1
BADANIA PRZESIEWOWE
Seminarium
Genetyczne badania przesiewowe polegają na:
•
Identyfikacji choroby genetycznej
•
Identyfikacji predyspozycji do wystąpienia choroby genetycznej
•
Identyfikacji genotypu, który zwiększa ryzyko posiadania dziecka z chorobą
uwarunkowaną genetycznie
Cel badań przesiewowych:
•
Badania przesiewowe polegają na testowaniu całej populacji w celu wykrycia osób
narażonych na wystąpienie choroby.
•
Identyfikacja tych osób umożliwia podjęcie skutecznego leczenia już od
pierwszych dni życia.
•
Nosiciele chorób genetycznych mogą zostać objęci opieka poradni genetycznej.
Badania przesiewowe w okresie noworodkowym
•
Populacyjne badania przesiewowe w kierunku fenyloketonurii (Polska)
•
Populacyjne badania przesiewowe w kierunku niedoczynności tarczycy (Polska)
•
Populacyjne badania przesiewowe w kierunku galaktozemii
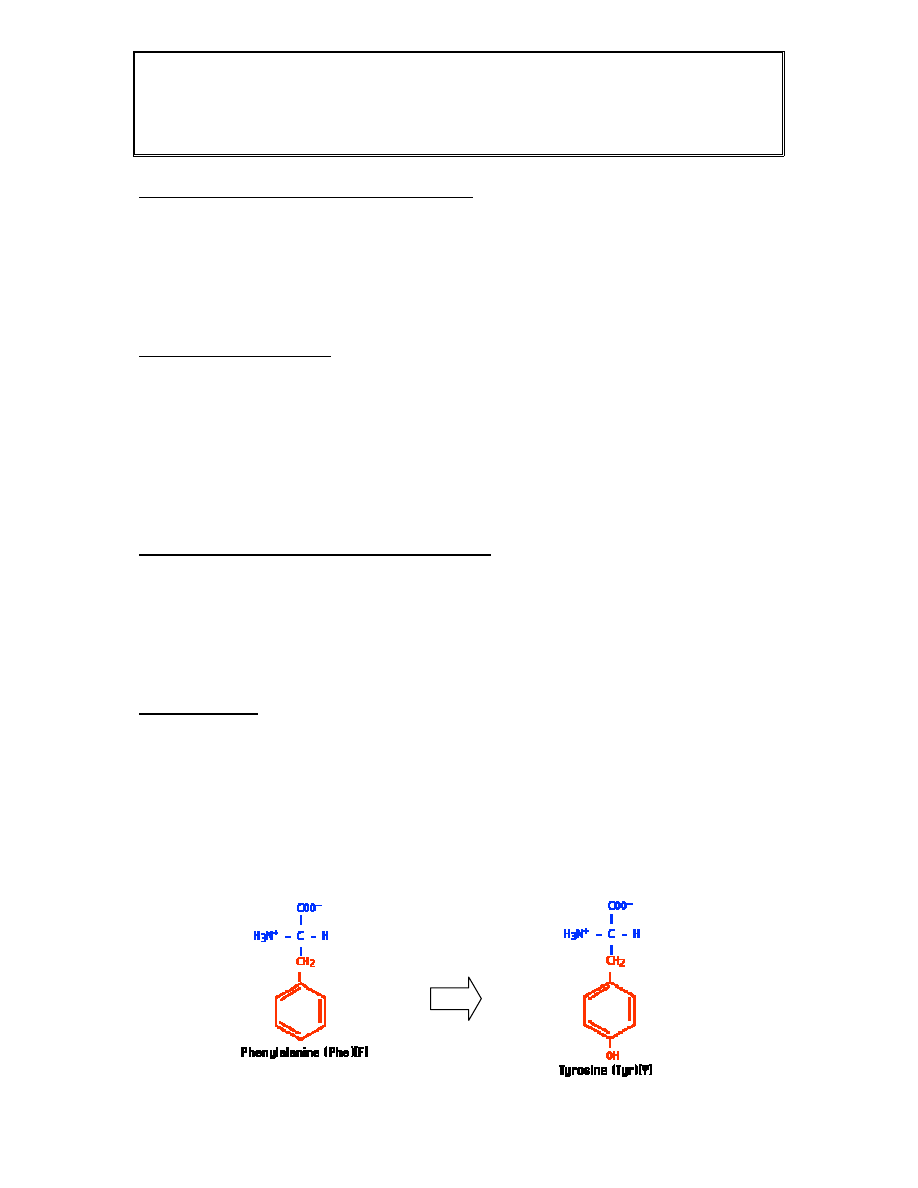
Fenyloketonuria
•
Choroba genetyczna, uwarunkowana autosomalnie recesywnie
•
Spowodowana mutacjami w genie hydroksylazay fenyloalaniny (PAH)
•
Efektem mutacji jest podwyższone stężenie fenyloalaniny we krwi i jej
metabolitów w moczu chorych
•
Częstość występowania od 1:10 000 do 1:20 000 żywych urodzeń

2
Fenyloketonuria – objawy
•
stopniowo rozwijające się opóźnienie rozwoju umysłowego
•
wymioty
•
jasna skóra i niebieskie oczy
•
stęchły mysi zapach
•
drgawki (25%)
•
zmiany w EEG (50%)
•
inne: małogłowie, wydatna szczęka górna, szerokie rozstawienie zębów,
hipoplazja szkliwa i opóźnienie wzrostu
Fenyloketonuria – objawy u dzieci chorych matek
•
opóźnienie rozwoju fizycznego i psychoruchowego
•
wady wrodzone
•
małogłowie
•
niska urodzeniowa masa ciała
Fenyloketonuria
•
Testy przesiewowe na fenyloketonurię opierają się na analizie poziomu stężenia
fenyloalaniny we krwi (metoda kolorymetryczna).
•
Krew powinna być pobrana rano, na czczo. Pobiera się ją z palca ręki, z pięty lub
z płatka ucha.
Fenyloketonuria – leczenie
•
Opiera się głównie na ograniczeniu spożycia fenyloalaniny. Głównym źródłem
białka dla osób chorych jest preparat stanowiący mieszaninę różnych
aminokwasów z wyłączeniem fenyloalaniny, wzbogacony o tyrozynę,
węglowodany, tłuszcz i składniki mineralne. W umiarkowanych ilościach
dozwolona jest żywność o niskiej zawartości fenyloalaniny, niedozwolona
ż
ywność wysokobiałkowa (mięso, ryby, ser, lody).
•
Skuteczność leczenia kontroluje się, analizując poziom fenyloalaniny we krwi.
Zawartość fenyloalaniny w produktach spożywczych
•
owoce – 2,7 %
•
warzywa – 3,5 % do ok.5 %
•
mleko i jego przetwory - ok. 5 - 5,5 %
•
chleb i produkty zbożowe – 5,6 %
•
mięso – 4,6 %

3
Niedoczynność tarczycy (hipotyreoza )
(
zwykle występuje sporadycznie ,niekiedy tylko jest
genetycznie uwarunkowana)
•
Niedoczynność tarczycy (hipotyreoza) jest chorobą wynikającą z niedoboru
hormonów tarczycy (tyroksyny) lub też z powodu oporności obwodowej tkanek
na działanie hormonów tarczycy.
•
Wrodzoną niedoczynność tarczycy (WNT) rozpoznaje się, jeżeli zaburzenia
syntezy tyroksyny (hormonu tarczycy) występują już od okresu życia płodowego.
WNT może mieć charakter trwały, gdy utrzymuje się przez całe życie chorego lub
przejściowy, kiedy niedoczynność tarczycy występuje w pierwszych miesiącach
ż
ycia, a potem poziom hormonów normalizuje się.
•
Częstość występowania wynosi w krajach europejskich od 1:3000 do 1:4000 żywo
urodzonych dzieci.
•
W Polsce w 1998 r. częstość WNT wynosiła 1 : 3810.
Niedoczynność tarczycy – objawy
•
przedłużająca się żółtaczka
•
trudności z karmieniem
•
duży język
•
obniżona temperatura ciała
•
ochrypły płacz
Niedoczynność tarczycy
•
W przypadku badań przesiewowych analizuje się poziom stężenia hormonu -
tyreotropiny (TSH).
•
Noworodki z pierwotna niedoczynnością tarczycy maja podwyższone stężenie
TSH.
•
Leczenie WNT polega na doustnym podawaniu soli sodowej L-tyroksyny.
•
Monitorowanie leczenia polega na ocenie rozwoju fizycznego i kontroli badań
hormonalnych w surowicy (FT4, TSH, ewentualnie T4).
Galaktozemia
•
Choroba genetyczna, uwarunkowana autosomalnie recesywnie
•
Mutacje w genie GALT
•
W moczu stwierdza się obecność cukrów (galaktozy), a we krwi brak aktywność
enzymu urydilotransferazy galaktozo-1-fosforanu
•
Częstość występowania w Europie 1:55 000 żywo urodzonych dzieci
•
Galaktozemia, wrodzony błąd metaboliczny uniemożliwiający przekształcenie
galaktozy w glukozę. Gromadzący się na skutek istnienia bloku metabolicznego
galaktozo-1-fosforan wywiera działanie toksyczne.

4
Galaktozemia –objawy
•
wymioty
•
biegunka
•
wzdęcie brzucha
•
brak apetytu
•
ubytek na wadze
•
pocenie, senność,
•
wrażliwość na infekcje
•
niedobór wzrostu
•
uszkodzenie wątroby
•
uszkodzenie nerek
•
uszkodzenie soczewki oka
•
uszkodzenie układu nerwowego
Galaktozemia – leczenie
•
Leczenie polega na szybkim wprowadzeniu diety bezgalaktozowej – preparaty
bez galaktozy i laktozy oparte na białku sojowym.
•
Powikłaniami w przypadku galaktozemii mogą być: opóźniony rozwój psychiczny i
dysfunkcja gonad
.
Podejrzewając zaburzenia przemiany galaktozy należy najpierw zaprzestać karmienia
mlekiem, a następnie diagnozować dziecko!!!
Badania przesiewowe na nosicielstwo mutacji
(Identyfikacja nosicieli )
•
Badania przesiewowe w rodzinach podwyższonego ryzyka urodzenia dziecka z
chorobą genetyczną.
•
Identyfikacja par małżeńskich, w których oboje partnerzy są heterozygotycznymi
nosicielami choroby.
•
Badania przesiewowe w rodzinach w których urodziło się dziecko z chorobą
genetyczną.
Badania przesiewowe na nosicielstwo mutacji koncentrują się na grupach etnicznych o
wysokim ryzyku wystąpienia określonych chorób uwarunkowanych mutacjami
pojedynczego genu.
•
anemia sierpowata ( Afrykańczycy, osoby z basenu Morza Śródziemnego, Indii i
Ś
rodkowego Wschodu)
•
choroba Taya-Sachsa (Żydzi Aszkenazyjscy)
•
talasemia ββββ (basen Morza Śródziemnego, południowo-wschodnia Azja)
•
mukowiscydoza (północna Europa, USA)
Badania przesiewowe kobiet ciężarnych
•
Ultrasonografia (USG).
•
Badania przesiewowe oparte na oznaczaniu specyficznych substancji pochodzenia
płodowego obecnych w surowicy krwi matki.

5
Wyszukiwarka
Podobne podstrony:
BADANIA PRZESIEWOWE 2
badania przesiewowe skuteczna profilaktyka
badania przesiewowe
Badania przesiewowe 2011, Badania przesiewowe:
PZ zal 2 Rozp MZ badania przesiewowe dzieci 0-6 lat
badania przesiewowe skuteczna p Nieznany
Badania przesiewowe
Badania przesiewowe w wykrywaniu raka szyjki macicy
Badania przesiewowe
ćw 6a badania przesiewowe
Badania przesiewowe (screeningowe)
ćw 7b badania przesiewowe
Badania przesiewowe 2011 id 766 Nieznany (2)
Badania przesiewowe w celu wykrycia raka jelita grubego
BADANIA PRZESIEWOWE CZĘŚĆ I
PZ zal 1 Rozp MZ Badania przesiewowe kobiety w ciazy
Prenatalne nieinwazyjne badania przesiewowe wad wrodzonych
Cw 10 Badania przesiewowe
więcej podobnych podstron