1
W
PROWADZENIE DO SPEKTROMETRII MAS
Opracownie: dr
Maciej Góra, dr Jarosław Wilamowski
1. ZASADA DZIAŁANIA SPEKTROMETRU MAS
Spektrometria mas (MS – ang. Mass Spectrometry) jest jedną z podstawowych technik
analitycznych stosowanych w chemii organicznej głównie do ustalania składu mieszanin oraz
identyfikacji związków organicznych. Pozwala ona na precyzyjny pomiar mas cząsteczkowych
analizowanych związków oraz mas fragmentów, na które rozpadają się podczas pomiaru, co dostarcza
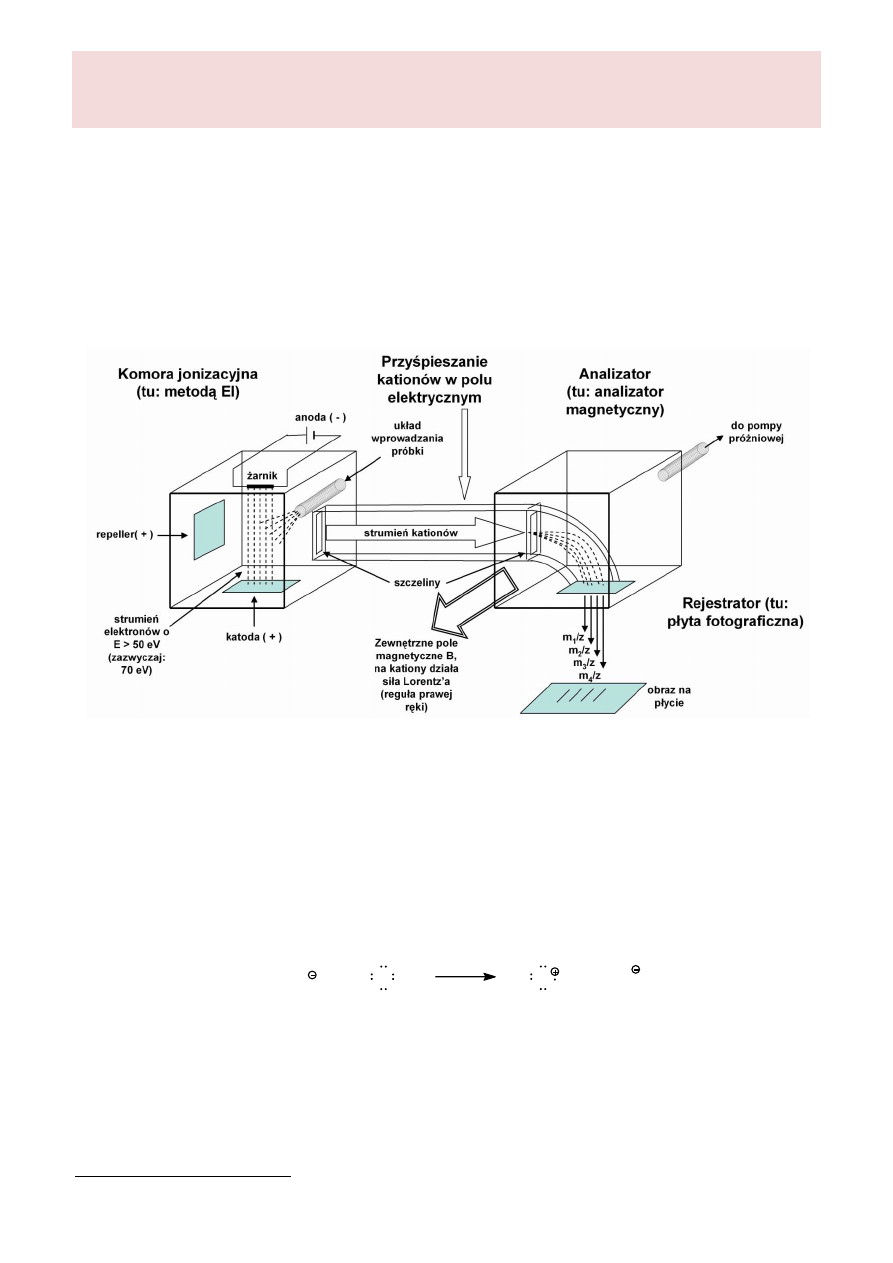
pośrednio informacji o strukturze badanych cząsteczek. Ogólny schemat budowy jednego z modeli
spektrometru mas przedstawia rysunek 1.
Rysunek 1. Schemat budowy spektrometru mas starszego typu
Próbka analitu wprowadzona zostaje pomiędzy elektrody, gdzie ulega jonizacji pod wpływem
strumienia elektronów (EI – ang. electron impact).
1
Zderzenie elektronu z cząsteczką organiczną (M)
powoduje wybicie elektronu (zwykle jednego) z powłoki walencyjnej cząsteczki, w wyniku czego
tworzy się tzw. kationorodnik (cząstka posiadająca ładunek dodatni i nieparzystą liczbę elektronów;
jeśli jej masa odpowiada masie cząsteczki zwana jest jonem molekularnym lub macierzystym i
oznaczana przez M
+
):
e
-
+ M → M
.+
+ 2e
-
np. dla metanu:
+
C
H
H
H
H
e
2e
+
C
H
H
H
H
kationorodnik
(z niesparowanym elektronem)
Podczas jonizacji mogą powstawać również aniony, gdy cząsteczka przyjmie elektron na jeden
z nieobsadzonych orbitali antywiążących, jednak jony te, podobnie jak cząstki nienaładowane
(niezjonizowane cząsteczki, rodniki) nie opuszczają zwykle komory jonizacyjnej. Kationy
1
Inne metody jonizacji i konstrukcje analizatorów scharakteryzowano w dalszym rozdziale.
2
molekularne (M
+
) są cząstkami o wysokiej energii i mogą ulegać rozpadowi (fragmentacji) tworząc
szereg innych kationów, rodników, kationorodników i cząsteczek obojętnych.
Powstające w komorze jonizacyjnej kationy są przyśpieszane w polu elektrycznym i po
przejściu przez szczeliny zapewniające spójność wiązki trafiają do analizatora. W przypadku
zamieszczonego na rysunku analizatora magnetycznego, tor lotu obdarzonych ładunkiem cząsteczek
ulega zakrzywieniu pod wpływem zewnętrznego pola magnetycznego. Odchylenie to jest
proporcjonalne do stosunku masy jonu do jego ładunku m/z. Przy założeniu, że wszystkie jony
trafiające do analizatora mają taki sam ładunek (zwykle +1), spowoduje to większe zakrzywienie toru
lotu jonów lżejszych, a mniejsze dla jonów cięższych. Efekt ten można zaobserwować w części
rejestrującej obecność jonów jako sygnały odpowiadające jonom o różnym m/z. Natężenie
rejestrowanego sygnału jest wprost proporcjonalne do ilości padających jonów, a co za tym idzie do
prawdopodobieństwa ich powstawania. W pierwszych spektrometrach do rejestracji jonów służyły
odpowiednio przygotowane płyty fotograficzne, na których padające jony powodowały powstawanie
(po wywołaniu i utrwaleniu obrazu) ciemnych prążków. Obecnie jako rejestratory stosuje się
powielacze elektronowe oraz fotopowielacze.
Klasyczne widmo MS (rys. 2) ma postać wykresu słupkowego: przy wartościach m/z (oś
odciętych) widoczne są pionowe kreski, odpowiadające jonom o określonym stosunku masy do
ładunku, o powstałym z analizowanego związku. Intensywność (wysokość) poszczególnych sygnałów
jest proporcjonalna do liczby jonów o danej masie, które dotarły do detektora. Miarą intensywności
sygnałów jest abundancja, czyli względna intensywność piku (I), wyrażana w procentach, w
odniesieniu do najwyższego piku na widmie. Najbardziej intensywny pik na widmie nazywany jest
pikiem głównym lub podstawowym i z definicji ma abundancję równą 100%. Pik, którego m/z
odpowiada masie cząsteczkowej analizowanego związku odpowiada kationorodnikowi powstającemu
na skutek wybicia jednego elektronu z powłoki walencyjnej cząsteczki i nosi nazwę piku
molekularnego (M
+
). W przypadku niektórych związków (np. niektórych alkoholi lub kwasów
karboksylowych, trzeciorzędowych halogenków alkilu) bywa, że przy standardowej technice jonizacji
EI pik molekularny ma małą intensywność lub jest niewidoczny na widmie masowym.
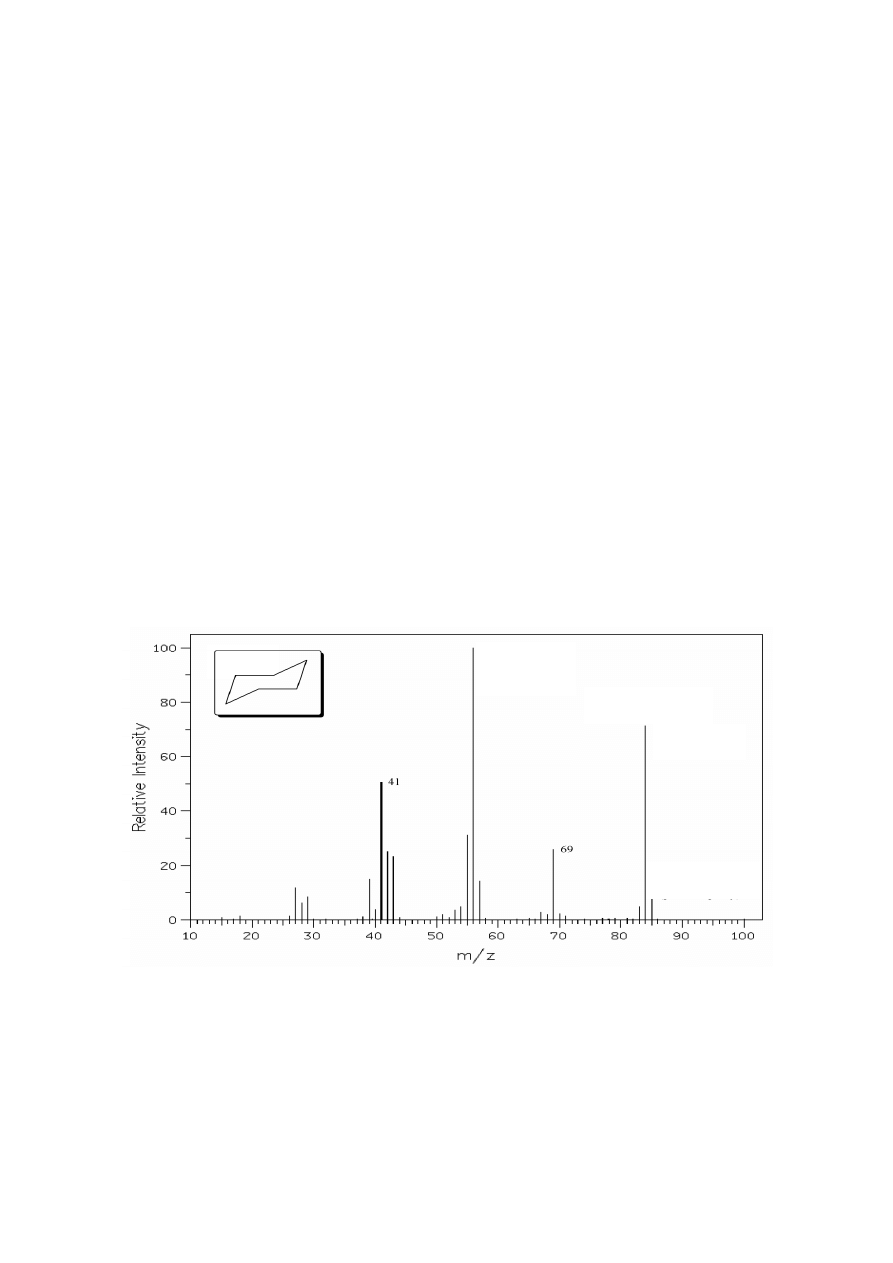
Rysunek 2. Widmo MS cykloheksanu.
Część cząsteczek związków trafiających do detektora może zawierać izotopy atomów o masach
większych niż izotopy podstawowe. Na przykład jedna na ok. dziewięćdziesiąt cząsteczek metanu
będzie zawierała izotop
13
C lub jeden atom
2
H i posiadała masę 17 u (a nie 16 u, jak można wyliczyć
dla wzoru CH
4
posługując się masami podstawowych izotopów). W przypadku cykloheksanu
prawdopodobieństwo, że cząsteczka będzie zawierać dokładnie jeden cięższy izotop jest jeszcze
większe i wynosi ok. 7%. W konsekwencji na widmach MS są widoczne piki przy wartościach m/z
większych niż pik molekularny, zwane pikami izotopowymi (np. pik M
+
+1 przy m/z = 85 w widmie
cykloheksanu). W przypadku widm MS związków zawierających wyłącznie atomy węgla, wodoru,
tlenu i azotu o niewielkich masach cząsteczkowych intensywność pików izotopowych o różniących się
m/z = 56
(pik główny,
podstawowy)
M
+
= 84
(pik molekularny)
M
+
+1 = 85
(pik izotopowy)

3
od piku molekularnego o dwie, trzy lub więcej jednostek (czyli piki M
+
+2, M
+
+3 itp.) mają znikomą
intensywność, ale ich wzajemne proporcje pozwalają określić wzór sumaryczny cząsteczki. Służą do
tego tzw. tablice Beynona, które opisane zostały w zaawansowanych podręcznikach do spektrometrii
mas. Porównanie względnej intensywności piku molekularnego (I
M
) oraz izotopowego I
M+1
pozwala
jednak w uproszczony sposób oszacować liczbę atomów węgla w cząsteczce (n
C
), wykorzystując
zależność:
Dobrą zgodność uzyskuje się jednak tylko w przypadku cząsteczek zawierających nie więcej
niż kilkanaście atomów węgla. Piki izotopowe mogą dostarczać jeszcze więcej informacji, np.
o składzie pierwiastkowym cząsteczek zawierających atomy siarki, chloru lub bromu – szczegóły
opisano w
Widmo MS często przedstawia się w postaci cyfrowej, wymieniając kolejno położenie i
abundancję najbardziej intensywnych sygnałów i zaznaczając, który z pików pochodzi od jonu
macierzystego. Przykładowo, powyższe widmo MS cykloheksanu można zapisać w formie:
MS [m/z, (% int.)]: 84 (72, M
+
), 69 (26), 56 (100), 55 (31), 41 (51).
Modułowa konstrukcja współczesnych aparatów pozwala na zmianę elementów
rozdzielających mieszaninę, jonizujących próbkę, odchylających jony i detektorów, a co za tym idzie
dostosowania aparatury do potrzeb eksperymentu. Niezależnie jednak od stopnia zaawansowania
aparatury i jej przeznaczenia, w standardowych pomiarach
spektrometr mas zawsze mierzy stosunek
masy kationu do jego ładunku m/z – nie są wykrywane np. rodniki i cząsteczki obojętne!
Przedstawiona powyżej technika jonizacji elektronami EI posiada szereg wad, jednak ze względu na
dużą powtarzalność wyników niezależną od stosowanego sprzętu, jest wciąż powszechnie stosowana.
Istnieje szereg baz widm związków organicznych zarejestrowanych w standardowych warunkach
(jonizacja elektronami o energii 70 eV) pozwalających na szybką identyfikację składników mieszanin.
2. ALTERNATYWNE TECHNIKI JONIZACJI ORAZ ANALIZY I REJESTRACJI
JONÓW
Aby otrzymać widmo MS związku chemicznego, należy uprzednio przeprowadzić jego
jonizację. Pierwszą zastosowaną w tym celu metodą (i do dziś jedną z najpopularniejszych) jest
jonizacja elektronami w komorze próżniowej – EI. Ponieważ jednak efektywna jonizacja wymaga w
tym przypadku zastosowania strumienia elektronów o relatywnie dużej energii, cząsteczki jonizowane
tą techniką najczęściej ulegają fragmentacji z utworzeniem tzw. jonów fragmentacyjnych (por. rozdz.
6). Prawdopodobieństwo fragmentacji jest tym większe im większa jest energia elektronów źródła
jonizującego. Do najważniejszych zalet tej metody należy zaliczyć szybkie otrzymywanie
powtarzalnych widm i możliwość ich wykorzystania do identyfikacji substancji w oparciu o istniejące
bazy danych. Wadami są natomiast: konieczność wprowadzania próbki w stanie gazowym, a zatem
utrudniona analiza substancji o małej lotności i trwałości (czyli zazwyczaj o dużej masie
cząsteczkowej), często niepożądana fragmentacja jonów i konieczność utrzymywania wysokiej próżni
w aparaturze.
Ze względu na te niedogodności, rozwinięto szereg innych, alternatywnych metody jonizacji
analitu:
CI (Chemical Ionization) – technika, w której bezpośredniej jonizacji ulega gaz pomocniczy.
Jony tego gazu (np. CH
4
, NH
3
) przekazują następnie energię cząsteczkom analitu na skutek
zróżnicowanych interakcji (np. wymiany elektronów i protonów). Zaletami tej metody jest
otrzymywanie wyraźnych pików molekularnych oraz niewielka fragmentacja.
Często spotykaną metodą jonizacji jest bombardowanie próbki szybkimi atomami - FAB (Fast
Atom Bombarding) np. argonu, ksenonu, cezu lub jonami tych pierwiastków - SIMS (Secondary Ion

4
Mass Spectroscopy). W obu technikach zjonizowane składniki próbki są wyrzucane z matrycy i
trafiają do dalszych sekcji spektrometru, tylko w niewielkim stopniu ulegając fragmentacji.
Do łagodnych metod jonizacji pozwalających na badania labilnych makrocząsteczek zaliczyć
można technikę adsorbcji/desorbcji laserem wspomaganą matrycą – MALDI (Matrix Adsorbed Laser
Desorbed Ionisation). Energia, którą w tym przypadku dostarcza laser pracujący najczęściej w
zakresie UV jest pochłaniana przez matrycę i następnie przekazywana próbce. W widmach
zarejestrowanych tą metodą najczęściej obserwuje się piki pochodzące od protonowanych jonów
molekularnych.
Rozseparowanie jonów w zależności od ich masy oraz ładunku następuje w analizatorze
wykorzystującym w tym celu pole magnetyczne (jak na rys. 1), elektryczne lub czas przelotu jonów.
Analizator magnetyczny rozdziela jony ze względu na ich ładunek i pęd. Krzywizna po której
porusza się dany jon jest uzależniona od przyłożonego z zewnętrznego pola magnetycznego.
W praktyce stosowane jest przemiatanie (skaning) magnetyczny. W celu otrzymania widma masowego
zmieniana jest wartość indukcji pola magnetycznego w taki sposób, aby tylko jony o odpowiednim
stosunku masa/ładunek (m/z) docierały do detektora.
Pozostałe jony w wyniku kolizji ze ścianami
analizatora ulegają rozładowaniu.
Analizator elektrostatyczny wykorzystuje
zjawisko zakrzywienia trajektorii przelotu jonów pod
wpływem pola elektrycznego przyłożonego do
elektrod o odpowiedniej krzywiźnie. Podobnie jak w
przypadku
skaningu
magnetycznego,
zmiana
przyłożonego do elektrod napięcia powoduje, że
tylko jony o odpowiednim m/z docierają do
detektora. Często spotykaną odmianą tego analizatora
jest analizator kwadrupolowy – jon przelatujący
pomiędzy elektrodami do których przyłożony jest
zmienny potencjał oscyluje pomiędzy nimi. Dobór
wartości U (napięcie stałe) i V (napięcie zmienne) pozwala tylko jonom o odpowiednim m/z dotrzeć
do detektora (Rys. 3).
Analizator czasu przelotu TOF (Time Of Flight) łączony jest zazwyczaj z impulsową
techniką jonizacji (np. MALDI). Przy założeniu, że wszystkie powstające jony posiadają jednostkowy
ładunek (nie są wielokrotnie zjonizowane) czas ich dotarcia do detektora będzie dłuższy dla jonów
cięższych i krótszy dla lekkich. Analizator TOF pozwala na rozdział cząsteczek różniących się masami
w bardzo szerokim zakresie co znajduje zastosowanie np. w analizie polimerów.
Detektory stosowane obecnie dzielą się na dwie klasy: powielacze elektronowe oraz
fotopowielacze. W obu przypadkach ich działanie polega zarówno na detekcji jonów opuszczających
analizator, jak i wstępnym wzmocnieniu sygnału generowanego przez jon. Detektory typu płyty
fotograficznej są już praktycznie niespotykane.
3. WYSOKOROZDZIELCZA SPEKTROMETRIA MAS (HRMS)
Wysokorozdzielcza spektrometria mas HRMS (ang. High Resolution Mass Spectrometry) jest
jedną z ważniejszych z punktu widzenia chemii organicznej technik, ponieważ pozwala nie tylko na
określenie masy, ale również na ustalenie wzoru sumarycznego jonu. Klasyczna spektrometria mas
posiada rozdzielczość 1 u i jony o zbliżonej masie nie są w niej rozróżniane. Przykładowo, gdy na
widmie MS jest widoczny pik przy m/z = 44, nie mamy pewności, który z podanych poniżej jonów
odpowiada za powstanie tego sygnału. Każdy z nich posiada w przybliżeniu tę samą masę (44 u),
jednak ze względu na różnice w składzie pierwiastkowym, różnią się one masami o kilka setnych lub
tysięcznych części u.
Rys 3. Schemat działania analizatora kwadrupolowego.
5
np. m/z = 44 może pochodzić od:
+
C
H
3
CH
2
CH
3
m/z = 44,06260
C
H
3
CHO
+
m/z = 44,02620
C
H
3
CH
NH
2
+
m/z = 44,05002
O
C
O
+
m/z = 43,98983
N
H
NH
CH2
+
m/z = 44,03740
W technice HRMS, dzięki zastosowaniu bardzo precyzyjnych analizatorów i rejestratorów,
możliwe jest rozróżnienie jonów z dokładnością do 0,0001 u. Zatem, gdy na widmie HRMS odczyta
się dla jonu np. wartość m/z = 44,0263, wiadomo, że pochodzi on od jonu o wzorze sumarycznym
[C
2
H
4
O]
+
.
4. ZASTOSOWANIE
SPEKTROMETRII
MAS
W
ANALIZIE
CHEMICZNEJ
(GC-MS, LC-MS)
Ze względu na możliwość łącznego stosowania MS wraz z innymi metodami analitycznymi,
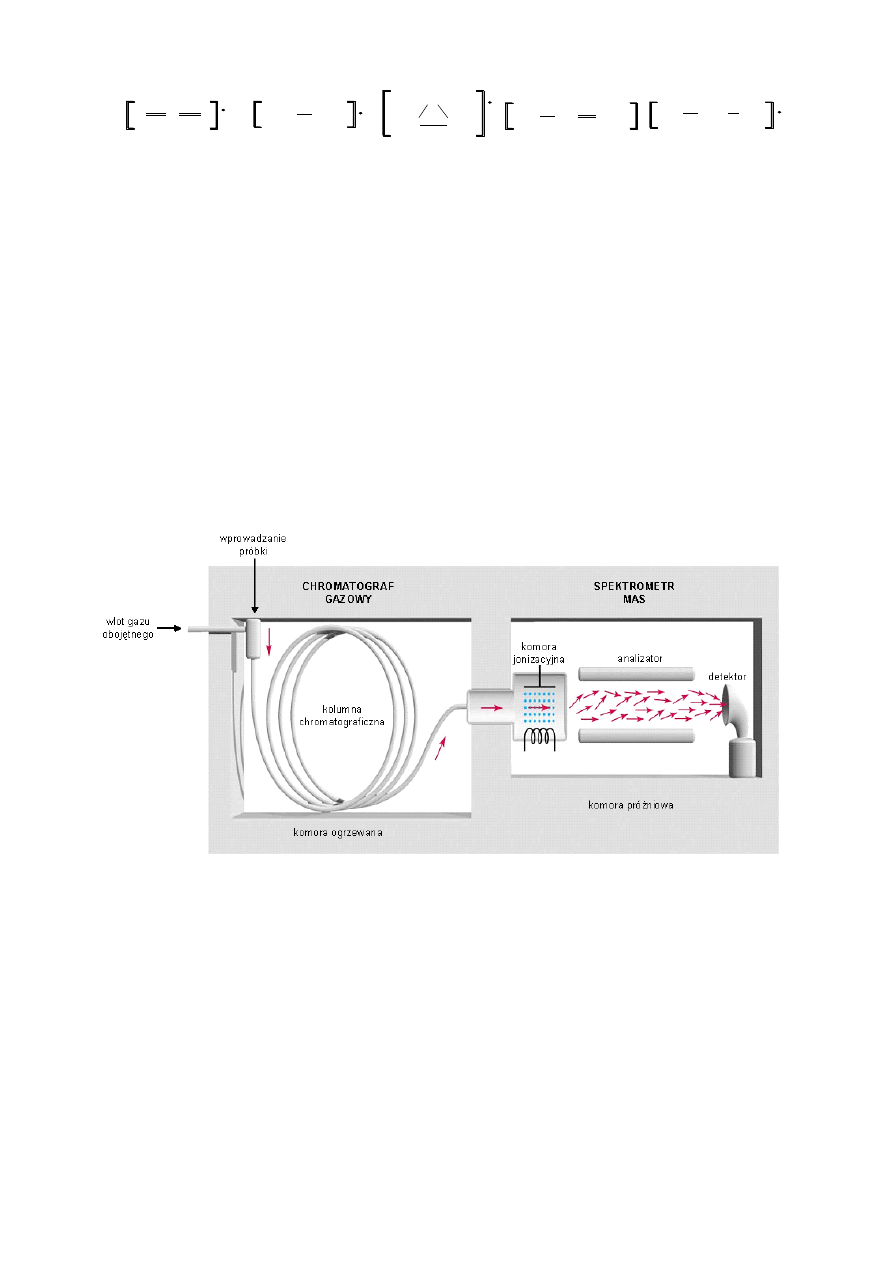
głównie technikami rozdziału, obserwuje się w tej dziedzinie dynamiczny rozwój. Doskonałym
narzędziem do tego celu jest chromatograf gazowy sprzężony ze spektrometrem mas (GC-MS ang.
Gas Chromatography – Mass Spectrometry). W tej technice rozdział mieszaniny następuje w
najczęściej kapilarnej kolumnie chromatografu gazowego, a każda frakcja opuszczająca
kolumnę jest
kierowana do komory jonizacyjnej spektrometru (Rys. 4).
Rysunek 4. Schemat chromatografu gazowego sprzężonego ze spektrometrem mas.
Takie rozwiązanie pozwala na szybką identyfikację związków obecnych w próbce. Oznaczenia
ilościowe poszczególnych składników są możliwe dzięki bezpośredniej zależności pomiędzy
stężeniem składnika analitu a odpowiedzią detektora spektrometru.
W analogiczny sposób działają również urządzenia typu LC-MS (ang. Liquid Chromatograpy –
Mass Spectrometry) chociaż stosowane rozwiązania techniczne są nieco inne. Różnice sprowadzają się
głównie do odpowiedniego systemu wprowadzania próbki do komory jonizacyjnej. W obu opisanych
technikach istnieje możliwość szybkiej identyfikacji związków wchodzących w skład mieszaniny
przez automatyczne porównywanie widm masowych poszczególnych składników próbki z bazami
danych zawierającymi widma substancji wzorcowych.
6
5. ROZPOZNAWANIE SKŁADU PIERWIASTKOWEGO NA PODSTAWIE WIDM MS
Jak wspomniano w rozdziale 1, ze względu na niejednorodność izotopową pierwiastków
wchodzących w skład związku (Tab. 1), w widmie są również obecne piki o m/z większej o jedną,
dwie lub nawet kilka jednostek od piku molekularnego, czyli tak zwane piki izotopowe. W przypadku
związków budowanych wyłącznie z C, H, O i N, mają one niewielką intensywność ze względu na
naturalny, niewielki udział cięższych izotopów tych pierwiastków (spójrz np. na
w oraz informację o możliwości
szacowania liczby atomów węgla w cząsteczce w
zawartą rozdz. 1). Piki izotopowe są jednak w wielu przypadkach
doskonałym narzędziem pozwalającym na ustalenie obecności atomów siarki, chloru i bromu w
badanych związkach. Pierwiastki te można rozpoznać na podstawie stosunku intensywności M
+
i
M
+
+2.
Tabela 1. Zawartość izotopów w naturalnie występujących pierwiastkach.
Pierwiastek
Rozpowszechnienie izotopów [%]
izotop podstawowy
(o masie m)
izotop o masie
m+1
izotop o masie
m+2
H
1
H – 99,984
2
H – 0,0016
C
12
C – 98,931
13
C – 1,069
N
14
N – 99,620
15
N – 0,380
O
16
O – 99,761
17
O – 0,039
18
O – 0,200
S
32
S – 95,018
33
S – 0,750
34
S – 4,215
Cl
35
Cl – 75,53±0,02
37
Cl – 24,47±0,02
Br
79
Br – 50,520
81
Br – 49,480
I
127
I – 100,000
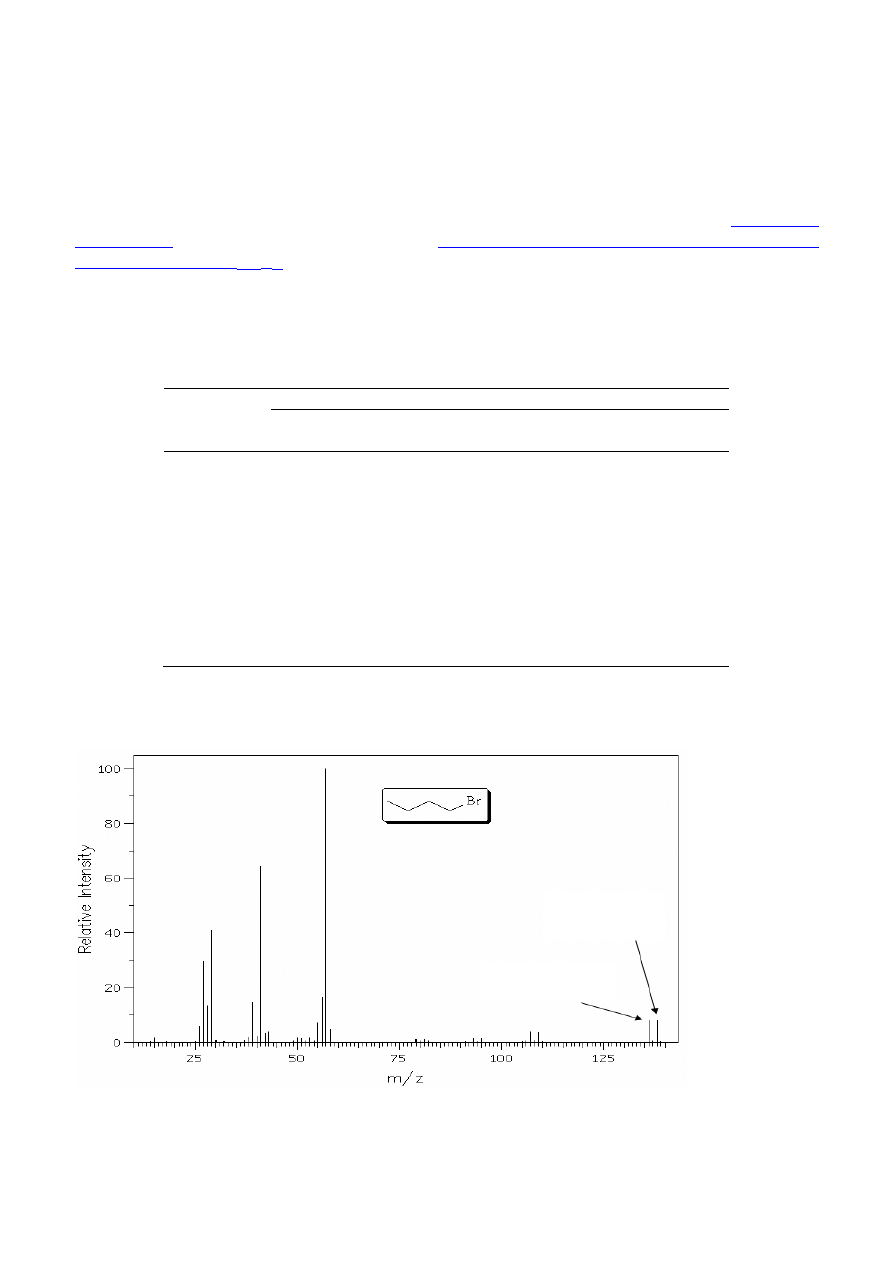
Obecność jednego atomu bromu w cząsteczce (rys. 5) powoduje, że na widmie obserwowany
jest pik molekularny M
+
pochodzący od związku z izotopem
79
Br oraz pik M
+
+2 o podobnej
intensywności pochodzący od związku z izotopem
81
Br (I
M
: I
M+2
≈ 1 : 1).
Rysunek 5. Widmo MS 1-bromobutanu.
W przypadku obecności jednego atomu chloru w cząsteczce (rys. 6) obserwowana jest
podobna zależność pomiędzy pikami M
+
(z izotopem
35
Cl) oraz M
+
+2 (z izotopem
37
Cl), jednak ze
M
+
= 136
(pik molekularny)
M
+
+2 = 138
(pik izotopowy)
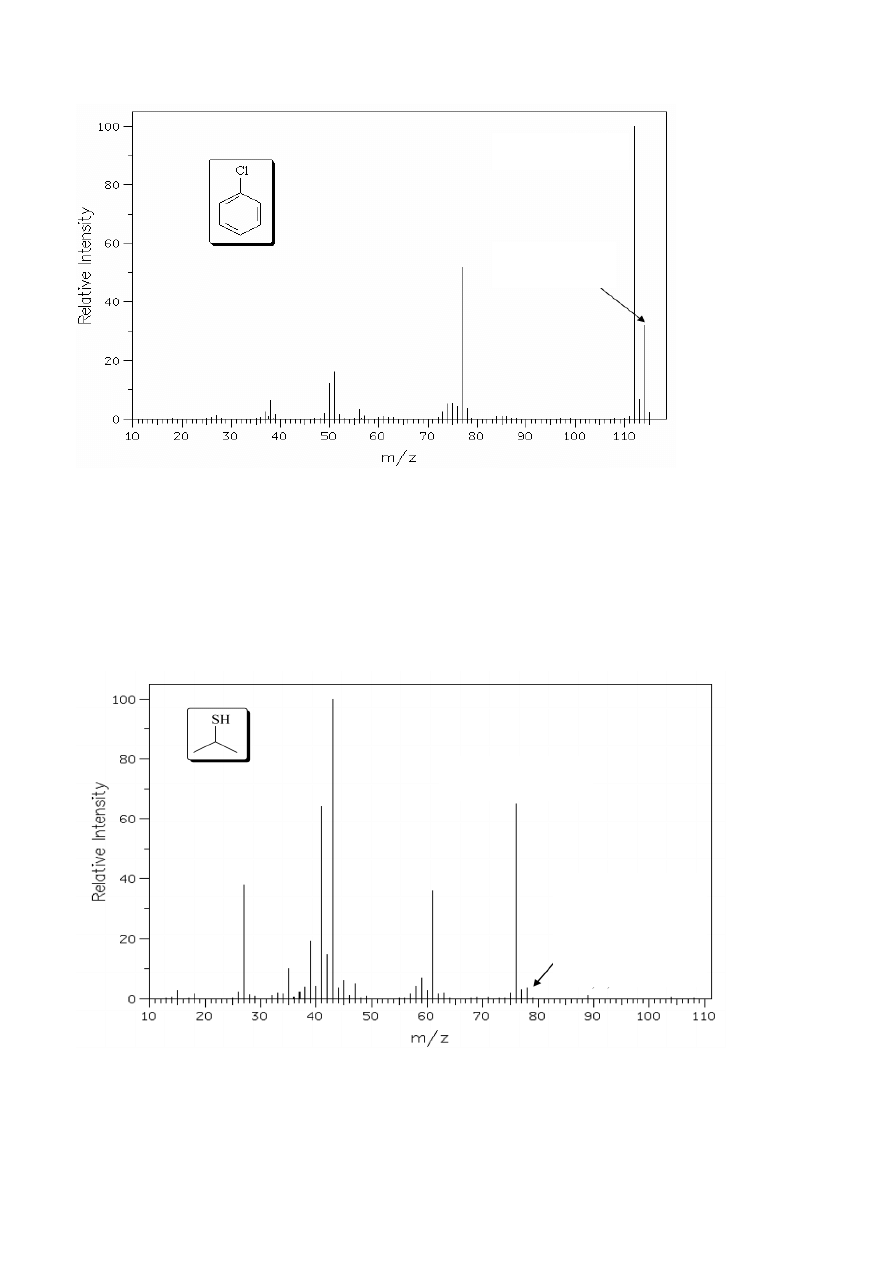
7
względu na inny stosunek izotopów wchodzących w skład naturalnie występującego pierwiastka
stosunek intensywności pików I
M
: I
M+2
≈ 3 : 1.
Rysunek 6. Widmo MS chlorobenzenu.
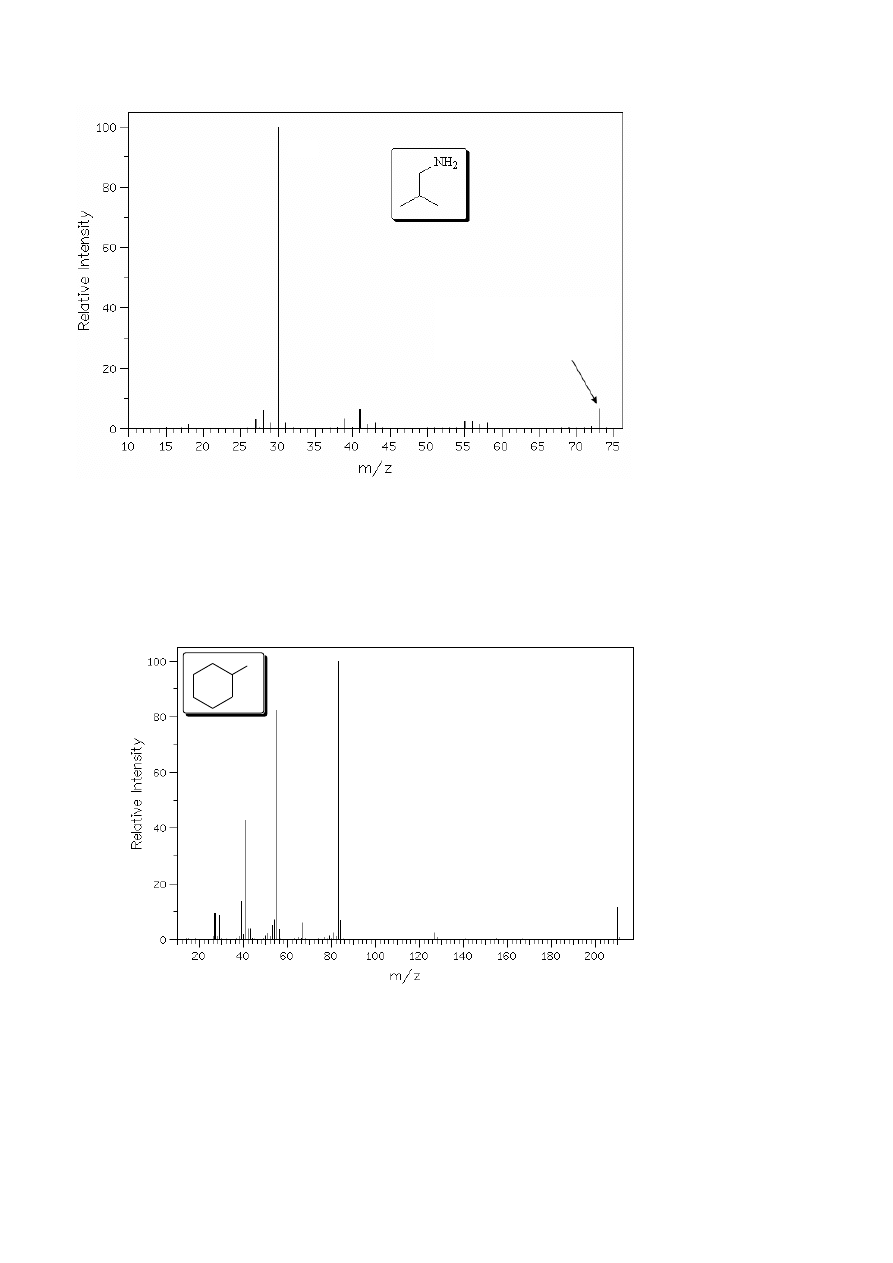
O obecności jednego atomu siarki w cząsteczce świadczy występowanie piku M
+
+2
pochodzącego od związku zawierającego izotop
34
S (rys. 7.). Sygnał ten ma niewielką abundancję (I
M
: I
M+2
≈ 24 : 1) jednak jest wyraźnie intensywniejszy od piku M
+
+1 co odróżnia go od obserwowanych
niekiedy bardzo słabych sygnałów M
+
+2 pochodzących od izotopów C, H, N i/lub O.
Jeśli cząsteczka zawiera więcej niż jeden spośród atomów Br, Cl lub S układ i wzajemne
proporcje piku molekularnego i pików izotopowych M
+
+2, M
+
+4 itd. jest dużo bardziej złożony i jego
analiza wykracza poza ramy tego opracowania.
Rysunek 7. Widmo MS propano-2-tiolu.
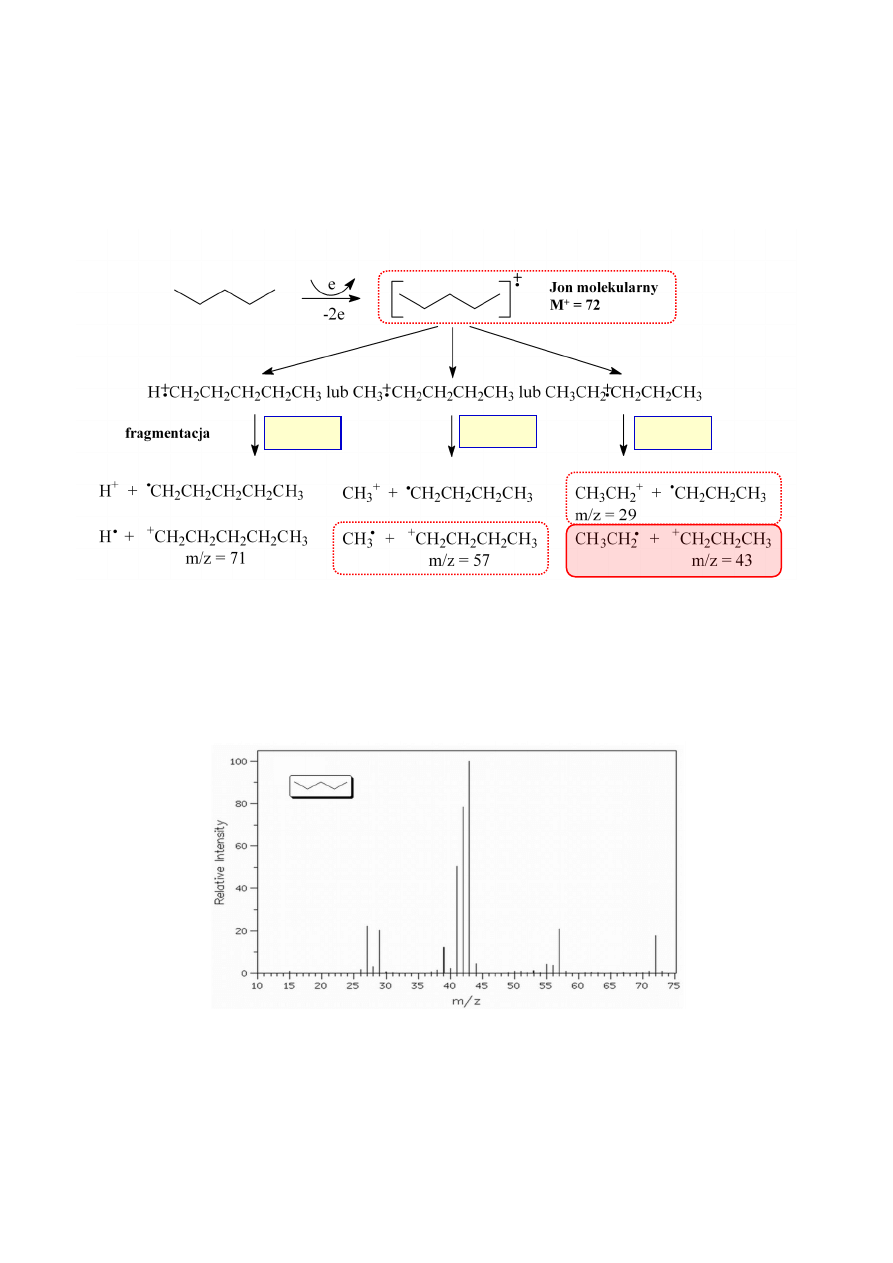
Widmo masowe związku chemicznego pozwala również na przewidywanie obecności atomów
azotu w cząsteczce. Zgodnie z tzw. regułą azotu nieparzysta masa jonu molekularnego M
+
dowodzi
obecności nieparzystej liczby atomów azotu w cząsteczce (rys. 8a). Jest to naturalną konsekwencją
występowania azotu na nieparzystym stopniu utlenienia w związkach organicznych, czyli wiązania się
z nieparzystą liczbą atomów węgla i/lub wodoru. Jeśli jednak pik molekularny występuje przy
M
+
+2 = 78
(pik izotopowy)
intensywniejszy niż
dla związków
zawierających tylko
C, H, O i/lub N
M
+
= 76
(pik molekularny)
M
+
= 112
(pik molekularny)
M
+
+2 = 114
(pik izotopowy)
8
parzystych wartościach m/z, nie można nic powiedzieć na temat obecności azotu w cząsteczce:
związek może nie zawierać azotu lub zawierać parzystą liczbę jego atomów.
Rysunek 8a. Widmo MS izobutyloaminy.
Relatywnie łatwo jest rozpoznać też obecność atomu jodu w cząsteczce analizowanego
związku. Ze względu na dużą masę tego atomu (127 u) i jego tendencję do odrywania się w postaci
rodnika, w widmie związków jodoorganicznych widoczny jest intensywny zazwyczaj pik przy m/z
mniejszym o 127 jednostek (tzw. M
+
-127) od położenia piku molekularnego i jednocześnie występuje
niewielka liczba innych pików między M
+
i M
+
-127 (rys. 8b). Dodatkowym potwierdzeniem może być
obecność piku przy m/z = 127 od kationu I
+
, jednak jego intensywność jest zwykle niewielka.
Rysunek 8b. Widmo MS
jodocykloheksanu.
M
+
= 73
(pik molekularny
o nieparzystej masie)
30
M
+
= 210
m/z = 83
(M
+
- 127)
m/z = 127
I

9
6. PODSTAWOWE SCHEMATY FRAGMENTACJI CZĄSTECZEK
6.1. Wprowadzenie
Podczas jonizacji dochodzi do wybicia elektronu z powłoki walencyjnej cząsteczki, w wyniku
którego powstają kationorodniki (rozdz. 1). Najłatwiej wybijane są elektrony z orbitali niewiążących
(wolnych par elektronowych), w dalszej kolejności elektrony z orbitali , a najtrudniej elektrony z
wiążących orbitali . Prawdopodobieństwo dalszego rozpadu (fragmentacji) takiego kationorodnika
jest związane z jego trwałością i trwałością powstających podczas tego rozpadu mniejszych jonów i
rodników. Fragmentacja cząsteczki jest przyczyną pojawiania się w widmach masowych szeregu
pików o zróżnicowanej intensywności, uwarunkowanej trwałością tworzących się drobin oraz
statystycznym prawdopodobieństwem ich powstawania.
Macierzysty kationorodnik o nieparzystej liczbie elektronów (M
•+
) może ulegać rozpadowi na
dwóch podstawowych drogach fragmentacji:
a) utworzyć rodnik (A
•
) oraz kation parzystoelektronowy (B
+
):
M
•+
→ A
•
+ B
+
b) odszczepić obojętną cząsteczkę (C) związku nieorganicznego (H
2
O, CO, CO
2
, HX) lub
organicznego (np. alken) i utworzyć kolejny nieparzystoelektronowy kationorodnik o mniejszej
masie (D
•+
):
M
•+
→ C + D
•+
Powstające kationorodniki (D
•+
) mogą ulegać dalszym rozpadom, zgodnie z podanymi wyżej
schematami, natomiast kationy parzystoelektronowe (B
+
) mogą przegrupowywać się do trwalszych
kationów lub odszczepiać obojętne cząsteczki i tworzyć kolejne kationy parzystoelektronowe o
mniejszych masach. W praktyce nie obserwuje się rozpadu kationów parzystoelektronowych na
kationorodniki !
Rodniki i obojętne cząsteczki związków nie są widoczne w widmie masowym, gdyż nie są
obdarzone ładunkiem, natomiast abundancja pików pochodzących od powstających kationów i
kationorodników jest bezpośrednio związana ze znanym z chemii organicznej szeregiem trwałości
kationów – w szczególności karbokationów. Największy udział we fragmentacji cząsteczki mają te
drogi rozpadu, w wyniku których będą powstawały względnie trwałe kationy (np. trzeciorzędowe,
allilowe, benzylowe, stabilizowane rezonansowo kationy oksoniowe i amoniowe). Pewne znaczenie
dla orientacji fragmentacji ma również trwałość powstających jednocześnie rodników.
6.2. Ogólne reguły fragmentacji (na przykładzie alkanów)
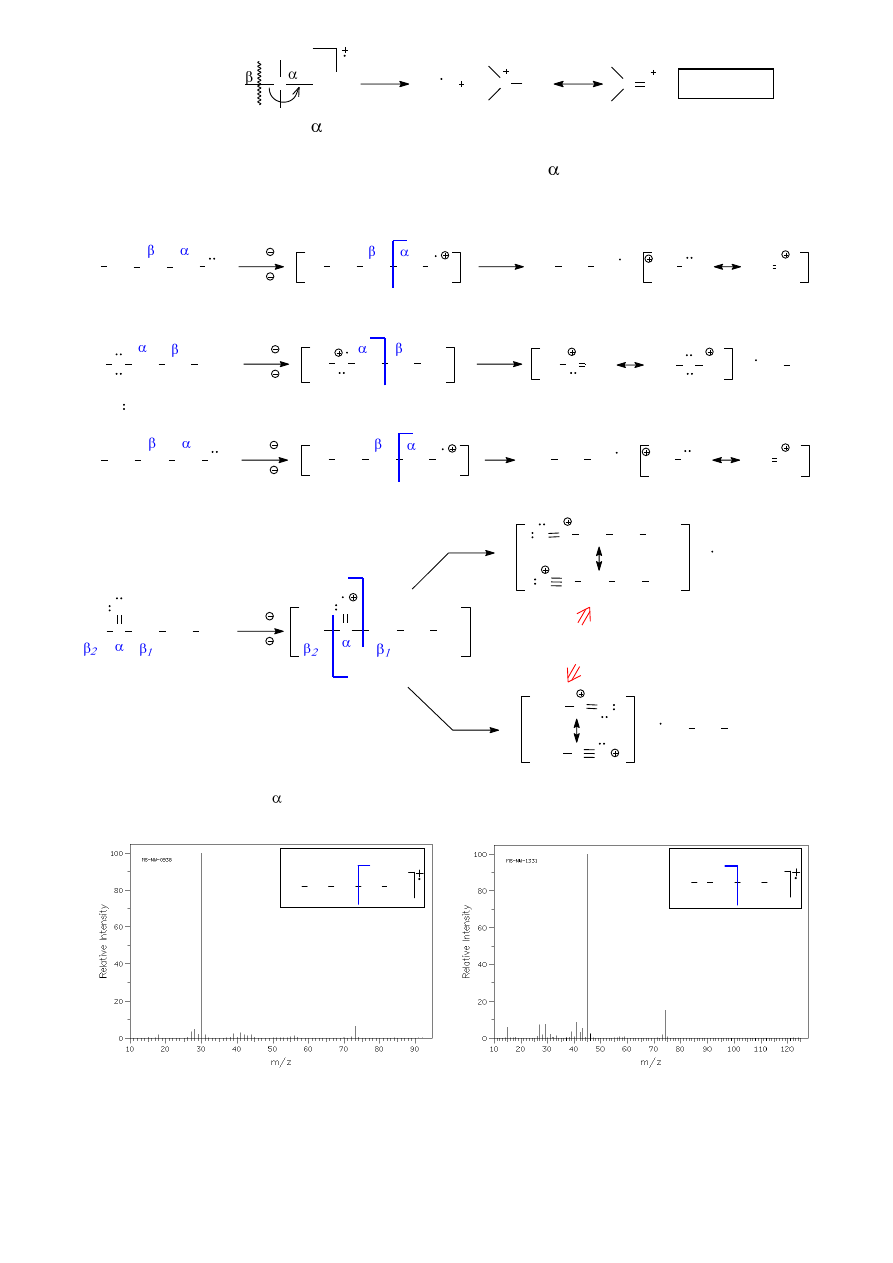
Spróbujmy przewidzieć możliwe drogi fragmentacji n-pentanu. Podczas bombardowania
elektronami może zostać wybity jeden z elektronów wiązań C-H lub C-C i powstaje kationorodnik
M
•+
(Schemat 1). Jon macierzysty M
•+
może teoretycznie:
a) odszczepić rodnik lub kation wodorowy tworząc odpowiednio karbokation (pierwszo- lub
drugorzędowy) lub rodnik alkilowy (przykładem może być droga A). Odszczepianie rodników lub
kationów wodorowych jest jednak możliwe tylko wtedy, gdy powstające kationy/rodniki organiczne
cechują się szczególnie wysoką trwałością, a ani karbokationy (rodniki) pierwszorzędowe ani
drugorzędowe nie są drobinami trwałymi, stąd prawdopodobieństwo takiej fragmentacji jest bardzo
małe.
b) rozpaść się z rozerwaniem wiązania C1-C2 (droga B), tworząc rodnik metylowy i kationu
n-butylowy (m/z = 57), względnie kation metylowy (m/z = 15) i rodnik n-butylowy. Z tych dwóch
możliwości pierwsza jest bardziej prawdopodobna, gdyż o orientacji rozszczepienia wiązania decyduje
w pierwszym rzędzie trwałość karbokationu (kation n-butylowy jest trwalszy niż metylowy). Co
więcej, droga B wydaje się bardziej prawdopodobna niż droga A, ze względu na wyższą trwałość
powstających drobin.
10
c) ulec fragmentacji z rozerwaniem wiązania C2-C3 (droga C). Efektem takiej przemiany jest
utworzenia kationu n-propylowego (m/z = 43) i rodnika etylowego, względnie rodnika n-propylowego
i kationu etylowego (m/z = 29). Wszystkie kationy i rodniki w obu wariantach są pierwszorzędowe
(nie powstają nietrwałe kationy i rodniki wodorowe, jak na drodze A, ani nietrwałe rodniki i kationy
metylowe, jak na drodze B), wydaje się więc można przewidywać, że fragmentacja n-pentanu na
drodze C będzie dominującą. Co więcej, można zauważyć, że podczas powstawania kation n-
propylowy może przegrupować się do trwalszego, drugorzędowego kationu izopropylowego i w
związku z tym rozpad prowadzący do powstania piku przy m/z = 43 będzie najbardziej
prawdopodobną drogą fragmentacji n-pentanu.
Schemat 1. Możliwe drogi fragmentacji n-pentanu.
Porównanie z widmem masowym rozważanego związku (rys. 9) wskazuje na dobrą zgodność
przewidywań opartych na trwałości kationów z rezultatem eksperymentu: pik podstawowy leży przy
wartości m/z = 43 (kation propylowy lub izopropylowy), a inne względnie intensywne piki są obecne
przy m/z = 72 (pik molekularny), m/z = 57 (kation butylowy), m/z = 29 (kation etylowy).
Rysunek 9. Widmo MS n-pentanu.
Obecność intensywnych pasm przy m/z = 42 można wytłumaczyć powstawaniem
kationorodnika propenylowego ( [CH
2
=CH
2
-CH
3
]
.
+
), powstającego z kationorodnika pentylowego
w wyniku eliminacji obojętnej cząsteczki etenu. Kationorodnik ten, wskutek odszczepienia rodnika
wodorowego, daje trwały kation allilowy (CH
2
=CH-CH
2
+
) o m/z = 41.
Dominującą drogę fragmentacji n-pentanu można zobrazować w sposób przedstawiony na
schemacie 2. W powszechnie przyjętej notacji rozpad kationorodnika może być ukazany przy
pomocy dwuodcinkowej łamanej: dłuższy odcinek łamanej przecina rozrywające się wiązanie, a
droga A
droga B
droga C

11
krótszy, prostopadły doń, wskazuje ten fragment, który po rozpadzie przyjmie ładunek dodatni. Nad
krótszym odcinkiem łamanej często zapisuje się masę powstającego kationu (schemat 2).
CH
3
CH
2
CH
2
CH
2
CH
3
CH
3
CH
2
CH
2
CH
2
CH
3
+ e
- 2 e
.
+
43
CH
3
CH
2
CH
2
CH
2
CH
3
+
m/z = 43
Schemat 2. Uproszczony zapis dominującej fragmentacji n-pentanu.
Widma masowe związków o prostym łańcuchu alkilowym charakteryzują się występowaniem
szeregu pików różniących się od siebie o m/z = 14, co wynika z nakładania się dwóch ścieżek rozpadu.
Jedna z nich rozpoczyna się od oderwania rodnika metylowego a druga – etylowego. Powstające
kationy ulegają dalszej fragmentacji zgodnie ze schematem:
R
CH
2
CH
2
CH
2
R
CH
2
CH
2
CH
2
Odrywający się eten posiada masę równą 28 u, jednak nakładanie się obu ścieżek rozpadów
prowadzi do pojawienia się na widmie szeregu pików odległych właśnie o m/z = 14.
Rysunek 10. Widmo MS n-dekanu.
W przypadku alkanów rozgałęzionych fragmentacja łańcucha rozpoczyna się od rozerwania
wiązania przy najwyżej rzędowym atomie węgla, dzięki czemu powstają możliwie najtrwalsze
karbokationy. Przykładowo, główną drogą fragmentacji 3-metylopentanu jest odszczepienie rodnika
etylowego (o masie 29 u) od trzeciorzędowego atomu węgla, w wyniku czego powstaje względnie
trwały karbokation drugorzędowy o m/z = 57, odpowiadający za pik podstawowy na widmie MS tego
węglowodoru. Oderwanie grupy metylowej od tego samego atomu węgla dałoby karbokation również
drugorzędowy, ale jednocześnie powstający rodnik metylowy byłby mniej trwały.
CH
3
CH
2
CH CH
2
CH
3
CH
3
CH
3
CH
2
CH CH
2
CH
3
CH
3
+ e
- 2 e
.
+
57
CH
3
CH
2
CH
CH
3
CH
2
CH
3
+
m/z = 57
(pik podstawowy: I = 100%)
M
+
: m/z = 88
Rozszczepienie (alkohole, tiole, etery, sulfidy, aminy, związki karbonylowe)
Homolityczne rozszczepienie wiązania pomiędzy atomami i względem heteroatomu
posiadającego wolne pary elektronowe (N, O, S) jest fragmentacją bardzo często obserwowaną w
przypadku związków posiadających podstawniki mogące stabilizować ładunek dodatni - alkoholi, tioli,
eterów, sulfidów, amin a także związków posiadających grupy karbonylowe (aldehydów, ketonów,
kwasów karboksylowych i ich pochodnych). Odszczepiana cząsteczka może być rodnikiem alkilowym
lub wodorowym, a powstający, trwały dzięki stabilizacji rezonansowej, kation jest obserwowany w
widmie masowym.
12
R
C
X
R
X
C
X
C
Przykładem rozpadów
mogą być fragmentacje amin, alkoholi, eterów i związków
karbonylowych przedstawione na schematach 3a-d, zilustrowane widmami na rysunkach 11a-d. O ile
w przypadku amin, eterów i związków karbonylowych rozpady są dominującą drogą fragmentacji,
to alkohole po jonizacji szybko ulegają również innym transformacjom (np. dehydratacji, por. rozdz.
6.5), co skutkuje bardzo niską intensywnością pików molekularnych (rys. 11c).
CH
3
CH
2
CH
2
CH
2
NH
2
CH
3
CH
2
CH
2
CH
2
NH
2
+ e
- 2 e
30
CH
3
CH
2
CH
2
CH
2
NH
2
+
m/z = 30
CH
3
O CH
2
CH
2
CH
3
CH
3
O CH
2
CH
2
CH
3
+ e
- 2 e
45
CH
3
O CH
2
CH
2
CH
3
+
m/z = 45
CH
2
NH
2
a)
m/z = 73
CH
3
O CH
2
b)
m/z = 74
CH
3
CH
2
CH
2
CH
2
OH
CH
3
CH
2
CH
2
CH
2
OH
+ e
- 2 e
31
CH
3
CH
2
CH
2
CH
2
OH
+
m/z = 31
CH
3
C CH
2
CH
2
CH
3
O
CH
3
C
CH
2
CH
2
CH
3
O
+ e
- 2 e
43
m/z = 43
CH
2
OH
c)
m/z = 74
d)
m/z = 86
71
C CH
2
CH
2
CH
3
O
C CH
2
CH
2
CH
3
O
C
CH
3
O
C
CH
3
O
CH
3
+
CH
2
CH
2
CH
3
+
m/z = 71
trwałe kationy
acyliowe
rodnik
metylowy
trwalszy rodnik
pierwszorzędowy
dominująca
droga fragmentacji
A
B
Schemat 3. Równania rozpadów : a) n-butyloaminy; b) 1-metoksypropanu; c) butan-1-olu; d) pentan-2-onu
a)
b)
X = O, N, S
CH
3
CH
2
CH
2
CH
2
NH
2
30
73, M
+
45
30
74, M
+
CH
3
O CH
2
CH
2
CH
3
45
13
c)
d)
Rysunek 11. Widma MS: a) n-butyloaminy; b) 1-metoksypropanu; c) butan-1-olu; d) pentan-2-onu
Efektem rozpadów są m. in. charakterystyczne dla alkoholi pierwszorzędowych piki przy
m/z = 31 (CH
2
=OH
+
, schemat 3c i widmo 11c) oraz typowe dla związków karbonylowych
stabilizowane rezonansowo kationy acyliowe (schemat 3d i widmo 11d). Ostatni przykład dobrze
ilustruje wpływ trwałości powstających drobin na orientację fragmentacji – jon molekularny pentan-2-
onu może ulegać rozpadowi na dwóch drogach (A i B), tworząc kationy acyliowe o porównywalnej
trwałości. Jak można przekonać się analizując widmo 11d, dominuje droga B, w wyniku której
powstaje kation acetylowy (m/z = 43) oraz pierwszorzędowy rodnik propylowy, trwalszy niż rodnik
metylowy, który towarzyszy rozpadowi na drodze A.
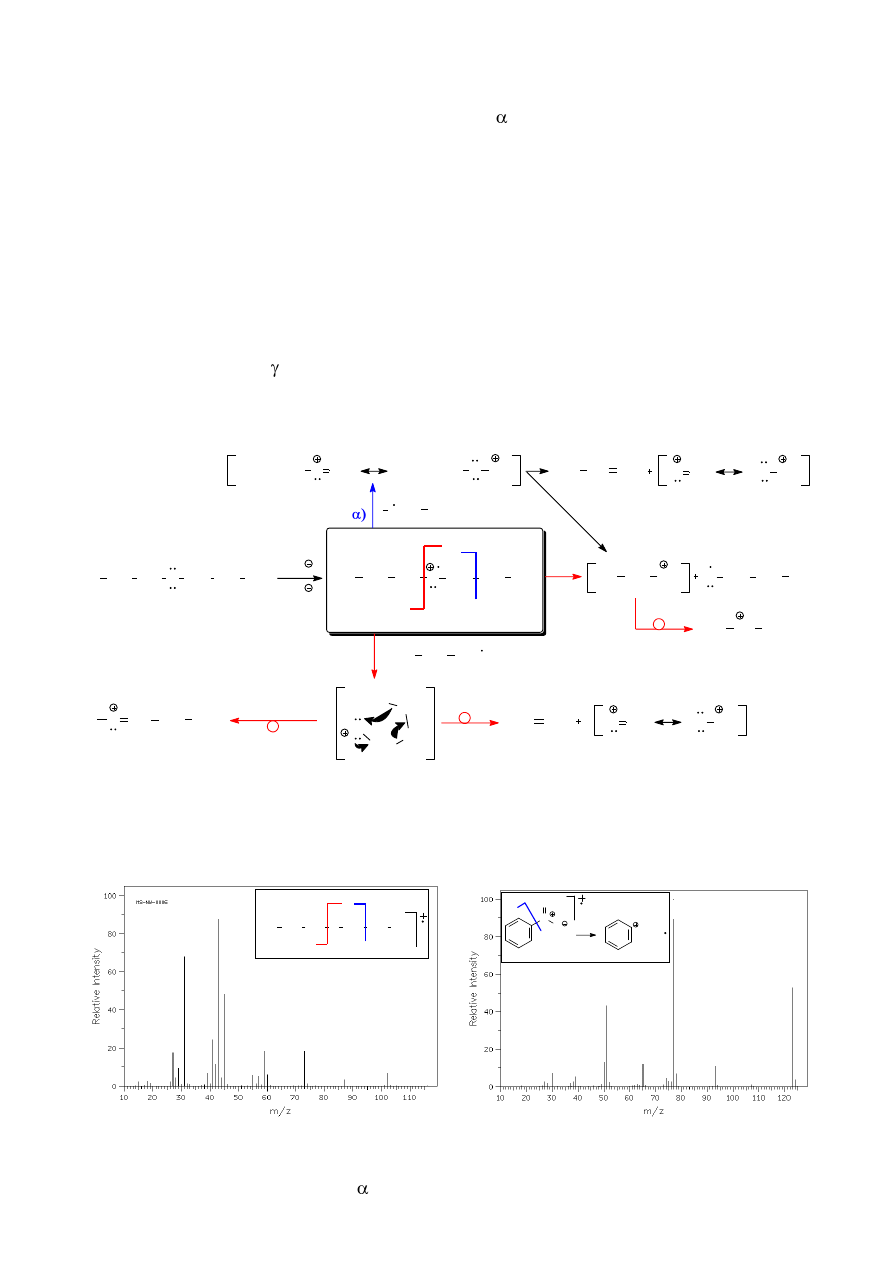
Należy pamiętać, że kationy acyliowe powstają nie tylko w wyniku fragmentacji ketonów, ale
również aldehydów, kwasów karboksylowych, estrów i innych pochodnych kwasów. Przykładowo,
podstawowe piki w widmach MS zarówno kwasu benzoesowego, jak i benzoesanu etylu są położone
przy m/z = 105 i pochodzą od kationu benzoilowego. Można przekonać się jednocześnie, że w
przypadku omawianego estru inny możliwy rozpad
, polegający na odszczepieniu rodnika
metylowego i utworzeniu kationu oksoniowego o m/z = 135 ma minimalny udział we fragmentacji
związku. Przyczyną jest niska trwałość tworzących się drobin: nietrwałego rodnika metylowego
i destabilizowanego przez grupę karbonylową kationu oksoniowego.
a)
b)
Rysunek 12. Widma MS i dominujące rozpady: a) kwasu benzoesowego; b) benzoesanu etylu.
Kationy acyliowe mogą ulegać dalszym transformacjom – jedną z typowych jest odszczepienie
obojętnej cząsteczki tlenku węgla. W wyniku takiej przemiany, zarówno z kwasu benzoesowego, jak
i jego estru tworzą się, poprzez kation benzoilowy, kationy fenyliowe (C
6
H
5
+
) o m/z = 77.
Rozszczepienie wiązań węgiel-heteroatom (aminy, etery, sulfidy, estry, amidy, związki
nitrowe, halogenopochodne związków organicznych)
Fragmentacja związków z wiązaniem C-O, C-S, C-N i C-X (X = F, Cl, Br, I) może przebiegać
również z rozpadem węgiel – heteroatom, pod warunkiem, że powstające kationy są względnie trwałe,
lub w wyniku przegrupowania mogą przekształcić się w formę stabilną. Tym często pozornie
CH
3
CH
2
CH
2
CH
2
OH
31
CH
3
C
CH
2
CH
2
CH
3
O
71
43
74, M
+
86, M
+
31
56
43
41
43
71
122, M
+
105
C
O
OH
105
C
O
+ OH
m/z = 105
150, M
+
105
C
O
OCH
2
CH
3
105
C
O
+ CH
3
CH
2
O
m/z = 105
77
77
122
14
trywialnym rozpadom często towarzyszą bardzo złożone przegrupowania, szczególnie w przypadku
tlenowych i azotowych związków alifatycznych. I tak, fragmentacja jonu molekularnego eteru
dipropylowego może przebiegać kilkoma drogami:
A) Zgodnie z opisanym w rozdz. 6.3 rozpadem . Powstały kation oksoniowy o m/z = 73
może odszczepiać następnie obojętną cząsteczkę propenu dając jon CH
2
=OH
+
(m/z = 31)
lub odszczepiać obojętną cząsteczkę formaldehydu tworząc kation propylowy, który
wskutek przegrupowania zyskuje stabilną postać kationu izopropylowego (m/z = 43).
Przedstawiając schemat fragmentacji fakt przegrupowania sygnalizujemy rysując kółko
przy strzałce pokazującej taką przemianę.
B) Rozszczepiając wiązanie węgiel-tlen w taki sposób, że niesparowany elektron pozostaje na
atomie tlenu i tworzy się jednocześnie kation propylowy (m/z = 43), który przekształca się
w trwalszy kation izopropylowy, jak opisano w punkcie A.
C) Rozszczepiając wiązanie węgiel-tlen w taki sposób, że niesparowany elektron pozostaje na
atomie węgla. Podczas takiej fragmentacji dochodzi do przeniesienia anionu wodorkowego
do atomu tlenu od sąsiedniego atomu węgla, lub przeniesienia anionu wodorkowego od
atomu węgla połączonego z eliminacją cząsteczki etenu. Siłą napędową tych
przegrupowań jest zapewnienie trwałości powstającego kationu (kation z atomem tlenu
posiadającym sekstet elektronów walencyjnych byłby skrajnie nietrwały).
CH
2
O CH
2
CH
2
CH
3
CH
2
CH
3
CH
2
O CH
2
CH
2
CH
3
CH
2
CH
3
+ e
- 2 e
73
CH
3
CH
2
CH
2
O CH
2
m/z = 73
CH
3
CH
2
CH
2
O CH
2
m/z = 102
(rozpad
CH
2
CH
3
O
CH CH
2
CH
3
H
B
C
A
m/z = 59
HO CH
2
CH
3
CH
CH
2
HO CH
2
m/z = 31
59
43
CH
2
CH
2
CH
3
O CH
2
CH
2
CH
3
CH
3
CH
CH
3
m/z = 43
- H
2
C=O
O
CH
2
CH
2
CH
2
H
CH
2
CH
2
- CH
3
B
C
A
HO CH
2
HO CH
2
m/z = 31
H
2
C
CH
2
przegrupowanie
[1,2] anionu
wodorkowego
SKRAJNIE NIETRWA£Y!
Schemat 4. Równania możliwych rozpadów eteru dipropylowego.
Potwierdzeniem poprawności przewidywanych dróg fragmentacji eteru dipropylowego jest
widmo MS tego związku (rys. 13 a), gdzie obecne są piki pochodzące od wszystkich postulowanych
kationów.
a)
b)
Rysunek 13. Widma MS: a) eteru dipropylowego; b) nitrobenzenu.
Rozpady wiązań węgiel-tlen i węgiel-azot w związkach alifatycznych często mają marginalne
znaczenie w porównaniu z rozpadami . Dowodem na to może być widmo MS izobutyloaminy (rys.
CH
2
O CH
2
CH
2
CH
3
CH
2
CH
3
73
59
43
102, M
+
73
59
43
31
N
O
O
77
+ NO
2
m/z = 77
77
123, M
+
15
8a), w którym pik pochodzący od rozpadu (m/z = 30) jest pikiem podstawowym, a intensywność
piku pochodzącego od kationu powstałego w wyniku rozszczepiania wiązania azot-węgiel nie
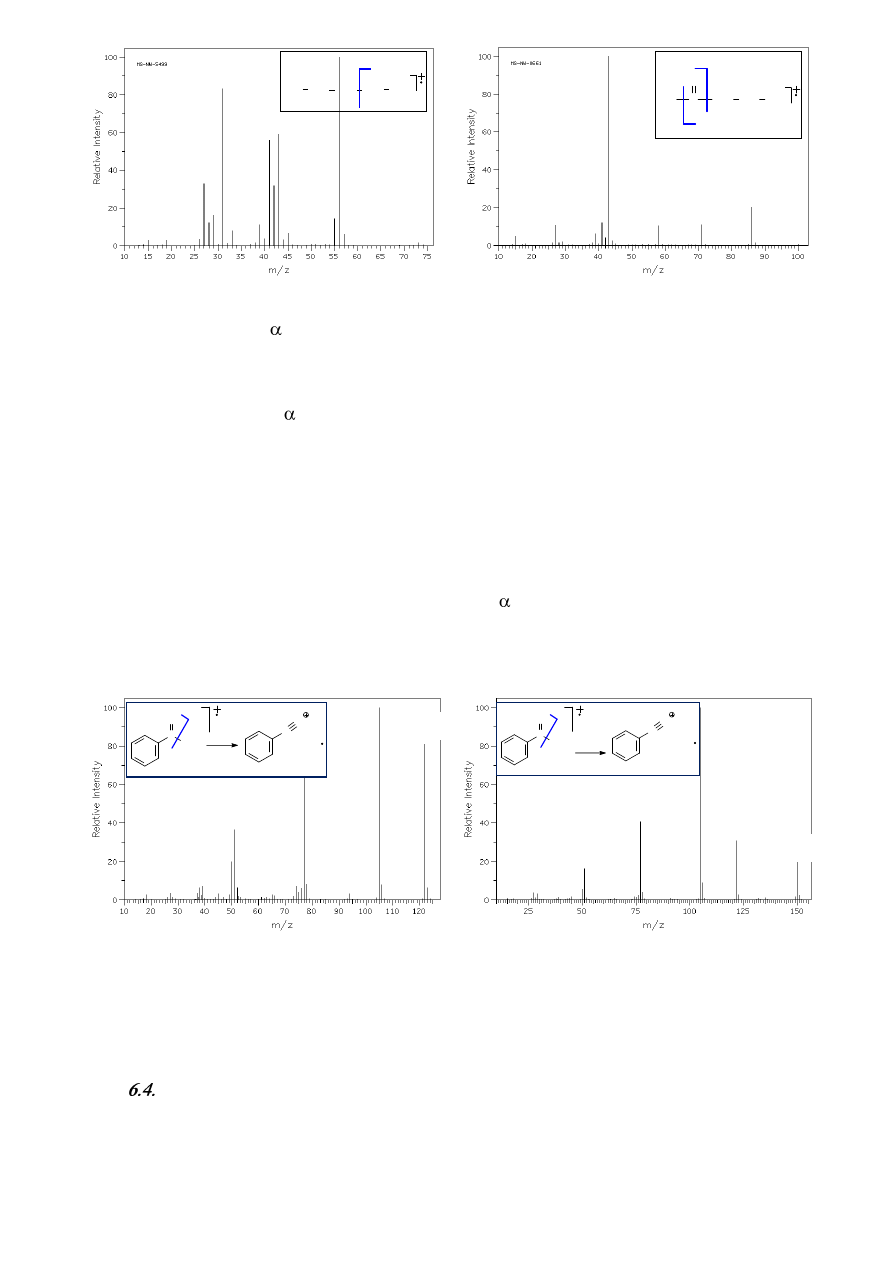
przekracza 5%. Większe znaczenie tego typu rozpady mają w przypadku fragmentacji pochodnych
związków aromatycznych, gdzie rozpad jest praktycznie niemożliwy – przykładem może być widmo
nitrobenzenu, gdzie pik podstawowy leżący przy m/z = 77 (kation fenyliowy) pochodzi właśnie od
rozpadu cząsteczki zachodzącego wskutek rozszczepienia wiązania węgiel-azot (rys. 13b).
6.5. Rozszczepienie allilowe (alkeny)
Proces rozszczepienia allilowego jest obserwowany podczas rejestracji widm MS alkenów a
jego mechanizm jest analogiczny do przedstawionego w rozdz. 6.3 rozszczepienia
(Schemat 5a).
Tendencja do odszczepienia podstawników od allilowych (a nie winylowych!) atomów węgla
w jonach molekularnych alkenów jest konsekwencją możliwości stabilizacji rezonansowej
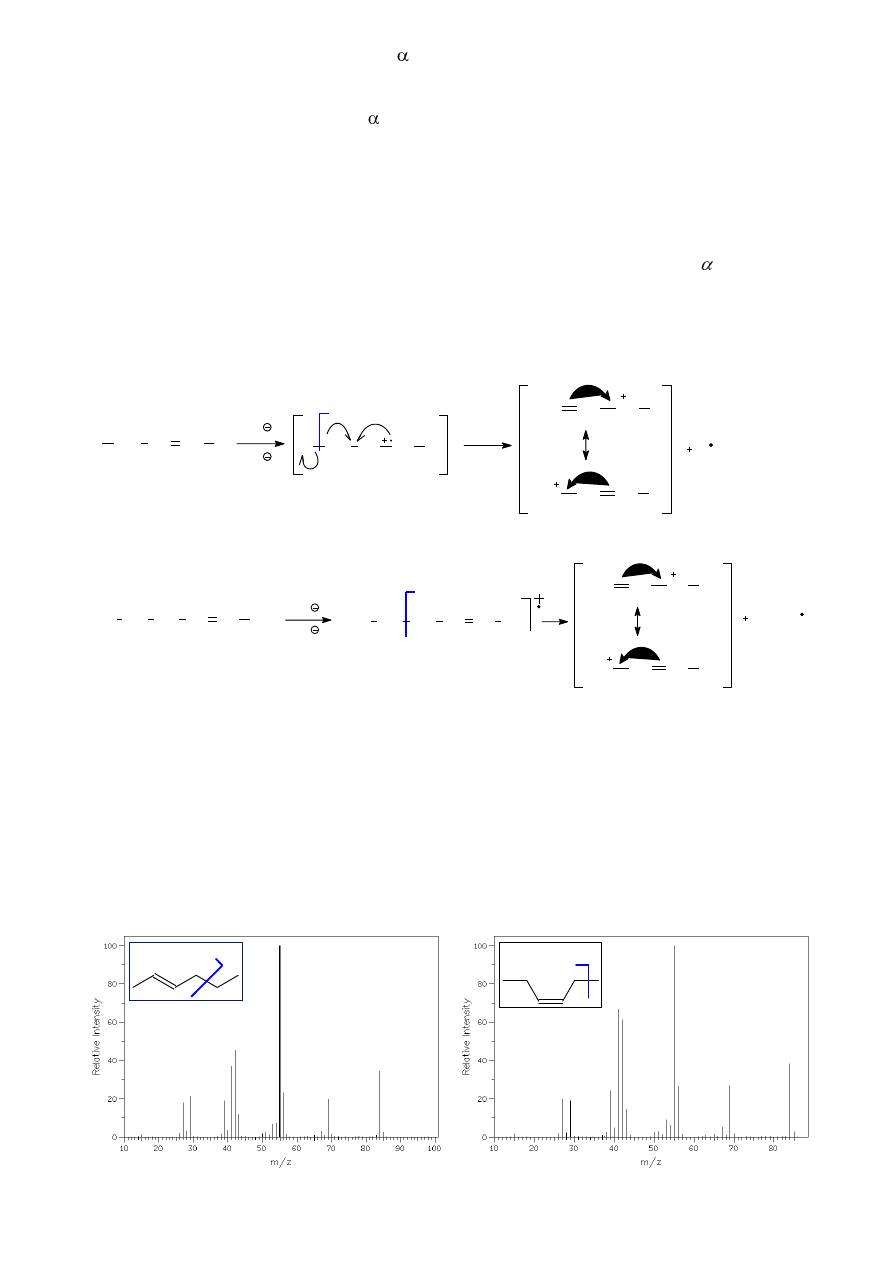
powstającego kationu allilowego. I tak, pikiem podstawowym w widmie MS heks-2-enu jest pik przy
m/z = 55, odpowiadający kationowi powstałemu w wyniku odszczepienia rodnika etylowego od
allilowego atomu węgla w jonie molekularnym heks-2-enu (Schemat 5b).
a)
R
CH
2
CH
CH
R'
R
CH
2
CH
CH
R'
CH
CH
H
2
C
R'
H
2
C
CH
CH
R'
R
+ e
- 2e
stabilizowany rezonansowo
kation allilowy
b)
CH
2
CH
2
CH
CH
CH
3
CH
3
CH
CH
H
2
C
CH
3
H
2
C
CH
CH
CH
3
CH
3
CH
2
+ e
- 2e
m/z = 55
CH
2
CH
2
CH CH CH
3
CH
3
55
Schemat 5. a) Ogólny schemat rozszczepienia allilowego i b) fragmentacja allilowa heks-2-enu.
Ten prosty schemat fragmentacji alkenów komplikuje się jednak, gdy porównamy widmo heks-
2-enu i heks-3-enu (rys. 14a i 14b). Układ i intensywności pików na obu widmach są zbliżone, a
przecież heks-3-en w wyniku rozszczepienia allilowego powinien dawać kation o m/z = 69! Okazuje
się, że względu na możliwość migracji wiązania wielokrotnego w alkenach jonizowanych techniką EI
jednoznaczne ustalenie położenia tego wiązania jest utrudnione (Schemat 6). Izomery położeniowe i
stereoizomeryczne alkeny mają zwykle niemal identyczne widma masowe.
Można w tym miejscu zaakcentować, że rozróżnienie stereoizomerów przy użyciu
spektrometrii mas jest w miażdżącej większości przypadków niemożliwe!
a)
b)
Rysunek 14. Widma izomerycznych: a) (E)-heks-2-enu oraz b) (Z)-heks-3-enu
55
123, M
+
55
55
M
+
, 84
M
+
, 84
69
69
69

16
CH
2
CH
CH CH
2
CH
3
CH
3
+ e
- 2e
CH
2
CH
CH CH
2
CH
3
CH
3
CH
2
CH
2
CH
CH CH
3
CH
3
CH
2
CH
2
CH
2
CH
CH
2
CH
3
55
69
41
Schemat 6. Izomeryzacja jonu molekularnego powstającego z heks-3-enu.
6.6. Reakcje eliminacji towarzyszące fragmentacji (alkohole, halogenki alkilu)
Dla niektórych związków główną drogą transformacji zjonizowanych cząsteczek mogą być
reakcje eliminacji obojętnych molekuł nieorganicznych lub organicznych. Typowym przykładem
takich związków są alkohole, dla których eliminacja cząsteczki wody z jonu molekularnego jest
głównym procesem, obok rozpadu , inicjującym fragmentację cząsteczek. Fragmentacja jest tak
szybka, że pik molekularny w widmach alkoholi o łańcuchach dłuższych niż trójwęglowe jest niemal
niewidoczny i pierwszym pojawiającym się sygnałem jest zwykle pik odpowiadający M
+
- 18 (masa
cząsteczki wody).
W przypadku alkoholi posiadających atomy wodoru w pozycji możliwe są dwie ścieżki
fragmentacji:
A) z utworzeniem nieparzystoelektronowego jonu fragmentacyjnego i cząsteczki wody,
B) z równoczesnym odszczepieniem cząsteczki wody i alkenu.
Alkohole nieposiadające wodorów mogą ulegać dehydratacji tylko na drodze A. Ponieważ
powstające po dehydratacji kationorodniki są identyczne jak w przypadku alkenów, to widma alkoholi
i alkenów o tym samym szkielecie są często bardzo podobne.
e
-
CH
2
CH
2
CH
2
CH
2
O
R
H
CH
CH
2
CH
2
CH
2
O
R
H
H
H
2
O
CH
2
CH
2
CH
CH
2
R
lub
H
2
O
R CH
CH
2
CH
2
CH
2
- 2e
-
CH
2
CH
2
CH
2
CH
2
O
R
H
dalsze rozpady
(typowe dla alkenów)
A
B
Schemat 7. Możliwe drogi dehydratacji alkoholi.
Omówiony schemat fragmentacji alkoholi można przeanalizować na przykładzie
3-metylobutan-1-olu (Schemat 8). Zjonizowana cząsteczka tego alkoholu może ulegać prostej
dehydratacji (droga A) dając kationorodnik o m/z = 70. Oderwanie rodnika metylowego od pozycji
allilowej daje kation parzystoelektronowy o m/z = 55. Dehydratacji jonu molekularnego może
towarzyszyć eliminacja etenu (droga B, zgodnie z mechanizmem przedstawionym na schemacie 7),
tworząc kation nieparzystoelektronowy o m/z = 42. Oderwanie rodnika wodorowego pozwala
przekształcić ten kation w kation allilowy o m/z = 41. Fragmentacja jonu macierzystego może
przebiegać również zgodnie ze schematem rozpadu (droga C) dając kation oksoniowy o m/z = 31.
Alternatywny sposób rozpadu kationorodnika 3-metylobutan-1-olu może polegać na odszczepieniu
kationu izopropylowego o m/z = 43 (nie przedstawiono na schemacie 8).
Omawiane rozpady są odpowiedzialne za najbardziej intensywne piki na widmie
3-metylobutan-1-olu (rys. 15a).

17
e
-
CH
3
CH
CH
2
CH
2
HO
CH
3
- H
2
O
CH
3
CH
CH
CH
2
CH
3
CH
2
CH
2
- 2e
-
CH
3
CH
CH
2
CH
2
O
H
CH
3
A
B
m/z = 88
niewidoczny
na widmie!
CH
2
CH
CH
3
- H
2
O
- CH
3
m/z = 42
m/z = 70
CH
CH
CH
2
CH
3
m/z = 55
rozpad
allilowy
CH
3
CH
CH
2
CH
3
C
rozpad
HO CH
2
HO CH
2
m/z = 31
- H
CH
2
CH
CH
2
m/z = 41
Schemat 8. Możliwe drogi fragmentacji 3-metylobutan-1-olu..
a)
b)
Rysunek 15. Widma MS: a) 3-metylobutan-1-olu; b) 2-bromo-2,3-dimetylobutanu.
Inną grupą związków, których cząsteczki w spektrometrze mas łatwo ulegają eliminacji są
halogenki alkilu (ale nie arylu!). Przykładowo, w widmie MS 2-bromo-2,3-dimetylobutanu pikiem
podstawowym jest jon pochodzący od kationorodnika 2,3-dimetylobutenowego (m/z = 85, rys. 15b),
który powstaje w wyniku dehydrohalogenacji jonu macierzystego (Schemat 9). Kolejny intensywny
pik leżący przy m/z = 43 świadczy o odszczepieniu z kationu izopropylowego o m/z = 43.
e
-
CH
3
CH
C
CH
3
CH
3
CH
3
Br
- 2e
-
M
+
: m/z = 164
niewidoczny na widmie!
CH
3
CH
C
CH
3
CH
3
CH
3
Br
- HBr
CH
3
C
C
CH
3
CH
3
CH
3
dalsze transformacje
typowe dla alkenów
m/z = 85
(M
+
-79)
43
Schemat 9. Schemat dehydrohalogencji jonu molekularnego 2-bromo-2,3-dimetylobutanu.
6.7. Przegrupowanie McLafferty’ego związków karbonylowych z protonami
(aldehydy, ketony, kwasy karboksylowe oraz ich pochodne – estry, amidy)
Zjonizowane związki karbonylowe w wyniku fragmentacji tworzą trwałe, stabilizowane
rezonansowo kationy acyliowe [R-C=O]
+
zwykle dobrze widoczne w widmie masowym (rozdz. 6.3),
wyjątkiem są łatwo odszczepiające cząsteczkę CO
2
kwasy karboksylowe. Dominującymi ścieżkami
rozpadu ich jonów jest przedstawiony wcześniej rozpad oraz, w przypadku związków alifatycznych,
odszczepienie grup alkilowych. W przypadku, gdy cząsteczka związku karbonylowego (aldehydu,
ketonu, estru, amidu) posiada proton w położeniu w stosunku do grupy karbonylowej, to utworzony
164, M
+
166, M
+
+2
88, M
+
85
70
55
43
42
31
OH
43
41
Br
18
z niej jon molekularny ulega przegrupowaniu McLafferty’ego zgodnie z podanym poniżej schematem
9. (UWAGA: tym razem położenie atomu wodoru jest określone jako względem grupy karbonylowej,
a nie heteroatomu! Atom wodoru względem grupy karbonylowej jest jednocześnie atomem
względem atomu tlenu)
kationorodnik
stabilizowany rezonansowo
(o masie parzystej, jeśli nie zawiera
nieparzystej liczby atomów azotu)
alken
O
H
R
R''
R'
R
O
R'
R''
H
O
R'
R''
O
R'
R''
H
Schemat 9. Ogólny schemat przegrupowania McLafferty’ego
Ujmując w sposób uproszczony, w wyniku przegrupowania Mc Lafferty’ego:
atom wodoru z pozycji przyłącza się to karbonylowego atomu węgla,
pęka wiązanie pomiędzy atomami i , a cząsteczka dzieli się na dwa fragmenty: alken
(lub inny związek z wiązaniem wielokrotnym między atomami i oraz kationorodnik
zawierający grupę karbonylową (w formie enolowej) połączoną z atomem
i
znajdującymi się przy nim podstawikami.
jeżeli utworzony w ten sposób kationorodnik wciąż posiada proton to może on ulec
ponownemu przegrupowaniu McLafferty’ego zgodnie z tym samym schematem.
Przykładem ilustrującym przegrupowanie McLafferty’ego może być fragmentacja jonu
molekularnego heksan-2-onu (Schemat 10). Zgodnie z przedstawionym powyżej schematem, w
wyniku tego przegrupowania powstaje propen oraz kationorodnik o m/z = 58. Innym sposobem
fragmentacji heksan-2-onu jest rozpad na jednej ze wskazanych dróg (A lub B), prowadzący do
kationów acyliowych o m/z = 43 oraz 85, przy czym pierwszy z rozpadów dominuje, ze względu na
wyższą trwałość powstającego jednocześnie rodnika. Fragmentacją o mniejszym znaczeniu jest rozpad
przedstawiony drogami C i D, dający kationy o m/z = 57 i 43.
H
2
C
CH
2
C
O
CH
3
HC
H
3
C
H
O
- 2e
+ e
H
2
C
C
O
CH
3
H
CH
CH
2
H
3
C
+
m/z = 58
M
+
, m/z = 100
przegrupowanie
McLafferty'ego
A
B
rozpad
rozpad
droga A
droga B
C
O
CH
3
CH
3
CH
2
CH
2
CH
2
+
m/z = 43
CH
3
CH
2
CH
2
CH
2
C
O
CH
3
+
m/z = 85
C
D
CH
3
CH
2
CH
2
+
CH
2
COCH
3
droga C
droga D
CH
3
CH
2
CH
2
+
CH
2
COCH
3
m/z = 43
m/z = 57
Schemat 10. Drogi fragmentacji heksan-2-onu.
Omawiane rozpady są odpowiedzialne za najbardziej intensywne piki na widmie heksan-2-onu
(rys. 16).
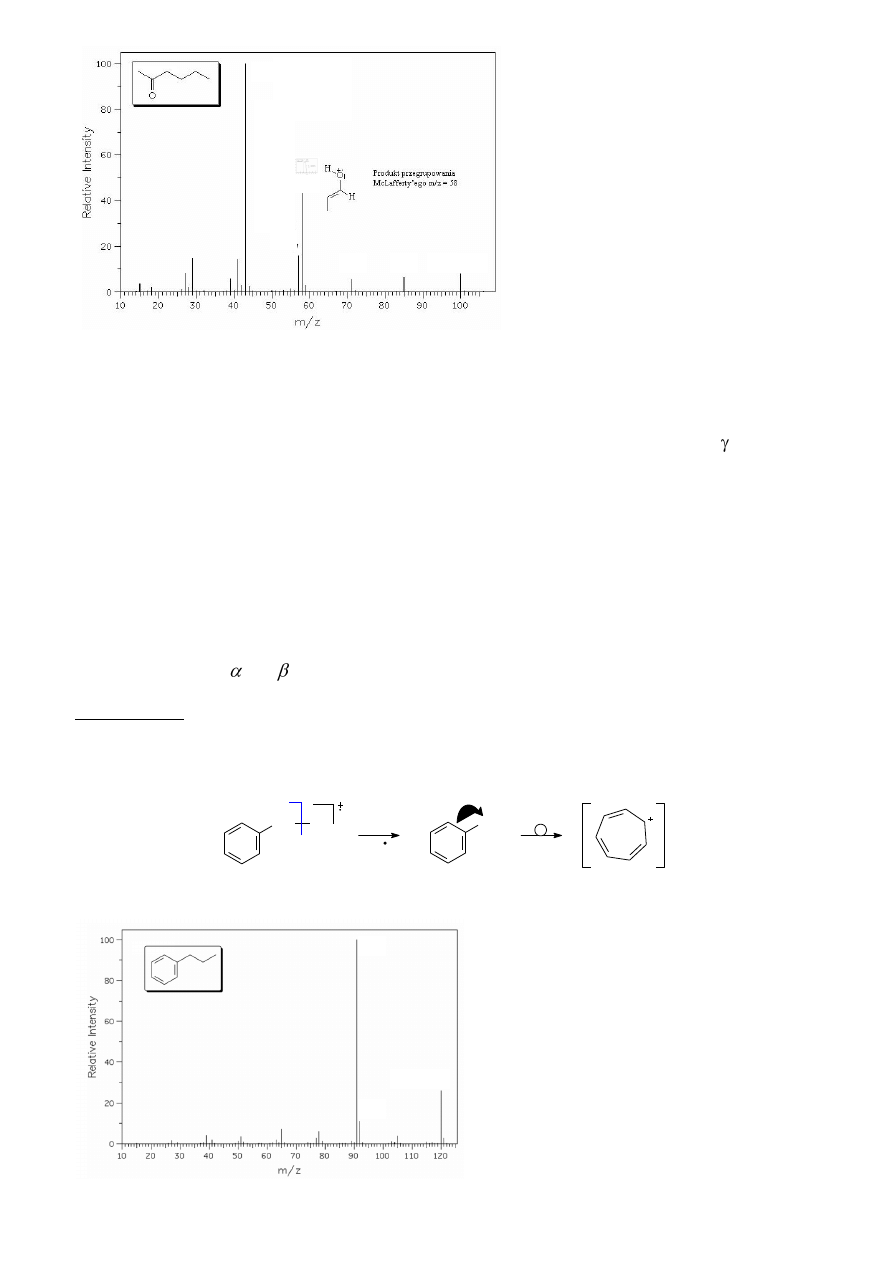
19
Rysunek 16. Widmo MS heksan-2-onu.
Analogicznym przegrupowaniom ulega podczas jonizacji szereg innych połączeń: aldehydów,
ketonów, pochodnych kwasów karboksylowych (np. benzoesan etylu - pik przy m/z = 122, widoczny
na widmie przedstawionym na rys. 12b) oraz innych związków, niezawierających grup
karbonylowych. Warunkiem ich zaobserwowania jest również występowanie protonów w stosunku
do grupy z wiązaniem wielokrotnym, mogącej stabilizować rezonansowo kationy, jednak powstające
produkty nie zawsze dają tak intensywne sygnały w widmach jak w przypadku klasycznego
przegrupowania McLafferty’ego.
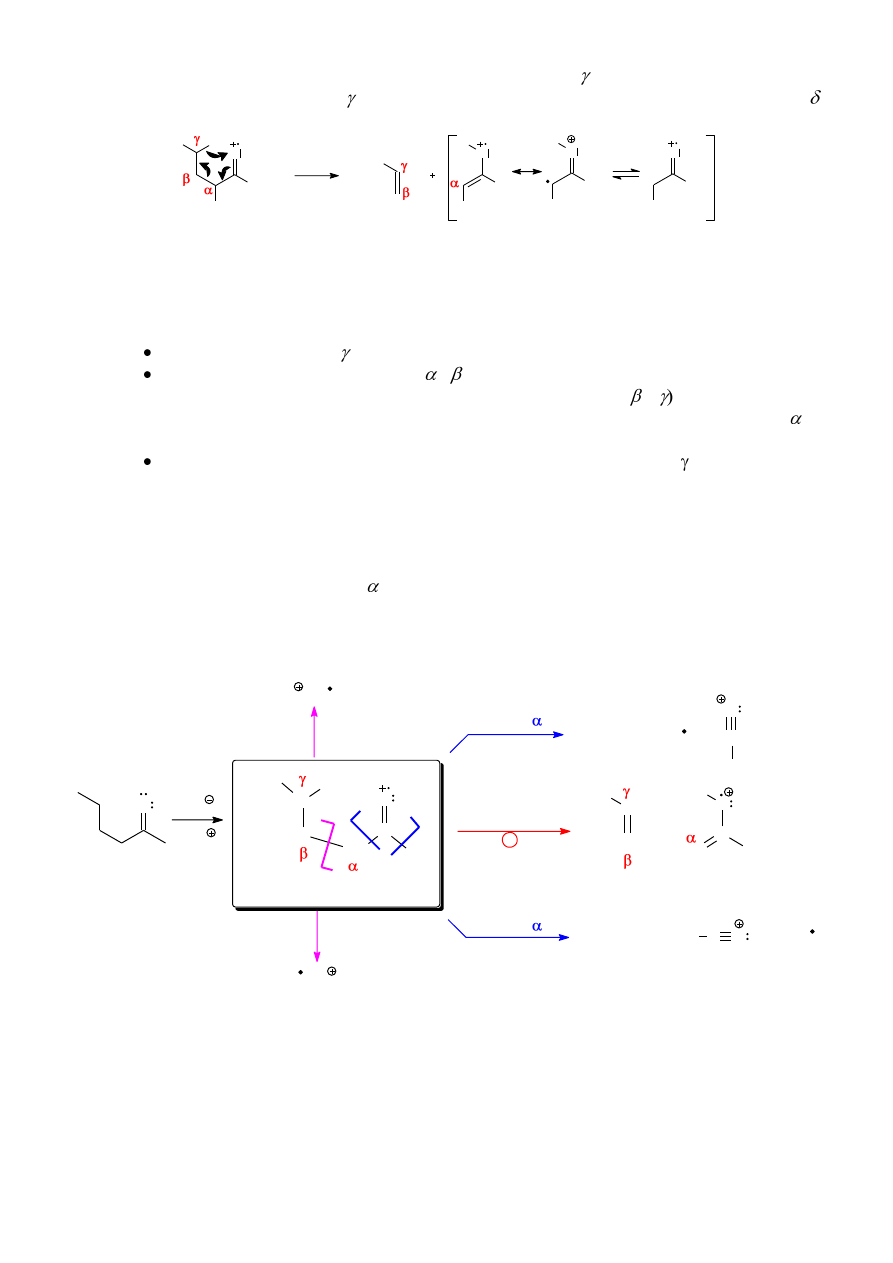
6.8. Przegrupowania alkiloarenów i inne fragmentacje związków aromatycznych
Jonizacja niepodstawionych związków aromatycznych prowadzi do powstania stabilnych
kationów często widocznych w widmach masowych jako piki główne (M
+
). W przypadku
węglowodorów podstawionych grupami alkilowymi jonizacji towarzyszy znacząca fragmentacja
jonów. Pierwotnie utworzony kationorodnik ulega fragmentacji polegającej na zerwaniu wiązania
pomiędzy atomami i w stosunku do fragmentu aromatycznego cząsteczki (schemat 11).
Powstający początkowo kation benzylowy ulega przegrupowaniu z utworzeniem trwalszego
aromatycznego siedmioczłonowego kationu tropyliowego o m/z = 91. O trwałości tego kationu może
świadczyć siedem możliwych do zaproponowania struktur rezonansowych. W wielu przypadkach
jonizacji alkilowych pochodnych benzenu pik kationu tropyliowego jest głównym pikiem
obserwowanym w widmie masowym (rys. 15).
CH
2
+
CH
2
R
m/z = 91
91
- R
kation benzylowy
kation tropyliowy
Schemat 11. Ogólny schemat fragmentacji alkiloarenów – przegrupowanie benzylowe.
Rysunek 15. Widmo n-propylobenzenu.
100, M
+
58
85
43
71
57
120, M
+
92
91
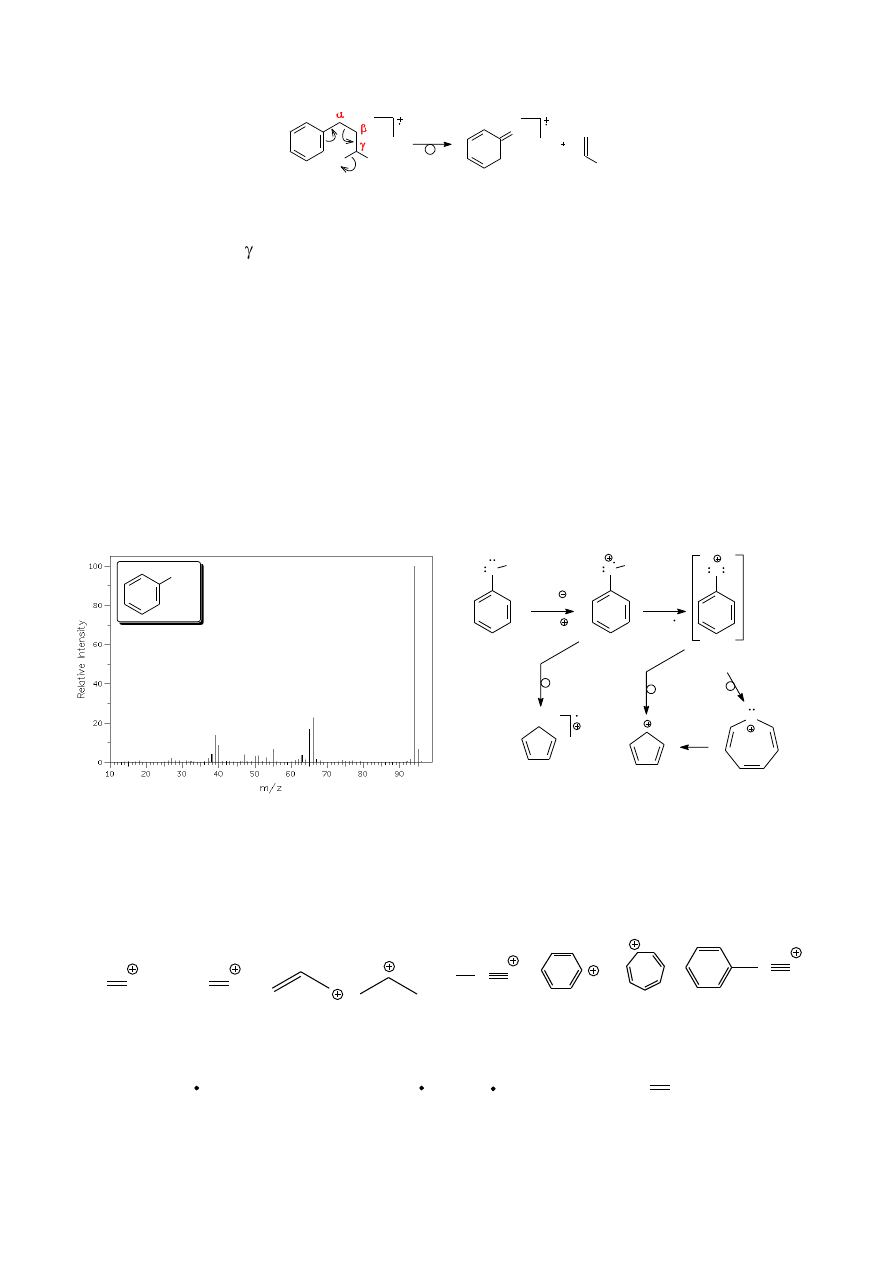
20
Zaznaczony na powyższym widmie pik o m/z = 92 jest rezultatem przegrupowania
prowadzącego do innego relatywnie trwałego kationu, tym razem nieparzystoelektronowego:
R
R
H
CH
2
m/z = 92
Jest to przegrupowanie analogiczne do przegrupowania McLafferty’ego, czyli wymagające
obecności protonu
w stosunku do pierścienia aromatycznego. Trójwęglowy łańcuch
n-propylobenzenu jest najprostszym przykładem alkiloarenu spełniającego ten warunek.
Oprócz wymienionych powyżej przegrupowań, w przypadku innych niż alkilowe pochodne
benzenu, obserwuje się często fragmentacje polegające na odrywaniu podstawników od pierścienia
benzenowego w postaci rodników. Tworzący się wówczas kation fenyliowy (w przypadku
monopodstawionych pochodnych benzenu o m/z = 77) jest widoczny na widmie jako jeden z bardziej
intensywnych pików (por. rys. 12a, 12b oraz 13b).
Przykładem bardziej złożonych transformacji jonów molekularnych pochodnych związków
aromatycznych mogą być procesy kontrakcji (zmniejszania) pierścienia. W widmie MS fenolu (rys.
15) pikiem podstawowym jest pik molekularny (m/z = 94). Pik pochodzące od przegrupowań typu
benzylowego (m/z = 93) ma znikomą intensywność. Jedyne istotne piki (poza pikiem głównym)
pochodzą od kationu i kationorodnika (m/z = 65 i 66) tworzących się wskutek oderwania od molekuły
cząsteczki tlenku węgla, której towarzyszy kontrakcja (zmniejszenie) pierścienia benzenowego
(schemat 12).
O
H
- 2e
+ e
O
H
- H
O
m/z = 94
O
m/z = 93
m/z = 65
skrajnie
nietrwały
- CO
m/z = 66
kontrakcja
pierścienia
kontrakcja
pierścienia
- CO
- CO
Rysunek 15. Widmo MS fenolu.
Schemat 12. Drogi fragmentacji fenolu z uwzględnieniem
kontrakcji pierścienia.
W interpretacji widm MS mo
że być pomocna znajomość mas typowych jonów
fragmentacyjnych oraz odrywających się rodników i cząsteczek:
TYPOWE JONY FRAGMENTACYJNE:
CH
2
OH
CH
2
NH
2
m/ z = 31
m/ z = 30
m/ z = 41
m/ z = 43
CH
3
C
O
m/ z = 43
m/ z = 77 m/ z = 91
C
O
m/ z = 105
CZĘSTO ODRYWAJĄCE SIĘ RODNIKI I CZĄSTECZKI:
CH
3
C
2
H
5
C
3
H
7
CO
CO
2
H
2
O
CH
2
CH
2
HCN
15
28
18
18
44
27
29
43
OH
65
94, M
+
66
Wyszukiwarka
Podobne podstrony:
MS 2011 1 spis tresci
Metody sprzedaży, ms 2011 1
MS 2011 1 spis tresci
MS 2011 4 10
Access2 Projektowanie bazy danych, Ogrodnictwo 2011, INFORMATYKA, Informatyka, MS Access
MS materialy dodatkowe gr D1 II rok 2011 12Z
MS GAS Pyt 2011
Przewodnik Relacyjne bazy danych 2008-2009, Ogrodnictwo 2011, INFORMATYKA, informatyka sgg, MS Acces
MS zadania gr D1 II rok 2011 12Z
prezentacja MS Project Szukański 2011 2012
1 GEN PSYCH MS 2014id 9257 ppt
2011 2 KOSZE
higiena dla studentów 2011 dr I Kosinska
więcej podobnych podstron