Prof. dr hab. med. Jacek Wachowiak
Klinika Onkologii, Hematologii i Transplantologii Pediatrycznej
II Katedry Pediatrii Uniwersytetu Medycznego
im. K. Marcinkowskiego w Poznaniu
CHOROBY ROZROSTOWE U DZIECI
I. CHOROBY ROZROSTOWE
UKŁADU KRWIOTWÓRCZEGO
1. BIAŁACZKI
EPIDEMIOLOGIA NOWOTWORÓW
U DZIECI I MŁODZIE
ś
Y DO LAT 18
CHOROBY ROZROSTOWE UKŁADU KRWIOTWÓRCZEGO (44,9%)
•
BIAŁACZKI (ALL, AML, CML, MDS)
33,6%
•
CHŁONIAKI (HD, NHL)
9,8%
•
HISTIOCYTOZA KOMÓREK LANGERHANSA
1,5%
GUZY LITE
(55,1%)
•
GUZY OUN
22,7%
•
NERCZAK ZARODKOWY
6,9%
•
ZWOJAK ZARODKOWY
6,7%
•
MI
Ę
SAKI TKANEK MI
Ę
KKICH
5,9%
•
GUZY KO
Ś
CI (osteosarcoma, mi
ę
sak Ewinga)
4,2%
•
SIATKÓWCZAK
3,3%
•
GUZY ZARODKOWE
2,3%
•
GUZY W
Ą
TROBY
1,0%
•
RAKI Z TKANEK NABŁONKOWYCH I INNE
2,1%
OSTRA BIAŁACZKA
LIMFOBLASTYCZNA
(acute lymphoblastic leukemia, ALL)

RÓ
ś
NICOWANIE ALL Z INNYMI CHOROBAMI
• Choroby tkanki ł
ą
cznej
• Niedokrwisto
ść
aplastyczna
• Małopłytkowo
ść
autoimmunologiczna
• Zaka
ż
enia (mononukleoza, cytomegalia)
• Choroby rozrostowe :
- zwojak zarodkowy (neuroblastoma)
- zespół mielodysplastyczny
- nieziarnicze chłoniaki zło
ś
liwe
- ostra białaczka szpikowa
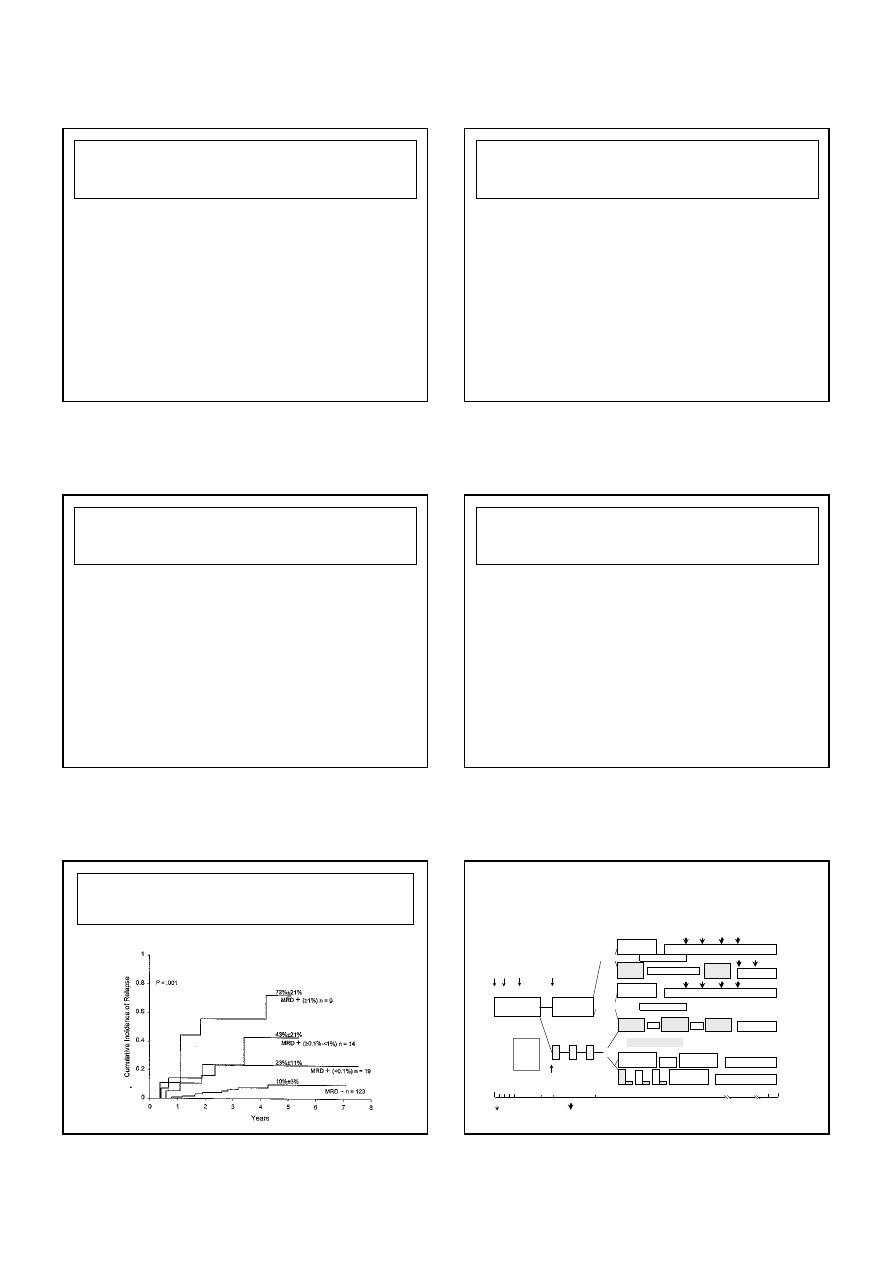
1. Wiek
≥
1 roku i
≤≤≤≤
6 lat;
2. Wst
ę
pna leukocytoza < 20 000/Ul;
3. Liczba blastów w krwi obwodowej
< 1000/ul w 8 dniu terapii;
4. Szpik M1 lub M2 w 15 dniu terapii
5. Szpik M1 w 33 dniu terapii
* Wszystkie warunki musz
ą
by
ć
spełnione
STRATYFIKACJA DZIECI Z ALL
(wg ALL IC-BFM 2002)
Kryteria standardowego ryzyka wznowy* (SR)
1. Liczba blastów w krwi obwodowej
< 1000/ul w 8 dniu terapii:
- oraz wiek <1 roku i >6 lat i/lub wst
ę
pna
leukocytoza < 20 000/Ul;
- oraz szpik M1 lub M2 w 15 dniu terapii
- oraz szpik M1 w 33 dniu terapii
2. Kryteria standardowego ryzyka, ale szpik M3
w 15 dniu i szpik M1 w 33 dniu terapii;
STRATYFIKACJA DZIECI Z ALL
(wg ALL IC-BFM 2002)
Kryteria po
ś
redniego ryzyka wznowy* (IR)
1. Liczba blastów w krwi obwodowej
< 1000/ul w 8 dniu terapii:
- oraz wiek <1 roku i >6 lat i/lub wst
ę
pna
leukocytoza < 20 000/Ul;
- oraz szpik M1 lub M2 w 15 dniu terapii
- oraz szpik M1 w 33 dniu terapii
2. Kryteria standardowego ryzyka, ale szpik M3
w 15 dniu i szpik M1 w 33 dniu terapii;
STRATYFIKACJA DZIECI Z ALL
(wg ALL IC-BFM 2002)
Kryteria po
ś
redniego ryzyka wznowy* (IR)
1. t (9; 22), BCR/ABL
2. t (4; 11), MLL/AF 4
3. oporno
ść
blastów na wst
ę
pn
ą
kortykosteroidoterapi
ę
(liczba blastów w krwi obwodowej > 1,0 G/L w 8 dniu terapii)
4. IR oraz szpik M3 w 15 dobie leczenia
4. Szpik M2 lub M3 w 33 dobie leczenia
5. wysoki poziom choroby resztkowej (MRD > 10
-3
)
w 12 tygodniu leczenia
* Przynajmniej jedno kryterium musi by
ć
spełnione
STRATYFIKACJA DZIECI Z ALL
(wg ALL IC-BFM 2002)
Kryteria wysokiego ryzyka wznowy* (IR)
Clinical importance of minimal residual disease in childhood acute
lymphoblastic leukemia
Coustan-Smith E, Sancho J, Hancock ML et al.
Blood 2000; 96: 2691-2696
H
R
3’
0
6-MP / MTX
10
12
H
R
1’
H
R
2’
'
BM sampling
12 Gy*
* presymptomatic cranial irradiation
#
selected indications for allo-SCT in all strata of HR
§ No randomization of AIEOP vs. BFM but choice by group according to previous
experience with one of the two high-risk strategies in trial 95
6-MP / MTX
II
HR:
PRED-PR
IR,M3 d15
t(9;22)
t(4;11)
NR d33
6-MP / MTX
BFM
Allo-SCT
#
III
12 Gy* only for T-ALL
III
III
6-MP / MTX
III
III
II
MP/MTX
4 wks
II
6-MP / MTX
II
AIEOP
I/I‘ **
d15
104 W
52
10 weeks interim maintenance
with 6-MP / MTX
H
R
1'
H
R
2'
H
R
3'
MP/MTX
4 wk s
MP/MTX
4 wk s
d33
SR – R
IR – R
IT MTX (in maintenance)
** Protocol I‘
DNR 30mg/m
2
x2 only for SR patients with BCP-ALL
dx
§
§
ALLIC BFM 2002 : TREATMENT
Version approved in Hannover on 23.02.2002
w12
6-MP / MTX
II
$
for BCP-ALL: MTX 2g/m
2
/24h x4, for T-ALL: MTX 5g/m
2
/24h x4
12 Gy* only for T-ALL
mM/M
$
12 Gy*
R
12 Gy*
LECZENIE ALL U DZIECI WEDŁUG PROGRAMU ALL-IC BFM 2002
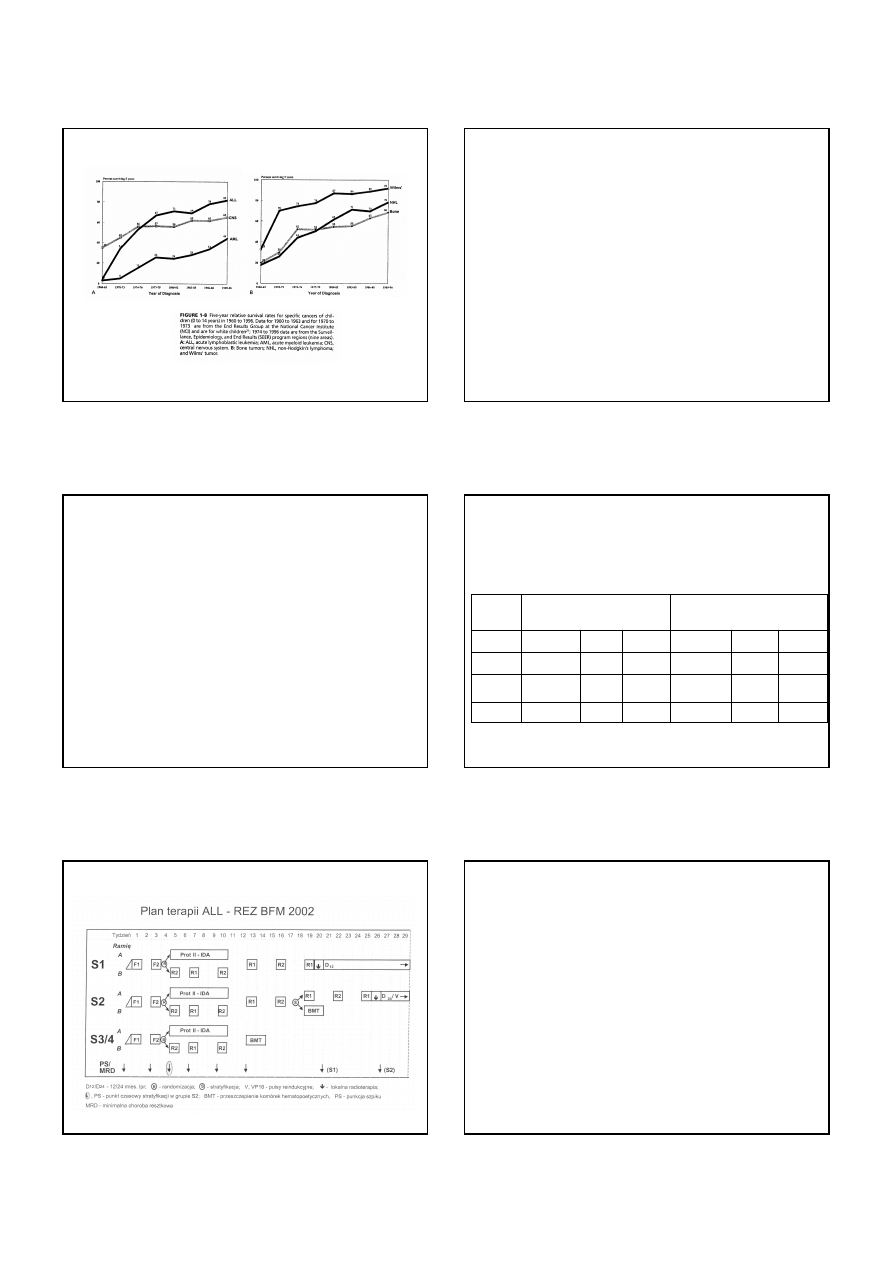
AKTUALNE WYNIKI LECZENIA ALL U DZIECI
W O
Ś
RODKACH PPGLBiC
• Grupa ryzyka standardowego
(Derwich i wsp, Ped Prakt 2002; 10: 15-17)
pEFS po 7,5 latach = 84%
• Grupa wysokiego ryzyka
(Kwieci
ń
ska i wsp, Ped Prakt 2002; 10: 19-22)
pEFS po 3,5 latach = 78%
WZNOWA ALL U DZIECI
• Cz
ę
sto
ść
wyst
ę
powania wznowy : 20-25%
• Rodzaje wznowy :
a. ze wzgl
ę
du na czas wyst
ą
pienia
-
bardzo wczesna – w 1 roku leczenia
- wczesna – do 6 m-ca po zako
ń
czeniu leczenia (74%)
- pó
ź
na – po 6 m-cach od zako
ń
czenia leczenia (26%)
b. ze wzgl
ę
du na lokalizacj
ę
- szpikowa (hematologiczna) (50%)
- pozaszpikowa (mózgowa 8-15%, gonadalna 20%)
- mieszana (15-20%)
Podział wznów ALL na grupy ryzyka
według kryteriów REZ-ALL-BFM-SG
Immunofenotyp: non- T
Immunofenotyp: pre-T
Lokalizacja
Czas
Pozaszpikowa
izolowana
Szpikowa
mieszana
Szpikowa
izolowana
Pozaszpikowa
izolowana
Szpikowa
mieszana
Szpikowa
izolowana
bardzo
wczesna
S2
S4
S4
S2
S4
S4
wczesna
S2
S2
S3
S2
S4
S4
pó
ź
na
S1
S2
S2
S1
S4
S4
WYNIKI LECZENIA WZNOWY ALL U DZIECI
• SZPIKOWA
- wczesna 8-18% !
- pó
ź
na
30-44%
• MIESZANA
- wczesna
25% !
- pó
ź
na
40%
• MÓZGOWA
- wczesna
41%
- pó
ź
na
83%
• GONADALNA
- wczesna
32%
- pó
ź
na
52%

OSTRA BIAŁACZKA
SZPIKOWA
(acute myeloblastic leukemia, AML)
OBJAWY KLINICZNE AML
• Objawy związane z wyparciem prawidłowego
krwiotworzenia przez blasty białaczkowe :
- niedokrwistość;
- skaza krwotoczna małopłytkowa;
- zakażenia;
• Powiększenie wątroby i/lub śledziony (ok. 50%)
• Objawy nacieczenia OUN (bóle głowy, nudności, wymioty,
zajęcie nerwów czaszkowych)
• Nacieki w skórze, kościach czaszki, kości żuchwowej, w
oczodole i w obrębie dziąseł
AML M0 wg FAB
AML M1 wg FAB
AML M2 wg FAB

AML M4 wg FAB
AML M5 wg FAB
AML M6 wg FAB
AML M7 wg FAB
AKTUALNE WYNIKI LECZENIA AML U DZIECI
W O
Ś
RODKACH PPGLBiC
(Dłu
ż
niewska i wsp, Ped Prakt 2002; 10: 29-32)
• Grupa ryzyka standardowego
pEFS po 3 latach = 79%
• Grupa wysokiego ryzyka
pEFS po 3 latach = 41%
1.
AML M0
2.
AML M1/M2 – bez pałek Auera
3.
AML M4 bez eozynofilii
4.
AML M5/M6/M7
5.
Obecno
ść
mutacji FLT3-ITD
6. Blasty >5% w 15 dobie terapii
KRYTERIA WYSOKIEGO RYZYKA WZNOWY
U DZIECI Z AML
WG AML-BFM 1998 / INTERIMPHASE 2004
BFM: AML-INTERIMPHASE 2004
W Balwierz, 2007
BFM: AML INTERIMPHASE 2004
W Balwierz, 2007
BFM: AML-INTERIMPHASE 2004
W Balwierz, 2007
PRZEWLEKŁA BIAŁACZKA
SZPIKOWA
(chronic myelocytic leukemia, CML)
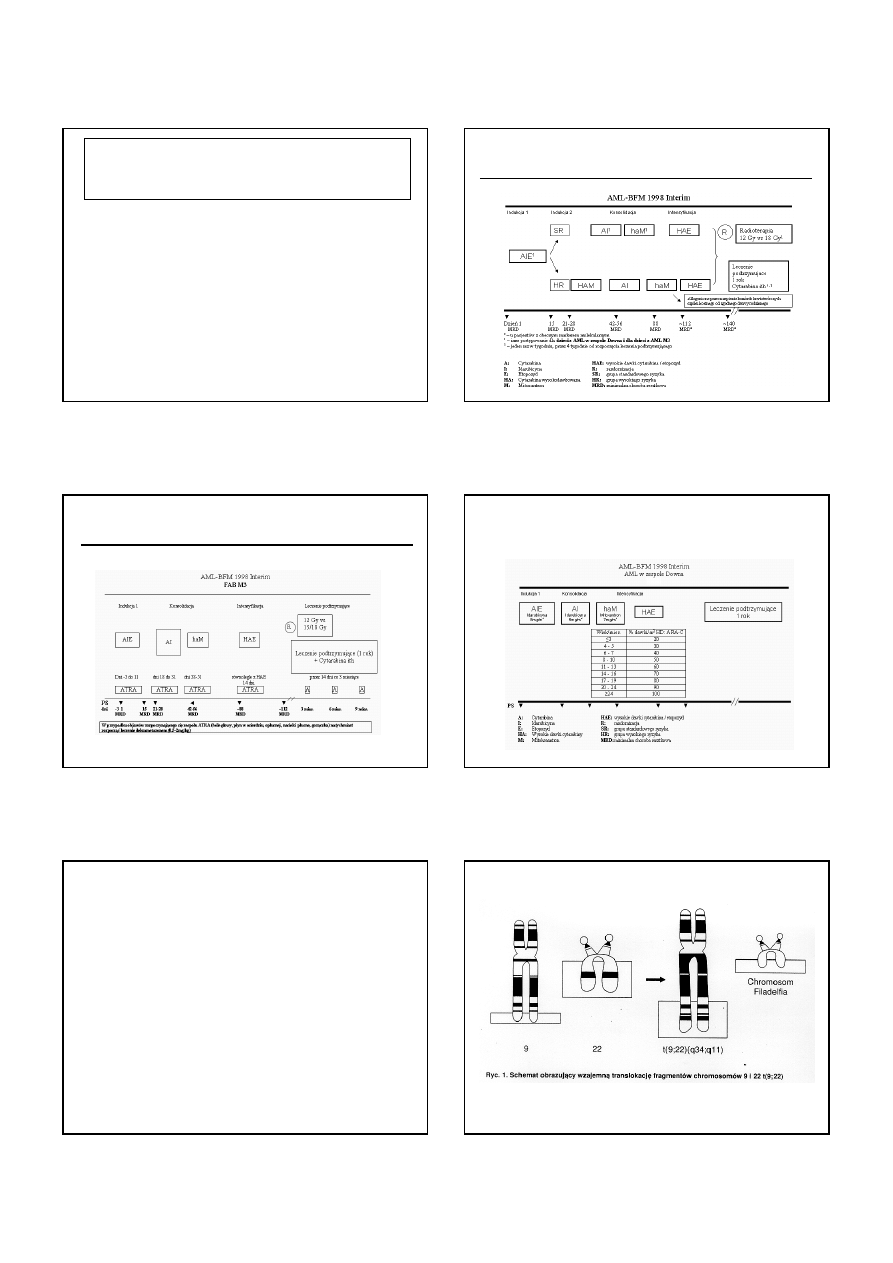
OBJAWY KLINICZNE CML
W FAZIE PRZEWLEKŁEJ U DZIECI
• OBJAWY PODMIOTOWE
- złe samopoczucie i zmęczenie (49%)
- dolegliwości brzuszne
(31%)
- bóle kości i stawów
(23%)
- utrata masy ciała
(15%)
- gorączka
(13%)
- priapism
(3%)
- brak dolegliwości
(18%)
• OBJAWY PRZEDMIOTOWE
- splenomegalia
(95%) !
- hepatomegalia > 3 cm
(33%)
CML w fazie przewlekłej
CML w fazie zaostrzenia
CML w kryzie blastycznej
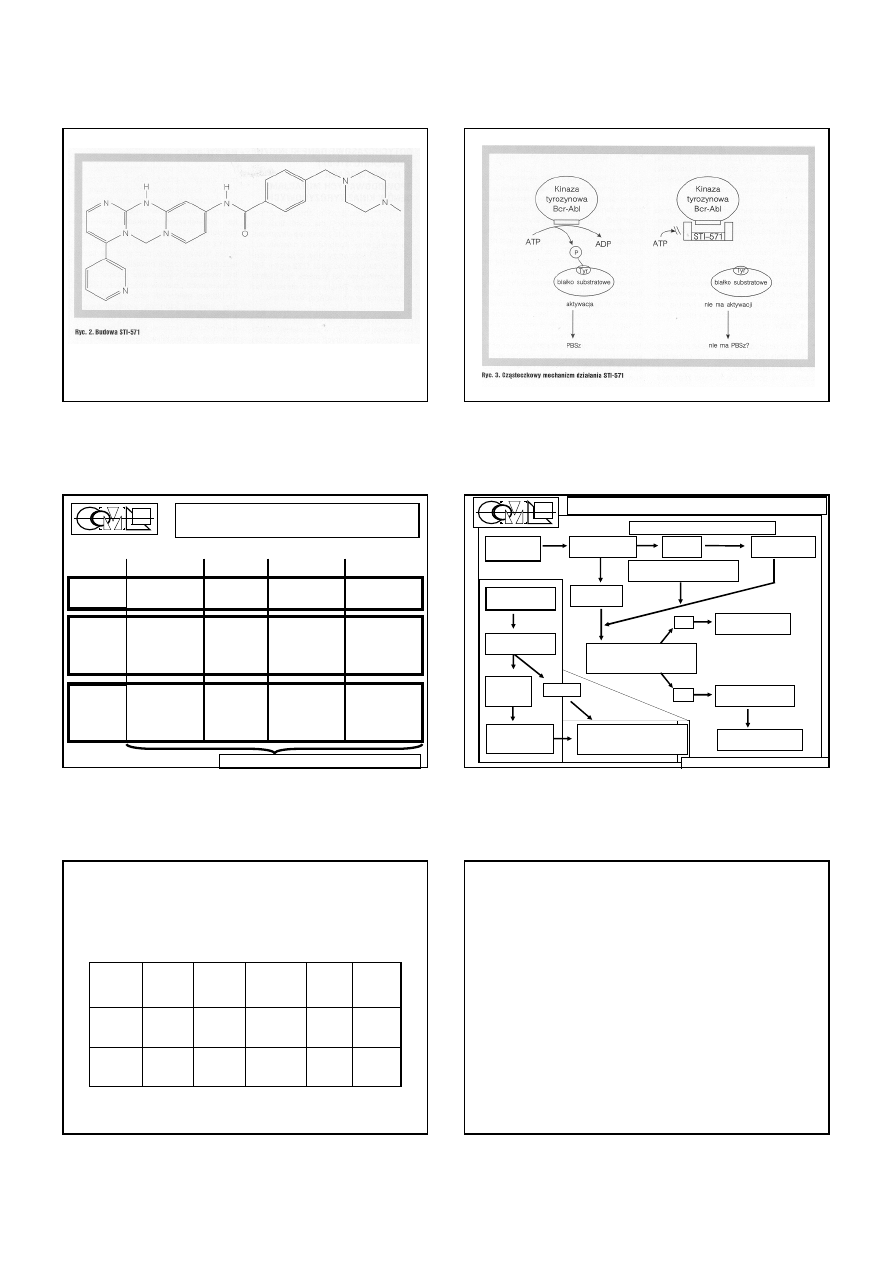
Criteria for response assessment of CML
treated with imatinib
Duration of imatinib therapy
Failure
Sub-
optimal
Response
Optimal
Response
3 Months
No haematol.
Response
No complete
haematological
remission (CHR)
1 – 2 log reduc-
tion in the
number of BCR-
ABL transcripts
6 Months
> 95 % Ph+
35 – 95 % Ph+
< 35 % Ph+
12 Months
> 35 % Ph+
1 – 35 % Ph+
< 0 % Ph+ (CCyR)
< 3 log reduc-
tion in the
number of BCR-
ABL transcripts
18 Months
< 35 % – 1 % Ph+
0 % Ph+ (CCyR)
< 3 log reduc-
tion in the
number of BCR-
ABL transcripts
0 % Ph+ (CCyR)
> = 3 log reduc-
tion in the
number of BCR-
ABL transcripts
No additional cytogenetic aberration except Ph+ !
PAED
II
Suttorp-M-EBMT Paed WP Poznan, 2008
CML in
chronic phase
standard dose
imatinib up-front
secondary failure
of imatinib
ongoing
response
perfectly HLA-matched donor
(either sibling or unrelated;
9-10/10 alleles, high res. typing)
yes
no
maximal duration of treatment
2 years
recommend stem
cell transplantation
continue imatinib as
long as effective
Interferon-alpha or 2nd
generation TK-inhibitor
primary failure
of imatinib
CML in advanced
phase (AC / BC)
medium / high dose
imatinib up-front
ALL- or AML-
based induction
therapy
primary
failure of
imatinib
response
urgent stem cell transplantation
accepting also partially HLA-
matched donors
!!! Monitoring !!!
Algorithms for combining Imatinib and SCT in pediatrics
Suttorp-M-EBMT Paed WP Poznan, 2.- 4. 06. 2008
PAED
II
Stem cell transplantation for chronic myeloid leukemia in children
Cwynarski K, Roberts IAG, Iacobelli S i wsp.
w imieniu EBMT PDs WP oraz EBMT CL WP
Blood 2003; 102: 1224-1231
95 o
ś
rodków
EBMT
OKRES
LICZBA
DZIECI
OKRES
OBSERWACJI
(mediana)
pLFS
po 3 latach
pOS
po 3 latach
MSD
1985-2001
182
1 – 202
miesi
ą
ce
(49 mies.)
CP1 63%
AP 35%
CP1 75%
AP 46%
MUD
1985-2001
132
1 – 193
miesi
ą
ce
(21 mies.)
CP1 56%
AP 34%
CP1 65%
AP 39%
ZESPÓŁ
MIELODYSPLASTYCZNY
(myelodysplastic syndrome, MDS)

DEFINICJA MDS
• MDS to niejednorodna grupa chorób układu
krwiotwórczego charakteryzujących się:
- narastającą pancytopenią (niedokrwistość, leukopenia,
małopłytkowść) w krwi obwodowej
- bogatokomórkowym szpikiem
( tj. w kontekście pancytopenii - nieefektywną hemopoezą )
- ponadto często spostrzegane są nieprawidłowości
morfologiczne, w tym zwiększony odsetek blastów w
szpiku i we krwi obwodowej
- zaburzenia w utylizacji żelaza
- progresja do ostrej białaczki
( stąd dawniej MDS = stan przedbiałaczkowy )
ETIOPATOGENEZA MDS
• w wyniku mutacji na poziomie macierzystej komórki
krwiotwórczej powstaje klon patologicznych komórek
posiadający zdolność do odnowy własnej populacji,
jednak z zaburzoną w różnym stopniu zdolnością
różnicowania i dojrzewania
• Komórki klonu posiadają przewagę wzrostową nad
prawidłowymi komórkami w szpiku, co prowadzi do
stopniowego wypierania przez nie prawidłowej tkanki
krwiotwórczej
PODZIAŁ ETIOPATOGENETYCZNY MDS
• Pierwotny MDS
(występuje bez uchwytnej przyczyny)
• Wtórny MDS
(występuje w następstwie chemioterapii i/lub
radioterapii chorób nowotworowych)
OBRAZ KLINICZNY MDS
• Związane cytopenią i zaburzoną czynnością krwinek
• Objawy niedokrwistości
• Skaza krwotoczna
• Nawracające zakażenia
• Rzadko powiększenie wątroby i śledziony
DIAGNOSTYKA MDS
• Badania cytologiczne
- krew obwodowa
- szpik
• Badanie histopatologiczne szpiku
• Badania cytogenetyczne
• Badania biochemiczne
KLASYFIKACJA ZESPOŁÓW MIELODYSPLASTYCZNYCH WG FAB
1. Niedokrwistość oporna na leczenie (refractory anemia, RA)
< 1% blastów we krwi obwodowej
< 5% blastów w szpiku
2. RA z obecnością syderoblastów pierścieniowatych (RA with ring
sideroblasts, RARS)
< 1% blastów we krwi obwodowej
< 5% blastów w szpiku
> 15% komórek erytroblastów stanowią syderoblasty
3. RA ze zwiększonym odsetkiem blastów (RA with excess of blasts, RAEB)
< 5% blastów we krwi obwodowej
5-20% blastów w szpiku
4. RAEB w okresie transformacji (RAEB in transformation, RAEB-t)
> 5% blastów we krwi obwodowej
< 20-30% blastów w szpiku i/lub pałeczki Auera w blastach
5. Przewlekła białaczka mielomonocytarna (CMML)
< 5% blastów we krwi obwodowej + monocytoza > 1 x 10
9
/l
5-20% blastów w szpiku

EPIDEMIOLOGIA MDS U DZIECI
• 0,5% chorób rozrostowych u dzieci
• U niemowląt i małych dzieci
- CMML
• U dzieci starszych
- RA
- RAEB
Młodzieńcza
białaczka mielo-
monocytarna
Młodzieńcza
białaczka mielo-
monocytarna
DIAGNOSTYKA RÓśNICOWA MDS U DZIECI
• Niedokrwistość aplastyczna
• Niedokrwistość megaloblastyczna
• Wrodzona niedokrwistość dyserytropoetyczna
• Małopłytkowość autoimmunologiczna
• Zespół dużej śledziony (hipersplenizm)
• Osteomielofibroza
• Ostra białaczka (ALL, AML)
• Przerzuty komórek nowotworowych do szpiku
kostnego
METODY STOSOWANE W LECZENIU MDS
• Metoda leczenia z wyboru
- allogeniczna transplantacja szpiku
od dawcy rodzinnego
od dawcy niespokrewnionego
• Intensywna chemioterapia
- nie daje zadowalających rezultatów
• Próby leczenia cytokinami
- erytropoetyną (EPO)
- GM-CSF
- G-CSF
- INF-alfa i gamma
• Leczenie immunosupresyjne (RA,RARS, RAEB)
- prednizon + ATG, CsA, talidomid, pentoxyfilina
Wyszukiwarka
Podobne podstrony:
Wyk. 02 Rozwój fizyczny, Lekarski, Propedeutyka pediatrii, Wykłady
Wyk 02 Pneumatyczne elementy
lekarski sem 02 niedokrwistosci
lekarski wyk 03 nowotwory
V Lekarski wyk ad 4
lekarski wyk 01 chloniaki
Mat Bud wyk 02
La rzut piłką lekarską w tył 02 09 18
Badania lekarskie pracowników 02 07 01
lekarski wyk 04 powiklania infekcyjne
Wyk1, Geologia - wyk 1 (02
Mat Bud wyk 02
RF.wyk.02.operacje
lekarski wyk 01 chloniaki
RK.wyk.02
wyk.02, PWR - MBM, SEMESTR IV, PODSTAWY AUTOMATYKI
więcej podobnych podstron