
Autor Elżbieta A.Gryz
Choroby demielinizacyjne.
Choroby z kręgu demielinizacyjnych to grupa schorzeń, w których procesem patologicznym leżącym u podstaw
objawów klinicznych jest demielinizacja.
Demielinizacja
Jest to rodzaj zmian patologicznych, polegających na uszkodzeniu i rozpadzie osłonek mielinowych. Proces ten
występuje głównie w Ośrodkowym Układzie Nerwowym, w postaci ognisk zwanych plakami. W demielinizacji tzw.
pierwotnej, której przyczyna jest niejasna, prawdopodobnie wywołana procesem immunologicznym, dochodzi do
rozpadu otoczki mielinowej, bez towarzyszących zmian w komórce nerwowej. Ponieważ osłonka mielinowa odgrywa
istotną rolę w przewodzeniu impulsów nerwowych, jej uszkodzenie powoduje upośledzenie funkcji neuronu i
powstanie objawów ubytkowych. W ślad za procesem rozpadu osłonki (demielinizacji), następuje proces jej
odtwarzania, przez komórki glejowe - remielinizacja. Demielinizacja może być również wywołana niedokrwieniem
lub zapaleniem tkanki, a także działania czynników toksycznych. We wszystkich tych przypadkach, obok
uszkodzenia mieliny występuje również uszkodzenie włókien nerwowych. Ten rodzaj demielinizacji jest nazywanym
wtórnym. Demielinizacji towarzyszy odczyn zapalny wokół plaqi (ogniska rozpadu mieliny)
Najbardziej znanymi chorobami, w których głównym procesem patologicznym jest demielinizacja są:
- stwardnienie rozsiane,
- zapalenie rdzenia i nerwów wzrokowych,
- centralna mielinoliza mostu,
- postępująca ogniskowa leukoencefalopatia,
- leukodystrofie.
Zmiany patomorfologiczne w CSN to:
- ogniska demielinizacji pełnej i częściowej o dużej nieregularności i rozmieszcze-
niu, szczególnie w pobliżu komór mózgowych (plaki),
- pierwotny odczyn zapalny z limfocytów, plazmocytów, makrofagów,
- ogniska remielinizacji.
S t w a r d n i e n i e r o z s i a n e
Stwardnienie rozsiane (sclerosis multiplex-SM) jest chorobą z kręgu demielinizacyjnych, w których powstawaniu
ognisk demielinizachi w CSN towarzyszą odpowiednie objawy kliniczne. Choroba ta często jest porównywana do
nieproszonego gościa, który nagle, bez zaproszenia pojawia się w naszym domu i ze wszystkimi bagażami,
problemami pozostaje z nami na zawsze. To porównanie uświadamia nas jaką chorobą jest stwardnienie rozsiane.
Niektórzy nazywają tę chorobę nieprzewidywalną, ponieważ w stosunku do danego pacjenta nie jesteśmy w stanie
przewidzieć jej przebiegu. Statystyka dostarczy nam jedynie prawdopodobnych danych co do przebiegu choroby,
niekoniecznie prawdziwych dla danego pacjenta. Choroba, jak wiele innych budzi lęk i strach, nie tylko wśród
pacjentów, ale również wśród lekarzy. Mimo, iż pierwszy opis pacjenta cierpiącego na SM pochodzi z XV wieku i
istnienia kilku hipotez nadal nie znamy jej przyczyny, nie potrafimy jak dotychczas skutecznie jej leczyć.
Stwardnienie rozsiane pozostaje główną przyczyną inwalidztwa neurologicznego wśród młodych ludzi, zwłaszcza w
obszarach o dużej zapadalności na tę chorobę, a więc w Ameryce Północnej, zachodniej Europie, Australii i Nowej
Zelandii, i nie jest ono zmniejszone poprzez stosowanie różnych leków, z jakimi wiązano, lub wiąże się nadzieję.
Nowe zasady diagnozowania (kryteria) pozwalają wyróżnić SM od innych możliwych przyczyn mogących
wywoływać rozsiane objawy uszkodzenia układu nerwowego (sarkoidoza,LE,etc)
Definicja:
Choroba ośrodkowego układu nerwowego o wielofazowym, nawrotowym i/lub postępującym przebiegu oraz
wieloogniskowych (rozsianych) objawach neurologicznych, wywołana specyficznym dla układu nerwowego
zaburzeniem immunologicznym powodującym pierwotną wieloogniskową i postępującą demielinizacją.

Epidemiologia:
Przyjmuje się, że na świecie żyje conajmniej 3000 000 osób ze stwierdzoną chorobą, a prawdopodobnie dalsze
6000000 nie wie, że ma tę chorobę.Wskaźnik rozpowszechnienia zależy od szerokości geograficznej, największy
wskaźnik zapadalności posiadają Europa Środkowa i Północna:40-75/100.000, Jak również Stany Zjednoczone
Ameryki Północnej, w Polsce okolice Poznania 50/100000. Prawie nie spotyka się zachorowań na SM w tropikach i
Japonii.
Etiopatogeneza:
W etiopatogenezie, złożonej i jak dotychczas, do końca niepoznanej bierze się pod uwagę następujące teorie, z
których dwie pierwsze jak się wydaje odgrywają kluczową rolę. Są to:
- teoria wirusowa,
- teoria autoimmunologiczna.
- element genetyczny (?)
Najlepiej poznaną i mającą najwięcej zwolenników jest teoria autoimmunologicznego pochodzenia
choroby.Dowodami na autoimmunologiczne pochodzenia choroby są:
-podobieństwo SM do eksperymentalnego alergicznego zapalenia mózgu i rdzenia,
- obecność następujących nieprawidłowości we krwi obwodowej:
spadek liczby supresorowych komórek T przed rzutem choroby;
czynnik limfotoksyczny i mielinotoksyczny;
obecność limfocytów uczulonych na mielinę podczas rzutu;
- nieprawidłowości w O.U.N.:
synteza immunoglobulin typu IgG i IgA w O.U.N.;
podwyższone miana przeciwciał przeciw wirusom, np. odry.
Teoria wirusowa, jest oparta na fakcie obecności podwyższonych mian przeciwciał przeciwko wielu virusom, nie
tylko w surowicy krwi, ale również i w płynie mózgowo-rdzeniowym. Istnieje jednak wiele przyczyn dla których
trudno udowodnić rolę wirusów w patogenezie SM. Przede wszystkim: inicjujący chorobę wirus /jeśli istnieje/ jest
obecny przed wystąpieniem objawów choroby, ale nie można go wykryć po wystąpieniu jej objawów. Po drugie
wirus lub wirusy mogą być wystarczająco częste, w populacji , ale wywołują SM tylko u osób genetycznie
wrażliwych. Trzecia możliwość to fakt, że wirus jest nieznany i dlatego nie może być wykryty. Jeśli jednak wirus
inicjujący powstanie choroby istnieje to prawdopodobnie działa w sposób przedstawiony na ryc 1.
Ryc.1 Prawdopodobne drogi działania wirusa wywołującego SM
typ działania
mechanizm
dowody
- bezpośrednia infekcja CSN
-śmierć oligodendrocytów
(produkujących mielinę)
- przewlekła infekcja powodująca
demielinizację
nigdy nie udowodniono wpływu
bezpośredniego wirusa na demielinizację
w SM
komórkowa odpowiedź na infekcję
wirusa w obwodowym systemie
nerwowym
- w odpowiedzi na infekcję wirusa
powstają przeciwciała które dzięki
krzyżowej reakcji ze składnikami
mieliny powodują demielinizację
- proces ten udowodniono w wielu
chorobach autoimmunologicznych
- badania na zwierzętach
-komórki immunologiczne od pacjentów z
SM, skierowane przeciw mielinie mogą
rozpoznawać pewne rodzaje wirusów in
Vitro
-SSPE jest chorobą demielinizacyjną
prawdopodobnie powstałą w takim
mechaniźmie

stymulacja klinicznego rzutu
- uważa się, że powodują pogorszenie - zwiększona liczba rzutów w czasie
infekcji gdo i innych wirusowych
Nie można pominąć od wielu lat toczącej się dyskusji na temat wpływu genetyki na rozwój SM.
Dowody na genetyczność SM ?
Stwierdzono, że bliżnięta jednojajowe częściej oba chorują na SM, w grupie chorych, ale jedynie w pewnej
określonej szerokości geografiznej częściej obecne pewne antygeny zgodności tkankowej. Do nich należą: HLA -
DRW, HLA- DW2, HLA- DRw53, HLA -Drw6, HLA -DQ. Okazało się, że miejsce na chromosomie 6 jest
wspónym kodującym dla układu HLA i zwiększające możliwość zapadnięcia na SM. Ponadto stwierdzono obecność
miejsca na chromosomie 14 kodującego Gm system ciężkich łańcuchów IgG, które jak wiemy powstają w SM.
Jak już wspomniano SM jest chorobą, w przebiegu której może wystąpić każdy z objawów neurologicznych.
Pierwszy rzut choroby może rozpoczynać się pojedynczym objawem np:ataksja kończyn górnych - (40%), jak
również od wielu objawów; np zaburzenia chodu wywołane ataksją i paraparezą - (50% przypadków) Są jednak
pewne objawy i zespoły kliniczne częściej występujące u chorych z SM.
Okazało się, że najczęstszym objawem poprzedzającym rozwój choroby czasem nawet na 2-3 dekady jest
pozagałkowe zapalenie nerwu wzrokowego. Ustalono, że z 50 % zapaleń n II rozwija się póżniej stwardnienie
rozsiane a 25 % stwardnienia rozsianego ropoczyna się zapaleniem nerwu wzrokowego. Ustalono również (na
podstawie grupy klinicznej pacjentów z neuritis retrobulbaris) czynniki ryzyka rozwoju SM. Okazało się, że u
chorych, którzy przebyli neuritis retrobulbaris częściej rozwija się SM w przypadku, gdy:
a. są to kobiety (kobiety:mężczyźni -1,7:1,0)
b. do zapalenia nerwu wzrokowego dochodzi w porze roku zimowej
c. stwierdzi się obecność antygenu HLA-DR2, HLA-B7
d. w płynie mózgowo-rdzeniowym wykaże się podwyższony poziom gamma-globulin i obecność prążków
oligoklonalnych
Inne objawy, jak już wspomniano, mogą być różnorodne, zarówno na początku, jak i w trakcie trwania choroby.
Klasycznie są to różnorodne zaburzenia widzenia, oczopląs, parestezje, zaburzenia czucia, dyzartria, drżenie
zamiarowe, niedowład lub porażenie jednej, dwu, trzech kończyn, spastyczność czy zaburzenia pęcherzowe.
Szczególnie wśród zaburzeń pęcherzowych należy podkreślić częste występowanie: trudności z rozpoczęciem mikcji,
potrzeby natychmiastowego oddania moczu, czy częstej potrzeby oddawania moczu. Do niezwykle rzadko
spotykanych należą napady padaczkowe.
Mówiąc o typowych objawach dla SM nie można pominąć objawu Lermitte’a (uczucie przepłwu prądu wzdłuż
kręgosłupa i/lub kończyn podczas zginania głową). Również objawem często podawanym przez pacjenta jest
nadmierna męczliwość, która sama, może na wiele lat wyprzedzić pojawienie się innych objawów SM.Większość
chorych skarży się na nadmierną męczliwość przez cały okres trwania choroby. Historycznie nie można pominąć
triady objawów opisywanych przez Charcota - oczopląs, drżenie, mowa skandowana. Rzeczywiście występowanie
tego zespołu należy obecnie do rzadkości.
Tab 1. przedstawia statystyczny przegląd najczęściej występujących objawów w SM, zarówno na początku jak i w
trakcie przebiegu.
Objawy
w początkowym
okresie
w późnym okresie
Niedowład kończyn
40 %
80 %
Zaburzenia widzenia (mroczek i podwójne widzenie)
36 %
66 %
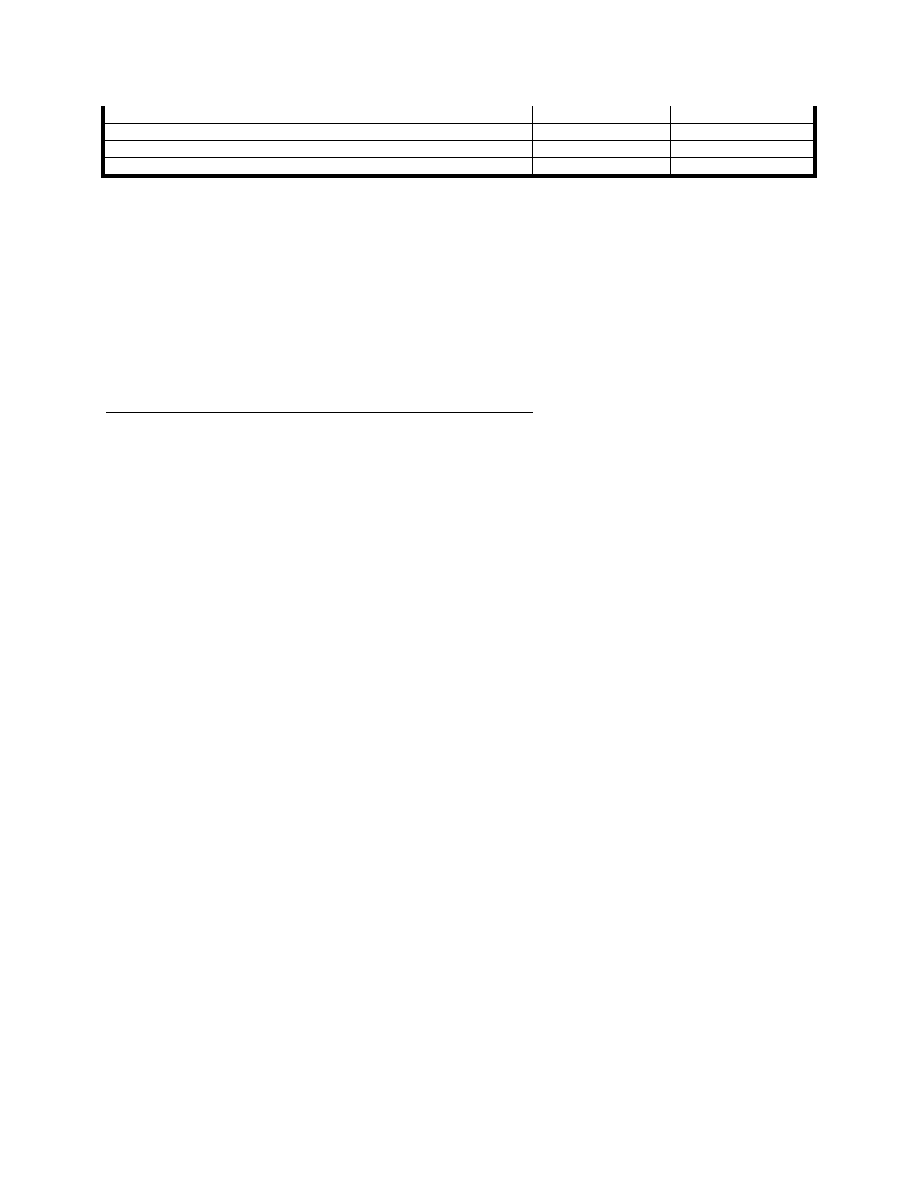
Zaburzenia czucia
21 %
73 %
Zaburzenia równowagi i koordynacji
10 %
77 %
Zaburzenia zwieraczy
9 %
56 %
Zaburzenia poznawcze
3%
60 %
Najczęściej choroba zaczyna się pomiędzy 20 a 40 rokiem życia. Wiek zachorowania, jak również szczegółowe
określenia objawów dokładnie określają kryteria Komitetu Posera (patrz poniżej).
W zależności od dominujących objawów klinicznych można wyróżnić następujące postaci SM:
- rozsiana (mieszana)
50 %
- rdzeniowa
20 %
- mózgowa
20 %
- skąpoobjawowa
7 %
- móżdżkowa
3 %
Przebieg choroby może być różny. Klinicznie może to przybrać postać:
- zwalniająca/relapsing-remitting/ 50 %

(przebiega rzutami, pomiędzy którymi są okresy bezobjawowe)
- powoli postępująca /progressive/ 5 %
(od początku narastają stopniowo objawy neurologiczne)
- ostra (szybko prowadzi do zgonu) 2 %
- złożona /RR-progresive/ 43 %
(początkowo przebiegająca rzutami staje się przewlekle postępująca)
- stabilna
(po jakimś czasie objawy nie narastają, ani też nie zmniejszają się)
- poronna (skąpoobjawowa)
Wśród typowo przebiegających postaci SM można spotkać rzadko nietypowe zespoły kliniczne. Do nich należa:
- zespół rzekomoreumatyczny z bólami mięśniowo-stawowymi,
- zespół korzeniowy (rwa kulszowa, neuralgia),
- zespół rzekomonerwicowy z dystonią neurowegetatywną
- zespoły psychiatryczne z psychozami,
- zespół poprzecznego uszkodzenia rdzenia kręgowego,
- zespół rzekomoguzowy z objawami wzmożonego ciśnienia śródczaszkowego
i objawami ogniskowymi z półkul mózgu.
Istnieje szereg badań dodatkowych, które powinny być wykonane u pacjenta podejrzewanego o SM. Są To:
1. Badanie płynu mózgowo-rdzeniowego, w którym stwierdzimy
:
- podwyższony poziom gamma globuliny (75 % chorych)
- wskaźniki:
- IgG /albumina -
powyżej 0,2
- wsk. Linka i Tibllinga
IgG płyn / IgG surowica
-------------------------------------------- powyżej 0,8
albumina płyn / albumina surowica
- obecność prążków oligoklonalnych
2. Badanie Rezonansu Magnetycznego mózgowia NMR, które wykaże:
- przynajmniej 3 ogniska o średnicy powyżej 3 mm, w tym dwa zlokalizowane okołokomorowo
Badanie CT nie jest wystarczające gdyż zmiany hypodensyjne wykaże w mniej niż
20% przypadków
Ct i MRI (mózgu, czy rdzenia) są bezwględnie konieczne do wykonania, gdy mamy objawy ogniskowe
3. Potencjały wywołane:
rodzaj potencjałów wywołanych
% nieprawidłowych wyników w SM
- wzrokowe potencjały wywołane
80 %
- słuchowe (pniowe)
50 %
- somatosensoryczne
70 %
Przełomem w badaniach nad SM stała się panelowa konferencja , która odbyła się w roku 1988 w USA na temat
stwardnienia rozsianego (Komitet Posera). Na konferencji tej ustalono dokładne kryteria diagnostyczne dla tej
choroby, jak również podano definicje dla poszczególnych określeń takich jak rzut, wiek zachorowania itd.
Kryteria Komitetu Posera
1.Wiek - początek pomiędzy 10 a 59 rokiem życia. (Bardzo rzadko chorują ludzie poza tymi granicami wiekowymi)

2. Atak (rzut) - pojawienie się objawów neurologicznych trwających dłużej niż 24 h. Parestezje, objaw Lermitte’a,
czy napad ton-kloniczny nie mogą być traktowane jako atak (rzut). Jednak występowanie ich w ciągu dni, tygodni
należy traktować jako rzut choroby.
3.Kliniczny dowód na uszkodzenie - wynik badania neurologicznego, lub obecność objawów neurologicznych, jeśli
pozostały po rzucie.
4.Parakliniczne dowody na istnienie uszkodzenia - nieprawidłowe potencjały wywołane, obecność zmian
charakterystycznych w obrazie rezonansu magnetycznego (MRI). Jeśli są obecne a nie były manifestowane klinicznie
to są one subkliniczne.
5. Remisja - wycofanie lub zmniejszenie się objawów trwających dłużej niż 24. Znacząca gdy trwa conajmniej
miesiąc.
6. Oddzielne uszkodzenia - (rozsiane) objawy muszą być wytłumaczone przez uszkodzenie różnych struktur
anatomicznych. Wyjątek stanowi zapalenie drugiego nerwu II. po upływie mniej niż 2 tygodni.
7. Laboratoryjne podparcie - podwyższony poziom gamma globulin, obecność prążków oligoklonalnych.
8. Objawy wynikają z obecności plaq i są z nimi związane. ( jeśli epi lub osłabienie mięśni-
dowód musi być ognisko w MRI (również i po podaniu kontrastu--gadolinium), czy nieprawidłowe potencjały
wywołane (EVP), lub obecność prążków)
Kryteria diagnostyczne
Klinicznie pewne SM -
1. dwa rzuty i kliniczne dowody na dwa odrębne miejsca uszkodzenia
2. dwa rzuty i kliniczne dowody na jedno ognisko, a także parakliniczne na inne ( np VEP)
Laboratoryjne poparcie dla k.p SM
1. dwa rzuty. kliniczne lub parakliniczne dowody na ognisko i prążki oligoklonalne.
2. jeden rzut - kliniczne dowody dwóch ognisk i prążki oligoklonalne
3. jeden rzut - kliniczne dowód jednego i parakliniczne drugiego ogniska, i prążki oligoklonalne
Klinicznie prawdopodobne SM -
1. dwa rzuty i dowód na jedno ognisko
2. jeden rzut i dowód na dwa ogniska
3. jeden rzut, jedno ognisko, parakliniczny dowód na drugie
Laboratoryjne poparcie dla klinicznie prawdopodobnego SM
1. dwa rzuty i obecność prążków oligoklonalnych
Przebieg Stwardnienia Rozsianego może być różnorodny. Stan chorego również jest różny; często dynamicznie
zmieniający się: od niewielkich ubytków neurologicznych (postać poronna) do poważnego inwalidztwa. Stan chorego
i stopień nasilenia choroby ocenia się za pomocą Skali Kurtzkego.
Ocena nasilenia choroby (skala Kurtzkego):
0 - stan prawidłowy
1 - drobne objawy neurologiczne, nieupośledzające sprawności

2 - niewielkie upośledzenie sprawności (zaburzenia chodu, widzenia, osłabienie lub wzmo-
żone napięcie kończyn)
3 - wyraźne upośledzenie czynności (niedowład jednej kończyny, niewielki niedowład
połowiczy, lekka niezborność, zaburzenia pęcherzowe)
4 - znaczne upośledzenie czynności, umożliwiające jednak w miarę normalne życie i pracę
zawodową
5 - niemożność wykonywania pracy zawodowej, samodzielne poruszanie się na dystansie
do 500 m
6 - chód możliwy na krótkich odcinkach za pomocą kul lub lasek
7 - przemieszczanie się tylko na wózku inwalidzkim
8 - całkowite unieruchomienie w łóżku z możliwością posługiwania się kończynami górnymi
9 - całkowite unieruchomienie w łóżku bez możliwości wykonywania jakichkolwiek
czynności
10 - śmierć na skutek stwardnienia rozsianego
Rokowanie w tej chorobie jest trudne, między innymi dlatego, że jest to często choroba nieprzewidywalna. Statystyki
podają, że przebieg łagodny ma 25 % chorych.Śmierć w pierwszych 20 - 30 latach następuje u 25 % chorych. Około
1/3 chorych po 25 latach może jeszcze pracować, 2/3 chorych może chodzić. Średnie przeżycie wśród chorych z SM
jest krótsze o 15 - 20 lat w porównaniu z populacją zdrowych.
Szczególnie w okresie pierwszego rzutu choroby należy pamiętać, że poszczególne objawy choroby, zwłaszcza jeśli
są jednoogniskowe, mogą być wywołane również przez inne przyczyny. Dlatego w diagnostyce różnicowej należy
wziąść pod uwagę i inne przyczyny:
- guzy, zwłaszcza rdzenia i pnia mózgu
- nieprawidłowości rozwojowe (np.zespół Arnolda Chiariego,wgniecenia podstawy
czaszki
- choroby rdzenia kręgowego (mielopatia szyjna, wypadnięcie krążka między-
kręgowego, niedobór Vit B-12, zapalenia rdzenia)
- choroby tkanki łącznej (toczeń układowy, guzkowe zapalenie tętnic)
- choroby zapalne O.U.N. (borelioza, sarkoidoza)
- choroby naczyniowe mózgu
- nerwica neurasteniczna.
Leczenie
Leczenie SM należy podzielić na leczenie
1.- rzutu choroby,
2.- między rzutami
3.- leczenie postaci przewlekłych
ad - ostrego rzutu (łagodzenie następstw rzutu, leczenie przeciwzapalne)
najczęściej stosuje się kortykosteroidy:
Metyloprednisolon (Solu-Medrol) 1 g dziennie we wlewie kroplowym przez 3-5 dni,
potem Prednison (Encorton) doustnie 60 mg dziennie przez tydzień, a następnie
w dawkach stopniowo zmniejszanych przez kolejne 2 tygodnie;
lub
Prednison (Encorton) początkowo 60 mg doustnie przez 10 dni, potem w dawce
zmniejszanej o 10 mg co 3 - 5 dni; dawka podtrzymująca 10 mg, a potem 5 mg
do zakończenia w 4 tygodniach
2 - hamowanie postępu choroby - immunosupresyjne
nie ma pewnych dowodów na to, by leczenie to skutecznie łagodziło przebieg

choroby. Polecane jest z reguły w przypadkach szczególnie szybkiego postępu
choroby.
Azatiopryna 1-2 mg na kg m.c. codziennie przez 6 - 12 miesięcy
Cyclofosfamid 0,5 g na m kw. ciała w 500 ml 5 % glukozy co 4 tygodnie przez
12 - 24 miesiące pod kontrolą leukocytozy,
W ostatnich latach do leczenia wprowadzono serię interferonów beta.
Są to między innymi następujące preparaty:
Interferon Beta - 1 Avonex (beta interferon - 1 a) i beta interferon - 1 b
Biogen inc - 30 ug/tydzień
Schering - 8 mln IU/co drugi dzień
Powodują one
1. zmniejszenie liczby rzutów
2. zmniejszenie liczby ognisk demielinizacji w MRI
3. zmniejszają nasilenie rzutów
Nie zatrzymują choroby
Stosowane jak dotychczas jedynie w postaci przebiegającej rzu
tami
3. W przypadkach przewlekle postępujących dużego znaczenia nabiera działanie objawowe, a więc leki
zmniejszające napięcie mięśniowe, zwalczające bóle, rehabilitacja (ćwiczenia) zapobieganie przykurczom i szeregu
objawom wynikającym z chorovy.
a) spastyczność: Relanium 5 - 10 mg dziennie lub Baklofen 30 - 60 mg
dziennie stopniowo zwiększając dawkę lub Mydocalm 3 x 150 mg, Sirdulad 3 x 2 mg
do 3 x 4 mg,
Myolastan 2-4 mg 3 x 1
b) ból korzeniowy: Karbamazepina (Amizepin) 400 - 1200 mg dziennie lub
Clonazepam 3 - 6 mg dz. lub Phenytoin 3 - 6 tbl dz.
c) zaburzenia pęcherzowe:
nietrzymanie z parcia (wzmożona pobudliwość wypieracza)
leki cholinolityczne: Bellapan, Scopolan, Ditropan, Spasmophen;
blokery kanałów wapniowych: Cordafen, Isoptin
nietrzymanie z osłabienia zwieraczy
leki sympatykomimetyczne: Efedryna
leki przeciwdepresyjne: Imipramina
zatrzymanie moczu z powodu niedoczynności wypieracza
leki parasypatykomimetyczne: Betanechol
leki przeciwcholinesterazowe: Polstigmin, Mestinon, Mytelase
zatrzymanie moczu z powodu nadczynności zwieracza:
leki alfa-adrenolityczne: Minipress, Regitin
leki obniżąjące napięcie mięśni: Relanium, Mydocalm, Baclofen
d) zespół zmęczenia - Amantadyna 3 x 50 mg, leki przeciwdepresyjne.
Rehabilitacja ruchowa.
Wyszukiwarka
Podobne podstrony:
4. CHOROBY DEMIELINIZACYJNE - STWARDNIENIE ROZSIANE, stwardnienie rozsiane
choroby demielinizacyjne, IV rok Lekarski CM UMK, Neurologia, Neurologia od Grzela, wykady, opracowa
03 CHOROBY DEMIELINIZACYJNE
Choroby demielinizacyjne (3)
Choroby demielinizacyjne (4)
choroby demielinizacyjne
Stwardnienie rozsiane (2), Medycyna, Neurologia, 11 choroby demielinizacyjne
Choroby demielinizacyjne (2)
Choroby demielinizacyjne
Choroby demielinizacyjne (2)
choroby demielinizacyjne
choroby naczyn i serca(1)
więcej podobnych podstron