
Choroby demielinizacyjne

Co to są choroby
demielinizacyjne?
Jakie to choroby?
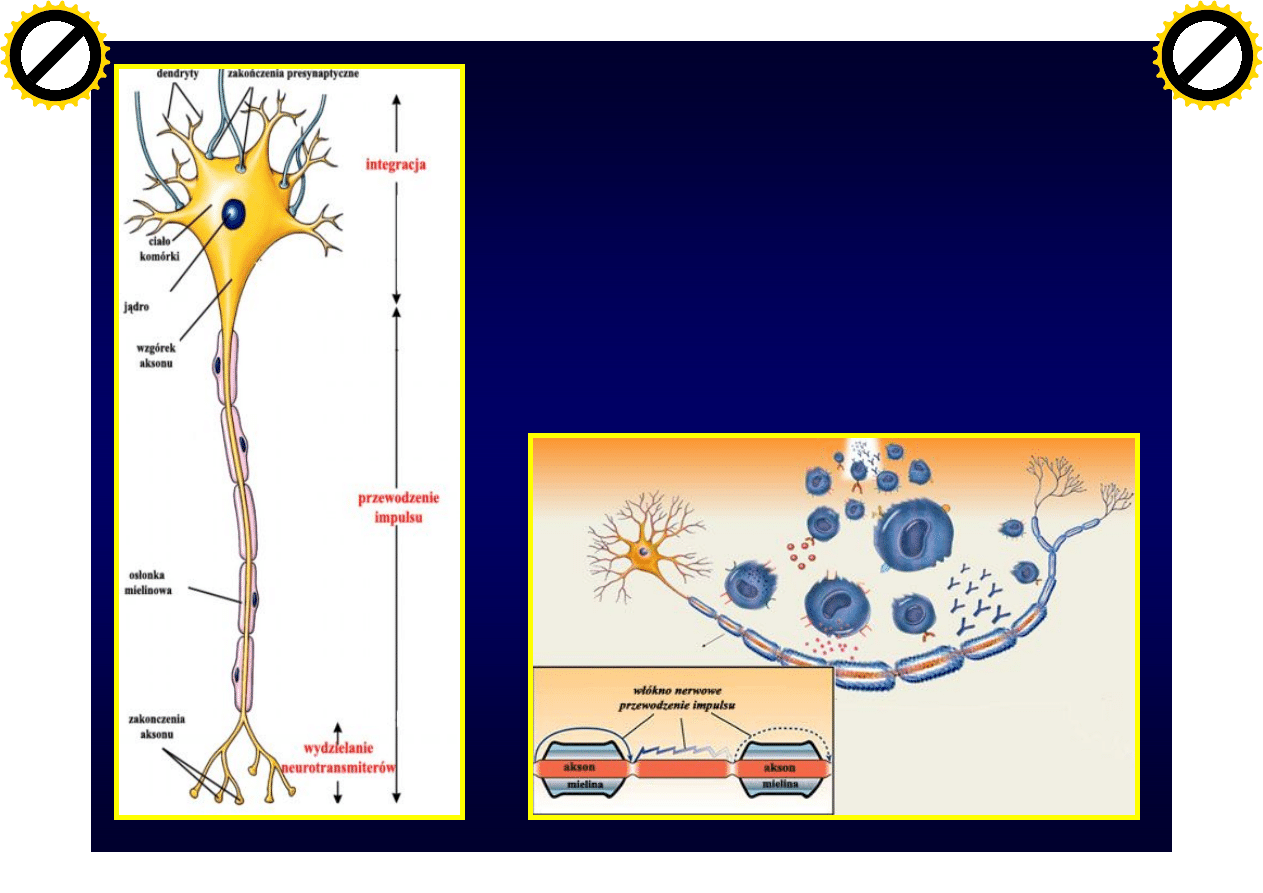
Uszkodzenie w SM
Demielinizacja dotyczy
ośrodkowego układu
nerwowego[ zaburzenia
przewodnictwa impulsów
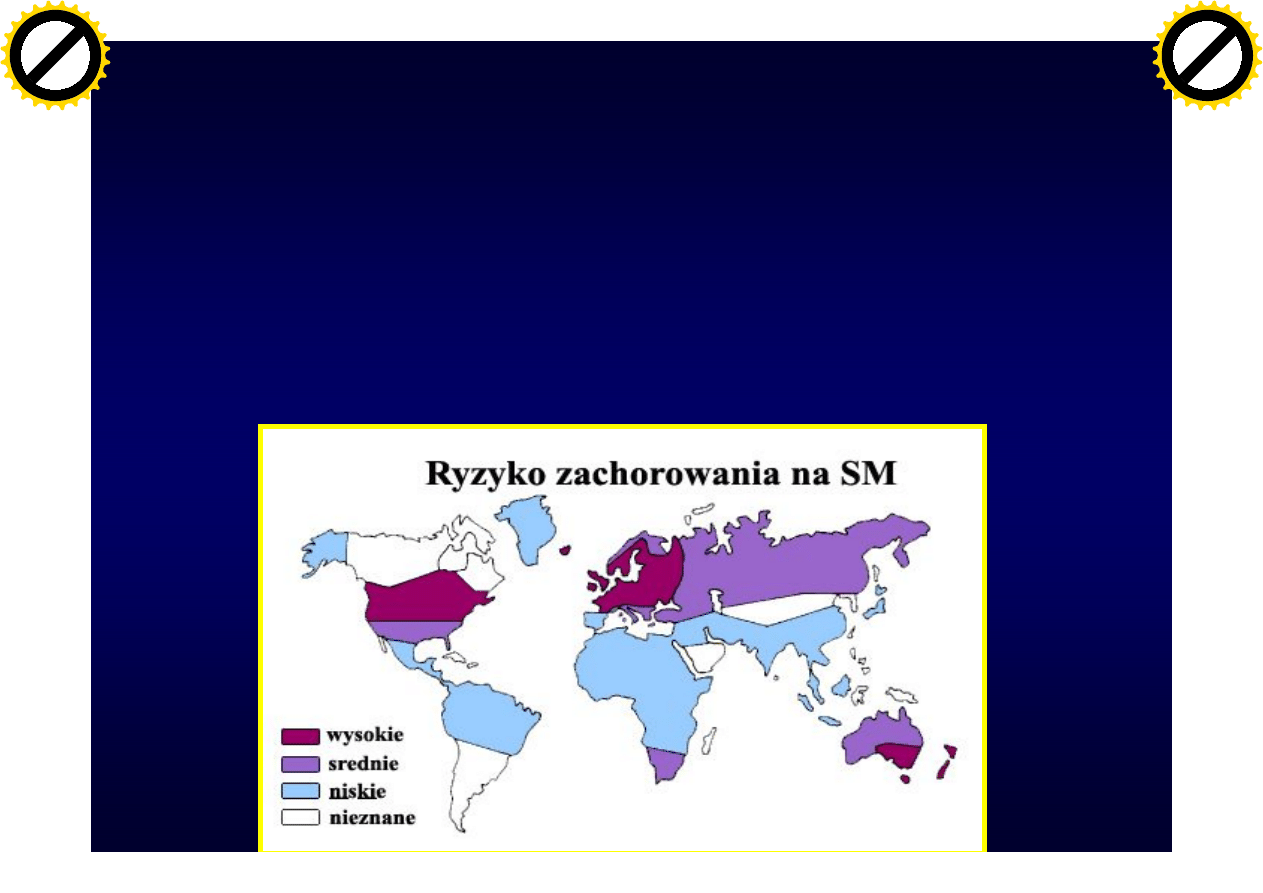
Stwardnienie rozsiane
• Jest jedną z najczęstszych chorób
neurologicznych osób między 20 a 40 rokiem
życia.
• Ogólna populacja 30-150/100tys. ludności

Czy stwardnienie rozsiane jest
chorobą genetyczną?

Podłoże genetyczne
• Postać rodzinna 10-15% przypadków
• Bliźnięta jednojajowe
– 25-30%
• Bliźnięta dwujajowe
– 2-5%
• HLA
– DRB1*1501, DQw6 (5-10x częściej) (rasa
biała)

Jakie procesy patologiczne
charakteryzują stwardnienie
rozsiane?
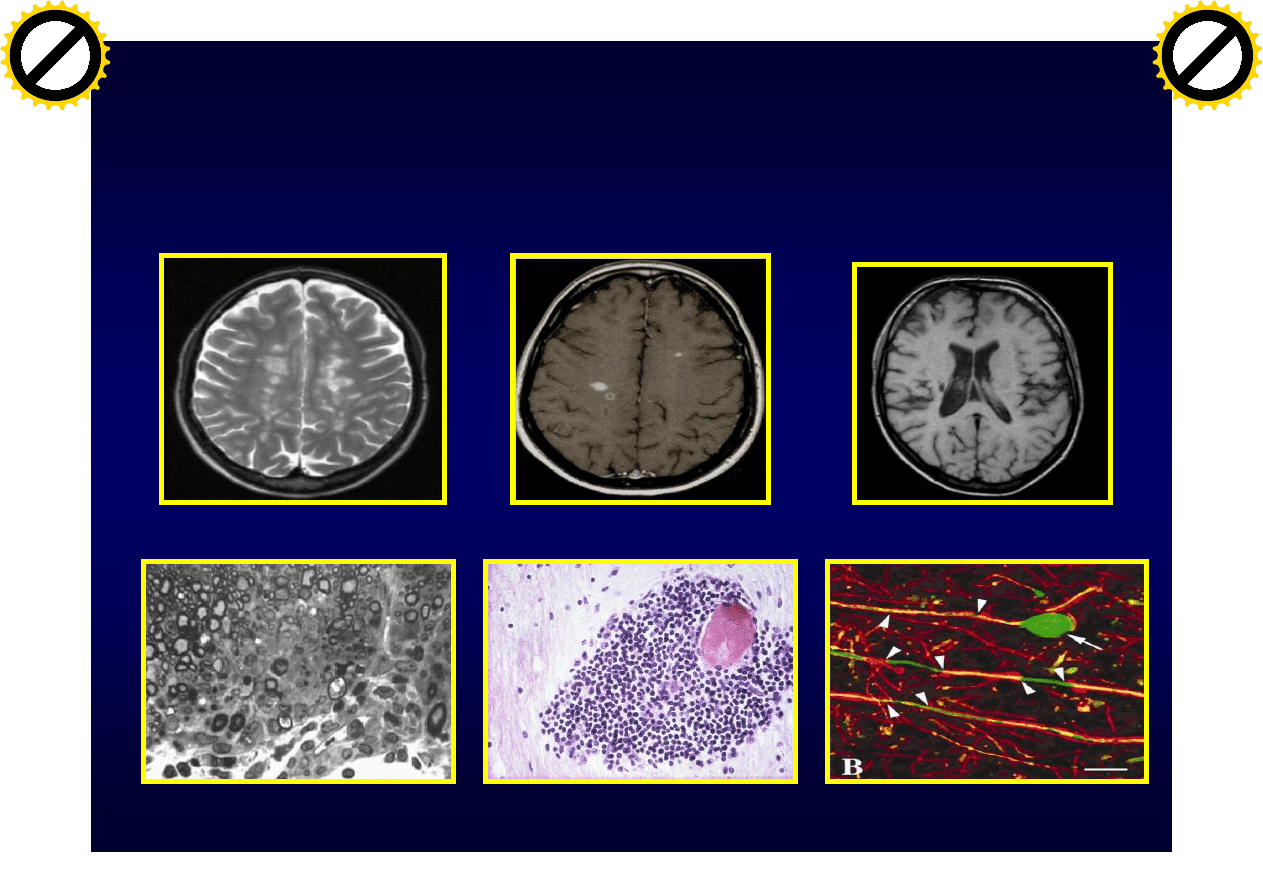
Stwardnienie rozsiane (SM)
Choroba
MRI: T2
MRI: T1 wzmocnienie
MRI: T1 atrofia
demielinizayjna
zapalna
neurodegeneracyjna
Trapp BD i wsp. NEJM, 1998:338:278-85
Gałązka G i wsp. J Immunol 2006:176;1588-99

Jakie procesy dominują na
początku choroby a jakie w
okresie zaawansowanym?

Rzuty
Gd(+)
Narastanie zmian w
obrazach T1-zale
żnych
Atrofia mózgu
Zapalenia
Neurodegeneracja
Przebieg choroby (lata)
Brak korelacji pomiędzy zapaleniem a
zmianami degeneracyjnymi
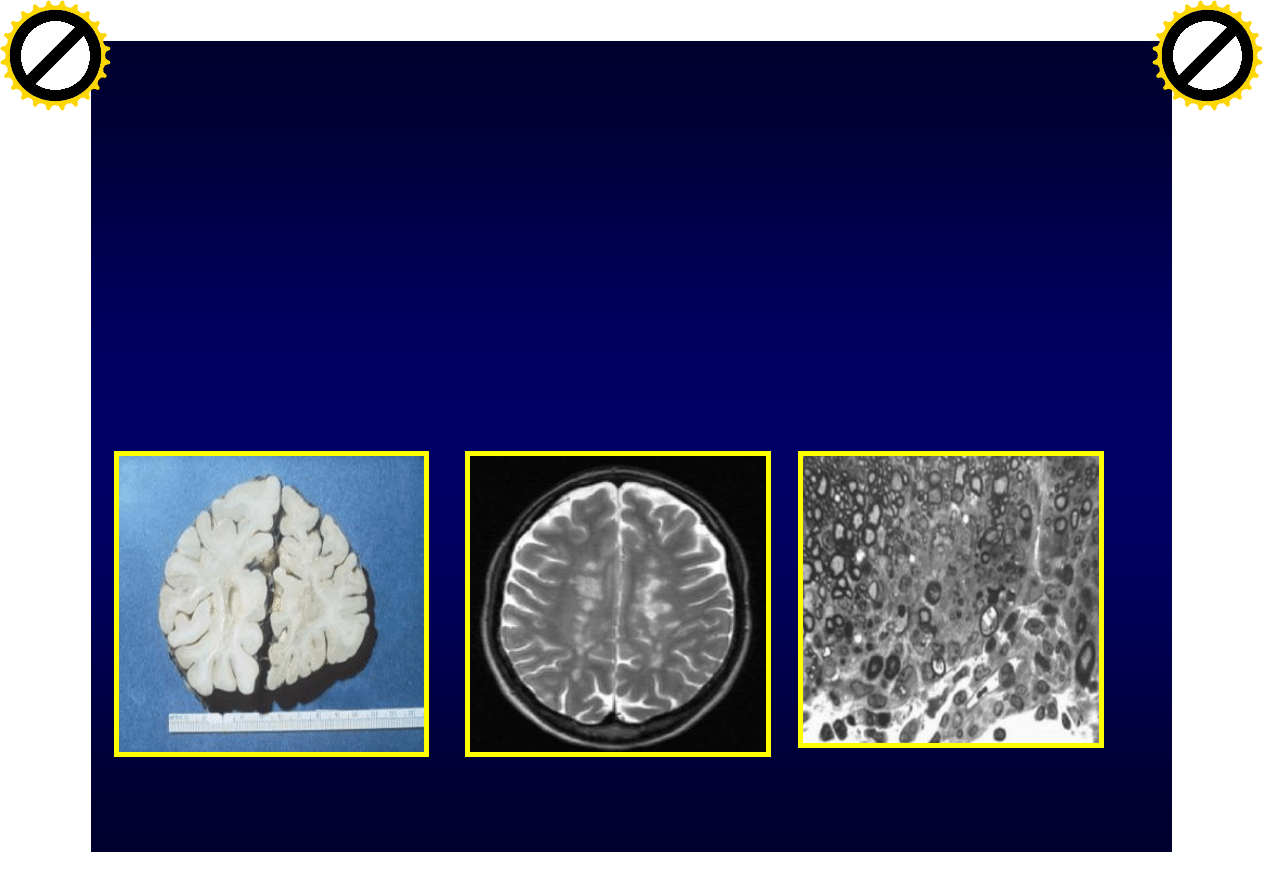
Demielinizacja ośrodkowego
układu nerwowego (plaki)
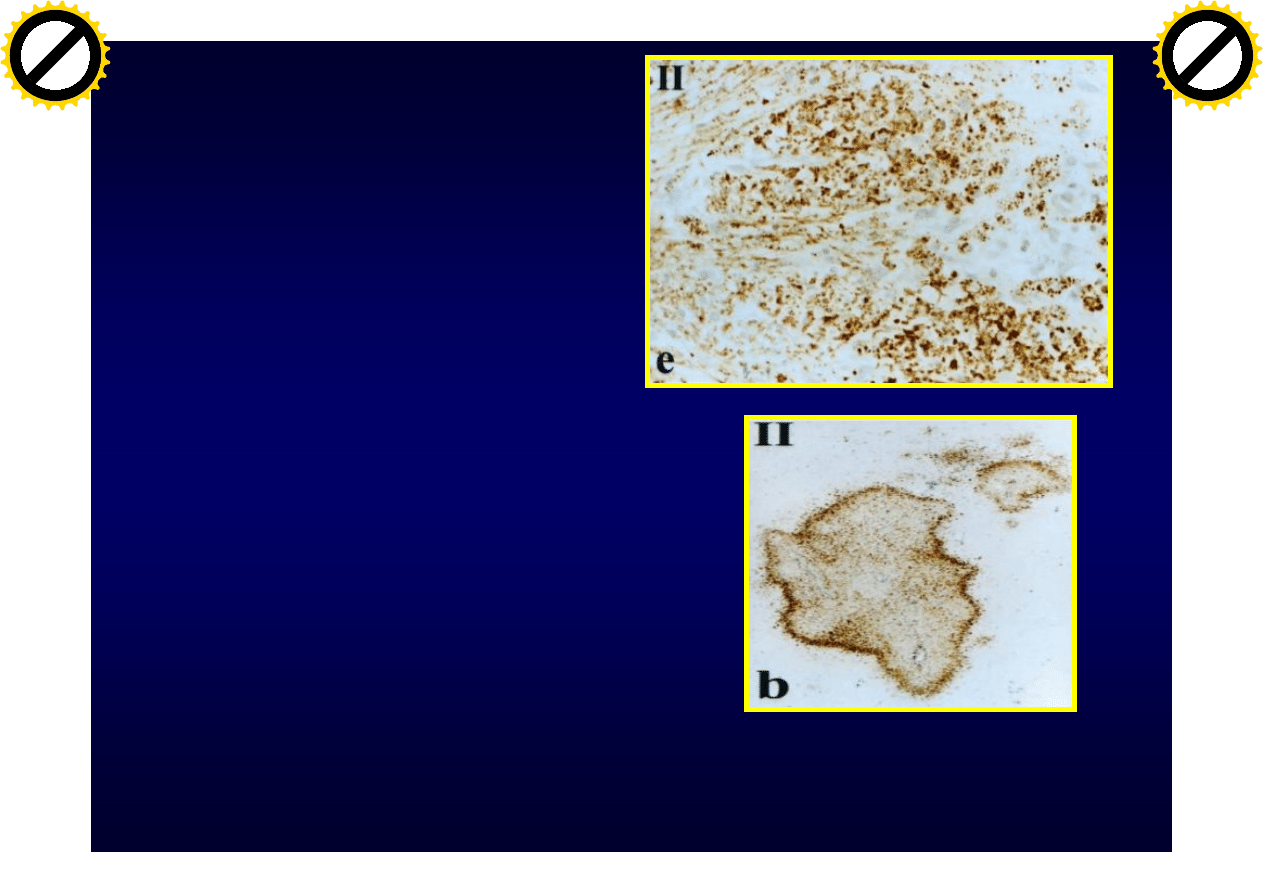
Typy plak
Typ I i II:
• Dobrze odgraniczone
• Okołonaczyniowe
• Naciek zapalny –
limfocyty i makrofagi
• Utrata wszystkich
białek mielinowych
• Istnieje remielinizacja
• W t.II złogi Ig i
komplementu
Lucchinetti C i wsp.: Ann Neurol. 2000,47:707-717

Typy plak
Typ III
• Nieostre granice
• Naciek zapalny –
limfocyty, makrofagi i
mikroglej
• Wokół naczyń
zachowana mielina
• Utrata głownie MAG
(Myelin Associated
Glycoprotein)
Lucchinetti C i wsp.: Ann Neurol. 2000,47:707-717
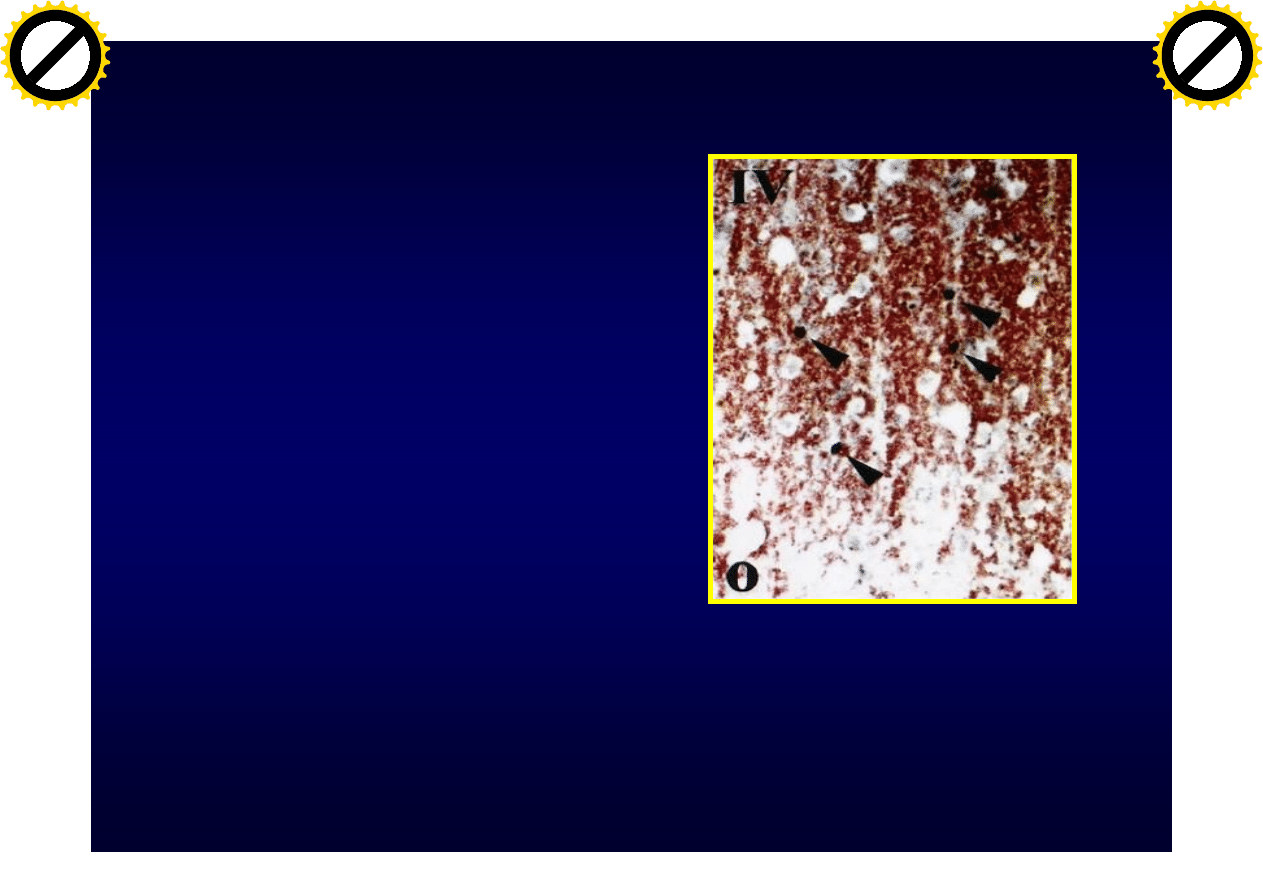
Typy plak
Typ IV
• Naciek zapalny –
limfocyty, makrofagi
• Apoptotyczne
oligodendrocyty
(fragmentacja DNA)
• Brak remielinizacji
Lucchinetti C i wsp.: Ann Neurol. 2000, 47:707-717
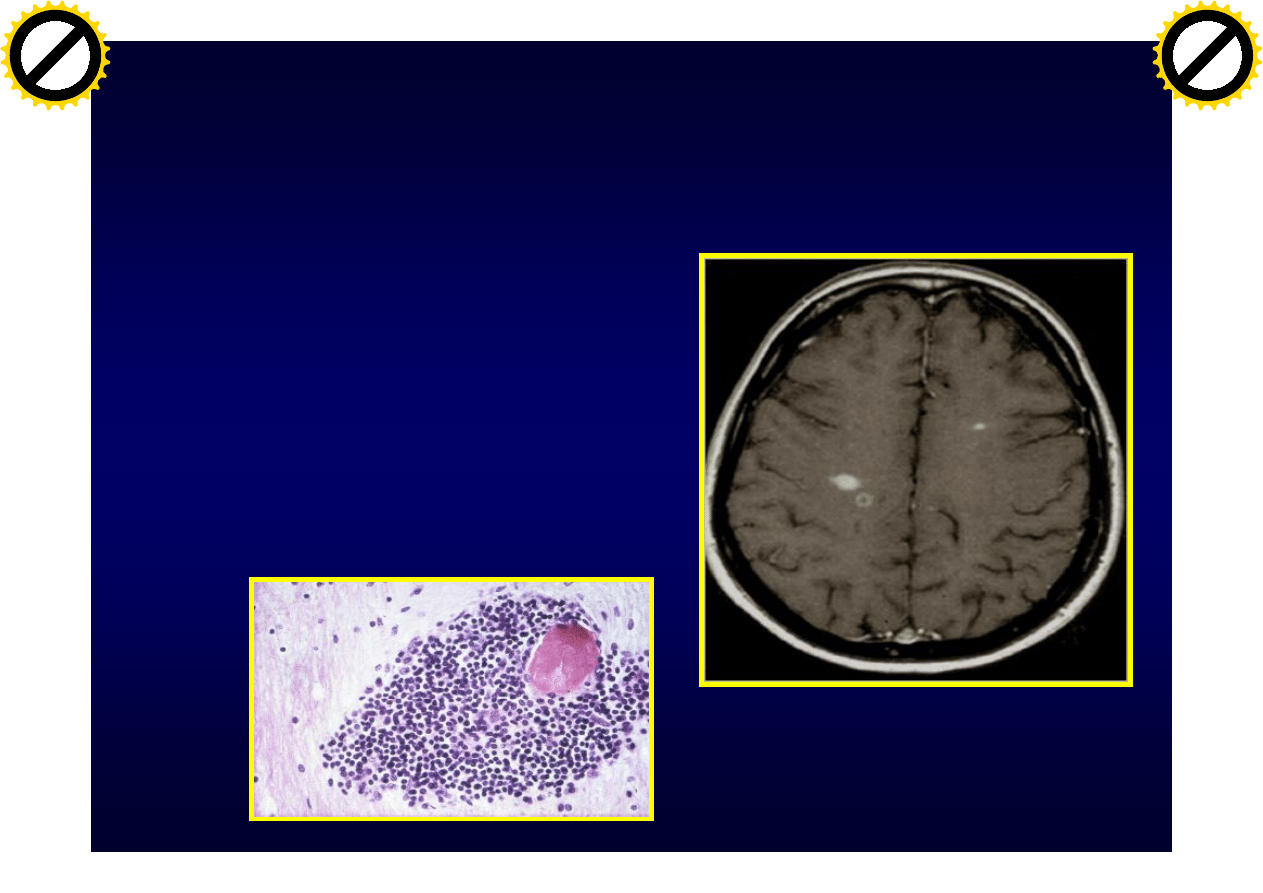
Zapalenie
• Nacieki zapalne
– limfocyty T (
ab: CD4+,CD8+ i
gd)
– limfocyty B
– mikroglej
– Makofagi
Rola komórek immunologicznych
• Autoreaktywne limfocyty T i B
– Molekularna mimikra
– Aktywacja przez inne procesy immunologiczne
bystander activation
– epitope spreading
– Degeneracja receptora TCR
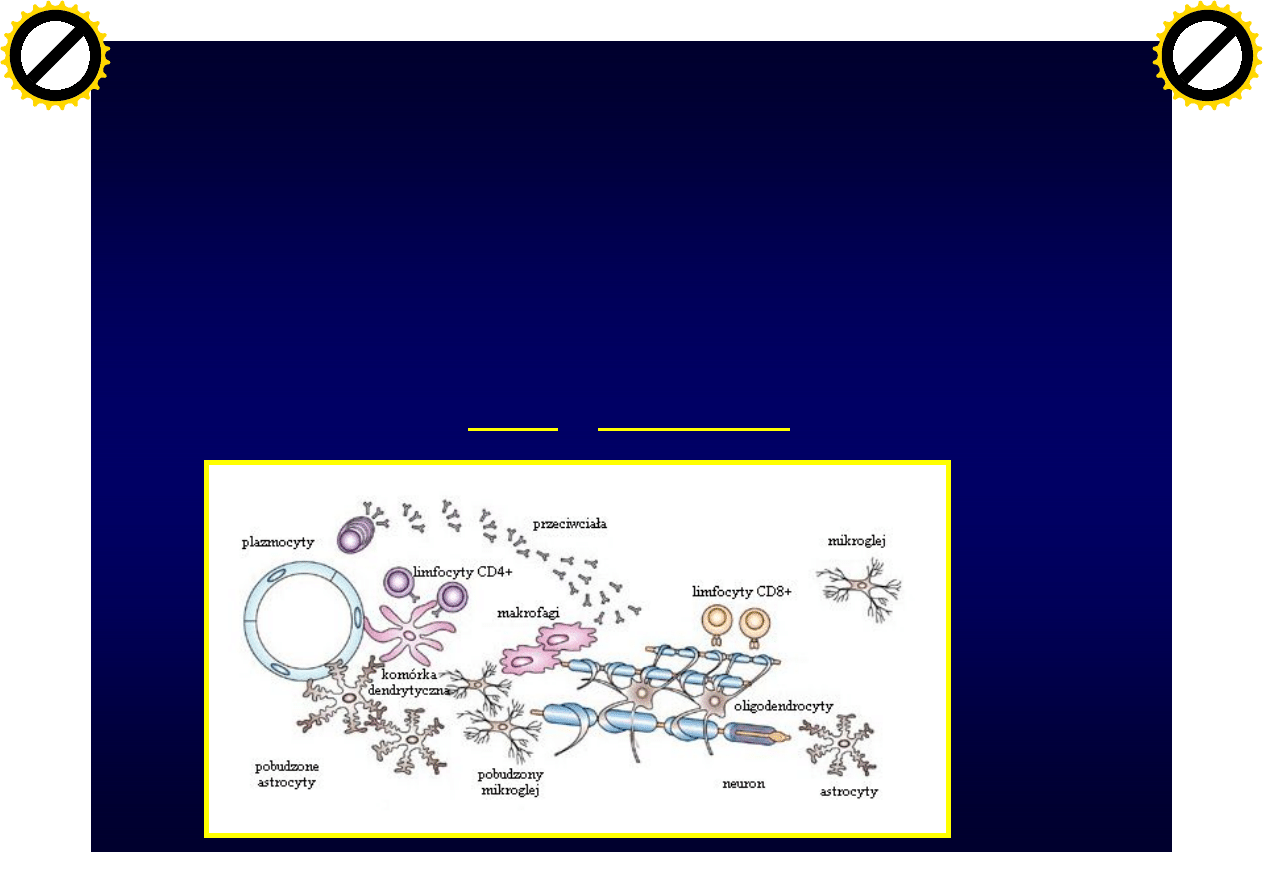
Rola komórek immunologicznych
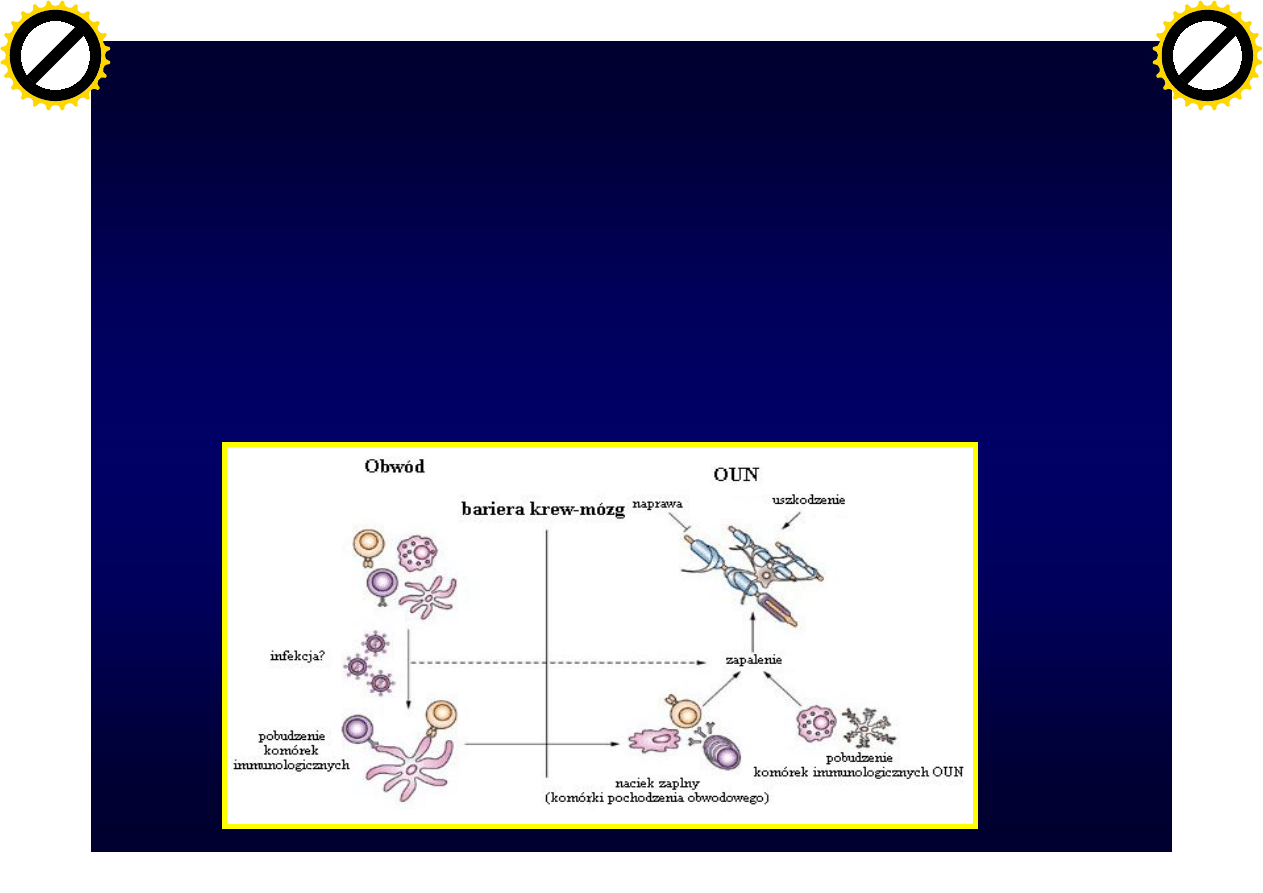
• Przejście przez barierę krew-mózg limfocytów
– Proliferacja limfocytów
– Wydzielanie cytokin prozapalnych
– Stymulacja mikrogleju i makrofagów
• EAE wywoływane przez limfocyty T rozpoznające antygeny
mielinowe (CD4+ i CD8+) i przeciwciała
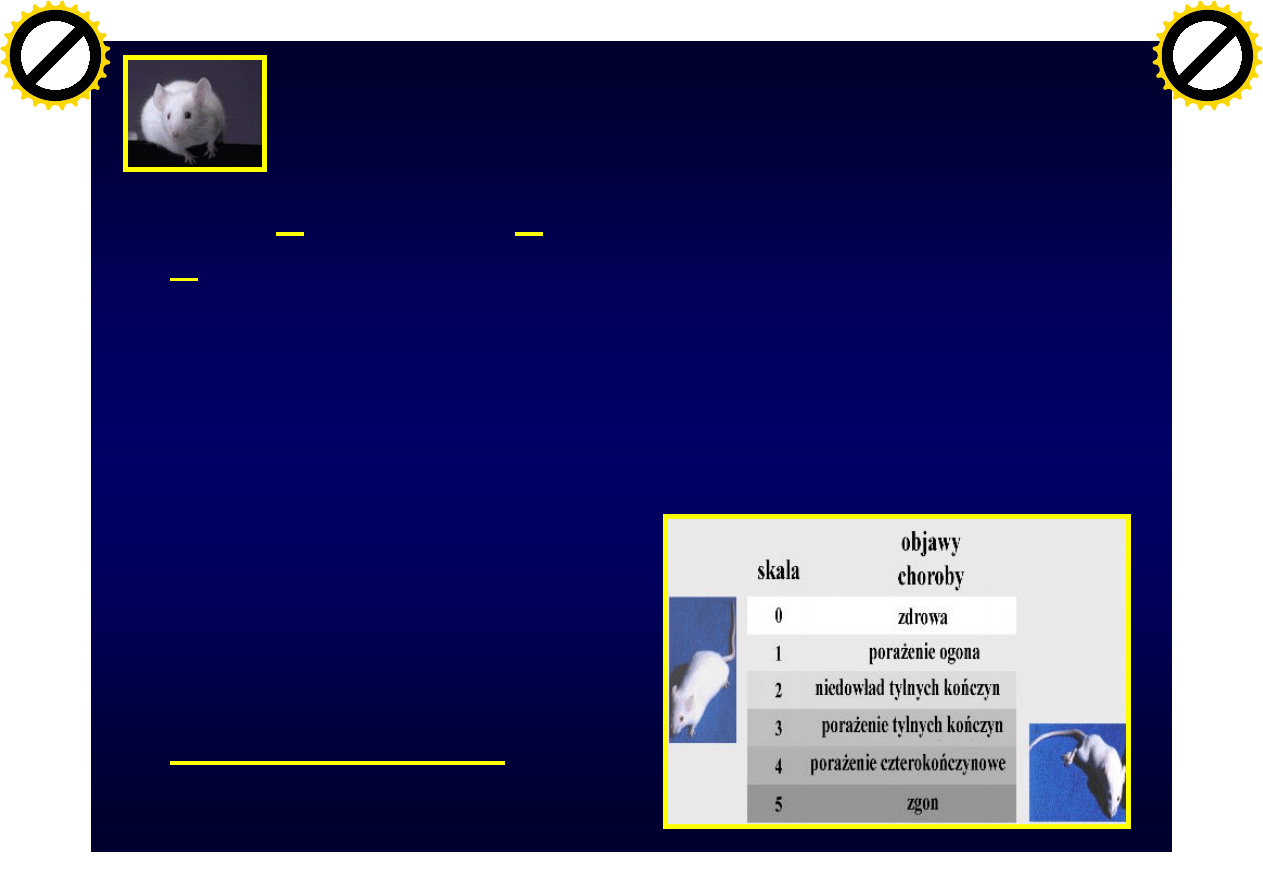
Modele zwierzęce
• EAE (Experimental Autoimmune
Encephalomyelitis) (mysz, szczur, małpy)
– uczulenie antygenami mielinowymi
– passive transfer przeniesienie choroby przez przeszczepinie
limfocytów mielinowo-reaktywnych
– Limfocyty CD4+, jak i CD8+
• Wirus Theiler TMEV
(Theiler Murine
Encephalomyelitis Virus)
– zakażenie oligodendrocytów –
odpowiedź - limfocyty T CD8+
• Toksyczna demielinizacja
– cuprizone, lysolecithine
• Różnice SM i EAE
– IFN
g i TNFa

Rola komórek immunologicznych
Ø Limfocyty T:
§
ab
hCD4+ Th1 (transfer choroby)
iCD4+ Th2, Th3
i funkcji Treg (CD4+CD25+FoxP3+) (hamowanie EAE, deplecja
zaostrzenie EAE)
CD8+ cytotoksyczne (KO dla perforyn)
CD8+ regulatorowe (deplecja)
§
gd cytotoksyczne (in vitro)
Ø NTK regulatorowa (stymulacja – hamowanie EAE)
Ø NK cytotoksyczne? regulatorowa? (deplecja
zaostrzenie EAE)
Rola komórek immunologicznych
• Limfocyty B
– prążki oligoklonalne w płynie
mózgowo-rdzeniowym
przeciwko ?
– przeciwciała przeciwko
antygenom mielinowym (MBP,
MOG, MAG, PLP)
– przeciwciała anty-lipidowe
– postać Devic’a (przeciwciała
przeciwko akwaporynie)
• Makrofagi
– Cytokiny
– NO
Neurodegeneracja
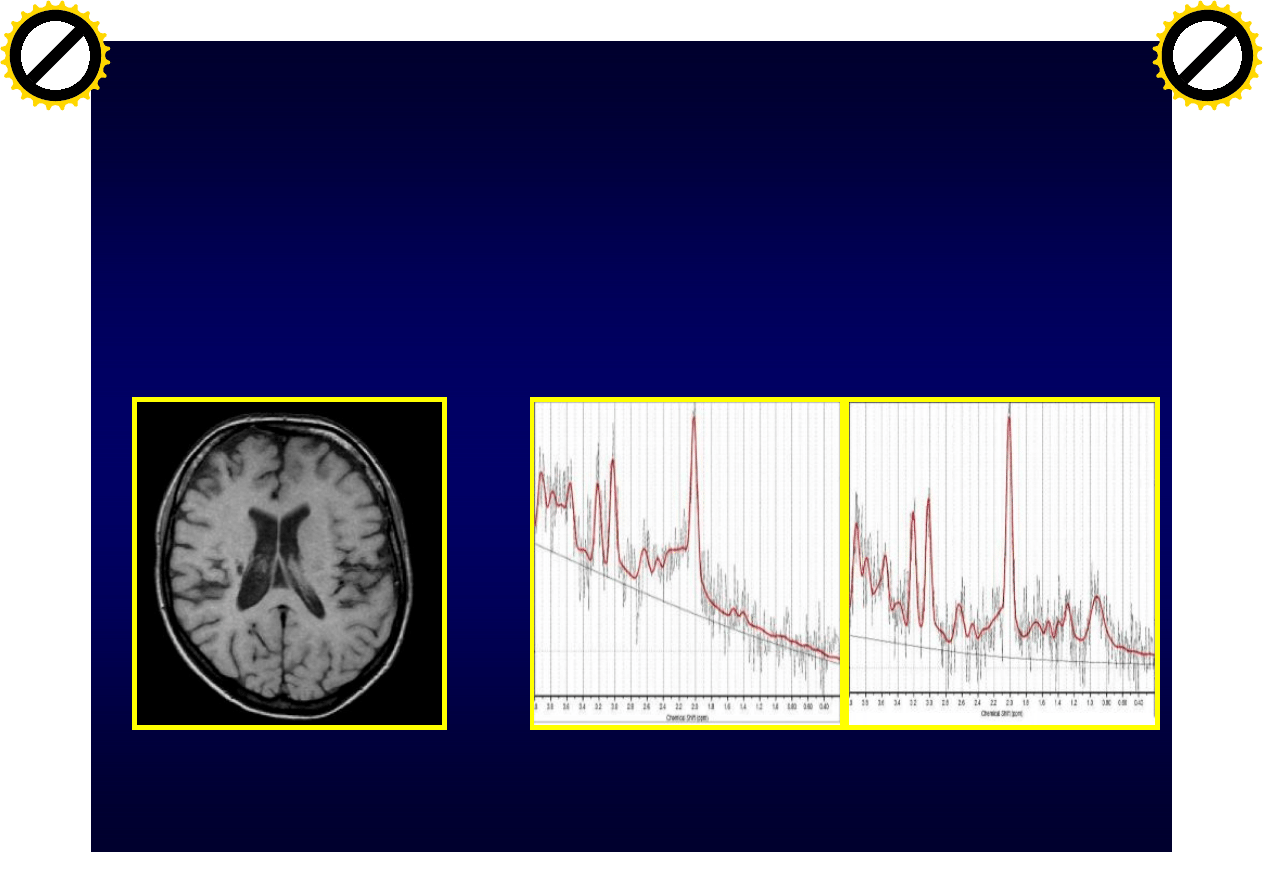
Postać pierwotnie i wtórnie postępująca SM
Pacjent z SM:Obniżony NAA,
podwyższona cholina
w stosunku do kreatyny
Spektroskopia
wodorowa
Zdrowa osoba
MRI:
T1-atrofia
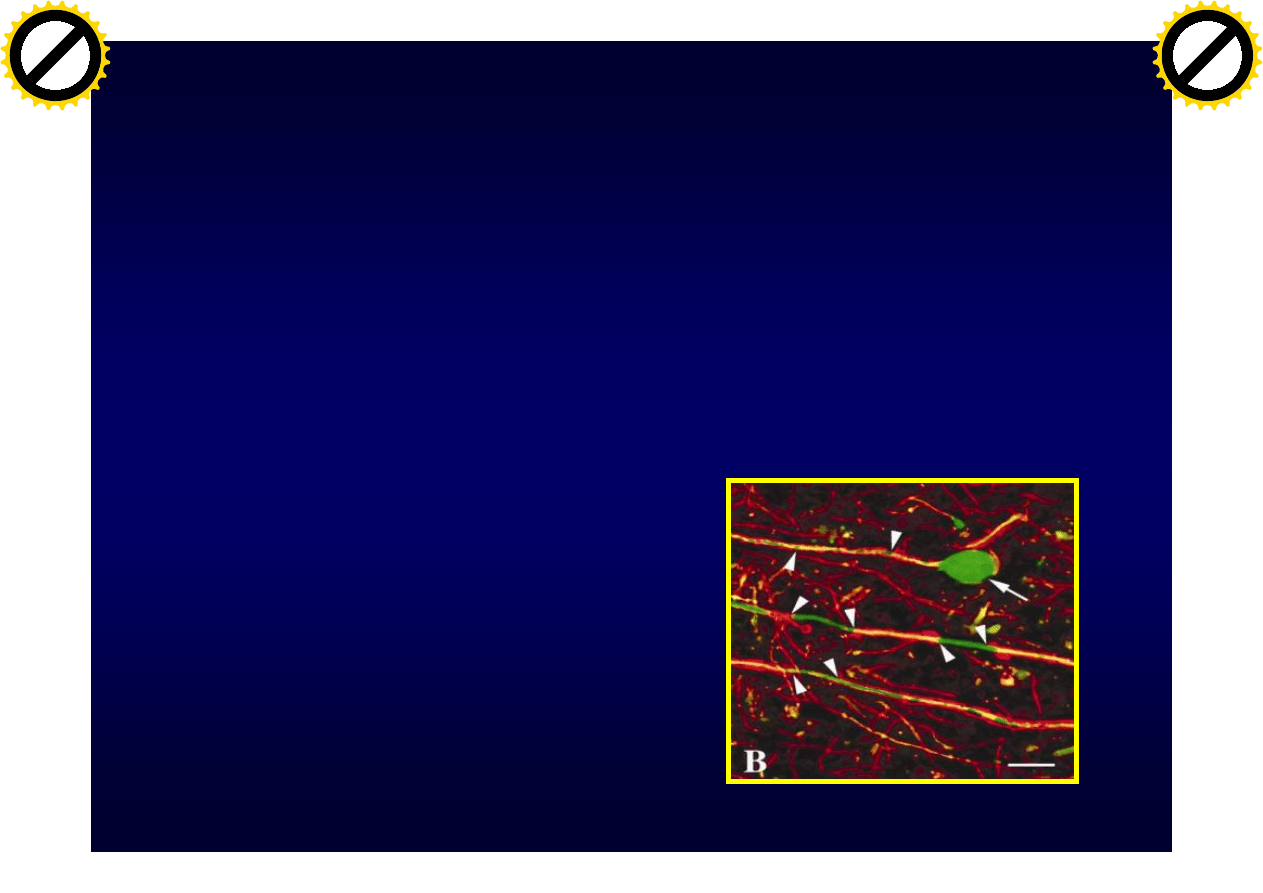
Uszkodzenie neuronów
• Demielinizacja
• Nieprawidłowa
mielina (MAG KO,
CNPase KO)
• limfocyty CD8+
(perforin KO)
• Makrofagi (ADCC)
• cytokiny
• przeciwciała
aksonopatia
Trapp BD i wsp. NEJM, 1998:338:278-85

Komórki OUN a zapalenie i
neurodegeneracja
• Neurony
– Indukują T reg FoxP3+
• Mikroglej
– Cytokiny (TNF)
– NO (uszkodzenie aksonów i oligodendrocytów) inhibitor
amniguanidyna zmniejsza objawy EAE
– Glutaminiany (AMPA receptor) antagoniści AMPA w
EAE –zmniejszają uszkodzenie aksonów i
oligodendrocytów (NMDA na oligodendrocytach)
• Astrocyty
– glioza

Czy zapalenie jest szkodliwe?
• Modele z uszkodzeniem oun
– Limfocyty MBP reaktywne zmniejszają
uszkodzenie neuronów zwojowych siatkówki
– Autoreaktywne limfocyty T powstają po
uszkodzeniu rdzenia i wywołują EAE po
podaniu „naiwnym” szczurom
• Czynniki troficzne BDNF

Związek zapalenia i neurodegeneracji
1. Zapalenie indukuje procesy neurodegeneracyjne
2. Neurodegeneracja indukuje zapalenie
3. Zapalenie i neurodegeneracja indukują się
wzajemnie
4. Zapalenie chroni przed neurodegeneracją
5. Nieznane czynniki indukują zapalenie i
neurodegenerację
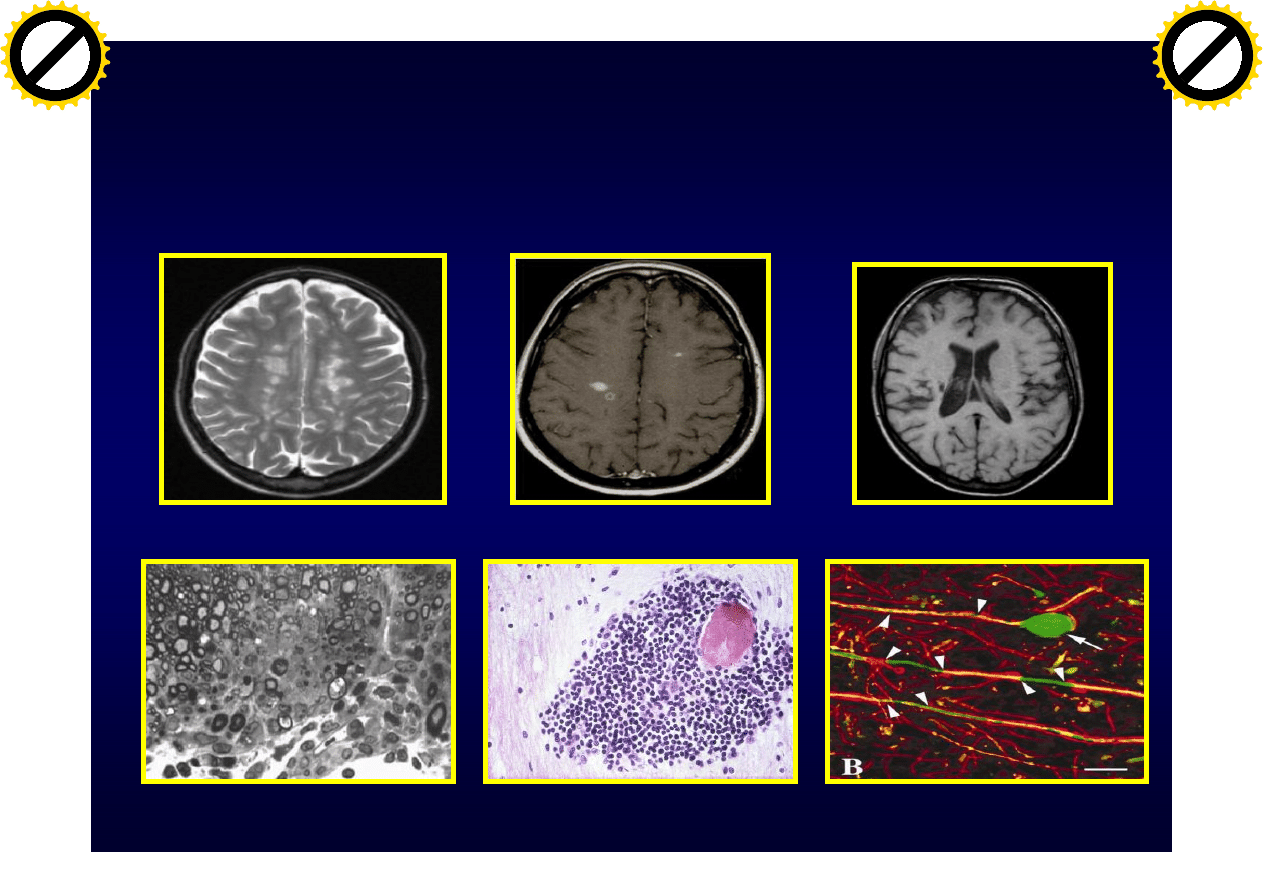
Stwardnienie rozsiane (SM)
Choroba
MRI: T2
MRI: T1 wzmocnienie
MRI: T1 atrofia
demielinizayjna
zapalna
neurodegeneracyjna
Trapp BD i wsp. NEJM, 1998:338:278-85
Gałązka G i wsp. J Immunol 2006:176;1588-99

Czy istnieje marker SM?
Czy istnieją markery aktywności
choroby?

Jakie są kryteria rozpoznania
stwardnienia rozsianego ?

Kryteria McDonalda (1)
• 2 rzuty z klinicznymi objawami 2 różnych
miejsc uszkodzenia
• 2 rzuty lub więcej z klinicznymi objawami
1 miejsca uszkodzenia
– „rozsianie miejsc” udowodnione badaniem
MRI
– 2 lub więcej zmian w MRI + PMR
– lub nowy dane na inne miejsce uszkodzenia

• 1 rzut kliniczne objawy z 2 lub więcej
miejsc uszkodzenia
– „rozsianie” w czasie potwierdzone MRI
– lub drugi rzut
• 1 rzut, 1 miejsce uszkodzenia (CIS -
clinically isolated syndrome)
– „rozsianie miejsc” potwierdzone MRI
– 2 lub więcej uszkodzeń w MRI + PMR
– „rozsianie” w czasie potwierdzone MRI
– lub drugi rzut
Kryteria McDonalda (2)

Kryteria McDonalda (3)
• Przebieg postępujący klinicznie sugerujący
SM
– PMR (nie w nowelizacji z 2005)
– i -„rozsianie miejsc” udowodnione badaniem
MRI lub VEP
– i - „rozsianie” w czasie potwierdzone MRI
– albo przebieg postępujący przez kolejny rok

Badanie płynu mózgowo-
rdzeniowego w świetle kryteriów
McDonalda i nowelizacji 2005r.
• Jest badaniem dodatkowym mającym
uwiarygodnić rozpoznanie SM

Dlaczego ważne jest wczesne i
pewne rozpoznanie SM?
• Wczesne włączenie leczenia przy pewnym
rozpoznaniu SM
– Zwolnienie przebiegu choroby
– Dłuższy okres sprawności chorego
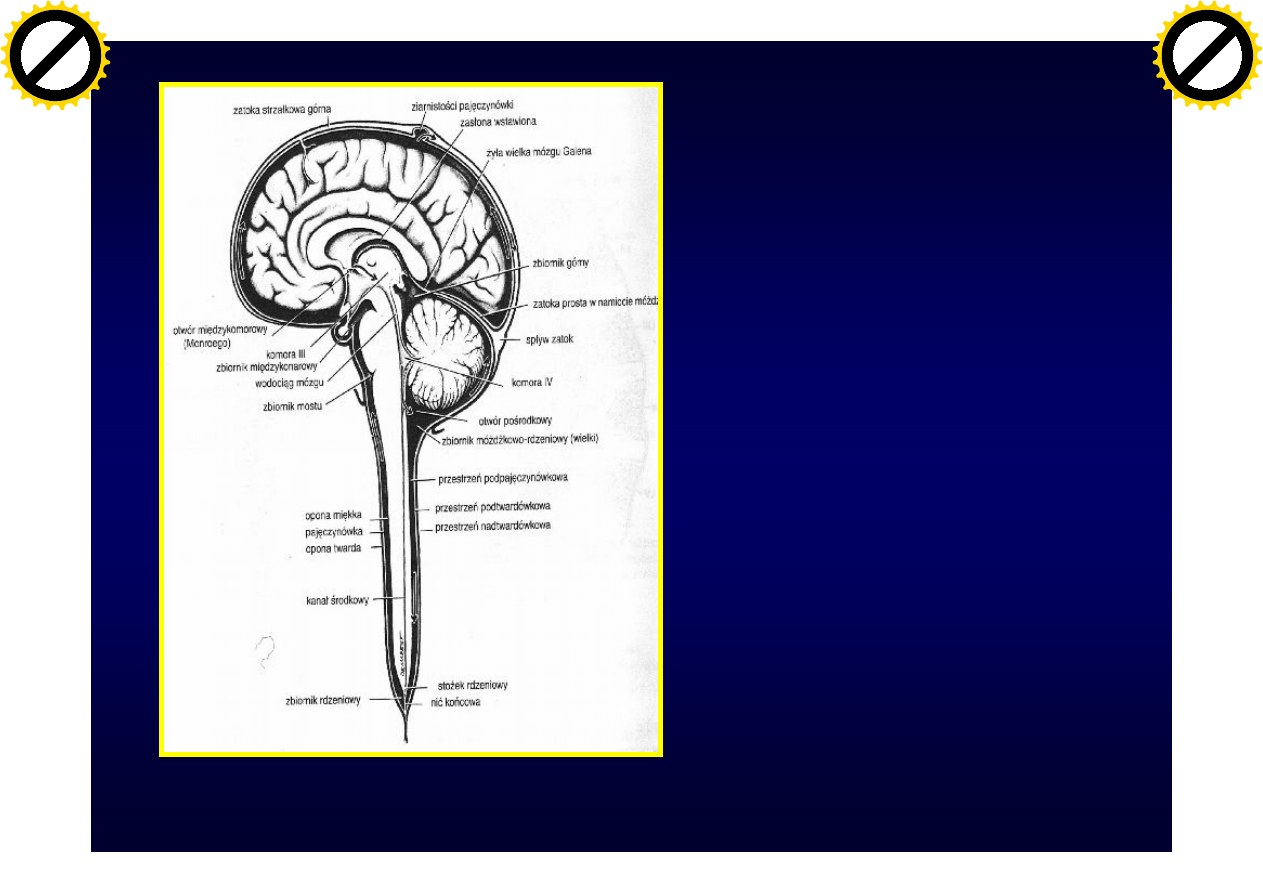
Płyn mózgowo-
rdzeniowy (PMR)
wytwarzany jest
splotach
naczyniówkowych
komór bocznych,
trzeciej i czwartej
(„gap junction”)
Neuroanatomia, Fix JD, Wrocław 1997
Krążenie
PMR w
obrębie oun
(gradient
stężenia
białek)
Neuroanatomia, Fix JD, Wrocław 1997
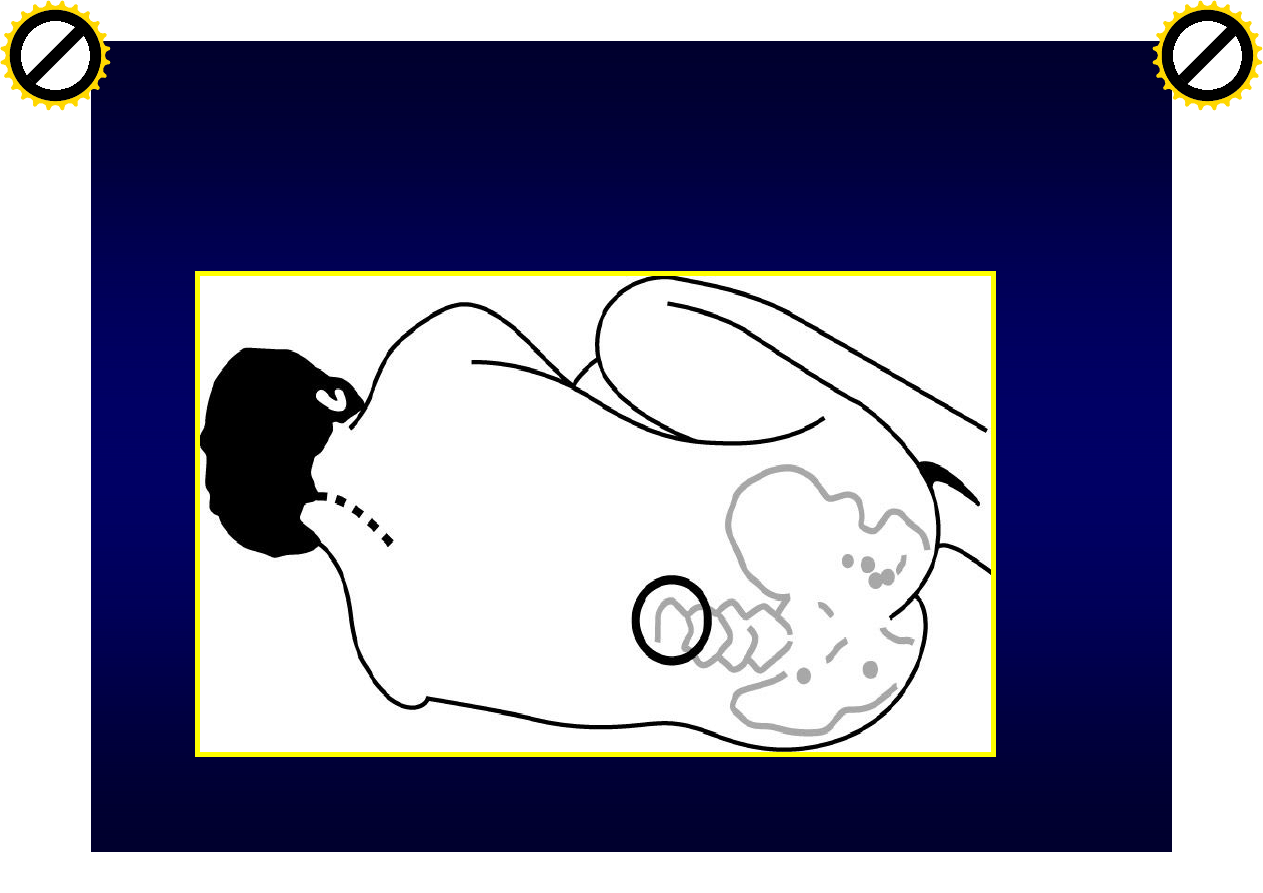
Punkcja lędźwiowa (PL)

Jakie są przeciwwskazania do
punkcji lędźwiowej?

Przeciwwskazania do PL
1. Stan zapalny w miejscu wkłucia
2. Wzmożone ciśnienie śródczaszkowe
3. Zaburzenia krzepnięcia
–
wrodzone
–
polekowe

Pochodzenie białek PMR
• Dyfuzja lub aktywny transport z surowicy
poprzez barierę krew-mózg i krew-płyn
mózgowo-rdzeniowy (ok. 90% białek)
(albumina produkowana wyłącznie w
wątrobie)
• Wytwarzane w obrębie oun (intrathectal
syntesis) np. transtyretyna, prostaglandyna
D, cystatyna C, MBP, białko tau, syntetaza
glutaminowa

Wzrost stężenia białek w PMR
1. Uszkodzenie bariery krew-mózg lub krew-
płyn mózgowo-rdzeniowy (wzrost stężenia
wszystkich białek, w tym albuminy)
•
W dużym stopniu – ropne zapalenie opon
•
W średnim stopniu – zespół Guillain-Barre,
neuroborelioza
•
W małym stopniu – wirusowe zapalenie opon,
polineuropatie
2. Synteza wewnątrzpłynowa (np.
immunoglobuliny)

Albumina w PMR
• Albumina wytwarzana jest tylko w wątrobie, w
związku z tym może służyć jako marker
„szczelności” bariery krew-mózg.
• Stężenie albuminy w PMR wynosi około 34mg/dl
w warunkach prawidłowych.
• Wskaźnik stężenia albuminy w PMR do stężenia
w surowicy zależy od wieku i wynosi
– 5,0 poniżej 15 roku życia,
– 6,5 pomiędzy 16 a 40 r.ż.,
– 8 pomiędzy 40 a 60 r.ż.
– 8-9 powyżej 60 r.ż.

Stężenie albuminy w PMR
• Stężenie albuminy w PMR
jest zależne
– od miejsca pobrania PMR
(gradient komory –
okolica lędźwiowa –
2,2x)
– od „szczelności” bariery
krew-mózg
Neuroanatomia, Fix JD, Wrocław 1997

Immunoglobuliny w PMR
• W obrębie rodziny immunoglobulin
wyróżnia się 5 klas: IgA, IgG, IgM, IgD,
IgE
• Dominującą klasą w PMR jest klasa IgG
• Stężenie immunoglobulin w PMR wynosi
do 40 mg/l

Wzrost stężenia
immunoglobulin w PMR
• Może wynikać
– ze zwiększonej przepuszczalności bariery krew-
mózg
– ze zwiększonego stężenia w surowicy
– ze zwiększonej syntezy wewnątrzpłynowej
• Do oceny pochodzenia immunoglobulin
stosuje się indeks IgG

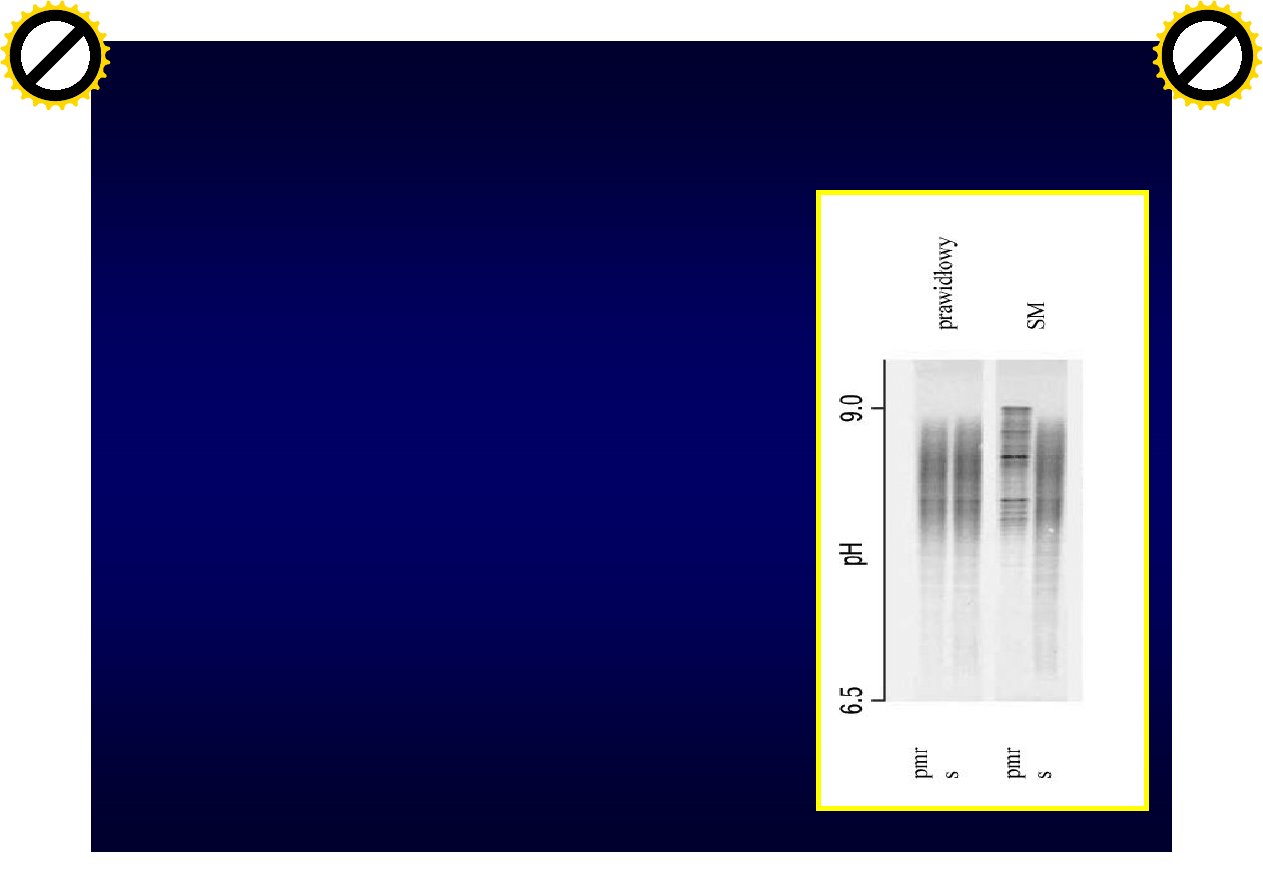
Uznane markery SM w PMR
ØIndeks IgG
ØPrążki oligoklonalne

Indeks IgG
Ø Prawidłowa wartość indeksu IgG < 0,72
Ø Podwyższony u 78% chorych na SM

Metoda oceny prążków
oligoklonalnych
• Zalecaną metodą oceny prążków
oligoklonalnych jest ogniskowanie
izoelektryczne
• Należy stosować niezagęszczony PMR
• Równolegle wykonywać badanie dla
PMR i surowicy pobranych w tym
samym czasie
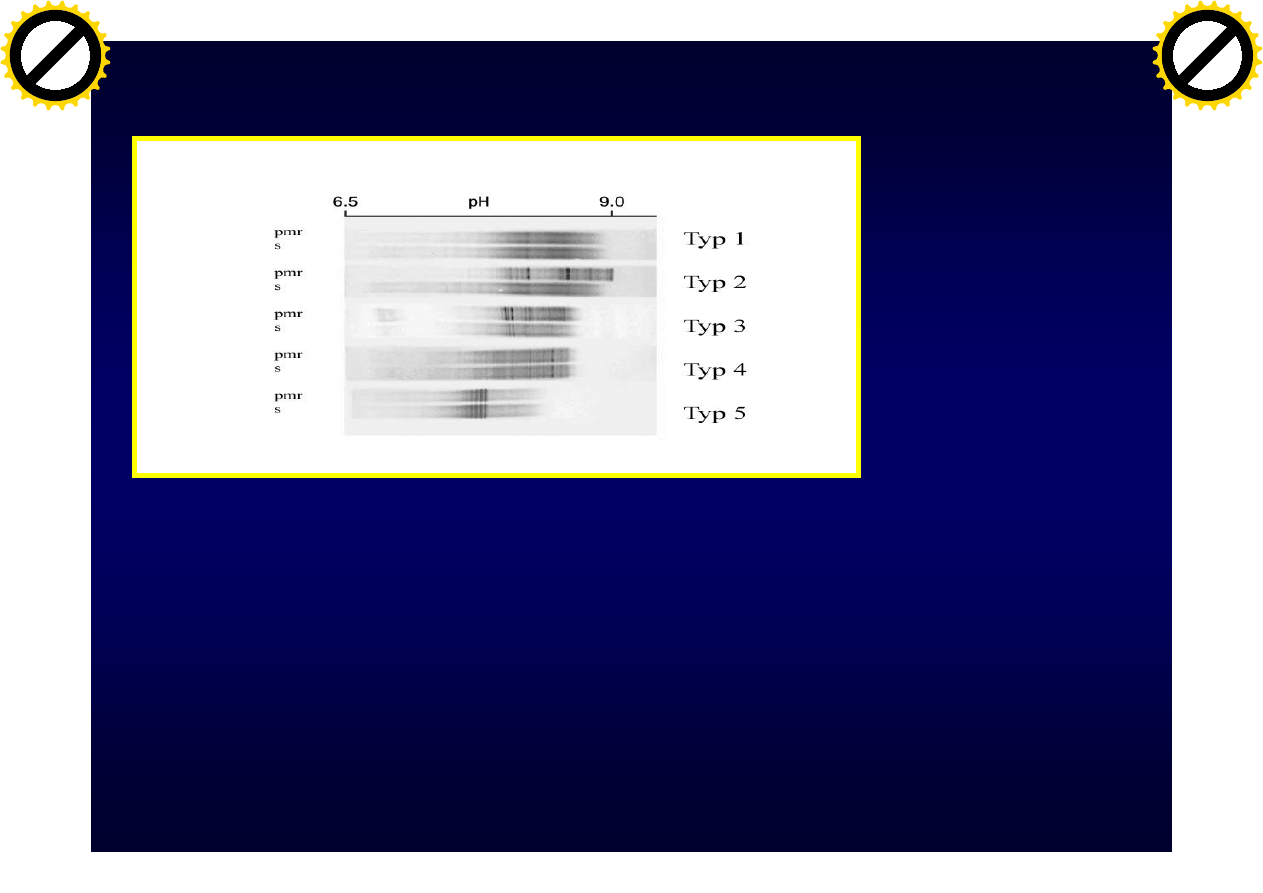
Prążki oligoklonalne w PMR
• Typ 1- prawidłowy, brak prążków oligoklonalnych
• Typ 2 - prążki oligoklonalne w pmr ale nie w surowicy
(synteza wewnątrzpłynowa)
• Typ 3 - prążki oligokonalne w pmr i dodatkowe w surowicy
(synteza wewnątrzpłynowa)
• Typ 4 - identyczne prążki oligoklonalne w pmr i surowicy
(nie wskazują na syntezę wewnątrzpłynową)
• Typ 5 - prążki monoklonalne w pmr i w surowicy
Ø Andersson i
wsp., J Neurol
Neurosurg
Psychiatry 1994

Indeks IgG i prążki oligoklonalne
üIndeks IgG jest podwyższony u 78%
chorych na SM
üPrążki oligoklonalne
• stwierdzane są u 98% chorych na SM
• ich występowanie jest stałe i niezmienne u
danego chorego na SM, w związku z tym nie
może służyć do oceny nasilenia procesu
chorobowego, ani efektu leczenia
immunomodulacyjnego

Produkcja immunoglobulin w PMR
ØLimfocyty B
– przechodzą przez barierę krew- mózg i
najprawdopodobniej ulegają niespecyficznej
stymulacji, przekształcają się w komórki
plazmatyczne produkujące immunoglobuliny
(synteza wewnątrz ośrodkowego układu
nerwowego)
– mogą produkować inne klasy immunoglobulin IgM,
IgA

Immunoglobuliny klasy M i A
• Mogą występować w PMR chorych na SM,
jednak są dość rzadkie przypadki a
dominująca synteza tych klas
immunoglobulin powinna budzić
wątpliwości rozpoznania SM
• Występowanie IgM, zwłaszcza
rozpoznających lipidy, ma świadczyć o
cięższym przebiegu SM

Przeciwciała przeciwko odrze,
różyczce i herpes zoster w PMR w SM
• Zwiększoną syntezę przeciwciał przeciwko
patogenowi ocenia się poprzez indeks
przeciwciał
• Indeks > 1,5 świadczy o wewnątrzpłynowej
syntezie przeciwko patogenowi.
• U chorych na SM stwierdza się
wewnątrzpłynową syntezę przeciwciał
przeciwko różyczce, odrze i herpes zoster
(objawy MRZ)

Osad PMR w SM
• Pleocytoza poniżej 30 komórek/ul
• Stwierdza się występowanie limfocytów T,
limfocytów B i komórek plazmatycznych
• W PMR stwierdza się komórki plazmatyczne u
79% chorych

Markery aktywności procesu
chorobowego

Markery zapalenia (1)
• Cytokiny
– prozapalne
• IL-2 rzut + ;
• IL-6 rzut +, progresja +, IFNβ $;
• IL-12 rzut +, progresja ++;
• IFN-γ rzut +, progresja ++, IFNβ $;
• TNF-α rzut +, progresja ++, sterydy $, IFNβ #/ $;
– hamujące zapalenia
• IL-10 remisja +, progresja +, IFNβ #;
• TGF-β rzut +, progresja ++, remisja #

Markery zapalenia (2)
• Receptory dla cytokin
– IL-2R rzut +,
– IL-6R rzut +, progresja +, IFNβ $;
– TNF-αR rzut +, progresja +;
• Molekuły adhezyjne
– ICAM-1 rzut +, progresja -, sterydy $;
– VCAM-1 rzut +, sterydy $;
– VLA-4 rzut +, progresja -, sterydy $;
– LFA-1 rzut +, progresja -, sterydy $;
– selektyna E rzut +, progresja ++, sterydy $;

Markery zapalenia (3)
• Immunoglobuliny (rozpoznające białka
mielinowe, lipidy) (w rzucie przeciwciała
anty-MBP)
• Składniki układu dopełniacza
• Chemokiny
• Limfocyty T, B i komórki plazmatyczne

Markery uszkodzenia bariery
krew-mózg
• Metaloproteazy
– MMP-9
• podwyższone stężenia także w innych chorobach
zapalnych oun
• rzut +, remisja +, progresja ++, IFNβ $;
• Inhibitory metaloproteaz (TIMP)

Markery demielinizacji
• Białka strukturalne osłonki mielinowej
– MBP (myelin basic protein) rzut +, progresja
++, sterydy +;
– PLP (proteolipid protein)
– MOG (myelin oligodendrocyte glycoprotein)
– MAG (myelin associated glycoprotein)

Markery neurodegeneracji
• Białka neuronalne
– Białko tau rzut +, remisja +, progresja ++;
– Białko 14-3-3
– Neurofilamenty (związek z rzutem choroby i
stopniem niesprawności)
– Enolaza specyficzna dla neuronów

Markery gliozy
• Białka astrocytarne
– GFAP (glial fibrillary acidic protein)
• wyższe stężenie koreluje z wyższą oceną w skali
EDSS
• progresja ++
– S-100b
• podwyższone stężenie w rzucie choroby (do 5
tygodni)
• rzut +, progresja +

Markery regeneracji
• Markery neuronów
– N-CAM (neuronal cell adhesion molecule) remisja +;
• Czynniki wzrostowe
– CNTF (ciliary neurotrophic factor) remisja +;
– BDNF (brain derived neurotrophic factor) remisja +;
– NT3 (neurotrophin 3)
– NGF (nerve growth factor)

Inne choroby demielinizacyjne
• ADEM (acute demyelinating encephalomyelitis)
– często podwyższona pleocytoza i podwyższone stężenie
białka (cechy ostrego zapalenia)
– rzadko prążki oligoklonalne
• Choroba Devic’a (Neuromyelitis optica)
– indeks IgG prawidłowy, prążki oligoklonalne
przejściowo słabo widoczne u 30-50% chorych
– stwierdzono ostatnio przeciwciała przeciwko kanałowi
wodnemu aquaporin-4, charakterystyczne dla choroby
Devic’a (65% chorych)

PMR w innych chorobach (1)
• Encefalopatie naczyniowe –PMR 30% #białko
(
różnicowanie z SM o późnym początku
)
• Udar niedokrwienny (faza ostra)- #pleocytozy
(granulocyty)
• Zapalenie naczyń OUN - #pleocytozy #białko
• Periarteritis nodosa – 30% objawy zajęcia oun,
niewielki#pleocytozy (limfocyty) i #białko
• Ziarniniak Wegenera - 25% objawy zajęcia oun,
niewielki #pleocytozy (limfocyty) i #białko,
mogą
pojawić się prążki oligoklonalne i przeciwciała
przeciwko odrze, różyczce i herpes zoster
,
dodatnie przeciwciała ANCA
(antineutrophil
cytoplasmic antibodies)

PMR w innych chorobach (2)
• Choroba Churg- Straussa - 20% objawy zajęcia oun,
niewielka pleocytoza (eozynofile)
• Choroba Behceta - 10% objawy zajęcia oun,
#pleocytozy (początkowo granulocyty, później
monocyty), wewnątrzpłynowa synteza
immunoglobulin
rzadko IgG, głównie IgA lub IgM,
55% prążki oligoklonalne – ustępują po leczeniu
sterydami
• SLE – 60% objawy zajęcia oun, 30%#pleocytozy,
wewnątrzpłynowa synteza immunoglobulin, głównie
IgG, rzadziej IgM, wyjątkowo IgA,
30-80% chorych
ma podwyższony indeks IgG i obecne prążki
oligoklonalne, ustępują po leczeniu sterydami

PMR w innych chorobach (3)
• Zespół Sjogrena - 20% objawy zajęcia oun, 50%
#pleocytozy (granulocyty, limfocyty),
70%
wewnątrzpłynowa synteza IgG, 90%
wewnątrzpłynowa synteza IgM, 90% - prążki
oligoklonalne ustępują po leczeniu sterydami
,
mogą być obecne przeciwciała przeciwko odrze,
różyczce i herpes zoster.
• Neurosarkoidoza - 80% ma zmiany w PMR,
#pleocytozy (limfocyty),
50-70% - prążki
oligoklonalne i # indeks IgG, ustępują po
leczeniu sterydami

PMR w innych chorobach (4)
• Neuroborelioza - #pleocytozy, #białka,
#limfocytów B i plazmocytów, charakterystyczne
przeciwciała anty-Borelia klasy IgM,
• Kiła ośrodkowego układu nerwowego -
#pleocytozy ( komórki jednojądrowe), #białka,
#limfocytów B i plazmocytów (głównie w
porażeniu postępującym), #IgG, IgM i IgA,
90%
prążki oligoklonalne i # indeks IgG,

SM jest chorobą heterogenną
• Heterogennośc kliniczna
• Heterogenność obrazu MRI
• Heterogenność obrazu patologicznego
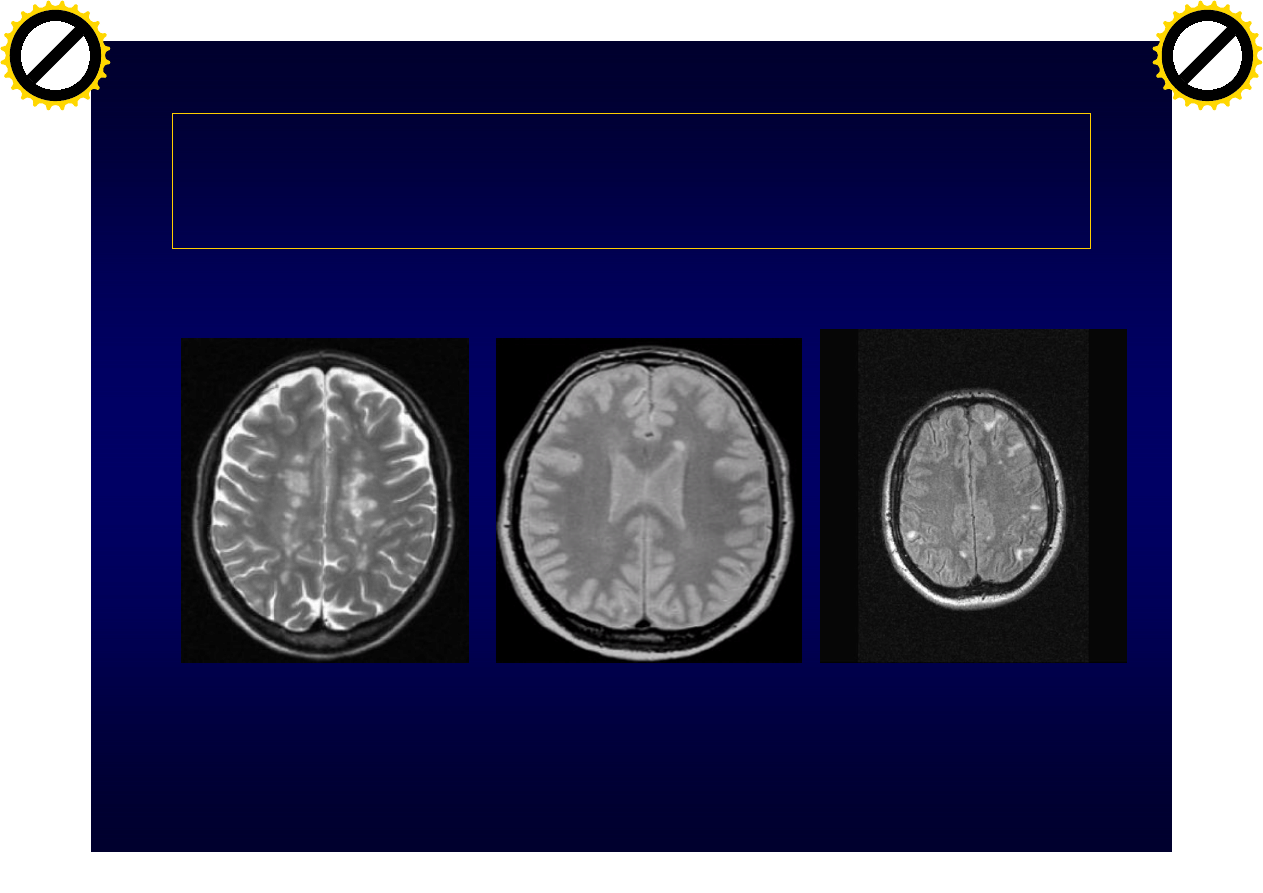
MRI heterogeneity

Kliniczne zróżnicowanie SM
• Postać łagodna
• Postać złośliwa
• Neuromyelitis optica
• Postać późna
• Postać Balo
• Postać rdzeniowa
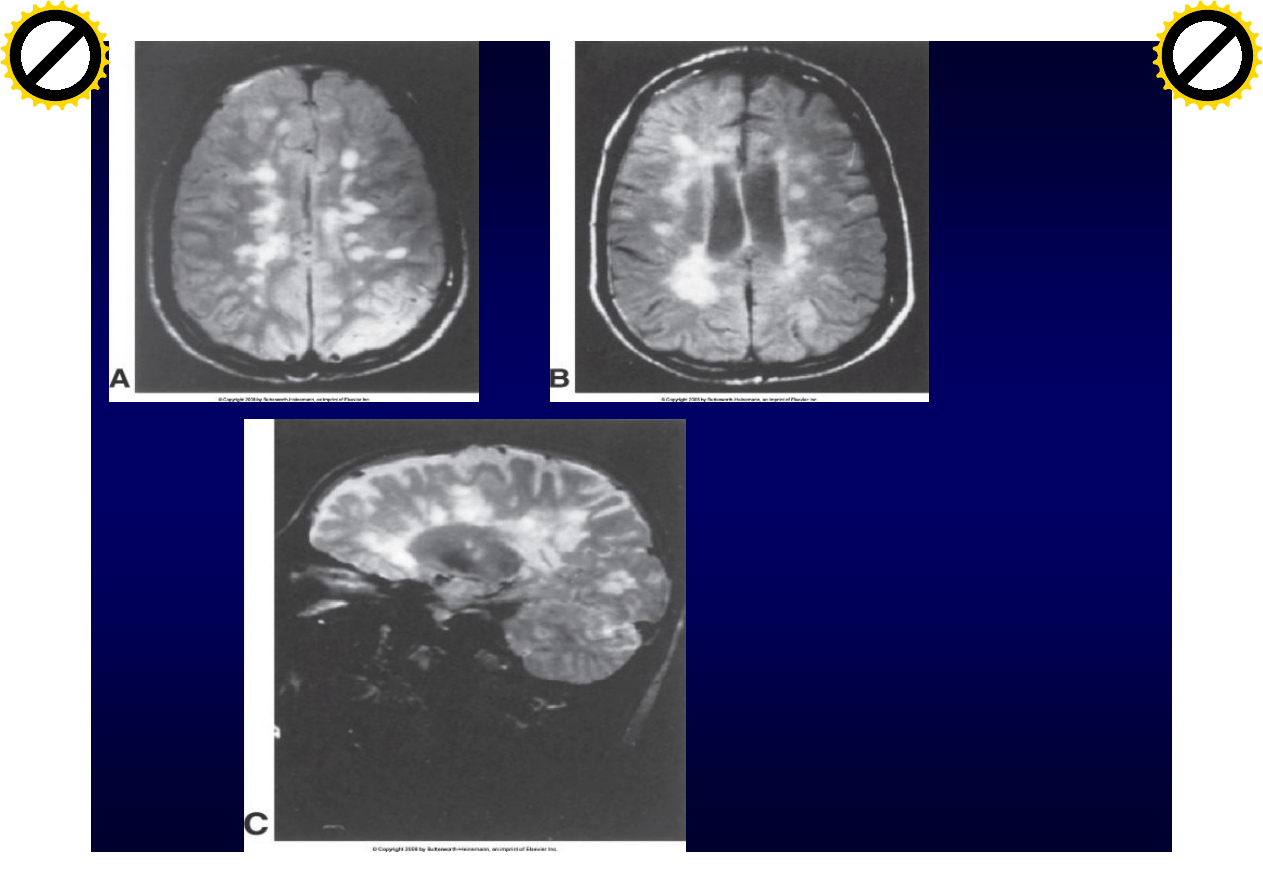
MRI
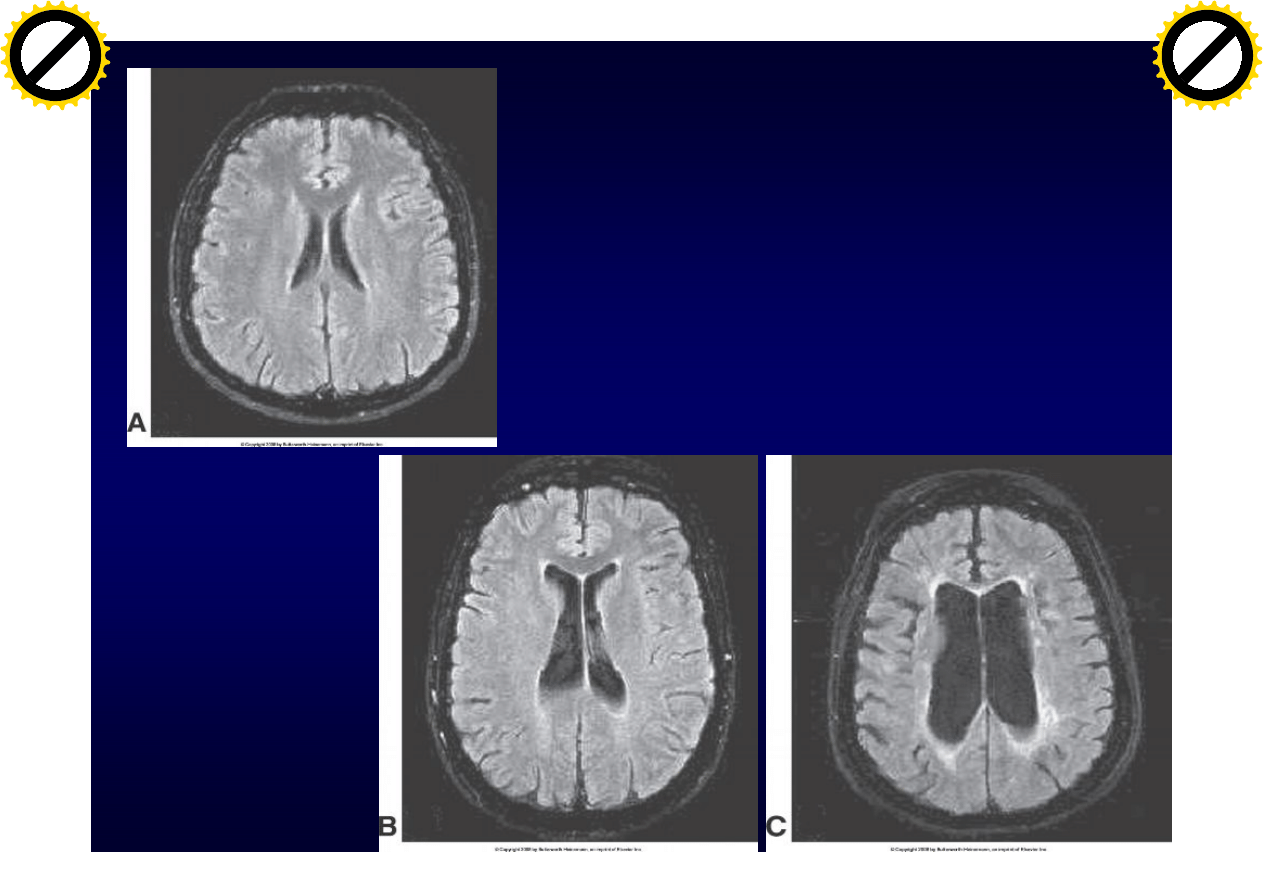
Atrofia

Ocena stanu neurologicznego w SM
Ø Kliniczna:
Ø Skala EDSS (od 0 do 10) - ocena:
ØUkład piramidowy
ØMóżdżek
ØPień mózgu
ØUkład czuciowy
ØZwieracze
ØWzrok
ØWyższe czynności mózgowe
Ø MSFC
Ø MRI
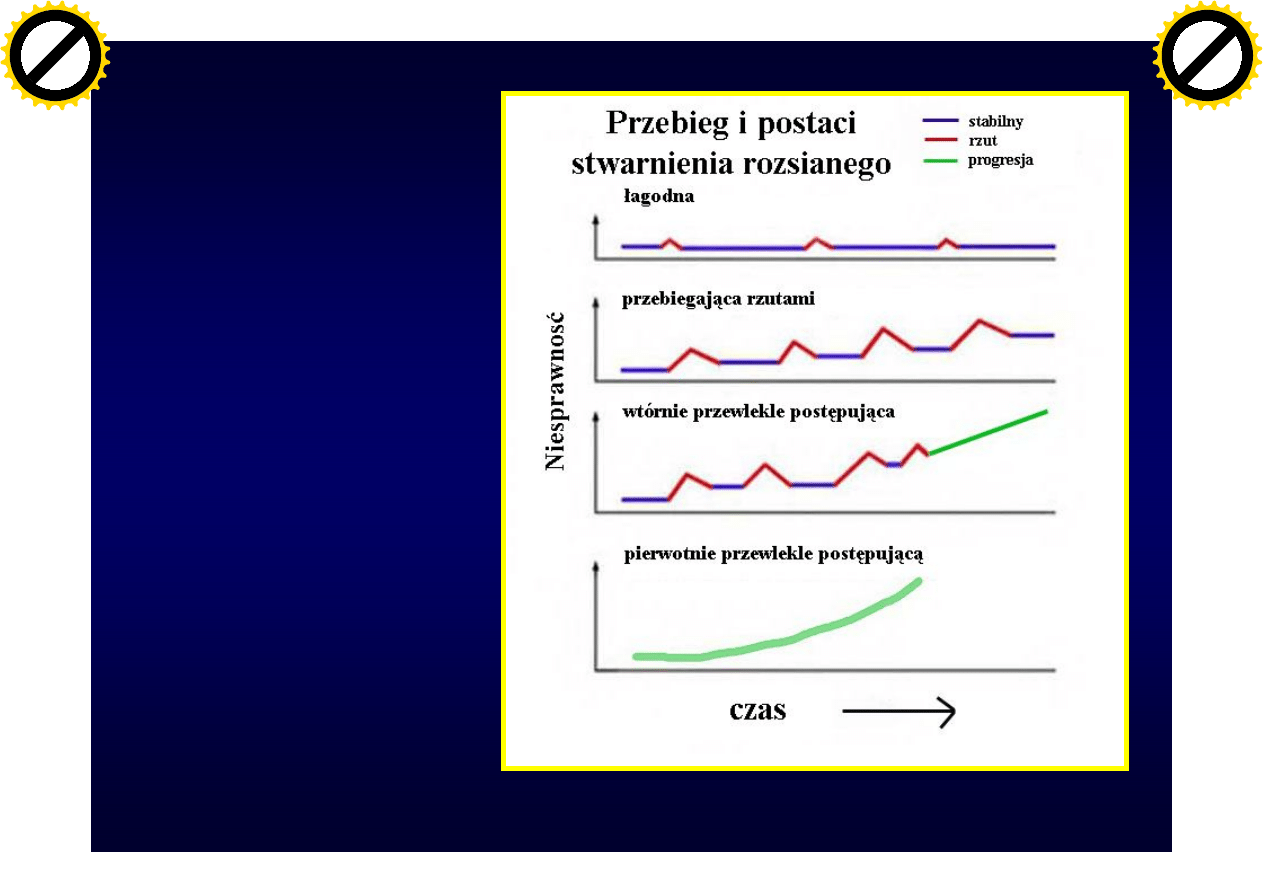
Przebieg
stwardnienia
rozsianego
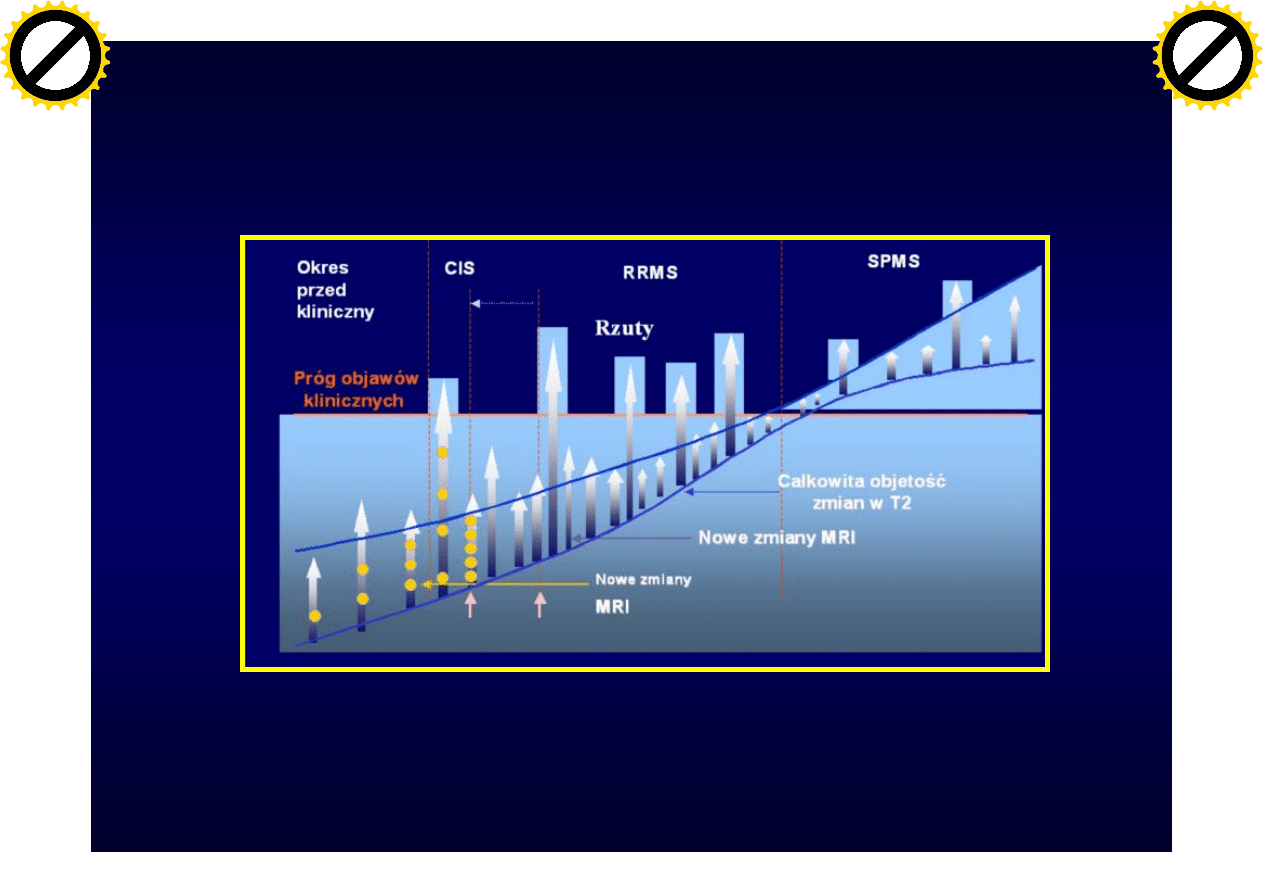
Ewolucja zmian w SM
Selmaj K.: Stwardnienie rozsiane 2006

Jak leczyć stwardnienie
rozsiane?

Leczenie
• Rzutu
• Immunomodulacyjne
• Objawowe

Leczenie rzutu choroby
• Cele:
– Zwalcza objawy rzutu
– Zwalnia postęp choroby
• Leki:
– Methylprednisolon (Solu-Medrol) i.v. 1,0g przez 3-7dni +
p.o. Medrol 32-16-8 mg 10-15dni (+leki osłaniające
błonę śluzową żołądka, + suplementacja potasu)
– Methylprednisolon (Medrol) p.o. 64-32-16-8 mg przez
20-28 dni (+leki osłaniające błonę śluzową żołądka, +
suplementacja potasu)
– Prednisolon p.o. 1mg/kg m.c. (dawka malejąca o ½ co 7
dni przez 28 dni (+leki osłaniające błonę śluzową
żołądka, + suplementacja potasu)
– Plazmafereza 5 zbiegów (co drugi dzień)

Wskazania do wczesnego leczenia
immunomodulującego w SM
• Zmiany zapalne dominują we wczesnym okresie
choroby
• Wczesna aktywność choroby, ilość i ciężkość
rzutów, wpływa na szybszą progresję choroby
• Nieodwracalność zmian aksonalnych
• Mechanizmy procesu chorobowego mogą być
trudniejsze do opanowania z czasem
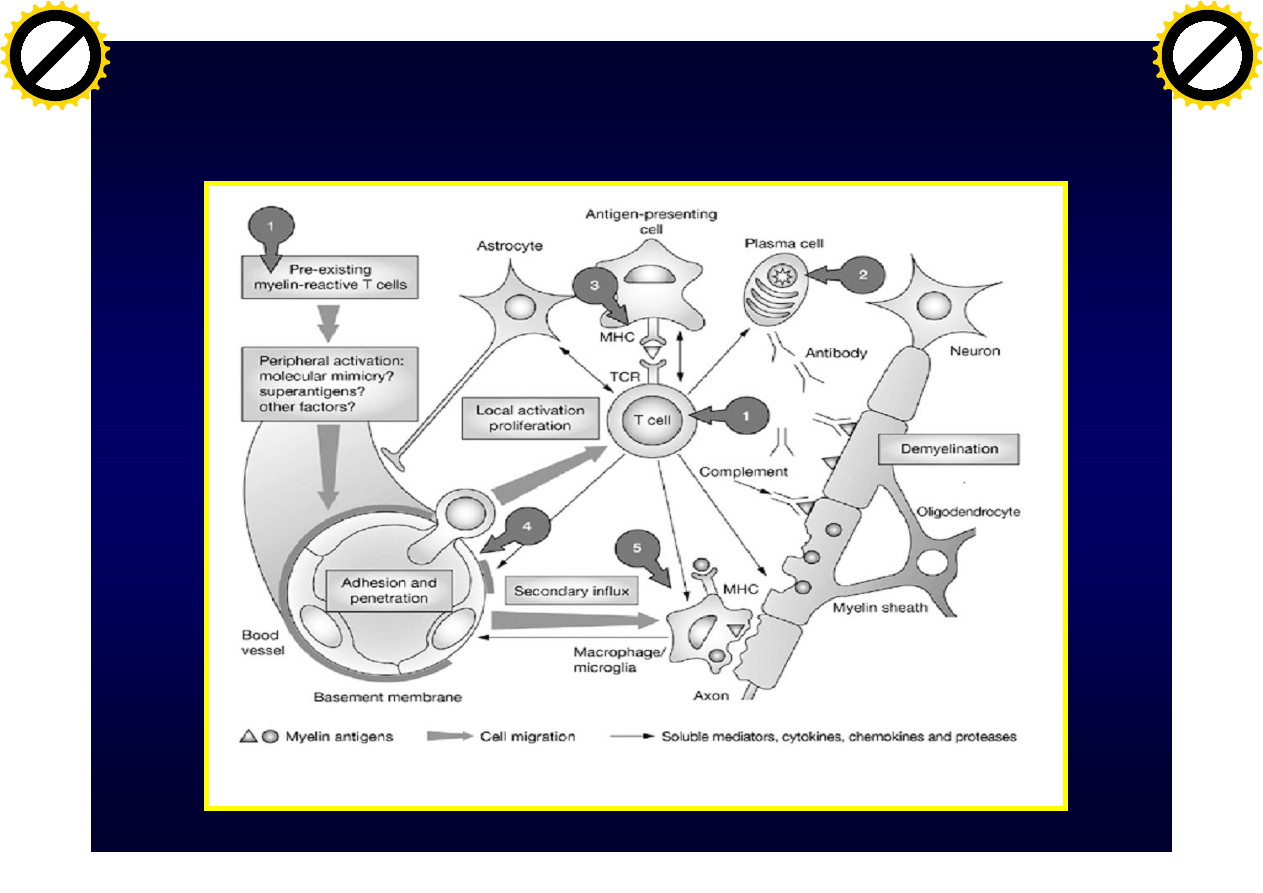
Leczenie immunomodulujące
Hohfeld R i wsp. Nat Clin Prac Neur 2005;1:34-44

Aktualna lista leków
immunomodulacyjnych
stosowanych w SM
• Interferon beta 1B
– Betaferon/Betaseron s.c. 8x10
6
MIU co drugi dzień
• Interferon beta 1A
– Avonex i.m. 30ug 1x w tyg.,
– Rebif s.c. 44 ug 3x w tyg.
• Glatiramer/Copaxone s.c. 20mg codziennie
• Natalizumab (Tysabri) i.v. 300mg co miesiąc
• Mitoxantrone i.v.12mg/m
2
co 3 miesiące (dawka
życiowa 140mg)
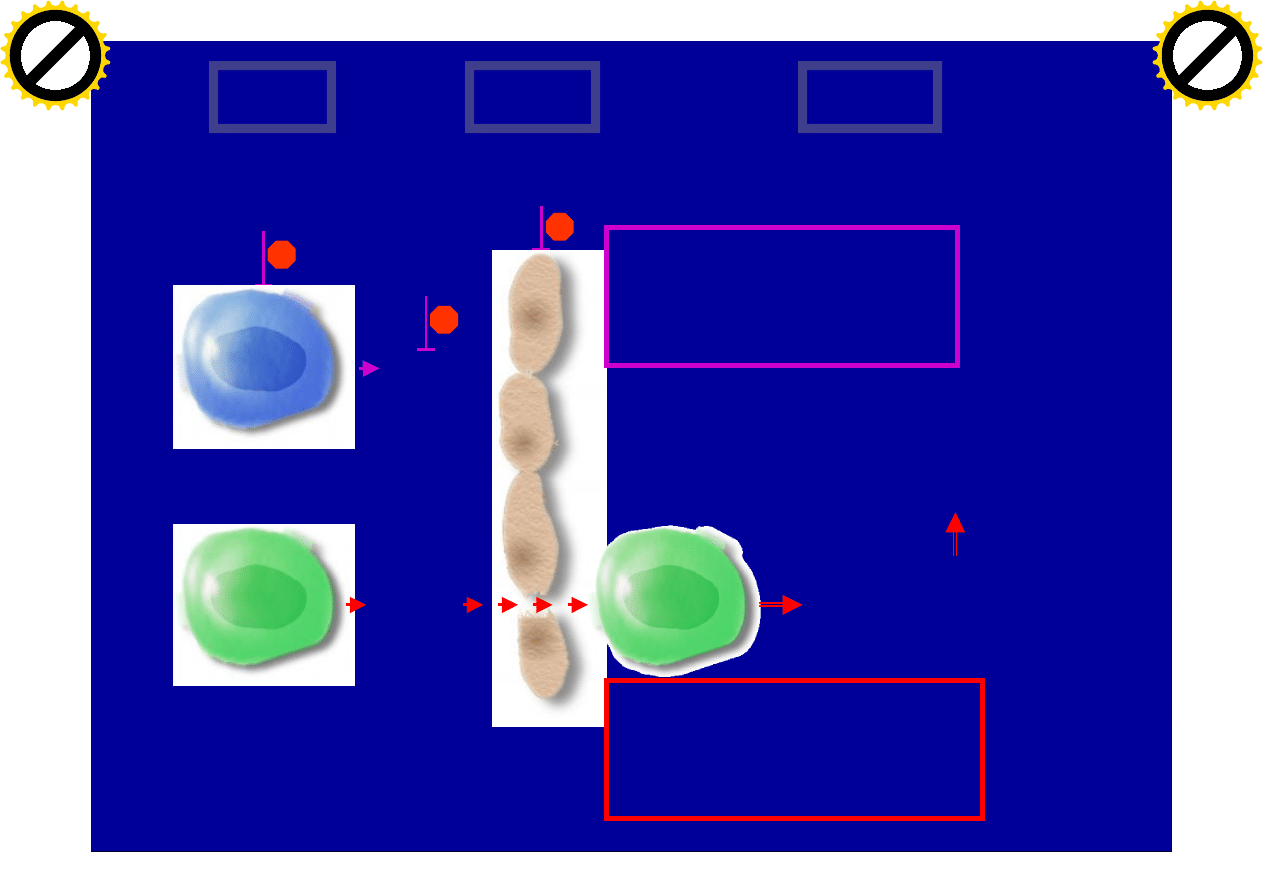
BBB
Blood
Brain
Effect on CNS function
• Bystander suppression
• Neuroprotection ?
Interferon-
b
Interferon-
b decreases
adhesion events
T cell
Delayed
â of Gd-
enhancing MRI activity
GA
T
H
2
MMPs
Interferon-
b
•
â influx of T cells
• Rapid
â of Gd-enhancing
MRI activity
GA
T
H
2
GA
• No
â of T cell migration
• No rapid
â of Gd-enhancing
MRI activity
No effect of
GA on MMPs
MMPs
Interferon-
b
From Wong et al., Neurology 2002
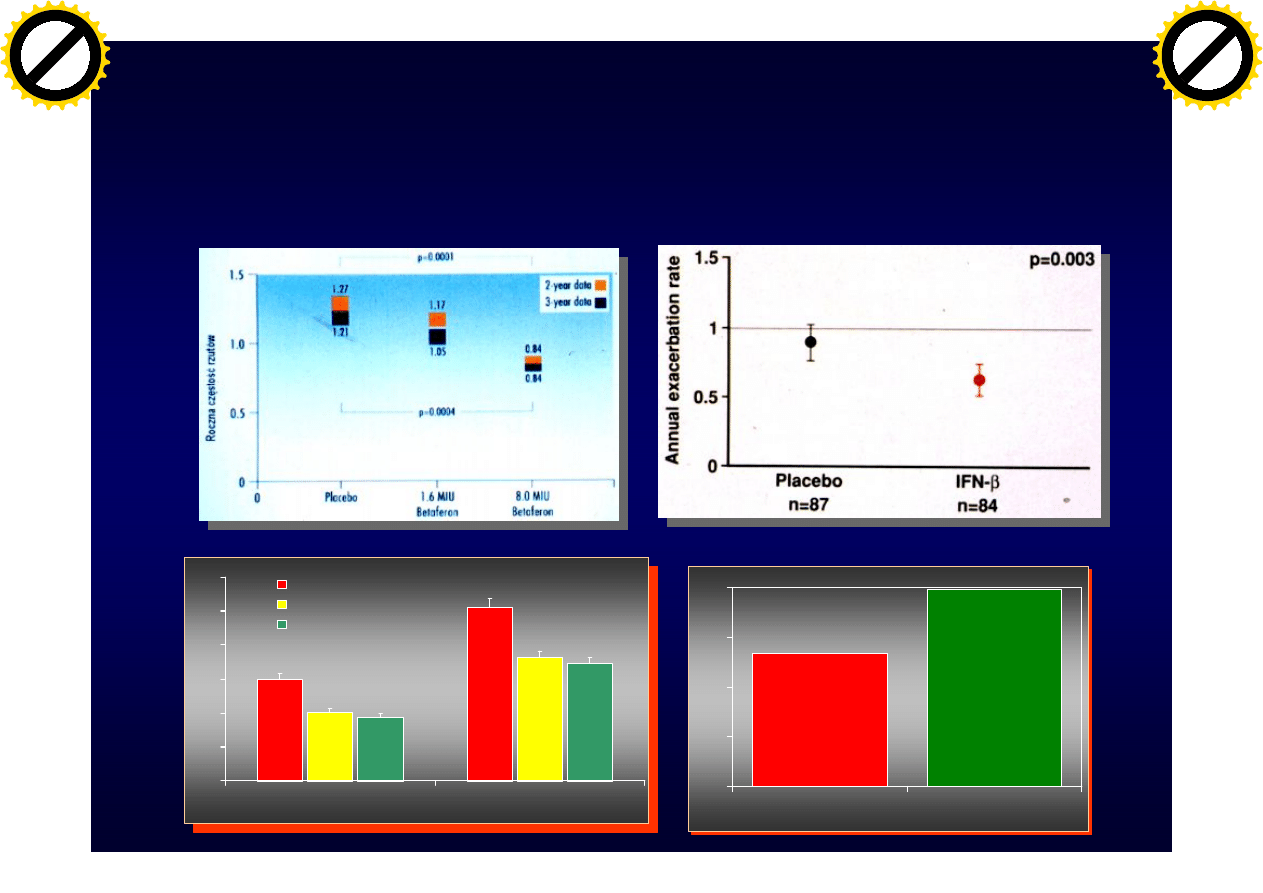
Wpływ IFN
b
i Glatirameru na
częstość rzutów SM
1.5
2.56
1.01
1.82
0.94
1.73
0
0.5
1
1.5
2
2.5
3
1 Year
2 Years
Placebo
22 mcg Rebif
44 mcg Rebif
1.5
2.56
1.01
1.82
0.94
1.73
0
0.5
1
1.5
2
2.5
3
1 Year
2 Years
Placebo
22 mcg Rebif
44 mcg Rebif
1.98
1.34
0
0.5
1
1.5
2
Cop 1
Placebo
1.98
1.34
0
0.5
1
1.5
2
Cop 1
Placebo
Betaferon
Avonex
Rebif
Glatiramer
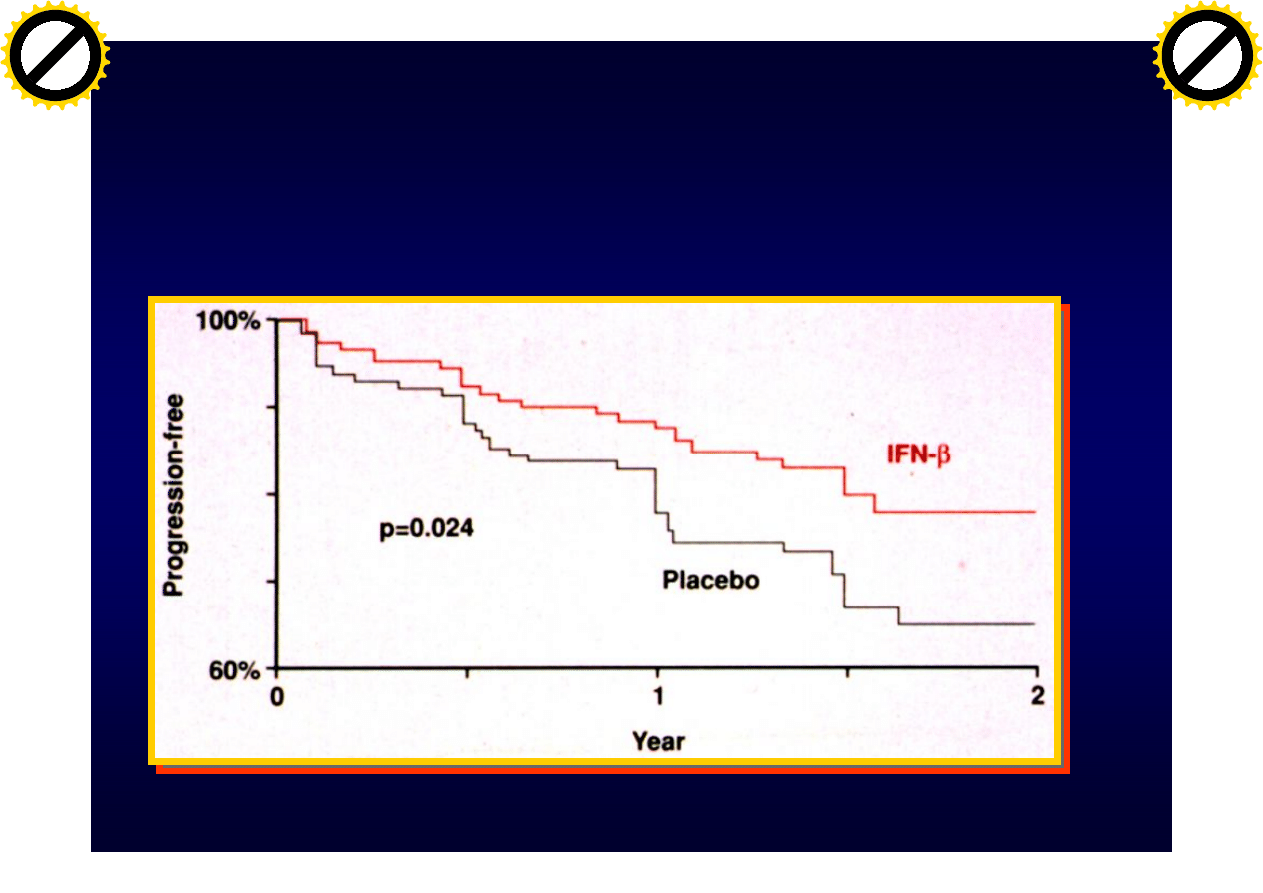
IFN
b
1A hamuje progresję
niesprawności
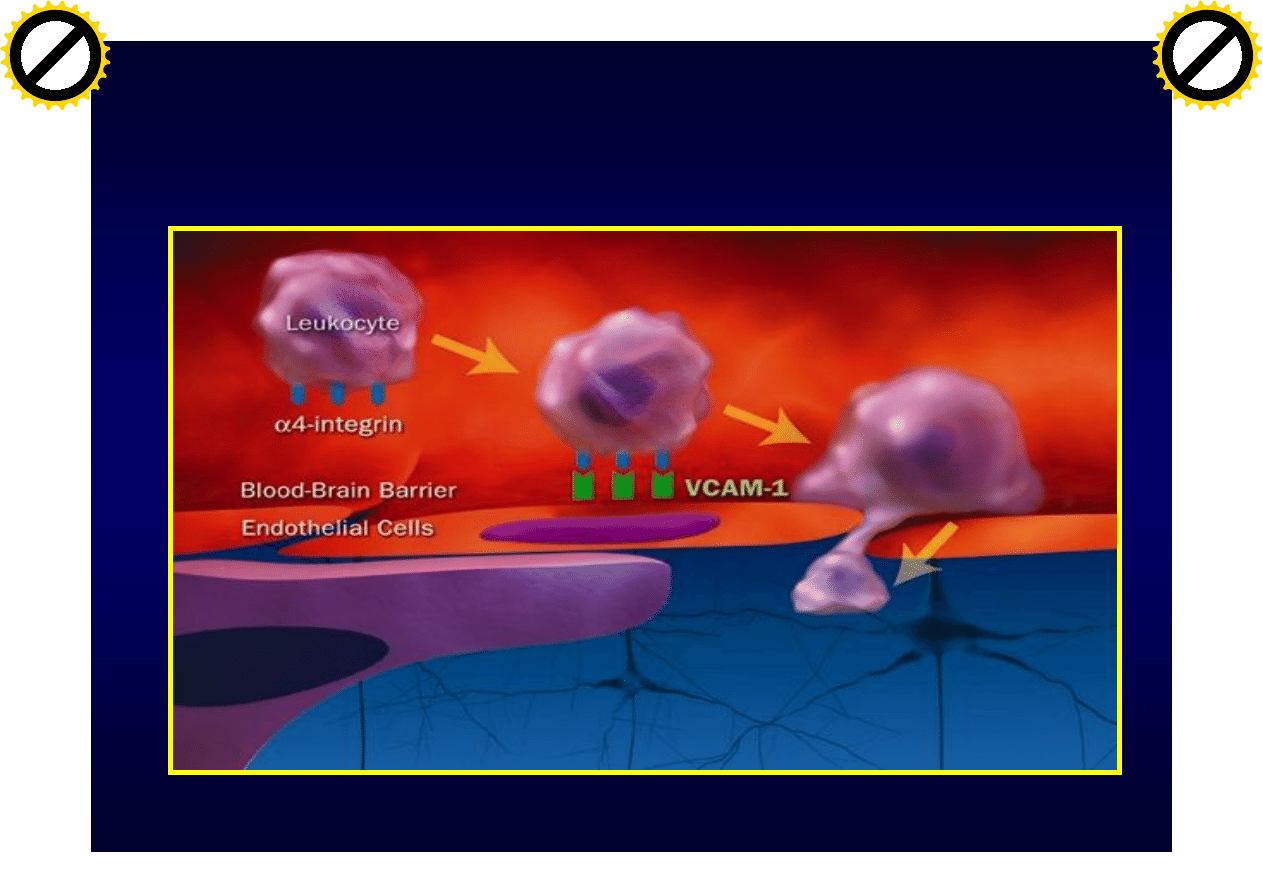
Lobb RR et al. J Clin Invest. 1994;94:1722-1728
.
Rola
a
4
b
1
integryny w patogenezie
SM
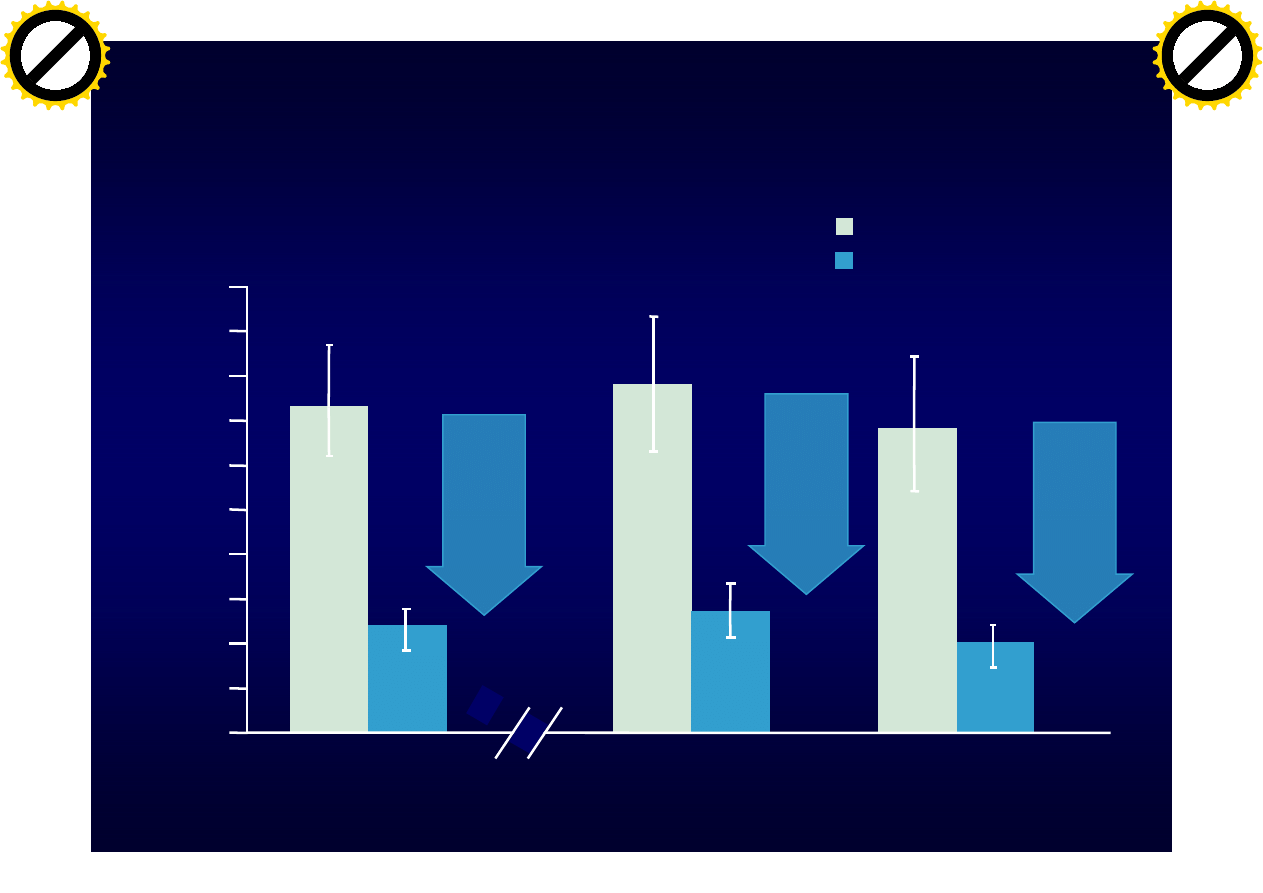
Placebo n=315
Natalizumab n=627
68%
reduction
70%
reduction
66%
reduction
Natalizumab – wpływ na liczbę
rzutów
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
A
nnual
iz
ed Rel
apse Rate (95%
CI)
P<0.001
0.73
0.23
Years 0-2
P<0.001
P<0.001
0.78
0.67
0.27
0.20
Year 0-1
Year 1-2
Polman CH, et al. N Engl J Med. 2006;354:899-910; Polman C, et al. Presented at the 57th Annual Meeting of the American Academy of
Neurology; April 12, 2005; Miami, FL..
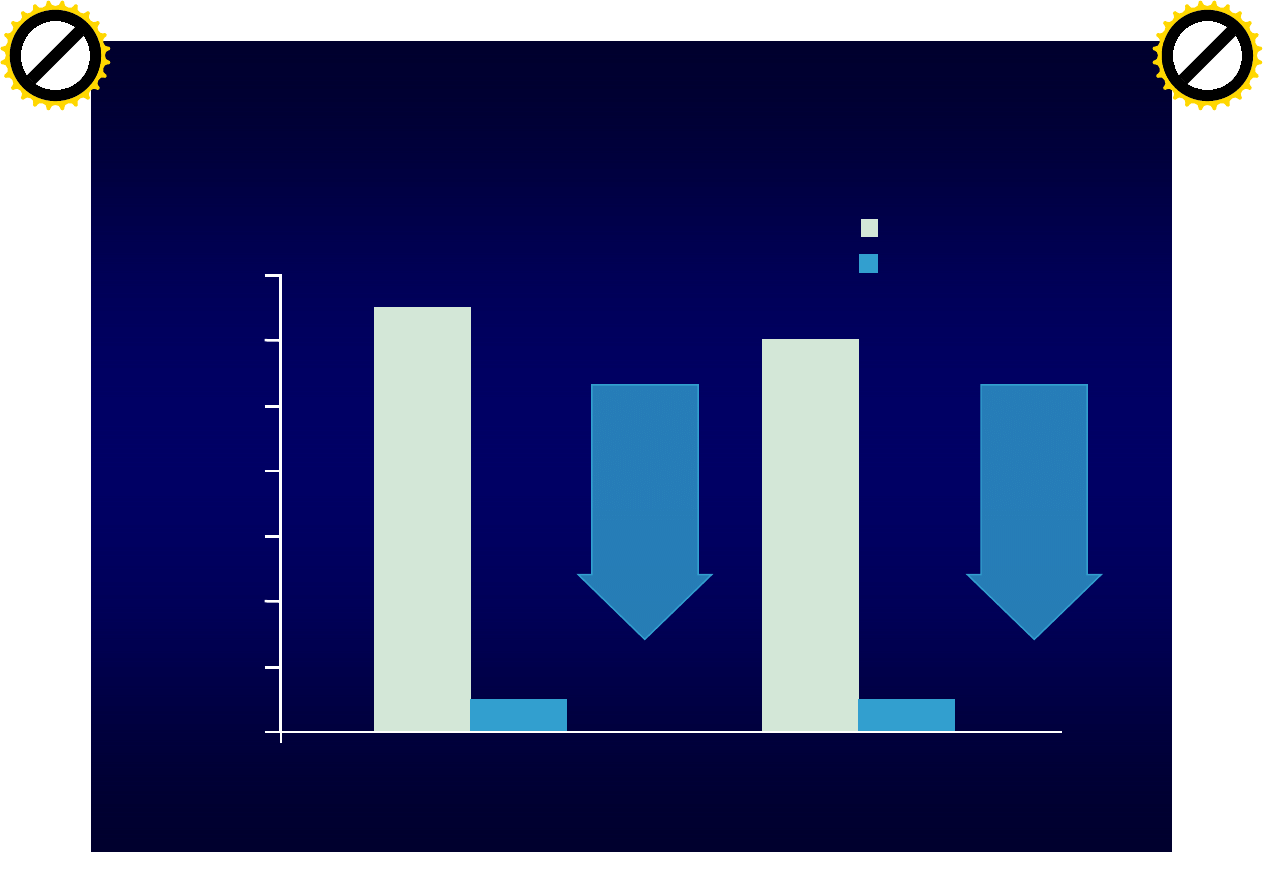
P<0.001
Mean N
u
mber
of Gd+
Lesio
ns
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
At Year 1
At Year 2
0.1
1.3
1.2
0.1
92%
reduction
92%
reduction
P<0.001
Placebo n=315
Natalizumab n=627
Natalizumab - zmiany Gd(+)
Polman CH, et al. N Engl J Med. 2006;354:899-910.
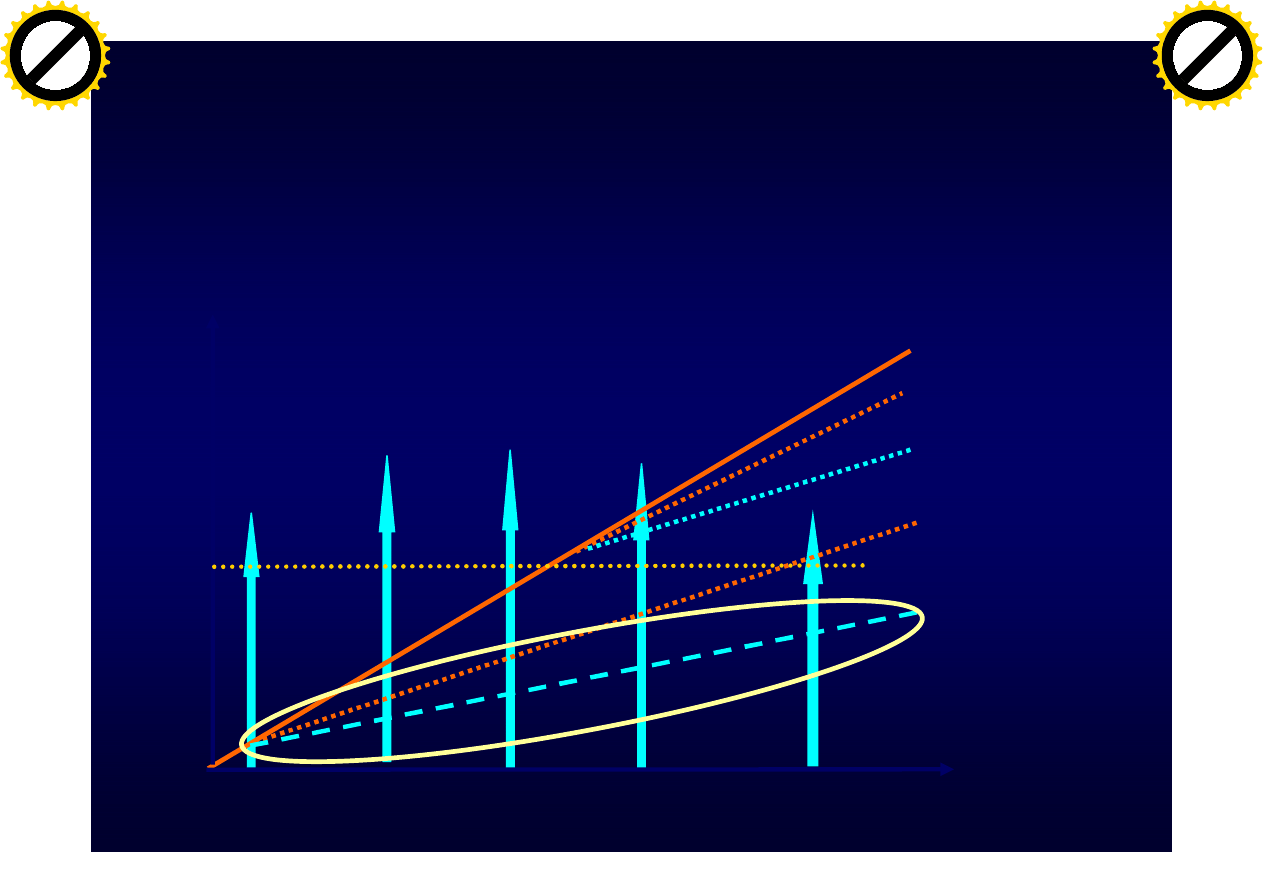
czas
Ax
onal los
s
RRMS
SPMS
Sta
ła niesprawność
Wczesne leczenie
(niskie dawkowanie)
Pó
źne leczenie
(niskie dawkowanie
)
Próg prezentacji
klinicznej
Wczesne leczenie
Wysokie dawki
Pó
źne leczenie
(wysokie dawkowanie
Skuteczność terapii IFNb w
zależności od dawki i czasu
rozpoczęcia leczenia

Powikłania leczenia immunomodulujcego
• Interferony (kontrola po tygodniu, miesiącu po
włączeniu leczenia i później co 3 miesiące)
– Objawy paragrypowe
– Skórne zmiany w miejscu wstrzyknięcia
– Leuko- i limfopenia
– Podwyższone próby wątrobowe
– Depresja?
• Glatiramer
– Skórne zmiany w miejscu wstrzyknięcia
• Natalizumab
– Postępująca wieloogniskowa encefalopatia (zakażenie wirusem JC)
• Mitoxantrone
– Kardiotoksyczność
– Leukopenia
– Uszkodzenie wątroby
– Zakażenie dróg moczowych
Niska dawka IFNß
Wysoka dawka IFNß
Tysabri
Opcje zmiany terapii leków
immunomodulujących
Copaxone
Mitoxantrone

Leczenie immunosupresyjne
• Cyklofosfamid
i.v. 500-700 mg/m
2
1x w
miesiącu (terapia ratunkowa)
– Kontrola morfologii, wydolności wątroby
– Krwotoczne zapalenie pęcherza
– Rak pęcherza
• Azathiopryna p.o.
2x50mg codziennie
• Metotreksat 7,5 mg 1x w tygodniu

Inne leki
• Amantadyna (antagonista receptorów NMDA)
działanie neuroprotekcyjne
– p.o. 2x 100mg
• Inhibitory fosfodiesterazy (hamują uwalnianie IL-
12, TNF)
– p.o. 2x 0,1-0,4 g
• Immunoglobuliny
– i.v. 0,2-04 g/kg 1 x w miesiącu
• Statyny (hamują aktywację limfocytów,
wydzielane cytokin)
– Simvastatyna 80mg/dz

Leczenie objawowe -Spastyczność
– Baklofen (analog GABA) 5-100mg/dz
– Tyzanidin (Sirdalud) (agonista receptora
a2-
adrenergicznego) 4-20mg/dz
– Tolperison (Mydocalm) 150-450mg/dz
– Tetrazepam (Myolastan) (benzodiazepina) 25-
150mg/dz
– Dantrolen 25-400mg/dz
– Toksyna botulinowa A
• Dysport 500-1000 j./ 3 mies.
• Botox 100-200 j./ 3 mies.

Leczenie objawowe - Zaburzenia zwieraczy
•
Leki antycholinergiczne
–
Oksybutynina (Ditropan) 5-15mg/dz
–
Tolterodyna (Detrusitol) 2-4mg/dz
•
Leki
a1-adrenomimetyczne
–
Midodryna (Gutron) 5-15mg/dz
•
Leki cholinergiczne
–
Distygmina (Ubretid) 5mg/dz
•
Leki
a1-adrenolityczne
–
Terazosyna (Hytrin) 1-10mg/dz
–
Doksazosyna (Cardura) 1-8mg/dz
–
Tamsulozyna (Omnic) 0,4mg/dz
•
Leki miorelaksujące
–
Baklofen
–
Tyzanidin

Leczenie objawowe –zaburzenia
koordynacji, drżenie, zaburzenia równowagi
• Drżenie
– Klonazepam 2-6mg/dz
– Izoniazyd 600-1200mg/dz
– Propranolol
– Toksyna botulinowa
– Lecznie operacyjne-stereotaktyczne
• Zaburzenia równowagi
– Betahistydyna (Betaserc) 24-48mg/dz
– Tietylperazyna (Torecan)
Wyszukiwarka
Podobne podstrony:
4. CHOROBY DEMIELINIZACYJNE - STWARDNIENIE ROZSIANE, stwardnienie rozsiane
choroby demielinizacyjne, IV rok Lekarski CM UMK, Neurologia, Neurologia od Grzela, wykady, opracowa
Choroby demielinizacyjne
03 CHOROBY DEMIELINIZACYJNE
Choroby demielinizacyjne (3)
choroby demielinizacyjne
Stwardnienie rozsiane (2), Medycyna, Neurologia, 11 choroby demielinizacyjne
Choroby demielinizacyjne (2)
Choroby demielinizacyjne
Choroby demielinizacyjne (2)
choroby demielinizacyjne
choroby naczyn i serca(1)
więcej podobnych podstron