceny klinicz-
nej oraz laboratoryjnej i w zamierzeniu nie mia-
ły charakteru podsumowania. Kładą one jedynie
nacisk na typowe oraz łatwo dostępne badania
i ich wyniki.
Diagnostyka różnicowa MS i IIDD; nozologia
i klasyfi kacja
Chociaż MS jest najprawdopodobniej najczęst-
szym ostatecznym rozpoznaniem w przypad-
kach objawów IIDD, to utrzymujące się przez
długi czas biomarkery kliniczne, radiologiczne
i immunologiczne mogą być pomocne w róż-
nicowaniu i defi niowaniu innych IIDD od MS.
Koncentrujemy się na dwóch najczęstszych roz-
poznaniach różnicowych IIDD: NMO i ADEM.
Panel ekspertów rozbudował niedawno zapro-
ponowane kryteria rozpoznania NMO, cechują-
ce się 90% czułością i swoistością w różnicowa-
niu NMO od MS [19, 20]. Jednakże kryteria rozpo-
znania ADEM nie były poddane dobrej walidacji
i w efekcie u 30% pacjentów spełniających kryte-
ria ADEM przy początkowej manifestacji klinicz-
nej w późniejszym czasie rozpoznane będzie MS
[21-24]. Wskaźnik konwersji z ADEM do MS bę-
dzie wyższy u dorosłych (u których ADEM wy-
stępuje rzadziej) niż u dzieci. W przypadku dzie-
ci u około 20% pacjentów z wstępnym rozpozna-
niem ADEM ostatecznie rozpoznane będzie MS.
Defi nicja i diagnostyka różnicowa NMO
NMO jest prawdopodobnie najczęściej spoty-
kanym IIDD, innym niż MS [25, 26]. W przeszło-
ści odróżniano to schorzenie od MS ze względu
na znacznie ograniczone objawy obejmujące za-
palenie nerwu wzrokowego i zapalenie rdzenia
kręgowego, a także z powodu przebiegu jed-
nofazowego, nienawracającego. Jednakże bar-
dziej współczesne badania sugerują, że NMO ma
zwykle charakter nawracający, co utrudnia róż-
Tabela III.
Charakterystyka kliniczna CIS i prawdopodobieństwo późniejszego rozpoznania MS
Objawy CIS typowe dla MS
Rzadsze objawy CIS mogące wystąpić w MS
Nietypowe objawy CIS, niespotykane w MS
Nerw wzrokowy
Jednostronne zapalenie nerwu
wzrokowego
Ból przy ruchach gałki ocznej
Częściowe i głównie centralne zatarcie
pola widzenia
Prawidłowy obraz tarczy nerwu wzroko-
wego lub łagodny jej obrzęk
Obustronne jednoczesne zapalenie nerwu
wzrokowego
Brak bólu
Brak poczucia światła
Obrzęk tarczy nerwu wzrokowego o nasileniu od
umiarkowanego do ciężkiego, przy braku wybro-
czyn krwawych
Zapalenie błony naczyniowej oka (łagodne, tylne)
Postępująca neuropatia nerwu wzrokowego
Ciężki, ciągły ból zlokalizowany w oczodole
Trwała, pełna utrata widzenia
Zapalenie nerwu wzrokowego i siatkówki – neu-
roretinitis (obrzęk tarczy nerwu wzrokowego
z gwiaździstymi wysiękami plamki)
Zapalenie błony naczyniowej oka (ciężkie,
przednie)
Pień mózgu/móżdżek
Obustronne porażenie międzyjądrowe
Ataksja z oczopląsem wielokierunkowym
Neuropatia nerwu odwodzącego
Drętwienie twarzy
Jednostronne porażenie międzyjądrowe, neuropatia
nerwu twarzowego, miokimie twarzy
Głuchota
Zespół jeden i pół
Neuralgia trójdzielna
Napadowe kurcze toniczne
Całkowita oftalmoplegia zewnętrzna; porażenie
skojarzonego spojrzenia w pionie
Zespoły terytoriów naczyniowych, np. boczny
opuszki
Neuropatia nerwu okoruchowego
Postępująca neuropatia czuciowa nerwu
trójdzielnego
Dystonia ogniskowa, kręcz karku
Rdzeń kręgowy
Częściowa mielopatia
Objaw Lhermitte’a
Odnerwienie dłoni
Niedoczulica
Naglące parcie na mocz, nietrzymanie
moczu, zaburzenia erekcji
Postępująca parapareza spastyczna
(asymetryczna)
Pełne poprzeczne zapalenie rdzenia kręgowego
Radikulopatia, arefl eksja
Segmentalna utrata czucia bólu i temperatury
Częściowy zespół Browna-Sequarda (oszczędzający
sznury tylne)
Nietrzymanie stolca
Postępująca parapareza spastyczna (symetryczna)
Uszkodzenie w obszarze unaczynienia tętnicy
rdzeniowej przedniej (oszczędzające jedynie
sznury tylne)
Zespół ogona końskiego
Wyraźny poziom zaburzeń czucia dla wszystkich
rodzajów czucia i zlokalizowany ból rdzeniowy
Pełny zespół Browna-Sequarda
Ostre zatrzymanie moczu
Postępująca ataksja czuciowa (sznury tylne)
Półkule mózgowe
Łagodne podkorowe upośledzenie funkcji
poznawczych
Niedowład połowiczy
Padaczka
Niedowidzenie połowicze
Encefalopatia (spowolnienie, splątanie, senność)
a
Ślepota korowa
a
Chociaż objawy encefalopatii wymagane są do rozpoznania ADEM, mogą być również spotykane jako manifestacja MS i/lub w późniejszym przebiegu MS.
Neurologia Praktyczna • 6/2011
25
nicowanie NMO z MS [27-31]. Prawidłowy obraz
mózgowia i długoodcinkowe zmiany w obrę-
bie rdzenia kręgowego w kontekście ostrego za-
palenia rdzenia kręgowego pomagają odróżnić
NMO od MS. Niedawno odkryto wysoce swoisty
i umiarkowanie czuły biomarker w surowicy krwi
(przeciwciało NMO-IgG), przydatny w diagnosty-
ce NMO [32, 33].
W różnicowaniu NMO od MS podgrupa
uzgodniła, że:
NMO powinno być różnicowane z MS, ponie-
waż ma inny przebieg kliniczny i rokowanie
[28, 31], a ponadto ze względu na przypusz-
czalne różnice w odpowiedzi na leczenie im-
munomodulacyjne [34-36].
NMO ma najczęściej charakter nawracający
i dlatego ta cecha nie jest przydatna w różni-
cowaniu NMO z MS.
Kluczową cechą kliniczną różnicującą NMO od
MS jest predyspozycja do ciężkich epizodów
zapalenia rdzenia kręgowego w NMO, które
często, ale nie zawsze manifestują się jako peł-
ne poprzeczne zapalenie rdzenia kręgowego
oraz do ciężkich zapaleń nerwu wzrokowego,
które często, ale nie zawsze wiążą się z niecał-
kowitą poprawą. Zapaleniu rdzenia kręgowe-
go w NMO, w przeciwieństwie do epizodów
pojawiających się w przebiegu MS, towarzy-
szą zwykle w fazie ostrej zmiany hiperinten-
sywne w sekwencjach T2-zależnych rozciąga-
jące się na trzy segmenty rdzeniowe lub wię-
cej (longitudinally extensive transverse myeli-
tis – LETM), które mogą być hipointensywne
w sekwencjach T1-zależnych MRI i ulegać róż-
nego stopnia wzmocnieniu kontrastowemu
po podaniu gadolinu
Kliniczne objawy zajęcia mózgowia w NMO
występują rzadko, a badanie MR mózgu jest
często prawidłowe [27, 37], szczególnie we
wczesnym stadium choroby [38, 39]. Zmia-
Ryc. 2.
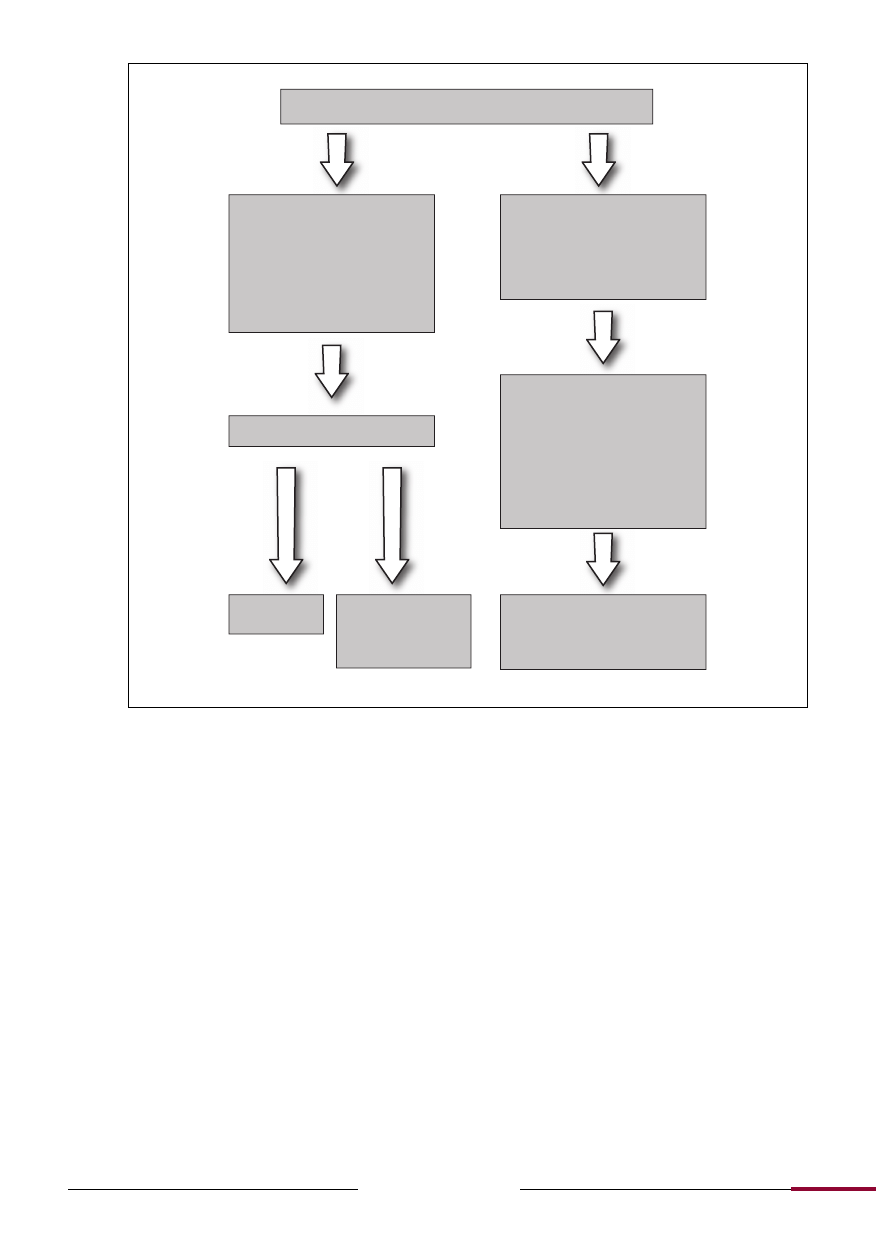
Diagnostyka różnicowa w przypadku wystąpienia demielinizacyjnego zapalenia nerwu wzrokowego.
Zapalenie nerwu wzrokowego
Typowe dla MS (jednostronna
utrata widzenia, ból, uszkodzenie
drogi dośrodkowej odruchu źre-
nic na światło, pozagałkowy lub
niewielki obrzęk tarczy nerwu
wzrokowego, utrata widzenia nie
postępuje w czasie dłuższym niż
2 tygodnie)
Nietypowe dla MS (brak bólu,
wysięki w siatkówce, krwotoki
siatkówkowe, ciężki obrzęk tarczy
nerwu wzrokowego, brak popra-
wy widzenia po miesiącu lub
obustronna utrata widzenia)
MRI mózgowia
• Niedokrwienna ON
• Dziedziczna ON
• Naciekowa ON
• Zapalna (sarkoid, toczeń)
• Infekcje (kiła, borelioza,
wirusowa,
neuroretinitis)
• Toksyczna/żywieniowa
• Schorzenia siatkówki
Rozważyć
inne rozpo-
znania
Nieprawi-
dłowe zmia-
ny odpowia-
dające de-
mielinizacji
Prawi-
dłowe
Niskie ryzyko
MS (20%)
Wysokie ryzyko MS
(60-90%), ponowna
ocena kryteriów
McDonalda
MRI, PMR, OCT
oraz inne badania neurofizjo-
logiczne, serologiczne w miarę
potrzeby
Neurologia Praktyczna • 6/2011
26
ny patologiczne mózgowia, jeśli występują,
zwykle nie spełniają typowych kryteriów Bar-
khofa/Tintoré rozproszenia w przestrzeni [40,
41]. Zmiany patologiczne mózgowia w NMO
mogą wykazywać predylekcję do obszarów
z wysoką ekspresją akwaporyny 4, tzn. pod-
wzgórza, rdzenia przedłużonego i innych ob-
szarów pnia mózgu [41, 42].
Prążki oligoklonalne lub podwyższony indeks
IgG w PMR stwierdza się u 10-20% pacjentów
z NMO, w porównaniu z 70-90% pacjentów
z MS (tab. IV) [28, 43].
U niektórych pacjentów z objawami IIDD wy-
stępuje wyłącznie nawracające poprzeczne za-
palenie rdzenia kręgowego, w przebiegu którego
stwierdza się długoodcinkowe zmiany w rdzeniu
kręgowym lub też wyłącznie nawracające zapa-
lenie nerwu wzrokowego, przy obecności prze-
ciwciał NMO-IgG. Populacja ta może reprezen-
tować ograniczone lub wstępne zespoły NMO.
Chociaż większość klinicystów uważa, że chorzy
ci powinni być leczeni tak jakby mieli NMO [26,
44, 45], do czasu lepszego poznania zależności
między izolowanym nawracającym poprzecz-
nym zapaleniem rdzenia kręgowego lub izolo-
wanym nawracającym zapaleniem nerwu wzro-
kowego i NMO, to członkowie Panelu doszli do
wniosku, iż te przestrzennie ograniczone zespo-
ły nie powinny być kwalifi kowane jako NMO, na-
wet przy obecności dodatnich odczynów serolo-
gicznych NMO-IgG w surowicy. Wystąpienie ob-
jawów zapalenia nerwu wzrokowego u pacjen-
ta z zapaleniem rdzenia kręgowego, i odwrotnie,
może umożliwić późniejsze rozpoznanie NMO.
Wyniki biopsji wskazujące na sarkoidozę lub
zapalenie naczyń, które stanowią rzadką przy-
czynę zapalenia nerwu wzrokowego, wyklucza-
ją NMO. Nierzadko spotyka się pacjentów z za-
paleniem nerwu wzrokowego, zapaleniem rdze-
nia kręgowego lub obiema patologiami rozwi-
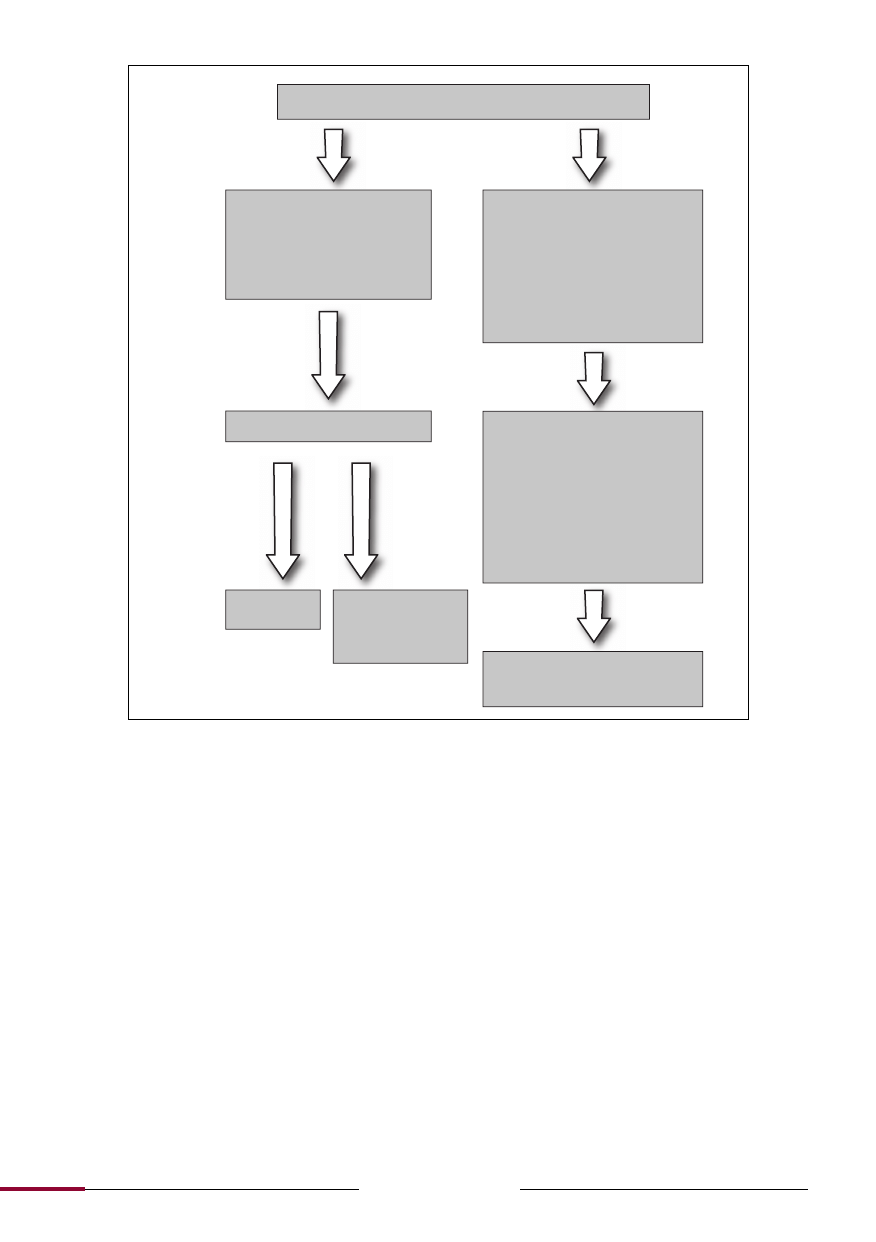
Ryc. 3.
Diagnostyka różnicowa w przypadku wystąpienia zespołu demielinizacji pnia mózgu.
Izolowany zespół pnia mózgu
Typowe dla MS
Porażenie międzyjądrowe,
neuropatia nerwu odwodzą-
cego, objawy wieloogniskowe
np.: niedoczulica twarzy i za-
wroty głowy lub utrata słuchu
Nietypowe dla MS
Nadostry początek zachorowania,
objawy z obszarów unaczynienia np.
zespół boczny opuszki, wiek powy-
żej 50 lat, izolowana neuralgia nerwu
trójdzielnego, fluktuujący niedowład
mięśni gałkoruchowych lub opusz-
kowych, objawy nieustępujące, go-
rączka, zespół oponowy
MRI mózgowia
• Niedokrwienie/krwotok
(naczyniak
jamisty)
• Zapalenie (sarkoid, toczeń)
• Infekcje
(kiła, listeria, borelioza, wirusowe)
• Toksyczne
• Żywieniowe
• Środkowa mielinoliza mostu
• Nerwowo-mięśniowe
(miastenia
rzekomoporaźna)
Rozważyć
inne rozpo-
znania
Nieprawi-
dłowe zmia-
ny odpowia-
dające de-
mielinizacji
Prawi-
dłowe
Niskie ryzyko
MS (20%)
Wysokie ryzyko MS
(60-90%), ponowna
ocena kryteriów
McDonalda
MRI, PMR oraz inne badania
neurofizjologiczne, serologicz-
ne w miarę potrzeby
Neurologia Praktyczna • 6/2011
27
jającymi się w kontekście lub poprzedzającymi
objawy tocznia trzewnego lub zespołu Sjögre-
na, u których stwierdza się dodatnie przeciw-
ciała NMO-IgG. Częstość występowania prze-
ciwciał NMO-IgG jest podobna w porównaniu
z „niepowikłanym NMO”. U tego typu pacjen-
tów NMO może współistnieć ze specyfi czną na-
rządowo lub niespecyfi czną chorobą autoimmu-
nologiczną [46]. Członkowie Panelu doszli jed-
nak do konserwatywnego wniosku, że do czasu
rozstrzygnięcia przyszłych badań kliniczne do-
wody potwierdzające układowy toczeń rumie-
niowaty lub zespół Sjögrena powinny wyklu-
czać rozpoznanie NMO. Obecność przeciwciał
przeciwjądrowych (ANA) lub przeciwciał zespo-
łu Sjögrena (SSA/SSB), wykrywanych często u pa-
cjentów w diagnostyce NMO, nie wyklucza roz-
poznania NMO, jeśli brak jest klinicznych dowo-
dów potwierdzających diagnozę tocznia lub ze-
społu Sjögrena.
Azjatycka postać wzrokowo-rdzeniowa MS
(optico-spinal MS – OSMS) może być mylo-
na z NMO. Nie jest jasne, czy różnice pomiędzy
OSMS i NMO w krajach Azji i Europy Zachod-
niej wynikają z odmienności biologicznych, czy
z różnic w nomenklaturze. W Azji pacjenci z za-
paleniem nerwu wzrokowego i zapaleniem rdze-
nia kręgowego klasyfi kowani są jako OSMS bez
względu na długość zmian w rdzeniu kręgowym.
W Europie Zachodniej tacy pacjenci bez zmian
typowych dla LETM w większości przypadków
klasyfi kowani są jako mający typowe MS. Ponad-
to w każdym przypadku zajęcia mózgowia po-
twierdzonego kliniczne lub radiologicznie (z wy-
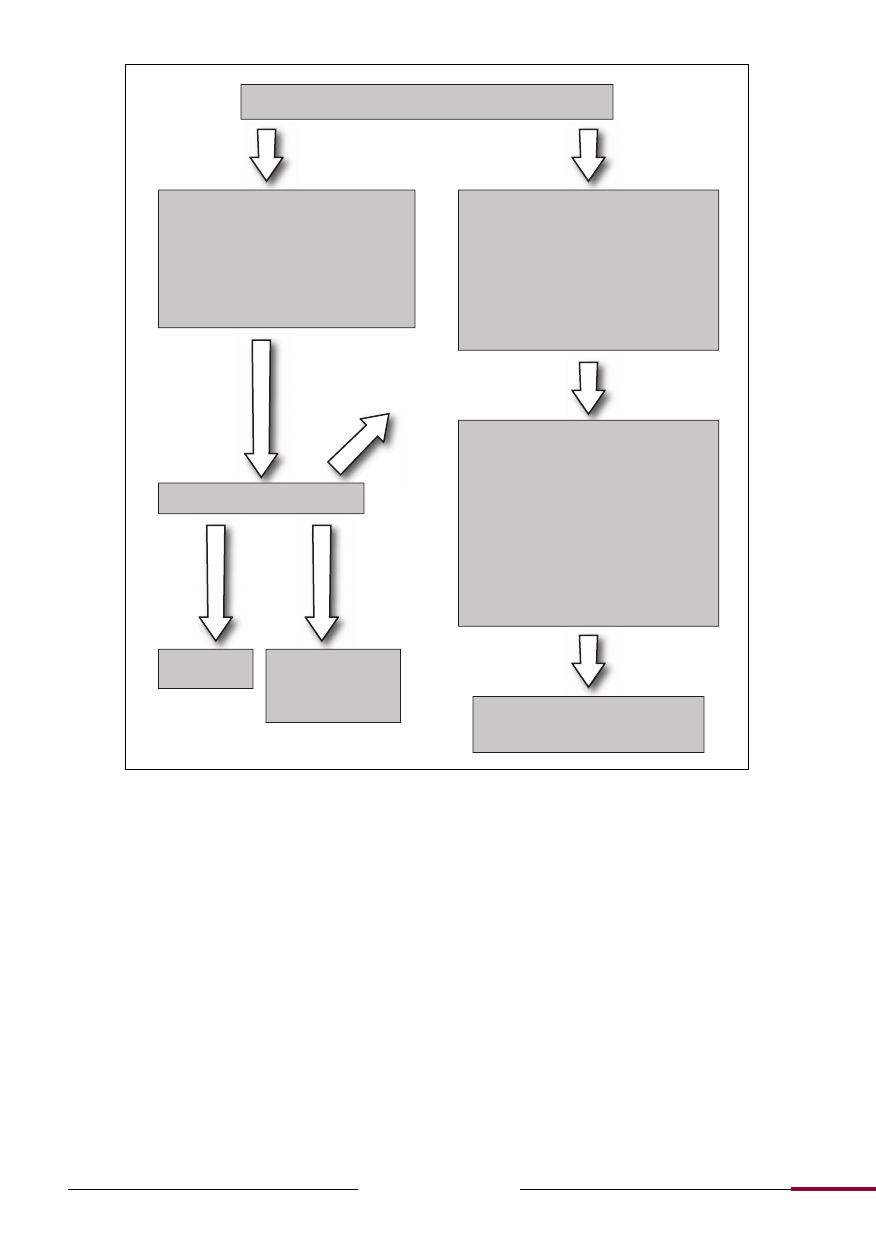
Ryc. 4.
Diagnostyka różnicowa w przypadku wystąpienia zespołu demielinizacji rdzenia kręgowego.
Izolowany zespół rdzeniowy
Typowy dla MS
• Ewolucja objawów w ciągu godzin-dni
• Częściowe zapalenie rdzenia kręgowego
• Czysto czuciowy
• Odnerwienie kończyny górnej
• Objaw Lhermitte’a
• Częściowy zespół Browna-Sequarda
• Spontaniczna remisja
Nietypowy dla MS
• Nadostry początek zachorowania
lub objawy podstępnie postępujące
• Pełne poprzeczne zapalenie rdzenia
kręgowego
• Wyraźny poziom czucia
• Ból korzeniowy
• Arefleksja
• Brak poprawy
Nietypowy dla MS
• Nadostry początek zachorowania
lub objawy podstępnie postępujące
• Pełne poprzeczne zapalenie rdzenia
kręgowego
• Wyraźny poziom czucia
• Ból korzeniowy
• Arefleksja
• Brak poprawy
MRI mózgu i rdzenia kręgowego
• Ucisk np. krążek międzykręgowy, guz
• Niedokrwienie/zawał
• Inne choroby zapalne, np. neuromyelitis
optica, sarkoid, toczeń, zespół Sjögrena
• Infekcje, np. kiła, borelioza, wirusowe,
gruźlica
• Toksyczne/żywieniowe/metaboliczne,
np. niedobór witaminy B12, toksyczność
tlenku azotu, niedobór miedzi
• Malformacje tętniczo-żylne
• „Maski” nierdzeniowe, np. zespół Guilla-
ina-Barrégo, miastenia rzekomoporaźna
Rozważyć inne
rozpoznania
Nieprawidłowe
Zmiany odpo-
wiadające de-
mielinizacji
MRI jasno wskazuje
na rozpoznanie inne
niż MS, np. ucisk
rdzenia kręgowego
Prawi-
dłowe
Niskie ryzyko
MS (20%)
Wysokie ryzyko MS
(60-90%), ponowna
ocena kryteriów
McDonalda
MRI, PMR, OCT
oraz inne badania neurofizjologiczne,
serologiczne w miarę potrzeby

Neurologia Praktyczna • 6/2011
28
jątkiem przypadków ograniczonych do pnia mó-
zgu lub podwzgórza) w Azji rozpoznaje się MS
[47, 48], podczas gdy w krajach zachodnich tacy
chorzy spełniający inne kryteria NMO są zwykle
klasyfi kowani jako NMO. Niezbędne jest prowa-
dzenie dalszych badań przebiegu naturalnego
choroby w tej grupie pacjentów w celu określe-
nia, czy założenia kliniczne odnoszące się do ich
fenotypu i odpowiedzi na leczenie są właściwie
przewidywane przez koncepcję diagnostyczną
ustaloną zgodnie z algorytmem azjatyckim i za-
chodnim.
Defi nicja i diagnostyka różnicowa ADEM
Historycznie ADEM różnicowano z MS ze wzglę-
du na jednofazowy przebieg i objawy encefa-
lopatii lub śpiączki występujące w kombina-
cji z objawami wieloogniskowymi (np. obja-
wami móżdżkowymi, mózgowymi objawami
ruchowymi lub czuciowymi, zapaleniem nerwu
wzrokowego lub zapaleniem rdzenia kręgowe-
go) charakterystycznymi dla IIDD, rozwijający-
mi się często w następstwie choroby infekcyjnej.
MRI mózgowia pokazuje zwykle symetryczne
zmiany wieloogniskowe lub rozlane [49]. Nawet
przy konserwatywnym podejściu wymagającym
obecności encefalopatii wstępne rozpoznanie
ADEM jest często zmieniane na prototypowy MS
Tabela IV.
Kryteria diagnostyczne neuromyelitis optica (NMO)
a
Kryteria główne (wymagane jest spełnienie wszystkich kryteriów głównych, ale mogą być one oddzielone okresem objawów niespecyfi cznych
Zapalenie nerwu wzrokowego jedno- lub obustronne
Poprzeczne zapalenie rdzenia kręgowego, kliniczne całkowite lub częściowe, związane jednak z radiologicznymi dowodami uszkodzenia rdzenia kręgowe-
go, rozciągającymi się na ponad trzy segmenty rdzeniowe w sekwencjach T2-zależnych MRI i hipointensywne w sekwencjach T1-zależnych wykonanych
w ostrej fazie zapalenia rdzenia kręgowego
Brak dowodów potwierdzających sarkoidozę, zapalenie naczyń, klinicznie jawnego układowego tocznia rumieniowatego lub zespołu Sjögrena czy innego
wyjaśnienia etiologii zespołu
Kryteria drugorzędowe (wymagane jest spełnienie przynajmniej jednego z nich)
Wykonane ostatnio badanie MR głowy musi być prawidłowe lub może wykazywać nieprawidłowości niespełniające kryteriów Barkhofa stanowiące ele-
ment kryteriów McDonalda, tzn
b
:
Niespecyfi czne zmiany w sekwencjach T2-zależnych nie spełniające kryteriów Barkhofa, podanych w kryteriach McDonalda
Zmiany w grzbietowej części opuszki, zachowujące ciągłość lub niełączące się ze zmianami w rdzeniu kręgowym
Zmiany w podwzgórzu i/lub pniu mózgu
„Linijne” zmiany sygnału obszarów okołokomorowych/ciała modzelowatego, jednak niemające kształtu owalnego i nie rozciągające się do parenchymy
półkul mózgowych w kształcie palców Dawsona
Dodatnie testy w surowicy lub PMR w kierunku przeciwciała NMO-IgG/akwaporyna-4
a
Kryteria te wykluczają ograniczone lub początkowe zespoły objawów mogące stanowić NMO, takie jak nawracające poprzeczne zapalenie rdzenia kręgowego z długood-
cinkowymi zmianami w rdzeniu kręgowym lub nawracające zapalenie nerwu wzrokowego; niezbędne są dalsze badania w celu wyjaśnienia zależności tych zespołów z NMO,
szczególnie w przypadku pozytywnego wyniku oznaczenia przeciwciał NMO-IgG/akwaporyna-4.
b
Okresowe wykonywanie badania MR głowy jest niezbędne w celu wykrycia nowych zmian mogących prowadzić do zmiany rozpoznania.
Tabela V.
Kryteria rozpoznania ostrego rozsianego zapalenia mózgu i rdzenia kręgowego (acute disseminated encephalomyelitis
– ADEM)
Podostra encefalopatia (zaburzenia stanu przytomności, zachowania lub funkcji poznawczych)
Ewolucja objawów w czasie od 1 tygodnia do 3 miesięcy; nowe objawy, obejmujące ogniskowe/wieloogniskowe zespoły demielinizacji, takie jak zapalenie
nerwu wzrokowego, zapalenie rdzenia kręgowego, mogą rozwijać się w ciągu pierwszych 3 miesięcy od zachorowania, pod warunkiem że nie są roz-
dzielone przez okres całkowitej remisji wyjściowych objawów (kiedy to rozpoznaje się MS)
Stan kliniczny poprawia się lub dochodzi do całkowitego wyzdrowienia, chociaż może utrzymywać się rezydualny defi cyt neurologiczny
MRI ujawnia przede wszystkim objawowe zmiany w obrębie istoty białej, które:
są zmianami świeżymi (odległe czasowo zmiany, którym towarzyszy encefalomalacja podają w wątpliwość rozpoznanie, jeśli nie można ich wytłuma-
czyć inaczej niż istnieniem odległej czasowo choroby demielinizacyjnej)
są zmianami mnogimi, rzadko pojawia się pojedyncza i duża zmiana ogniskowa
są zlokalizowane nad-, podnamiotowo lub w obu obszarach
zwykle obejmują przynajmniej jedną dużą zmianę (1-2 cm średnicy)
wykazują różnego stopnia wzmocnienie po podaniu gadolinu (wzmocnienie gadolinowe nie jest wymagane)
a
mogą współistnieć ze zmianami zlokalizowanymi w obrębie zwojów podstawy, jednak nie jest to wymagane
a
Jednoczesne wzmocnienie kontrastowe zmian może wystąpić, ale nie jest konieczne; jeśli jest obecne, stanowi czynnik zwiększający prawdopodobieństwo ADEM, jednak
powinno wzbudzić podejrzenie innych możliwych chorób (np. zapalenia naczyń, chłoniaka).

Neurologia Praktyczna • 6/2011
29
po wykazaniu ciągłej aktywności klinicznej od-
powiadającej MS [50]. W ostatnim czasie pojawi-
ły się opinie, że tradycyjne wymagania jednofa-
zowego przebiegu ADEM mogą być zbyt rygory-
styczne, a niektórzy pacjenci mogą doświadczać
nawrotów ADEM z ponownym pojawieniem się
w MRI zmian identycznych jak podczas pierwot-
nego zachorowania (nawracające ADEM) [21-24].
Chociaż kliniczna charakterystyka encefalopa-
tii z objawami wieloogniskowymi typowymi dla
IIDD czyni rozpoznanie ADEM bardziej prawdo-
podobnym niż MS, brak jest kryteriów klinicz-
nych, paraklinicznych czy obrazowych wiary-
godnie różnicujących początkowy epizod pioru-
nującego MS i ADEM (tab. V) [51].
Najnowsze defi nicje dziecięcego MS i ADEM
opracowano tak, aby uniknąć nakładania się tych
dwóch schorzeń, wymagając obecności ence-
falopatii jako niezbędnego elementu defi nicji
ADEM [52]. Chociaż te kryteria, osiągnięte na za-
sadzie konsensusu utworzono w oparciu o po-
pulacje dziecięce, rozsądne wydaje się ich sto-
sowanie również w populacjach dorosłych, pod
warunkiem prowadzenia dalszych obserwa-
cji odmienności pomiędzy przypadkami ADEM
u dzieci i dorosłych. Kryteria podkreślają względ-
ną swoistość encefalopatii dla ADEM, chociaż
nie różnicują stopnia nasilenia encefalopatii,
która może obejmować spektrum zaburzeń od
podrażnienia do śpiączki; ciężka encefalopatia
może być bardziej swoista dla ADEM w porów-
naniu z encefalopatią łagodniejszą.
Panel ekspertów zaproponował, aby rozpo-
znanie ADEM stawiano u pacjentów z pierwszym
epizodem odpowiadającym chorobie demielini-
zacyjnej rozpoczynającym się w sposób ostry lub
podostry (w ciągu dni lub tygodni), o przebiegu
stabilnym lub falującym, jednak wyłącznie przy
obecności dodatkowych cech charakterystycz-
nych. Obecna powinna być encefalopatia mani-
festująca się jako zaburzenie przytomności, zmia-
na zachowania lub zaburzenie funkcji poznaw-
czych. Nowe objawy mogą pojawiać się w ciągu
do 3 miesięcy od zachorowania, bez remisji (jed-
nak nie po upływie 3 miesięcy). Niemniej, jeśli po
ustąpieniu początkowych objawów nowe obja-
wy rozwijają się w czasie do 1 miesiąca, MS jest
bardziej prawdopodobne niż ADEM. Chociaż MRI
nie jest badaniem swoistym dla ADEM, charakte-
rystyczna jest obecność mnogich zmian zlokali-
zowanych nad- lub podnamiotowo, w kombina-
cji ze zmianami w obrębie jąder podkorowych
i przynajmniej jedną zmianą o średnicy większej
niż 1-2 cm. Zmiany w obrębie rdzenia kręgowego
mogą być obecne lub nie, jednak jeśli są obecne
mają charakter długoodcinkowy (tab. V).
W bardzo rzadkich przypadkach w przebie-
gu ADEM pacjenci mogą doświadczać nawro-
tu pierwszych objawów po upływie 3 miesięcy,
bez powstania nowych zmian obrazowych. Ist-
niejące zmiany mogą się jednak powiększać lub
ponownie wykazywać wzmocnienie po poda-
niu kontrastu gadolinowego [52, 53]. Innymi sło-
wy przebieg ma charakter nawrotowy, w odróż-
nieniu od wielofazowego przebiegu obejmują-
cego nowe objawy i nowe zmiany. Jeśli po wła-
ściwej ocenie klinicznej żadne inne rozpoznanie
nie wydaje się właściwe, uzasadnione jest rozpo-
znanie „nawrotowego ADEM”. Pojawienie się no-
wych zmian wraz z innymi objawami klinicznymi
po upływie 3 miesięcy od pierwszego zachoro-
wania wskazuje na MS bez względu na specyfi cz-
ny obraz kliniczny. Panel ekspertów nie aprobuje
rozpoznania „wielofazowego ADEM”, jeśli nowe
zmiany i inne objawy kliniczne rozwijają się wraz
z upływem czasu [52, 53], ponieważ nie jest moż-
liwe odróżnienie takiego obrazu klinicznego od
MS, a postawienie powyższej diagnozy opóźni
zastosowanie specyfi cznej dla MS immunotera-
pii. Członkowie Panelu podsumowali, że u więk-
szości pacjentów będzie się utrzymywała aktyw-
na choroba zapalna, co jest charakterystyczne
dla MS.
Klasyfi kacja idiopatycznych chorób zapalnych
Skutkiem poznania różnic między MS, NMO,
ADEM oraz ich wariantami było opracowanie kla-
syfi kacji IIDD, która powinna być uwzględnio-
na, kiedy rozważa się rozpoznanie IIDD (tab. VI).
MS rozpoznaje się, jeśli spełnione zostaną kryte-
Tabela VI.
Klasyfi kacja idiopatycznych zapalnych chorób demieli-
nizacyjnych
Przy pierwszych objawach
CIS
ADEM
Jednofazowe NMO
Niesklasyfi kowana (do czasu dalszej ewolucji choroby) choroba jednofazo-
wa, obejmująca manifestację piorunującą (wariant Marburg), stwardnienie
koncentryczne Balo i prezentację rzekomoguzową
Po kolejnym epizodzie klinicznym lub radiologicznym
MS
a
Nawrotowe NMO
Nawracające ADEM
Niesklasyfi kowana (do czasu dalszej ewolucji choroby); na przykład nawraca-
jące zapalenie nerwu wzrokowego lub poprzeczne zapalenie rdzenia krę-
gowego bez rozproszenia objawów w przestrzeni; lub klinicznie jednoogni-
skowe objawy bez bezobjawowych zmian w MRI (MRI może dawać wynik
prawidłowy) plus wywiad sugerujący odrębny epizod objawów z OUN bez
objawów obiektywnych
a
MS obejmuje jakiekolwiek IIDD spełniające ostatecznie kryteria McDonalda zakładające
rozproszenie objawów w czasie i przestrzeni, włączając wstępne manifestacje CIS i ADEM,
ewoluujące w kierunku MS; nie obejmuje NMO i nawrotowego ADEM; obejmuje również
rzekomoguzową chorobę demielinizacyjną i wariant Marburg MS po spełnieniu kryteriów
rozproszenia w czasie i przestrzeni.

Neurologia Praktyczna • 6/2011
30
ria rozproszenia objawów w czasie i przestrze-
ni [3, 4]. W obrębie MS wyróżnia się typ: rzuto-
wo-remitujący (relapsing-remitting), wtórnie po-
stępujący (secondary progressive), pierwotnie
postępujący (primary progressive) i postępują-
co-rzutowy (progressive-relapsing) [54]. Pacjen-
ci z nietypowymi zespołami IIDD, takimi jak wa-
riant Marburg MS [55, 56], stwardnienie koncen-
tryczne Balo [57] i inne rzekomoguzowe formy
choroby demielinizacyjnej [58, 59] traktowani
są jako pacjenci z chorobą „niesklasyfi kowaną”
w znaczeniu możliwości rozpoznania MS przy
pierwszej prezentacji klinicznej. Jednak po speł-
nieniu kryteriów rozproszenia w czasie i prze-
strzeni właściwe może być rozpoznanie MS po-
mimo nietypowej prezentacji wyjściowej lub
późniejszej. Należy również poważnie rozważyć
alternatywne diagnozy, jak na przykład chłoniak
mózgu, glejakowatość mózgu lub zapalenie na-
czyń. Niektóre wieloczasowe nawracające ma-
nifestacje kliniczne, jak na przykład nawracające
poprzeczne zapalenie rdzenia kręgowego, mogą
być również „niesklasyfi kowane” w znaczeniu
potencjalnego rozpoznanie MS przy pierwszych
objawach klinicznych, w sytuacji braku kliniczne-
go lub obrazowego potwierdzenia rozproszenia
w przestrzeni. NMO i ADEM stanowią odrębne
schorzenia w kategorii IIDD, mogące przebiegać
zarówno jednofazowo, jak i nawracająco. Niektó-
re z początkowych manifestacji CIS są prawdzi-
wie jednofazowymi zaburzeniami o charakterze
zapalno-demielinizacyjnym, jednak u wielu, jeśli
nie u wszystkich pacjentów z zespołami CIS, roz-
winie się MS.
Dyskusja/wnioski
Rozpoznanie MS wymaga wykluczenia szero-
kiego zakresu chorób. Chociaż wyłączenie bar-
dziej prawdopodobnych schorzeń alternatyw-
nych stanowi zasadniczy aspekt diagnostyki
MS, niewiele uwagi poświęca się opracowaniu
wytycznych dla lekarzy klinicystów. Panel eks-
pertów wyróżnił dwie obszerne kategorie cho-
rób, które powinny być uwzględnione w proce-
sie diagnostyki różnicowej chorób OUN sugeru-
jących MS: nie-IIDD z objawami naśladującymi
IIDD (w tym MS) oraz IIDD inne niż MS, spośród
których pewne schorzenia mogą przebiegać
w sposób rzutowo-remitujący. Zasugerowaliśmy
klasyfi kację prezentacji klinicznych, rozróżnia-
jąc objawy typowe, sugerujące MS, i nietypowe
dla MS, opisując jednocześnie całą gamę „czer-
wonych fl ag” mogących wskazywać na bardziej
prawdopodobną diagnozę alternatywną. Wnio-
ski i zalecenia płynące z naszej pracy wynikają
w dużej mierze z osiągniętego konsensusu, dla-
tego powinny być poddane ocenie w prospek-
tywnych badaniach klinicznych, najlepiej w for-
mie dużych badań wieloośrodkowych, obejmu-
jących zarówno specjalistyczne ośrodki MS, jak
i niewyspecjalizowane ośrodki kliniczne. Celem
tych działań ma być sprawdzenie ich przydatno-
ści w każdym z tych środowisk. Badania te mogą
doprowadzić do ustalenia algorytmów, pozwala-
jących na dokładne rozpoznanie zespołów OUN
sugerujących MS.
Badania prospektywne powinny dotyczyć
wszystkich zespołów CIS zgodnie z podany-
mi defi nicjami, obejmując zarówno pacjentów
z prawidłowym wynikiem badania MRI, jak i tych
z bezobjawowymi zmianami silnie sugerującymi
MS, wykrywanymi przypadkowo. Pierwsze opi-
sowe badanie retrospektywne, obejmujące pa-
cjentów z przypadkowo wykrywanymi w MRI
zmianami bezobjawowymi wykazało, że u części
z tych chorych dochodzi do szybkiej, wczesnej
konwersji do wczesnego klinicznie ewidentne-
go MS [18]. Ocena naturalnego przebiegu scho-
rzenia w tej grupie pacjentów wymaga przepro-
wadzenia długoterminowej obserwacji. U około
30% pacjentów, ze skądinąd typowymi izolowa-
nymi zespołami OUN sugerującymi demielini-
zację, badanie MR jest prawidłowe z wyjątkiem
zmian objawowych [16, 40, 60]. Obserwacja
prowadzona w okresie od 10 do 14 lat wykaza-
ła, że u około 20% spośród tych osób rozwinę-
ło się klinicznie ewidentne MS [13, 15]. U niektó-
rych pacjentów z CIS i negatywnym badaniem
MRI w płynie mózgowo-rdzeniowym stwierdza
się obecność prążków oligoklonalnych typo-
wych dla MS, natomiast u innych one nie wystę-
pują [61]. U pacjentów bez prążków oligoklonal-
nych zespół kliniczny jest rzeczywiście izolowa-
ny w czasie i przestrzeni, dlatego jedynym możli-
wym rozpoznaniem jest CIS [62].
Na diagnostykę różnicową i ocenę pacjentów
wpływają czynniki zarówno kliniczne jak i para-
kliniczne. Zaprezentowaliśmy całą gamę „czer-
wonych fl ag” wybranych na zasadzie kompro-
misu, które zgodnie z opinią członków Panelu
wskazują na duże prawdopodobieństwo scho-
rzenia innego niż MS w chwili zachorowania.
Czułość, swoistość i dokładność tych „czerwo-
nych fl ag” nie były badane. Każda z „czerwonych
fl ag” oceniana była przez członków Panelu od-
dzielnie, bez uwzględniania szerszego kontek-
stu klinicznego i/lub paraklinicznego; taka sytu-
acja rzadko ma miejsce w przypadku lekarza kli-
nicysty stawiającego rozpoznanie. Diagnostyka
różnicowa może być inna, jeśli „czerwona fl aga”
jest jedynym objawem lub współistnieje z inny-
mi istotnymi objawami czy nieprawidłowościami
paraklinicznymi i laboratoryjnymi. Wartość i zna-
czenie izolowanej „czerwonej fl agi” można czę-

Neurologia Praktyczna • 6/2011
31
sto wyjaśnić w kontekście danych demografi cz-
nych, klinicznych i paraklinicznych. Przykłado-
wo, zachorowanie poniżej 20. roku życia wymie-
nione jest jako drugorzędowa „czerwona fl aga”.
Jednak wobec stwierdzenia rozlanych i syme-
trycznych zmian sygnału istoty białej sugerują-
cych leukodystrofi ę, zachorowanie w młodym
wieku stanowić będzie główną „czerwoną fl agę”,
wskazującą na rozpoznanie inne niż MS. Jednak-
że w przypadku niedoczulicy twarzy lub zapale-
nia nerwu wzrokowego nie jest to w ogóle „czer-
wona fl aga”. Określenie zbioru objawów i „czer-
wonych fl ag” zwiększających pewność diagno-
styczną przy rozpoznaniu MS w przeciwieństwie
do innych schorzeń stanowi ważny cel badań na-
ukowych.
W diagnostyce różnicowej najtrudniej wyeli-
minować najczęstsze schorzenia alternatywne
naśladujące MS. W procesie diagnostycznym na-
leży brać pod uwagę względne rozpowszech-
nienie chorób w specyfi cznym obszarze geogra-
fi cznym lub populacji. Jednakże, brak dokład-
nych danych na temat rozpowszechnienia wielu
chorób alternatywnych w stosunku do MS (oraz
związanych z nimi „czerwonych fl ag”) utrud-
nia określenie ich znaczenia w oparciu o rozpo-
wszechnienie.
Pomimo faktu, iż nasza dyskusja obejmuje
szczegółową ocenę MS w populacjach azjatyc-
kich, a także charakterystykę azjatyckiego OSMS
wobec NMO i MS, większość danych odnoszą-
cych się do diagnostyki różnicowej IIDD wywodzi
się z badań przeprowadzonych w populacjach
zachodnich. Zaproponowany schemat postępo-
wania może mieć mniejsze zastosowanie w po-
pulacjach nie-zachodnich, co należałoby ocenić
w badaniach prospektywnych. Regionalne, uwa-
runkowane etnicznie różnice defi nicji chorób
dodatkowo utrudniają określenie podobieństw
i różnic pomiędzy NMO i azjatycką OSMS, kom-
plikując interpretację danych dotyczących sero-
pozytywności dla NMO-IgG w NMO i azjatyckim
OSMS [32, 33, 48]. Ogólnoświatowy konsensus
dotyczący defi nicji ułatwiłby dalsze badania kli-
nicznie i poszukiwania biomarkerów.
Biomarkery chorób w ogromny sposób uła-
twią diagnostykę różnicową. Precyzyjne i czu-
łe markery chorobowe – o charakterze obrazo-
wym lub laboratoryjnym – mogą w nieinwazyj-
ny sposób wspomagać diagnostykę różnicową.
Dobrym przykładem jest odkrycie przeciwcia-
ła NMO-IgG wspierającego różnicowanie NMO
od MS. Znaczące postępy diagnostyki obrazo-
wej mogą obejmować niekonwencjonalne tech-
niki MR, pozwalające na ilościową ocenę zmian
względnie specyfi cznych dla MS w obrębie pra-
widłowo wyglądającej istoty białej oraz MRI
o wysokiej sile pola magnetycznego pozwala-
jące lepiej zobrazować palce Dawsona [17] lub
specyfi czne dla MS zmiany korowe.
Panel ekspertów zaadaptował kryteria dia-
gnostyczne NMO zmodyfi kowane w oparciu
o kryteria zaprezentowane w przeszłości [19],
które powinny być traktowane jako tymczasowe
i podlegać dalszym międzynarodowym bada-
niom i walidacji. Inne grupy badawcze podawały
czułość i swoistość porównywalną z wartościa-
mi oryginalnymi, co można traktować jako nie-
zależną walidację [20]. Kryteria mogą wymagać
rewizji w kontekście przyszłych badań zależności
między objawami NMO i układowymi chorobami
autoimmunologicznymi, takimi jak układowy to-
czeń rumieniowaty [46]. Nie jest jasne, czy ogra-
niczona prezentacja NMO (np. nawroto we zapa-
lenie nerwu wzrokowego, nawrotowe poprzecz-
ne zapalenie rdzenia kręgowego z długoodcin-
kowymi zmianami morfologii rdzenia) u osób
z przeciwciałami przeciw akwaporynie-4 nale-
ży do kręgu chorób NMO [44]. Chociaż NMO ma
zwykle charakter nawracający, może mieć rów-
nież przebieg jednofazowy; szczegóło wa i dłu-
gotrwała obserwacja takich przypadków może
pomóc w zrozumieniu różnic patologicznych
między obiema jednostkami chorobowymi.
Zalecane kryteria diagnostyczne dla ADEM
(wymagające obecności encefalopatii i ogra-
niczonej ramy czasowej dla nawrotu objawów
ADEM) i MS [4] powinny ułatwić szybkie różnico-
wanie i w konsekwencji formułowanie różnych
zaleceń odnoszących się do poradnictwa i tera-
pii uwarunkowanej rokowaniem. Zaleca się pro-
wadzenie prospektywnych badań obserwacyj-
nych w celu oceny efektywności tych kryteriów.
Szczegółowe rozważanie problemów diagno-
styki różnicowej przy pierwszej prezentacji suge-
rującej MS powinno ułatwić spełnienie kluczo-
wego warunku stawianego przez kryteria dia-
gnostyczne McDonalda – konieczności wyklu-
czenia innych bardziej prawdopodobnych niż
MS wyjaśnień objawów klinicznych. Nasze reko-
mendacje opierają się w dużej mierze na kom-
promisie i brak jest obecnie danych wspierają-
cych te zalecenia. Potwierdzenie użyteczności
lub udoskonalenie algorytmów i innych zaleceń
odnoszących się do procedur diagnostycznych
wymaga sprawdzenia w badaniach prospektyw-
nych, w odpowiednim spektrum międzynarodo-
wych ośrodków klinicznych.
Podziękowania
Autorzy są wdzięczni i składają podziękowania za
konstruktywną krytykę podczas recenzji wcześ-
niejszej wersji manuskryptu następującym oso-
bom: dr. Stenowi Fredriksonowi (Kopenhaga),

Neurologia Praktyczna • 6/2011
32
dr. Andrew Goodmanowi (Rochester, NY), dr Ca-
therine Lubetzki (Paryż), dr. Johnowi Newsom-Da-
visowi (Oksford; nieżyjący), dr. Paulowi O’Conno-
rowi (Toronto), dr. Richardowi Rudickowi (Cleve-
land) i dr. Jackowi Simonowi (Portland, OR). Prace
Grupy Roboczej były wspierane przez Nati onal
Multi ple Sclerosis Society (US).
Tłumaczenie: dr n. med. Jacek Jaworski
P i ś m i e n n i c t w o
Schumacher, FA, Beeve, GW, Kibler, FR, et al. Problems
1.
of experimental trials of therapy in multiple sclerosis. Ann
NY Acad Sci 1965; 122: 552-568.
Poser, CM, Paty, DW, Scheinberg, LC, et al. New diagnos-
2.
tic criteria for multiple sclerosis: guidelines for research
protocols. Ann Neurol 1983; 13: 227-231.
McDonald, WI, Compston, A , Edan, G, et al. Recom-
3.
mended diagnostic criteria for multiple sclerosis: guide-
lines from the International Panel on the diagnosis of
multiple sclerosis. Ann Neurol 2001; 50: 121-127.
Polman, CH, Reingold, SC, Edan, G, et al. Diagnostic cri-
4.
teria for multiple sclerosis: 2005 revisions to the “McDon-
ald Criteria”. Ann Neurol 2005; 58: 840-846.
Charil, A, Yousry, TA, Rovaris, M, et al. MRI and the diag-
5.
nosis of multiple sclerosis: expanding the concept of
“no better explanation”. Lancet Neurol 2006; 5: 841-852.
Jacobs, LD, Beck, RW, Simon, JH, et al. Intramuscular in-
6.
terferon beta-la therapy initiated during a first demye-
linating event in multiple sclerosis. CHAMPS Study
Group. N Engl J Med 2000; 343: 898-904.
Comi, G, Filippi, M, Barkhof, F, et al. Effect of early inter-
7.
feron treatment on conversion to def inite multiple scle-
rosis: a randomized study. Lancet 2001; 357: 1576-1582.
Kappos, L, Polman, CH, Freedman, MS, et al. Treatment
8.
with interferon beta-1b delays conversion to clinically
def inite and McDonald MS in patients with clinically
isolated syndromes. Neurology 2006; 10: 1242-1249.
Swanton, JK, Rovira, A, Tintore, M, et al. MRI criteria for
9.
multiple sclerosis in patients presenting with clinically
isolated syndromes: a multicentre retrospective study.
Lancet Neurol 2007; 6: 677-686.
Ormerod, IE, Miller, DH, McDonald, WI, et al. The role of
10.
NMR imaging in the assessment of multiple sclerosis and
isolated neurological lesions. A quantitative study. Brain
1987; 110: 1579-1616.
Uitdehaag, BMJ, Kappos, L, Bauer, L, et al. Discrepancies
11.
in the interpretation of clinical symptoms and signs in
the diagnosis of MS: a proposal for standardization. Mult
Scler 2005; 11: 227-231.
Mastronardo, G, Rocca, MA , lannucci, G, Pereira,
12.
C , Filip pi, M. A longitudinal MR study of the presymp -
tom-atic phase in a patient with clinically defi nite multiple
sclerosis. Am J Neuroradiol 1999; 20: 1268-1272.
Brex, PA, Ciccarelli, 0, O’Riordan, JI, Sailer, M, Thompson, AJ,
13.
Miller, DH. A longitudinal study of abnormalities on MRI
and disability from multiple scle rosis. N Engl J Med 2002; 246:
158-164.
Minneboo, A, Barkhof, F, Polman, CH, Uitdehaag, BM,
14.
Knol, DL, Castelijns, JA. Infratentorial lesions predict long-
term disability in patients with initial fi ndings sug gestive of
multiple sclerosis. Arch Neurol 2004; 61: 217-221.
Optic Neuritis Study Group. Long-term brain magnetic res-
15.
onance imaging changes after optic neuritis in patients
without clinically defi nite multiple sclerosis. Arch Neurol
2004; 61: 1538-1541.
Tintore, M, Rovira, A, Rio, J, et al. Baseline MRI predicts fu-
16.
ture attacks and disability in clinically isolated syn dromes.
Neurology 2006; 67: 968-972.
Kangarlu, A, Bourekas, EC, Ray-Chaudhury, A, Rammohan, KW.
17.
Cerebral cortical lesions in multiple sclerosis detected by
MR imaging at 8 Tesla. Am J Neuror adiol 2007; 28: 262-266.
Lebrun, C, Bensa, C, Debouverie, M, et al. Unexpected
18.
multiple sclerosis: follow-up of 30 patients with mag netic
resonance imaging and clinical conversion profi le. J Neurol
Neurosurg Psychiatry 2008; 79: 195-198.
Wingerchuk, DM, Lennon, VA, Pittock, SJ, Lucchinetti, CF,
19.
Weinshenker, BG. Revised diagnostic criteria for neu romyelitis
optica. N e uro l o g y 2006; 66: 1485-1489.
Saiz, A, Zuliani, L, Blanco, Y, et al. Revised diagnostic crite-
20.
ria for neuromyelitis optica: application in a series of suspect-
ed patients. J Neurol 2007; 254: 1233-1237.
Cohen, O, Steiner-Birmanns, B, Biran, I, Abramsky, O, Honig-
21.
man, S, Steiner, I. Recurrence of acute dissemi nated en-
cephalomyelitis at the previously aff ected brain site. Arch
Neurol 2001; 58: 797-701.
Hartung, HP, Grossman, RI. ADEM: distinct disease or part
22.
of the MS spectrum. N e u r o l o g y 2001; 56: 1257-1260.
Anlar, B, Basaran, C, Kose, G , e t al . Acute disseminated en-
23.
cephalomyelitis in children: outcome and prognosis. N e u -
r o p e d i a t r i c s 2003; 24: 194-199.
Morimatsu, M. Recurrent ADEM or MS. I n t M e d 2004; 43:
24.
647-648.
Jacob, A, Matiello, M, Wingerchuk, DM, Lucchinetti, CF, Pit-
25.
tock, SJ, Weinshenker, BG. Neuromyelitis optica: changing
concepts. J Neuroimmunol 2007; 187: 126-138.
Wingerchuk, DM, Lennon, VA, Lucchinetti, CF, Pittock, SJ,
26.
Weinshenker, BG. The spectrum of neuromyelitis optica.
Lancet Neurol 2007; 6: 805-815.
O’Riordan, JI, Gallagher, HL, Thompson, AJ, et al. Clini cal, CSF
27.
and MRI findings in Devic’s neuromyelitis optica. J Neurol
Neurosurg Psychiatry 1996; 60: 382-387.
Wingerchuk, DM, Hogancamp, WF, O’Brien, PC, Weinshen-
28.
ker, BG. The clinical course of neuromyelitis optica (Devic’s
syndrome). Neurology 1999; 53: 1107-1114.
de Seze, J. Neuromyelitis optica. Arch Neurol 2003; 60: 1336-
29.
1338.
de Seze, J, Lebrun, C, Stojkovic, T, Ferriby, D, Chatel, M, Verm-
30.
ersch, P. Is Devic’s neuromyelitis optica a separate disease?
A comparative study with multiple sclerosis. Muit Scler
2003; 9: 521-525.
Ghezzi, A, Bergamaschi, R, Martinelli, V, e t al . Clinical cha-
31.
racteristics, course and prognosis of relapsing Devic’s
Neuromyelitis Optica. J Neurol 2004; 251: 47-52.
Lennon, VA, Wingerchuk; DM, Kryzer, TJ, et al. A serum au-
32.
toantibody marker of neuromyelitis optica: distinction from
multiple sclerosis. Lancet 2004; 364: 2106-2112.
Jarius, S, Franciotta, D, Bergamaschi, R, et al. NMO-IgG in the
33.
diagnosis of neuromyelitis optica. Neurology 2007;
Keegan, M, Pincada, AA, McClelland, RL, Darby, CH, Rodri-
34.
guez, M, Weinshenker, BG. Plasma exchange for severe at-
tacks of CNS demyelination: predictors of response. Neu-
rology 2003; 58: 143-146.
Papeix, C, Vidal, JS, de Seze, J, et al. Immunosuppressive the-
35.
rapy is more eff ective than interferon in neuromyeli tis opti-
ca. Muit Scler 2007; 13: 256-259.
Warabi, Y, Matsumoto, Y, Hayashi, H. Interferon beta-1b exa-
36.
cerbates multiple sclerosis with severe optic nerve and
spinal cord demyelination. J Neurol Sci 2007; 252: 257-261.
Mandler, RN, Davis, LE, Jeff ery, DR, Kornfeld, M. Devic’s neu-
37.
romyelitis optica: a clinicopathological study of 8 pa-
tients. Ann Neurol 1993; 34: 162-168.
Filippi, M, Rocca, MA, Moiola, L, et al. MRI and magneti-
38.
zation transfer imaging changes in the brain and cervical
cord of patients with Devic’s neuromyelitis optica. Neu-
rology 1999; 53: 1705-1710.
Rocca, MA, Agosta, F, Mezzapesa, DM, et al. Magnetiza tion
39.
transfer and diff usion tensor MRI show gray matter damage
in neuromyelitis optica. N euro lo g y 2004; 10: 476-478.
Barkhof, F, Filippi, M, Miller, DH, et al. Comparison of MR
40.
imaging criteria at first presentation to predict con version
to clinically definite multiple sclerosis. B r a i n 1997; 120:
2059-2069.
Pittock, SJ, Lennon, VA, Wingerchuk, DM, Lucchinetti, CF,
41.
Weinshenker, BG. Brain abnormalities in neuromye litis opti-
ca. Arch Neurol 2006; 63: 390-396.
Pittock, SJ, Weinshenker, GB, Lucchinetti, CF, Wingerchuk,
42.
DM, Corboy, JR, Lennon, VA. Neuromyeli tis optica brain le-

Neurologia Praktyczna • 6/2011
33
sions localized at sites of high aqua porin 4 expression. Arch
Neurol 2006; 63: 964-968.
B ergamas chi, R , Tonie t ti, S, Fr anciot t a , D, et al.
43.
Oligoclo
nal bands in Devic’s neuromyelitis optica and
multiple sclerosis. Differences in repeated cerebrospinal
fluid examinations. Mult Scler 2004; 10: 2-4.
Weinshenker, BG, Wingerchuk, DM, Vukusic, S, et al. Neuro-
44.
myelitis optica IgG predicts relapse after longitudi nal exten-
sive transverse myelitis. Ann Neurol 2006; 59: 566-569.
Matiello, M, Lennon, VA, Jacopb, A, et al. NMO-IgG pre dicts
45.
the outcome of recurrent optic neuritis. Neurology 2008; 70:
2197-2220.
Pittock, SJ, Lennon, VA, de Seze, J, et al. Neuromyelitis opti-
46.
ca and non-organ-specifi c autoimmunity. Arch Neurol 2008;
65: 78-83.
Kira, J. Multiple sclerosis in the Japanese population. Lan-
47.
cet Neurol 2003; 2: 117-127.
Matsuoka, T, Matsushita, T, Kawano, Y, et al. Heterogene ity of
48.
aquaporin-4 autoimmunity and spinal cord lesions in multi-
ple sclerosis in Japanese. Brain 2007; 130: 1206-1223.
Kesselring, J, Miller, DH, Robb, SA, et al. Acute dissemi nated
49.
encephalomyelitis. MRI findings and the distinc tion from
multiple sclerosis. B r a i n 1990; 113: 291-302.
de Seze, J, Deborrverie, M, Zephir, H, et al. Acute fulmi nant
50.
demyelinating disease: a descriptive study of 60 patients.
Arch Neurol 2007; 64: 1426-1432.
Banwell, B, Shroff , M, Ness, JM, et al. MRI features of pedi atric
51.
multiple sclerosis. Neurology 2007; 68(Suppl. 2): S46-S53.
Krupp, LB, Banwell, B, Tenembaum, S. for the Interna tional
52.
Pediatric MS Study Group. Consensus defi nitions proposed
for pediatric multiple sclerosis and related disorders. Neu-
rology 2007; 68(Suppl. 2): S7-S12.
Brinar, VV, Poser, CM. The spectrum of disseminated en-
53.
cephalomyelitis. Clin Neurol Neurosurg 2006; 108: 2295-
2310.
Lublin, FD, Reingold, SC. Defi ning the clinical course of mul-
54.
tiple sclerosis: results of an international survey. National
Multiple Sclerosis Society (USA) Advisory Committee on
Clinical Trials of New Agents in Multiple Sclerosis. Neuro-
logy 1996; 40: 469-479.
Mendez, MF, Pogacar, S. Malignant monophasic multiple
55.
sclerosis or “Marburg’s disease”. Neurology 1988; 38: 1153-
1155.
Johnson, MD, Lavin, P, Whetsell, WO. Fuminant mono-
56.
phasic multiple sclerosis, Marburg’s type. J Neurol Neuro surg
Psychiatry 1990; 53: 918-921.
Moore, GR, Neumann, PE, Suzuki, K, Lijtmaer, HN, Traugott, U,
57.
Raine, CS. Balo’s concentric sclerosis: new observations on le-
sion development. Ann Neurol 1985; 17: 604-611.
Hunter, SB, Ballinger, WE Jr., Rubin, JJ. Multiple sclerosis mim-
58.
icking primary brain tumor. Arch Pathol Lab Med 1987; 111:
464-468.
Kepes, J. Large focal tumor-like demyelinating lesions of the
59.
brain: intermediate entity between multiple sclerosis and
acute disseminated encephalomyelitis: a study of 31 pa-
tients. Ann Neurol 1993; 22: 18-27.
Morrissey, SP, Miller, DH, Kendall, BE, et al. The signifi cance of
60.
brain magnetic resonance imaging abnormali ties at pre-
sentation with clinically isolated syndromes suggestive
of multiple sclerosis. A 5-year follow-up study. Brain 1993;
116: 135-146.
Söderström, M, Ya-Ping, J, Hillert, J, Link, H. Optic neuri tis.
61.
Prognosis for multiple sclerosis from MRI, CSF and HLA
fi ndings. Neurology 1998; 50: 708-714.
Weinshenker, BG, Gilbert, JJ, Ebers, GC. Some clinical and
62.
pathologic observations on chronic myelopathy: a va-
riant of multiple sclerosis. J Neurol Neurosurg Psychiatry
1990; 53: 146-149.
Wyszukiwarka
Podobne podstrony:
Post⌐powanie diagnostyczne w przypadku podejrzenia zatorowoÿci pêucnej u kobiety w ciÑ╛y Zalecenia A
procedura postepowania w przypadku podejrzenia stosowania przemocy wobec dziecka, organizacja-pracy
04. Diagnostyka różnicowa bólów brzucha u dzieci, Uczelnia, rodzinna
diagnostyka referat praca kontrolna 2 diagnostyka roznicowa wymiotow
Diagnoza indywidualnego przypadku, Studia pedagogiczne rok III
programAktywnościRóżnychSfer, arkusze,analiza przypadku,diagnozy,analiza przypadku
Diagnostyka roznicowa zoltaczek
Diagnostyka różnicowa niedokrwistości
DIAGNOSTYKA ROZNICOWA ZAPALENIA Nieznany
Diagnostyka różnicowa chorób układu oddechowego bydła, Zootechnika, Choroby
Diagnostyka laboratoryjna diagnostyka różnicowa
NIETRZYMANIE MOCZU diagnostyka różnicowanie(2)
DIAGNOSTYKA RÓŻNICOWA BIEGUNEK, DIAGNOSTYKA R??NICOWA BIEGUNEK
więcej podobnych podstron