ćwiczenie 2
IZOLACJA CAŁKOWITEGO RNA Z ROŚLIN
Izolacja RNA jest pierwszym etapem prac mających na celu poznanie ekspresji badanych genów. RNA po izolacji może być
wykorzystany do następujących celów.
1. Ustalenia czy dany gen ulega ekspresji w badanym układzie biologicznym. Ekspresję badanych genów można stwierdzić
wykorzystując amplifikacji typu RT-PCR lub test hybrydyzacji.
2. Klonowania cDNA. Cząsteczki RNA, po przepisaniu na cDNA w reakcji RT-PCR, mogą zostać wklonowanie do wybranego
wektora genetycznego. Pula cząsteczek cDNA reprezentujących wszystkie mRNA danej tkanki stanowi bibliotekę cDNA.
3. Poznania budowy badanego genu. Sekwencja cDNA danego genu, może zostać porównana z sekwencją genomową tego
genu, co umożliwia identyfikację pozycji eksonów i intronów a także końca 3’ transkryptu. Koniec 5’ transkryptu można
zlokalizować wykorzystując test primer-extension. Długość cząsteczek mRNA badanych genów można ustalić za pomocą
hybrydyzacji typu Northern-blot.
4. Ustalenia poziomu ekspresji badanych genów za pomocą real time PCR lub hybrydyzacji typu Northern-blot.
Stosowane metody izolacji RNA powinny umożliwiać uzyskanie RNA niezdegradowanego, o wysokim stopniu czystości.
Izolacja mRNA przebiega zasadniczo w podobnych etapach jak izolacja DNA. Główne różnice wynikają z następujących faktów.
1. RNA jest znacznie bardziej narażony na degradację niż DNA. W materiale biologicznym oraz w środowisku zewnętrznym
obecna jest duża ilości bardzo aktywnych i stabilnych rybonukleaz. Nawet po zastosowaniu wysokiej temperatury, np. 100
o
C,
a także w obecności detergentów, tj. SDS, enzymy te zachowują wysoką aktywność. Ponadto, wiele rybonukleaz nie wymaga
dla swej aktywności kofaktorów, np. jonów dwuwartościowych, w związku z czym aktywności tych enzymów nie możemy
zablokować stosując EDTA. Dlatego podczas izolacji RNA do inaktywacji RNaz należy stosować bardzo silne związki
degradujące białka, takie jak chlorowodorek guanidyny lub izotiocjanian guanidyny. Związki te, oprócz degradacji
rybonukleaz, umożliwiają również zniszczenie struktur komórkowych. RNazy powinny zostać również usunięte ze sprzętu
używanego do izolacji. Do tego celu stosuje się inhibitory RNaz, tj. DEPC (dietylopirowęglan). W postaci 0,1% roztworu
wykorzystujemy go do płukania sprzętu mającego kontakt z RNA oraz do przygotowywania niektórych buforów. Roztwór
ten po autoklawowaniu inaktywującym DEPC, używany jest również do przechowywania RNA jako tzw. H
2
O
DEPC
. Należy
pamiętać, że 0,1% DEPC nie poddany inaktywacji inhibuje aktywność enzymów służących do prac z RNA, np. odwrotnej
transkryptazy. Kolejnym, często stosowanym inhibitorem RNaz, jest tzw. RNazin - białko które inaktywuje RNazy wiążąc
się z nimi. Inhibitor ten może towarzyszyć RNA podczas przechowywania oraz podczas reakcji enzymatycznych, gdyż nie
powoduje inhibicji innych enzymów.
2. Podczas izolacji RNA należy usunąć towarzyszący mu DNA. W tym celu stosuje się między innymi ekstrakcję fenolem o
niskim pH. Kwaśny fenol powoduje usunięcie nadmiaru DNA z roztworu. Dokładne usunięcie resztek DNA z preparatu
można natomiast uzyskać wykonując trawienie DNA-zą wolną od RNA-az. RNA pozbawiony DNA można również uzyskać
wytrącając RNA w LiCl lub stosując wirowanie w dwustopniowym gradiencie tiocianianu guanidyny i CsCl.
3. W komórkach eukariotycznych cząsteczki rRNA, tRNA oraz RNA niskocząsteczkowy stanowią łącznie ok. 80-98% RNA
komórkowego. Dlatego, jeżeli interesuje nas tylko pula mRNA, należy zastosować dodatkowy etap izolacji, podczas którego
usuwamy z preparatu pozostałe kwasy rybonukleinowych. Stosowane w tym celu techniki wykorzystują fakt, że cząsteczki
mRNA na 3’ końcach zawierają sekwencje poli(A). Sekwencje te mogą zostać związane z cząsteczkami poli(T)
przyłączonymi do stałego podłoża np. w kolumnach chromatograficznych lub na kuleczkach magnetycznych. Cząsteczki
RNA nie związane z podłożem są następnie odmywane a oczyszczone cząsteczki mRNA poddawane są elucji.
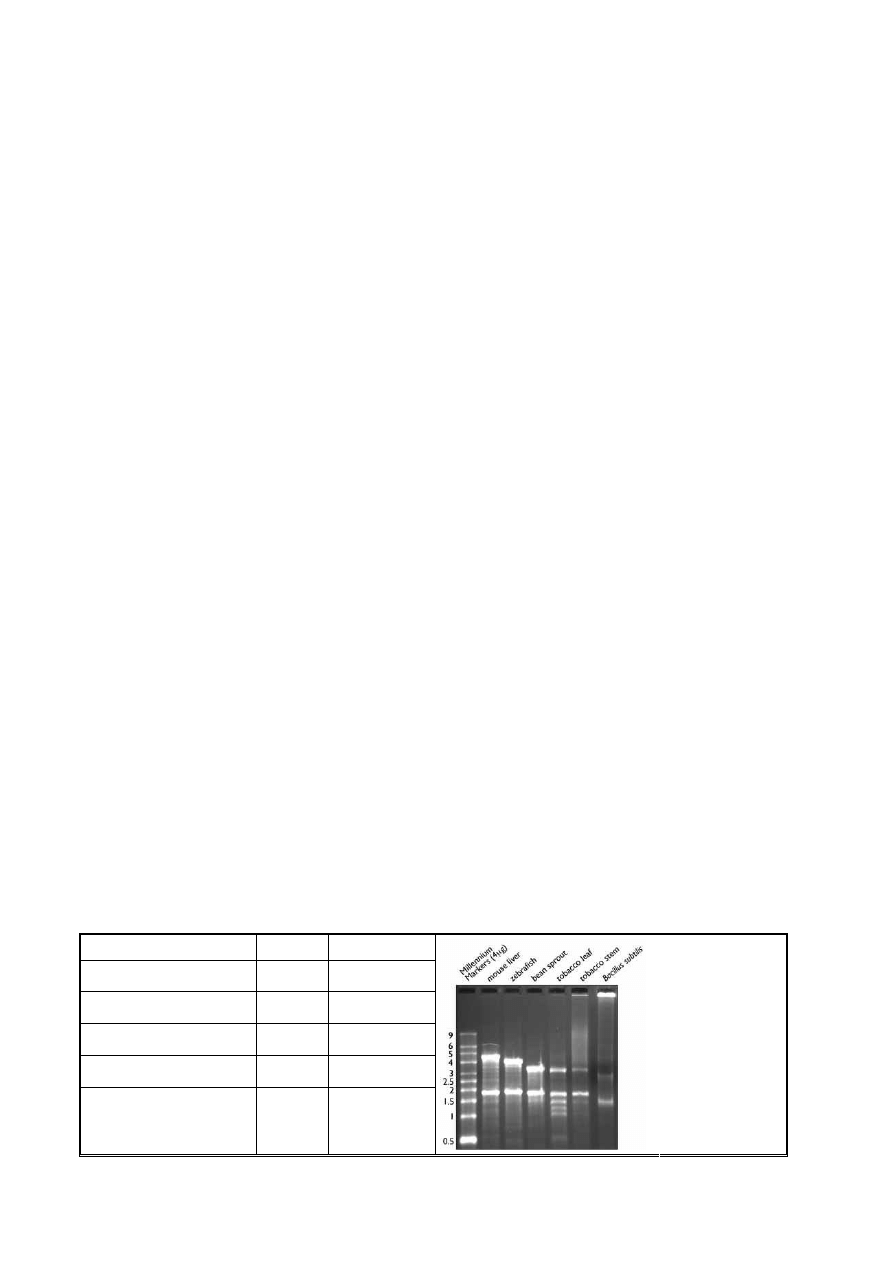
Po rozdziale elektroforetycznym całkowitego RNA w żelu agarozowym najlepiej widoczne są frakcje rRNA. Ich obraz może być
wykorzystany jako orientacyjny marker mas cząsteczkowych, gdyż znane są wielkości podstawowych frakcji rRNA
występujących u poszczególnych organizmów (Tab.1). Ponadto intensywność prążków rRNA, np. 23S, 18S, informuje nas o
jakości preparatu, gdyż ich zanikanie świadczy o postępującej degradacji RNA. Frakcje mRNA, ze względu na ich niewielką ilość
w preparacie oraz zróżnicowaną wielkość, charakterystyczną dla poszczególnych genów, nie są widoczne na żelach po rozdziale
RNA całkowitego.
Tab. 1 Wielkości cząsteczek rRNA różnych gatunków
Organizm rRNA
Wielkość (kb)
Mysz (Mus musculus) 18
S
28 S
1,9
4,7
Człowiek (Homo sapiens) 18
S
28 S
1,9
5,0
Drożdże (S.cerevisiae) 18
S
26 S
2,0
3,8
E. coli
16 S
23 S
1,5
2,9
Tytoń (Nicotiana tabacum)
16 S
18 S
23 S
25 S
1,5
1,9
2,9
3,7
Rys. 1
Fotografia
przedstawia rozdział
elektroforetyczny
RNA pochodzący z
różnych
organizmów.
Widoczne prążki
reprezentują dojrzałe
cząsteczki rRNA.

Materiały
Tab.2 Bufory do izolacji i rozdziału elektroforetycznego RNA.
Bufor homogenizacyjny
Buf. elektroforetyczny
10 x MOPS
Przygotowanie buforu
denaturujący do RNA
4M tiocjanian guanidyny
0,2 M MOPS pH 7,0
37 % formaldehyd - 187
μl
25 mM cytrynian sodu pH 7,5
50 mM octan sodu
100 % formamid – 562
μl
0,5 % sarkozyl
10 mM EDTA
10 x MOPS – 112
μl
0,1 M
β-merkaptoetanol - dodać
bezpośrednio przed użyciem
10 mg/ml Bromek etydyny – 10
μl
H
2
O
DEPC
- 129
μl (do 1 ml)
guanidine thiocyanate
DEPC
Odczynniki i sprzęt
- bufory tab. 2
- octan sodu pH 4,0; 2 M
- Fenol kwaśny pH 4,0
(nasycony H
2
O
DEPC
)
- Chloroform
- 75 % etanol
- izopropanol
- H
2
O
DEPC
- 37 % formaldehyd
- 0,1 % DEPC
- Bufor LB do RNA
- rękawiczki
- nożyczki
- moździerz
- probówki eppendorfa
- tipsy
- blok grzejny
- sprzęt i odczynniki do
elektroforezy agarozowej
Postępowanie
1. 100mg materiału roślinnego, roztartego w ciekłym
azocie, zawiesić w 400
μl buforu
homogenizacyjnego. Uwaga - izotiocjanian
guanidyny obecny w buforze jest substancją żrącą.
Pracę z nim należy wykonywać tylko w
rękawiczkach i przy zachowaniu dużej
ostrożności!
2. Zawiesinę inkubować w ciemnym miejscu, w
temp. pokojowej przez ok. 0,5 godz.
3. Do mieszaniny dodać 1/10 obj. 2M octanu sodu
pH 4,0 (40
μl) i 1 obj. kwaśnego fenolu (440μl).
Całość wytrząsać przez 5 min.
4. Do próby dodać 200
μl chloroformu i
kontynuować homogenizację przez 5 min. Jeżeli
preparat nie ulega rozwarstwieniu na dwie fazy,
należy dodać niewielką ilość chloroformu.
5. Całość wirować w mikrowirówce przez 10 min
przy 12 tys. rpm.
6. Fazę wodną, która zawiera RNA, przenieść do
nowej probówki eppendorfa i dodać do niej 1 obj.
chloroformu. Całość wytrząsać przez 1 min. a
następnie wirować 5 minut przy 12 tys. rpm.
7. Fazę wodną przenieść do nowej probówki
eppendorfa. RNA wytrącić dodając 1 obj.
izopropanolu.
8. Wytrącony RNA odwirować przy 12 tys. rpm
przez 10 min w +4
o
C.
9. W celu usunięcia resztek buf. homogenizacyjnego
osad RNA przemyć 100
μl 75% etanolu i
odwirować przez 3 min.
10. Dokładnie usunąć resztki etanolu i osuszyć osad.
11. Rozpuścić RNA w 20
μl H
2
O
DEPC.
12. Ilość i jakość wyizolowanego RNA sprawdzić
elektroforetycznie. Pozostałą część preparatu
można przechowywać w –20
o
C.
13. Do 5
μl preparatu dodać 1 obj. buforu
denaturującego do RNA. Uwaga: bufor ten
zawiera bromek etydyny! Całość denaturować
przez 5 min w +65
o
C.
14. Schłodzić preparat w lodzie a następnie dodać 2
μl
buforu LB i całość nałożyć na żel denaturujący.
15. Nałożyć preparat na 1,5% denaturujący żel
agarozowy.
16. Elektroforezę prowadzić w buforze 1x MOPS przy
napięciu 100 V.
17. Po zakończeniu elektroforezy żel analizujemy w
świetle UV.
Przygotowanie żelu denaturującego
1. Przygotowanie aparatu do elektroforezy RNA. W
celu pozbycia się RNA-az aparat do elektroforezy
należy umyć detergentem, przepłukać
0,1%
roztworem DEPC i wytrzeć do sucha ligniną z
etanolem.
2. Przygotowanie 1,5% żelu agarozowego (50
ml). Do 0,7g agarozy dodać do 42,5 ml wody i
zagotować. Po schłodzeniu agarozy do ok. +65
o
C
dodać 5 ml 10x st. buforu MOPS oraz 2,5 ml 37%
formaldehydu a następnie całość wylać do
uprzednio przygotowanego aparatu
elektroforetycznego. Uwaga - wszystkie te
czynności wykonać w dygestorium przy
włączonym nawiewie. Opary formaldehydu są
trujące.

3
Wyszukiwarka
Podobne podstrony:
Izolacja calkowitego DNA id 221 Nieznany
calkowanie 1 opis matematyczny Nieznany
Antyutleniacze pochodzenia rosl Nieznany (2)
Izolacja całkowitego RNA - KONSPEKT, studia - biotechnologia, biologia molekularna
gik mnu calkowanie id 190983 Nieznany
Sprawozdanie z izolacji genomowego DNA z komórki roślinnej
izolacja kofeiny id 221168 Nieznany
IZOLACJA izohan id 221166 Nieznany
calkowanie 1 id 108054 Nieznany
Calkowanie graficzne iloczynu d Nieznany
Lab5 calkowanie id 773752 Nieznany
IZOLACJA SCIAN2 id 221176 Nieznany
Izolacja genomowego DNA
Izolacja DNA plazmidy Genetyka Nieznany
więcej podobnych podstron