
Ćwiczenie 4
Inwertaza (II)
Kinetyka reakcji enzymatycznych
ENZYMOLOGIA
Wydział Nauk o Żywności i Rybactwa
Centrum Bioimmobilizacji
i Innowacyjnych Materiałów
Opakowaniowych
ul. Klemensa Janickiego 35

2
Ćwiczenie 4
Inwertaza (II). Kinetyka reakcji enzymatycznych
Inwertaza (sacharaza, β-fruktofuranozydaza) - enzym należący do grupy hydrolaz, został po
raz pierwszy wyizolowany z ekstraktu drożdży przez Berthelota w 1860 roku. Hydrolizuje
wiązania glikozydowe utworzone z udziałem grupy hydroksylowej w położeniu β, związanej
z węglem anomerycznym (C2) pierścienia fruktofuranozy. Sacharaza powoduje hydrolizę
dwucukru sacharozy do glukozy i fruktozy, trójcukru rafinozy do fruktozy i dwucukru
melibiozy. Głównym, naturalnym substratem β-fruktofuranozydazy jest sacharoza (Rys.1).
Inwertazy występują w bakteriach i innych mikroorganizmach, w roślinach wyższych (w
ścianie komórkowej) i u zwierząt. Przez pszczoły sacharaza wykorzystywana jest do
hydrolizy sacharozy podczas produkcji miodu. U ludzi inwertaza, podobnie jak laktaza,
występuje na wewnętrznej powierzchni komórek nabłonka wyściełającego jelito cienkie.
Bogatym źródłem inwertazy są drożdże. Inwertaza drożdżowa wykazuje optimum działania w
pH 4.0—5.5. Aktywność enzymatyczną inwertazy można oznaczyć różnymi metodami np.
metodą kolorymetryczną lub polarymetryczną.
Rys.1. Sacharoza (α-D-glukopiranozylo-β-D-fruktofuranozyd) z zaznaczonym miejscem działania
inwertazy (Dubin i Turyna, 1999)
Sacharoza to disacharyd zbudowany z α-D-glukozy i β-D-fruktozy połączonych wiązaniem
glikozydowym 1-2. W większych ilościach występuje w burakach cukrowych i trzcinie
cukrowej. Nie posiada właściwości redukujących.
Zastosowanie sacharazy w technologii żywności
Sacharaza otrzymywana jest głównie z drożdży Saccharomyces cerevisiae. Enzym ten może
być stosowany w produkcji cukierniczej, do przygotowania syropów cukru inwertowanego
używanego do wyrobu sztucznego miodu, cukierków, marmolad, konfitur, likierów itp.
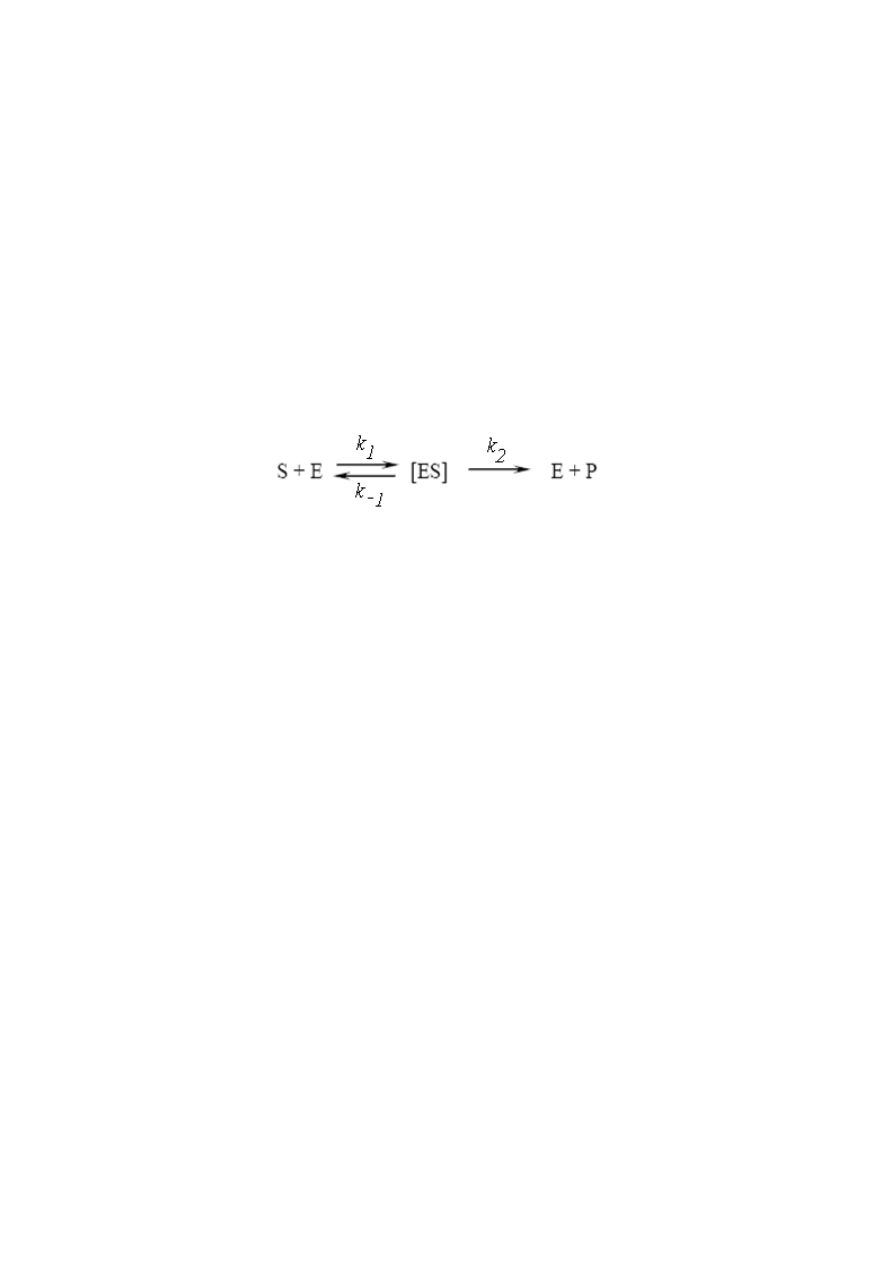
3
Kinetyka reakcji chemicznych zajmuje się badaniem szybkości przebiegu reakcji
w zależności od różnych czynników (stężenia reagujących substancji, temperatury, obecności
aktywatorów i inhibitorów). Zadaniem kinetyki jest ustalenie charakteru reakcji
i matematyczne ujęcie zależności między szybkością reakcji, a czasem jej trwania. Poznanie
tych zależności pozwala na określenie szybkości, z jaką będzie przebiegać reakcja
w dowolnie obranym czasie i dowolnym stężeniu substratu.
Szybkość reakcji enzymatycznej mierzona jest najczęściej przyrostem produktu lub ubytkiem
substratu w jednostce czasu.
Zasadniczym założeniem teorii Michaelisa-Menten, jako podstawy rozważań
kinetycznych, jest konieczność wytworzenia odwracalnego kompleksu między enzymem
i substratem (1)
(1)
S – substrat,
E – enzym,
[ES] – kompleks enzym – substrat,
P – produkt
k – stała szybkości reakcji
Kompleks ten podlega przemianie nieodwracalnej do produktu (lub produktów) P
z uwolnieniem enzymu. Dla pełnego scharakteryzowania kinetyki tych przemian niezbędne są
stałe szybkości reakcji. Ponieważ w porównaniu ze stężeniem substratu i produktu, stężenie
enzymu jest wielokrotnie mniejsze, więc w przybliżeniu można przyjąć, że stężenie
kompleksu ES jest stałe (szybkość jego powstawania jest zrównoważona szybkością
powstawania produktu) i zależy tylko od stężenia enzymu w roztworze. Oczywiście,
im większe jest stężenie kompleksu ES (a więc i enzymu), tym więcej produktu powstanie
w jednostce czasu.
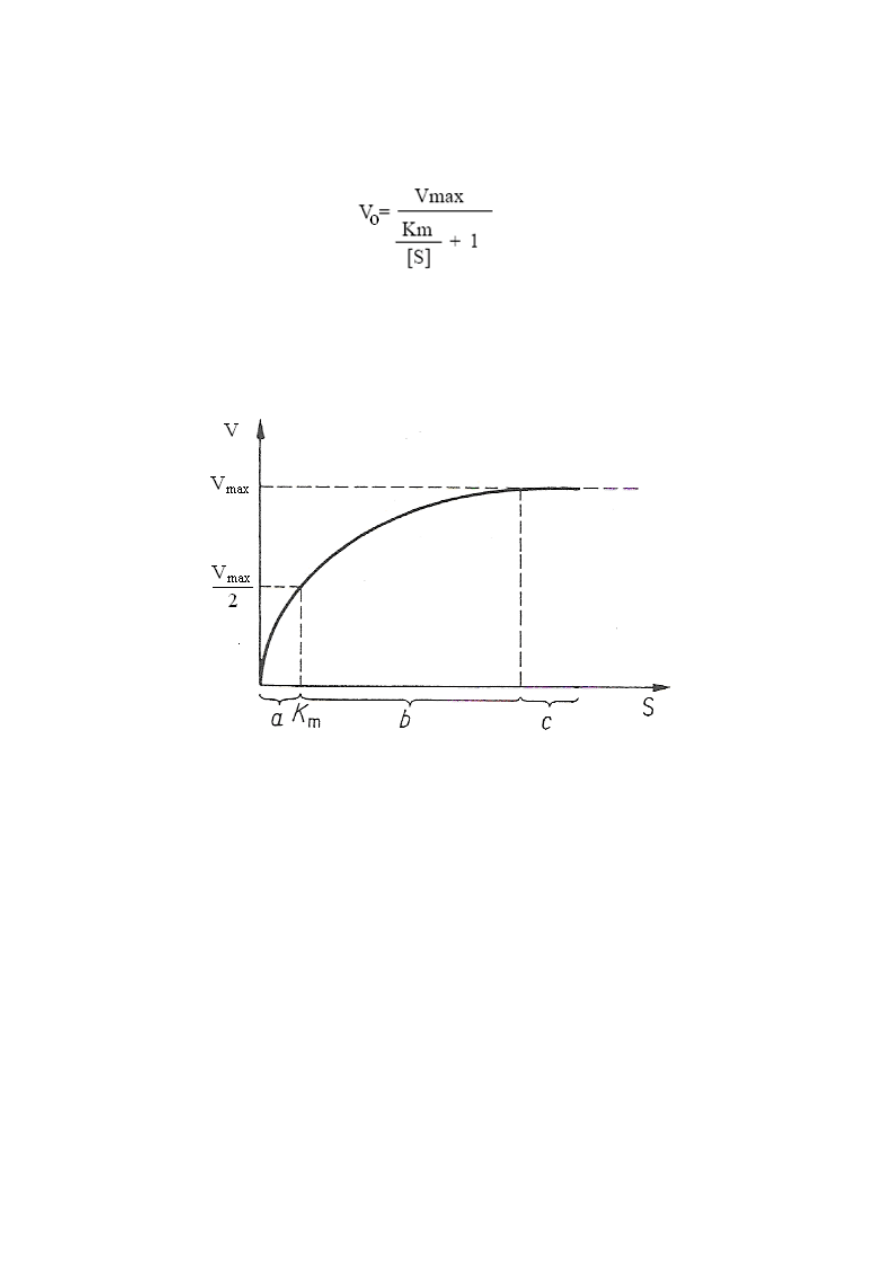
Przy stałym stężeniu enzymu szybkość reakcji enzymatycznej w pewnych granicach
zależy od stężenia substratu (rys. 1). Jak wynika z przedstawionej krzywej, przy małym
stężeniu substratu w stosunku do stężenia enzymu zwiększenie szybkości reakcji wraz ze
zwiększeniem stężenia substratu jest do niego w przybliżeniu wprost proporcjonalne
(zależność liniowa) i odpowiada tzw. kinetyce pierwszego rzędu.
Natomiast przy dużym stężeniu substratu szybkość reakcji ma wartość maksymalną
V
max
i nie zależy od dalszego zwiększania jego stężenia. W tym przypadku reakcja podlega
kinetyce zerowego rzędu. Tego typu kinetykę obserwuje się przy osiągnięciu pełnego
wysycenia cząsteczek enzymu przez substrat. Wtedy dalsze zwiększanie stężenia substratu nie
może już powodować zwiększenia szybkości reakcji, gdy stężenie enzymu pozostaje stałe;
4
reakcja przebiega ze stałą szybkością. Zależność szybkości reakcji od stężenia substratu
przedstawiona na rys. 1 jest zgodna z następującym równaniem Michaelisa-Menten
:
(2)
gdzie:
V
o
– szybkość reakcji, V
max
– szybkość maksymalna, K
m
– stała Michaelisa, [S] – stężenie
substratu; graficznie równanie to przyjmuje postać hiperboli:
Rys. 1. Wpływ stężenia substratu na szybkość reakcji przy stałym stężeniu enzymu – krzywa
wg Michaelisa-Menten; V
max
-szybkość maksymalna, S-stężenie substratu, K
m
-stała
Michaelisa-Menten, a – reakcja pierwszego rzędu, b – reakcja o mieszanej kinetyce, c –
reakcja zerowego rzędu (Kączkowski 2005)
Jeżeli wartość [S] = K
m,
równanie (2) przybierze postać V = V
max
/2, czyli reakcja osiągnie
połowę szybkości maksymalnej. Takie stężenie substratu, przy którym szybkość reakcji
stanowi połowę szybkości maksymalnej, nosi nazwę stałej Michaelisa (K
m
).
Odwrotność stałej (l/K
m
) jest nazywana powinowactwem enzymu do substratu. Z tej
zależności wynika, że mała wartość K
m
oznacza duże powinowactwo enzymu do danego
substratu i dużą szybkość procesu a duża wartość K
m
oznacza odwrotnie. Wartości stałej
Michaelisa, oznaczone dla wielu enzymów, wynoszą przeważnie od 10
-3
do 10
5־
M.
Wartość K
m
można wyznaczyć graficznie z wykresu, jak podano na wykresie 1. Podana
metoda graficzna jest jednak niezbyt dokładna, nie daje bowiem pewności, czy została
osiągnięta szybkość maksymalna reakcji V
max
. O wiele dogodniejszym sposobem
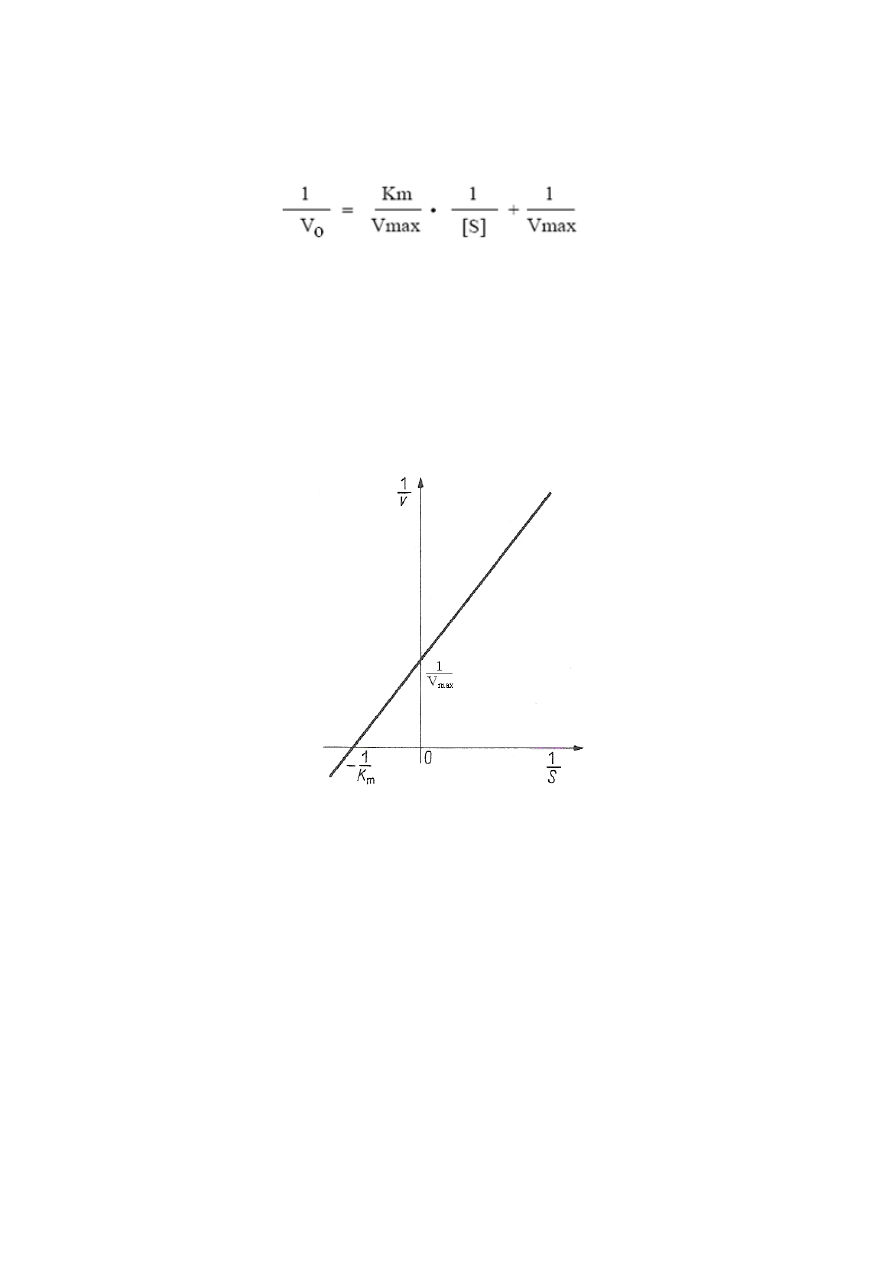
5
wyznaczania wartości Km jest wykorzystanie równania Lineweavera – Burka, które jest
przekształceniem równania Michaelisa – Menten:
(3)
Przekształcenie to ma na celu uzyskanie równania linii prostej (y = ax + b). Wartości K
m
/V
max
(a) i 1/V
max
(b) są wielkościami stałymi, stąd zależność 1/v od 1/[S] jest linią prostą. W
układzie współrzędnych wartości 1/v odkłada się na osi rzędnych a wartości 1/[S] na osi
odciętych. Wówczas równanie (3) daje linię prostą, której nachylenie określa stosunek
K
m
/V
max
(współczynnik regresji a) i która przecina oś rzędnych w punkcie wyznaczającym
1/V
max
(współczynnik regresji b), a oś odciętych w punkcie 1/K
m
(rys. 2). Na podstawie tego
wykresu można jednoznacznie wyznaczyć wartości stałej K
m
i V
max
.
Rys. 2. Graficzne przedstawienie równania Lineweavera – Burka; V
max
-szybkość
maksymalna, S-stężenie substratu, K
m
-stała Michaelisa-Menten (Kączkowski 2005)
Pomiary stałej Michaelisa dokonywane przy użyciu różnych substratów pozwalają
wnioskować o sposobie wiązania enzymu z substratem, a zastosowanie do badań
kinetycznych różnych aktywatorów i inhibitorów pozwala na wyciągnięcie wniosków
dotyczących budowy centrum katalitycznego enzymu oraz warunków powstawania
kompleksów enzym-substrat.

6
Część doświadczalna
Ćwiczenie 4
Inwertaza (II). Kinetyka reakcji enzymatycznych
Zasada: Inwertaza (sacharaza, β-fruktofuranozydaza) należy do hydrolaz rozszczepiających
wiązania β-glikozydowe w sacharozie, hydrolizując ją do glukozy i fruktozy. Optymalne pH
działania enzymu wynosi ok. 4,7; przy pH 10 jest on już zupełnie nieaktywny.
Aktywność inwertazy można mierzyć metodą redukcyjną, ponieważ w miarę postępowania
hydrolizy sacharozy wzrasta stężenie cukrów redukujących. Do oznaczenia ilości cukrów
redukujących, w ćwiczeniu wykorzystano reakcję z kwasem pikrynowym, który przez cukry
redukowany jest do kwasu pikraminowego. Powstający w środowisku alkalicznym
pikraminian sodu daje czerwono-brązowe zabarwienie. W ćwiczeniu ilość cukrów
redukujących oznaczono metodą kolorymetryczną w przeliczeniu na mg glukozy uwolnione
z zastosowanego do analizy enzymatycznej substratu (sacharozy).
Metoda pozwala wyznaczyć szybkości początkowe reakcji przy różnych stężeniach sacharozy
i z wykresu Michaelisa-Menten lub Lineweavera – Burka wyznaczyć stałą Michaelisa.
I. Wykreślanie krzywej kalibracyjnej
1. Przygotować 6 probówek i oznaczyć je numerami od 0 do 5.
2. Do probówek odważyć odpowiednią ilość roztworu wzorcowego (0,25% roztwór
glukozy), a następnie uzupełnić wodą do 1 ml.
Zawartość probówek przygotować według schematu zamieszczonego w tabeli poniżej:
Nr probówki
0
(próbka zerowa)
1
2
3
4
5
0,25% wzorzec glukozy [g]
0
0,2
0,4
0,6
0,8
1,0
Woda destylowana [g]
1,0
0,8
0,6
0,4
0,2
0
Końcowa ilość glukozy [mg]
0
0,5
1
1,5
2
2,5
3. Do probówek dodać po 1,5 ml 10% NaOH oraz 1,5 ml 0,5% roztworu kwasu
pikrynowego, następnie zawartość probówek wymieszać i wstawić na 5 minut do wrzącej
łaźni wodnej.
4. Po wyjęciu probówek z wrzącej łaźni, wstawić je do łaźni lodowej również na 5 minut.
7
5. Po ochłodzeniu oznaczyć wartość absorbancji przy λ = 530 nm wobec próbki zerowej
i wykreślić krzywą zależności wartości absorbancji (A) od ilości glukozy [mg].
Uwaga! Próbki zerowej proszę nie wylewać!
Krzywa wzorcowa (kalibracyjna) przedstawia zależność między absorbancją i stężeniem.
W tym przypadku roztwór stosuje się do prawa Lamberta-Beera, więc ma postać linii prostej
przechodzącej przez punkt przecięcia osi współrzędnych.
W celu przygotowania krzywej na podstawie wykonanych pomiarów absorbancji dla kilku
stężeń roztworu wzorcowego i wykreśla się krzywą odkładając na osi X mg glukozy, a na osi
Y odpowiednie wartości absorbancji. Należy tak dobierać podziałkę na osiach żeby krzywa
była nachylona do osi pod kątem mniej więcej 45°. Ilość glukozy [mg] w próbie badanej
odczytuje się ze sporządzonej krzywej wzorcowej.
II. Wyznaczanie szybkości początkowych przy różnych stężeniach substratu
1. Przygotować dwa zestawy po 5 probówek. Pierwszy zestaw oznaczyć numerami t
0
, t
5
, t
10
,
t
15
, t
20
(probówki w drugim zestawie ponumerować analogicznie dopisując symbol ’ czyli
t
0’,
t
5’,
itd.).
2. Do probówek t
0
, t
0’
, wprowadzić po 1,5 ml 10% NaOH.
3. Następnie przygotować 2 mieszaniny inkubacyjne o różnym stężeniu substratu według
schematu zamieszczonego w tabeli poniżej i natychmiast po wymieszaniu (t = 0) z każdej
probówki pobrać po 1 ml mieszaniny inkubacyjnej i dodać do probówek zawierających
1,5 ml 10% NaOH i włączyć minutnik na 5 minut.
4. W międzyczasie do pozostałych (przygotowanych w pkt. 1) probówek ponalewać
po 1,5 ml 10% NaOH i wprowadzać do nich pobrane po 5, 10, 15, 20 minutach próbki
inkubacyjne o obj. 1 ml. Uwaga! Próbki mieszaniny inkubacyjnej pobrane z probówki nr
1 dodawać kolejno do probówek t
0
, t
5
, t
10
itd., natomiast próbki mieszaniny inkubacyjnej
pobrane z probówki nr 2 dodawać kolejno do probówek t
0’
, t
5’
, t
10’
itd.,
Nr probówki
1
2
0,2 M roztwór sacharozy
w buforze octanowym
6
3
bufor octanowy [ml]
0
3
inwertaza [ml]
6
6
Końcowe stężenie
sacharozy [M]
0,1
0,05
8
5. Po pobraniu ostatniej próbki do wszystkich 10 probówek dodać po 1,5 ml 0,5% roztworu
kwasu pikrynowego, następnie zawartość probówek wymieszać i wstawić na 5 minut do
wrzącej łaźni wodnej. Schemat doświadczenia przedstawiono w tabeli poniżej:
Nr probówki
t
0
(próbka
kontrolna)
t
5
t
10
t
15
t
20
Czas inkubacji [min.]
0
5
10
15
20
NaOH [ml]
1,5
1,5
1,5
1,5
1,5
Próbka mieszaniny
inkubacyjnej [ml]
1
1
1
1
1
0,5% roztwór kwasu
pikrynowego [ml]
1,5
1,5
1,5
1,5
1,5
6. Po wyjęciu probówek z wrzącej łaźni, wstawić je do łaźni lodowej również na 5 minut.
7. Po ochłodzeniu oznaczyć wartość absorbancji przy λ = 530 nm wobec próbki zerowej (tej
samej, której używano przy wyznaczaniu krzywej kalibracyjnej). Uwaga! Próbki zerowej
proszę nie wylewać!
8. Z krzywej kalibracyjnej odczytać ilości mg glukozy odpowiadającym poszczególnym
wartościom absorbancji uzyskanym dla próbek pobieranych po różnych czasach
z mieszanin o różnym stężeniu substratu.
9. Po odjęciu ilości mg glukozy przypadających na próbę kontrolną, wyniki przedstawić
w postaci wykresu zależności między ilością glukozy [mg], a czasem inkubacji (na osi
odciętych – czas w minutach, na osi rzędnych – ilość glukozy w mg). Wykresy
te należy sporządzić dla obydwu stężeń sacharozy (przykład na rys. 3).
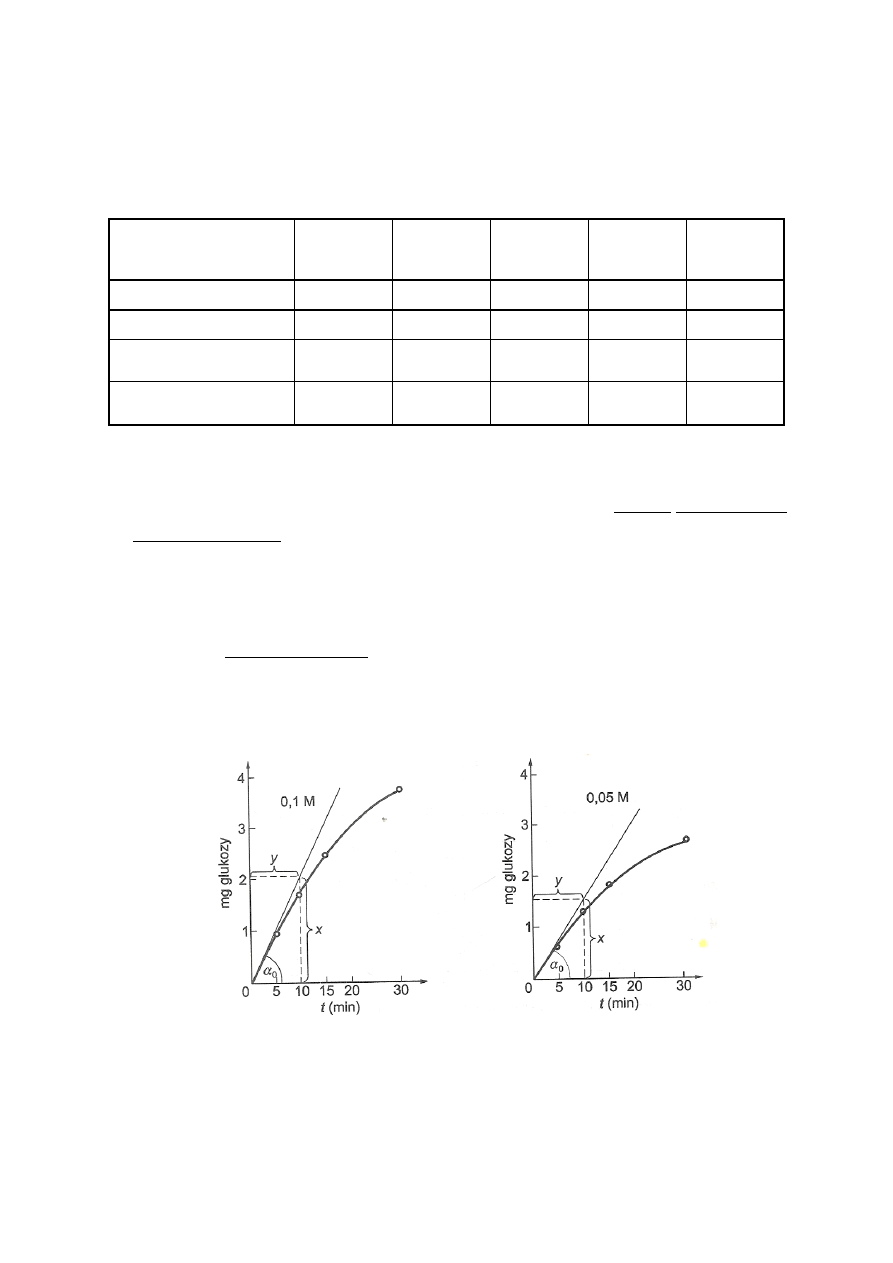
Rys. 3. Wyznaczanie szybkości początkowej reakcji przy różnych stężeniach substratu
(Kłyszejko-Stefanowicz 2003)
10. Z wykresów należy wyznaczyć szybkości początkowe (v
o
) reakcji enzymatycznej (przykład
na rys. 3). W tym celu należy przeprowadzić styczną do krzywej w punkcie t = 0. Styczna z
9
osią odciętych utworzy kąt α, którego tangens jest miarą szybkości początkowej (v
o
).
Wartości tg α nie wolno odczytywać z tablic na podstawie wartości kąta w stopniach,
ponieważ nanoszone na osi odciętych i rzędnych wartości nie są tej samej wielkości.
Oblicza się go ze stosunku wielkości przyprostokątnej przeciwległej do kąta α, podanej w
wartościach odkładanych na osi rzędnych (mg glukozy), do przyprostokątnej przyległej do
kąta α, podanej w wartościach odkładanych na osi odciętych (czas w minutach).
11. Z szybkości początkowych v
o
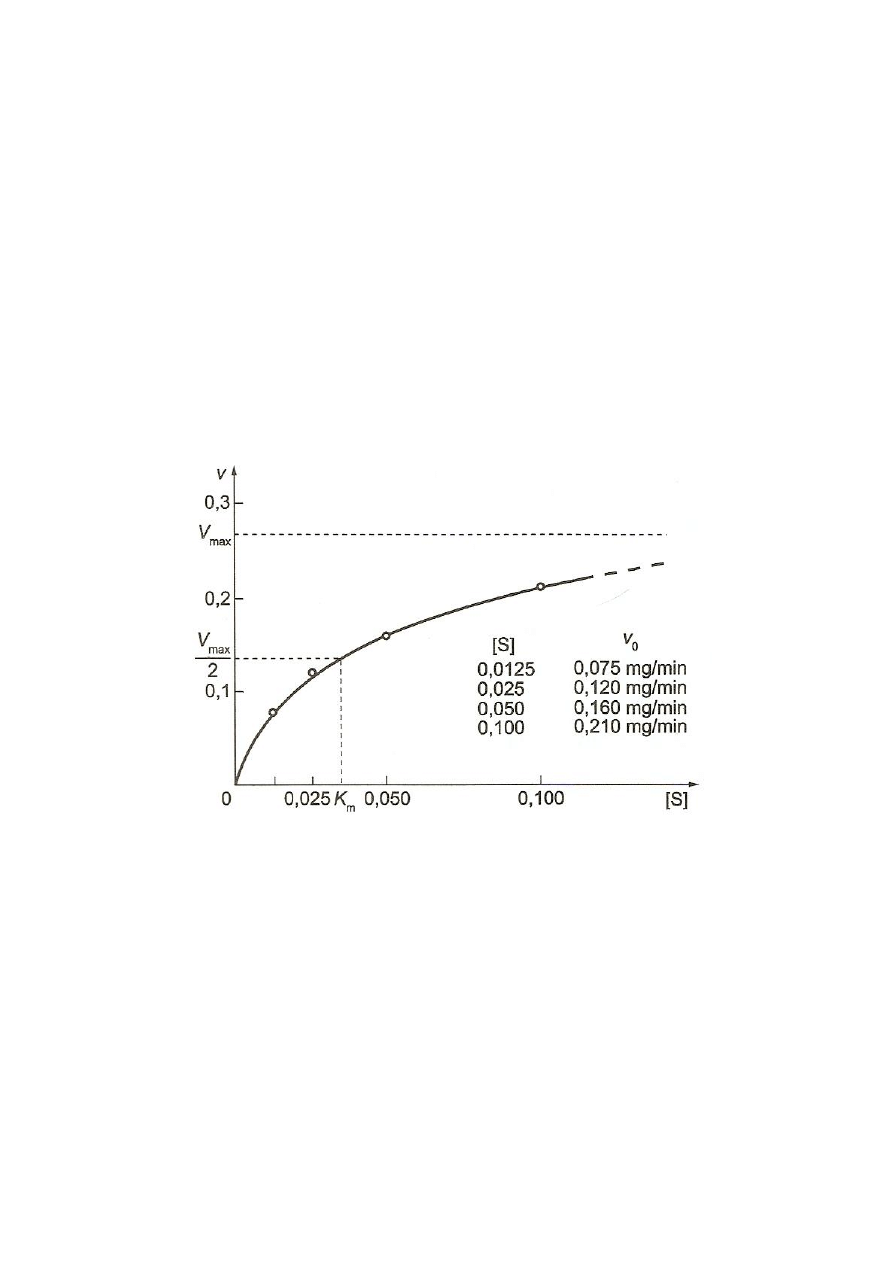
, wyliczonych dla obydwu stężeń substratu, wykreślić
zależność tych szybkości od stężenia substratu (krzywa Michaelisa-Menten) (rys. 4).
Ze względu na ograniczenia czasowe doświadczenie wykonywane jest jedynie dla dwóch
stężeń substratu, co sprawia, że wykreślona krzywa (hiperbola) ma przebieg jedynie
orientacyjny.
Rys. 4. Graficzne przedstawienie równania Michaelisa-Menten (Kłyszejko-
Stefanowicz 2003)
12. Wyznaczyć prostą dla równania Lineweavera – Burka – zależności 1/v od 1/[S] (Rys 5).
Wyznaczyć stałą Michaelisa K
m
oraz szybkość maksymalną V
max.
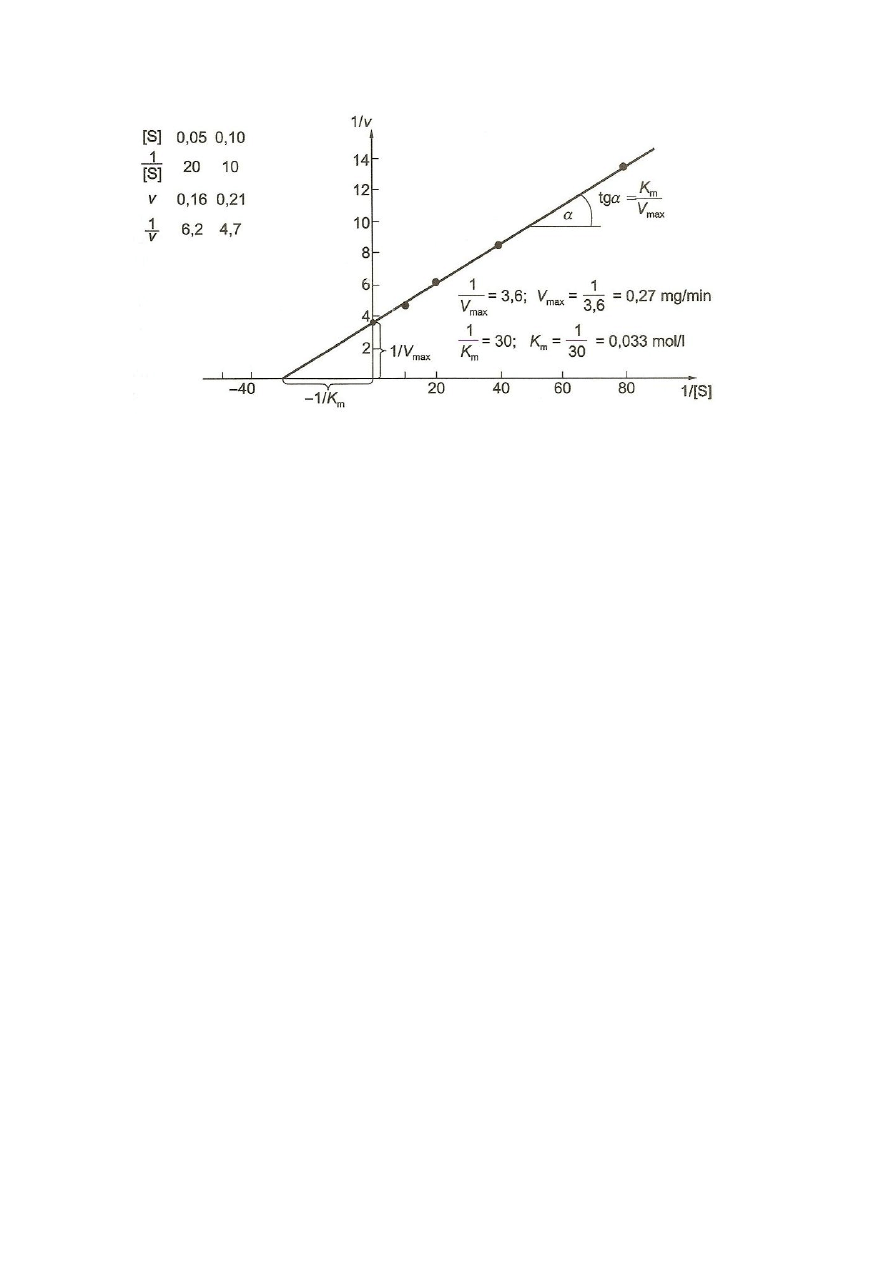
10
Rys. 5. Graficzne przedstawienie równania
Lineweavera – Burka
(Kłyszejko-
Stefanowicz 2003)
Literatura:
1) Podstawy biochemii. Kączkowski J. Wydawnictwa Naukowo-Techniczne, Warszawa,
2005.
2) Praktikum z biochemii. Praca zbiorowa pod red. Dubina A. i Turyny B. Instytut Biologii
Molekularnej Uniwersytetu Jagiellońskiego, Kraków 1999.
3) Przepisy do ćwiczeń z biochemii. Praca zbiorowa pod red. Stryjeckiej-Zimmer M.
Akademia Medyczna im
.
prof
.
F
.
Skubiszewskiego w Lublinie, Lublin, 2004.
4) Ćwiczenia z biochemii. Praca zbiorowa pod red. Kłyszejko-Stefanowicz L. Wydawnictwo
Naukowe PWN, Warszawa, 2003.
5) Ogólna technologia żywności. Pijanowski E., Dłużewski M., Dłużewska A., Jarczyk A.
Wydawnictwa Naukowo-Techniczne, Warszawa, 2004.
Wyszukiwarka
Podobne podstrony:
ĆWICZENIE 4 kinetyka reakcji enzymatycznej
06 Kinetyka reakcji enzymatycznych
kinetyka reakcji enzymatycznych I
Wyznaczanie parametrów kinetyki reakcji enzymatycznej za pomocą metod polarymetrycznych 5x
KINETYKA REAKCJI ENZYMATYCZNYCH
Kinetyka reakcji enzymatycznej m poteraj
Kinetyka reakcji enzymatycznych Nieznany
Ćwiczenie 10.Kinetyka reakcji, I rok, chemia
5 Kinetyka reakcji enzymatycznych
Kinetyka Reakcji Enzymatycznych
trusek hołownia, procesy membranowe, KINETYKA REAKCJI ENZYMATYCZNYCH
ENZYMY KINETYKA REAKCJI ENZYMATYCZNYCH
Kinetyka Reakcji Enzymatycznych
Kinetyka reakcji enzymatycznych
I Wyznaczanie parametrow kinetyki reakcji enzymatycznej polarymetr
kinetyka reakcji enzymatycznych
więcej podobnych podstron