Dziedziczenie
Dziedziczenie
jednogenowe
jednogenowe
sprzężone z płcią
sprzężone z płcią
Przygotowali:
Sylwia Kamińska
Andrzej Bieńkowski
Katarzyna Blus-Bieńkowska

Większość chorób dziedziczących się
Większość chorób dziedziczących się
jednogenowo spowodowana jest
jednogenowo spowodowana jest
mutacjami punktowymi w obrębie
mutacjami punktowymi w obrębie
genów. Choroby dziedziczone
genów. Choroby dziedziczone
autosomalnie dominująco,
autosomalnie dominująco,
autosomalnie recesywnie i sprzężone z
autosomalnie recesywnie i sprzężone z
płcią są najczęściej wynikiem
płcią są najczęściej wynikiem
mutacji genów kontrolujących syntezę
mutacji genów kontrolujących syntezę
białek strukturalnych.
białek strukturalnych.

Choroby sprzężone z płcią
Choroby sprzężone z płcią
są
są
determinowane przez obecność alleli na
determinowane przez obecność alleli na
chromosomie X. Przy chorobach
chromosomie X. Przy chorobach
recesywnie dziedziczonych, aby
recesywnie dziedziczonych, aby
choroba ujawniła się u kobiety, oba
choroba ujawniła się u kobiety, oba
chromosomy X muszą posiadać recesywny
chromosomy X muszą posiadać recesywny
allel (kobieta musi być homozygotą,
allel (kobieta musi być homozygotą,
heterozygotyczna kobieta jest nosicielką),
heterozygotyczna kobieta jest nosicielką),
natomiast choroba ujawni się u mężczyzn
natomiast choroba ujawni się u mężczyzn
gdyż posiadają oni tylko jeden
gdyż posiadają oni tylko jeden
chromosom X (samiec jest hemizygotą).
chromosom X (samiec jest hemizygotą).

Czasem u kobiet heterozygot
Czasem u kobiet heterozygot
obserwuje się wystąpienie takiej
obserwuje się wystąpienie takiej
choroby bez pełnych objawów
choroby bez pełnych objawów
klinicznych ze względu na lionizację
klinicznych ze względu na lionizację
czyli losową inaktywację jednej kopii
czyli losową inaktywację jednej kopii
chromosomu X.
chromosomu X.
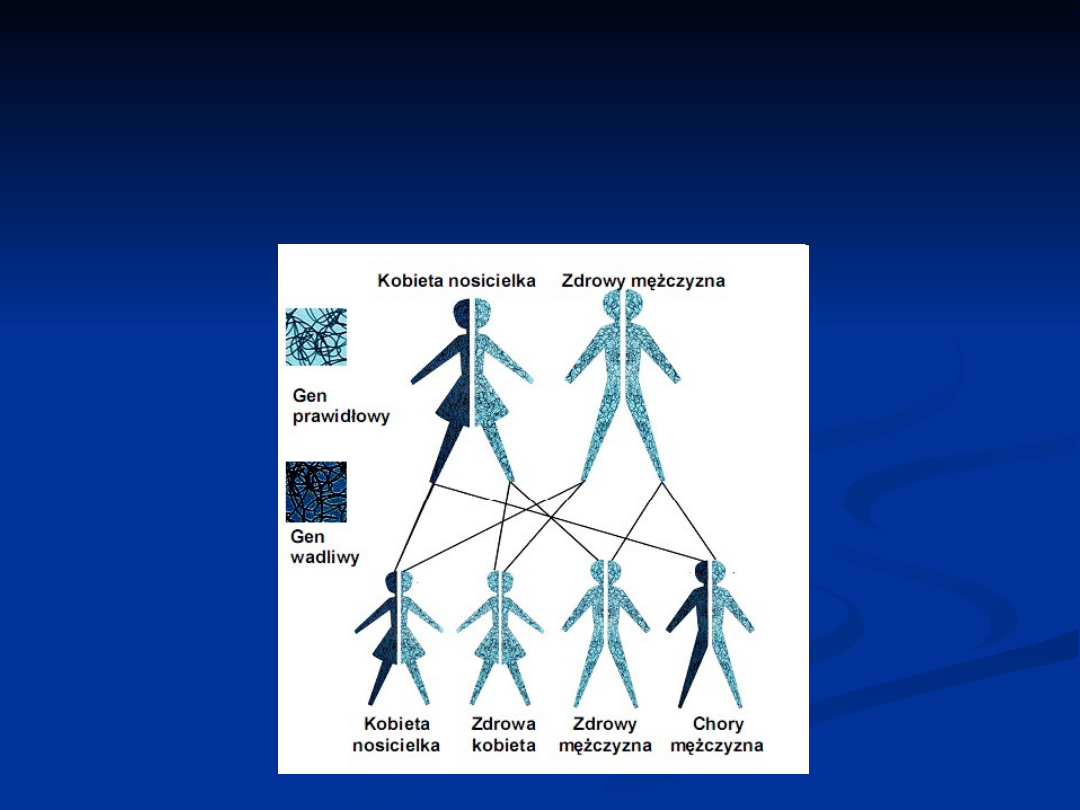
W jaki sposób choroby sprzężone
W jaki sposób choroby sprzężone
z chromosomem X
z chromosomem X
są przekazywane przez kobiety
są przekazywane przez kobiety
będące nosicielkami
będące nosicielkami

Jeśli kobieta będąca nosicielką ma syna
Jeśli kobieta będąca nosicielką ma syna
, może
, może
przekazać mu albo chromosom X z prawidłowym
przekazać mu albo chromosom X z prawidłowym
genem albo chromosom X ze zmienionym genem.
genem albo chromosom X ze zmienionym genem.
Zatem dla każdego z synów ryzyko odziedziczenia
Zatem dla każdego z synów ryzyko odziedziczenia
zmienionego genu i wystąpienia choroby wynosi
zmienionego genu i wystąpienia choroby wynosi
50% (1 na 2 przypadki). Szansa, że syn odziedziczy
50% (1 na 2 przypadki). Szansa, że syn odziedziczy
prawidłowy gen wynosi również 50%
prawidłowy gen wynosi również 50%
(1 na 2 przypadki). Jeśli tak się stanie,
(1 na 2 przypadki). Jeśli tak się stanie,
nie będzie on dotknięty chorobą.
nie będzie on dotknięty chorobą.
Ryzyko pozostaje takie samo dla każdego
Ryzyko pozostaje takie samo dla każdego
z synów.
z synów.

Jeśli kobieta będąca nosicielką ma córkę
Jeśli kobieta będąca nosicielką ma córkę
,
,
może przekazać jej albo chromosom X z
może przekazać jej albo chromosom X z
prawidłowym genem albo chromosom X ze
prawidłowym genem albo chromosom X ze
zmienionym genem. Zatem dla każdej z córek
zmienionym genem. Zatem dla każdej z córek
ryzyko odziedziczenia zmienionego genu wynosi
ryzyko odziedziczenia zmienionego genu wynosi
50% (1 na 2 przypadki). Jeśli tak się stanie,
50% (1 na 2 przypadki). Jeśli tak się stanie,
córka będzie nosicielką, tak samo jak
córka będzie nosicielką, tak samo jak
jej matka. Istnieje też 50% szans (1 na 2
jej matka. Istnieje też 50% szans (1 na 2
przypadki), że córka odziedziczy prawidłowy gen.
przypadki), że córka odziedziczy prawidłowy gen.
Jeśli tak się stanie, nie będzie ona nosicielką i
Jeśli tak się stanie, nie będzie ona nosicielką i
zupełnie nie będzie dotknięta chorobą.
zupełnie nie będzie dotknięta chorobą.
Ryzyko pozostaje takie samo dla każdej z
Ryzyko pozostaje takie samo dla każdej z
córek.
córek.
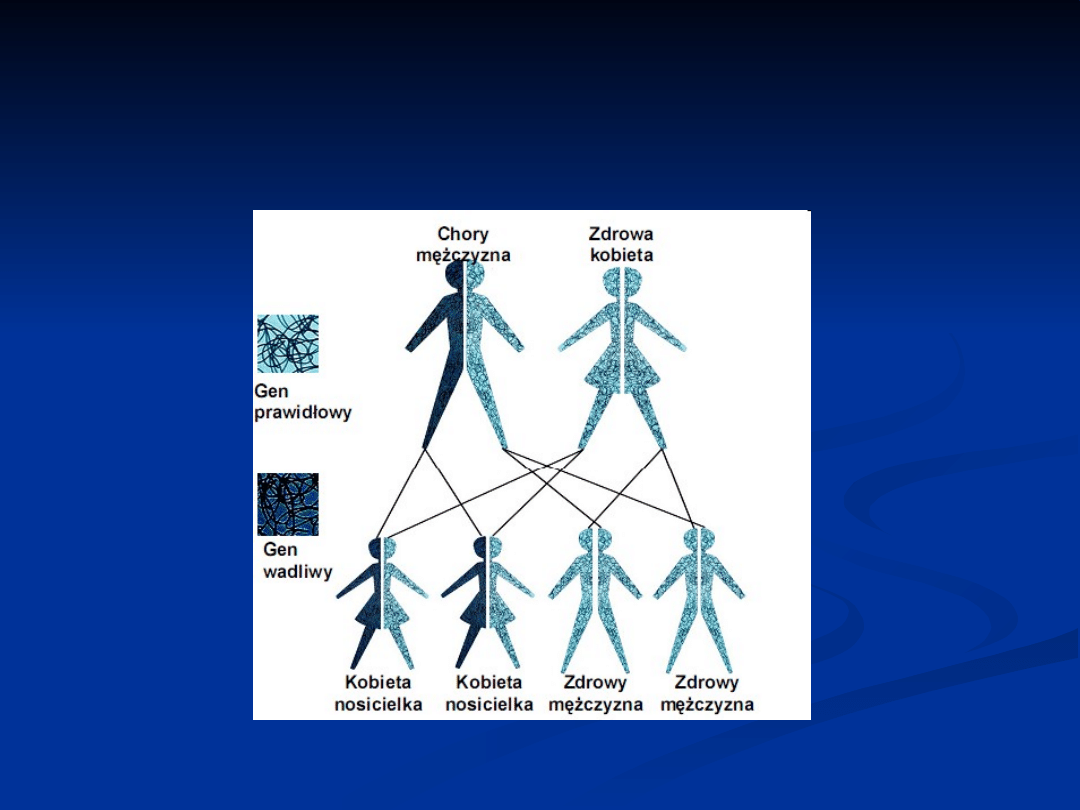
W jaki sposób choroby sprzężone z
W jaki sposób choroby sprzężone z
chromosomem X są przekazywane przez
chromosomem X są przekazywane przez
chorych mężczyzn
chorych mężczyzn

Jeśli mężczyzna dotknięty chorobą sprzężoną
Jeśli mężczyzna dotknięty chorobą sprzężoną
z chromosomem X ma córkę
z chromosomem X ma córkę
, to
, to
zawsze
zawsze
przekaże jej zmieniony gen. Dzieje się tak, ponieważ
przekaże jej zmieniony gen. Dzieje się tak, ponieważ
mężczyźni mają tylko jeden chromosom X i zawsze
mężczyźni mają tylko jeden chromosom X i zawsze
przekazują go swoim córkom. Zatem wszystkie jego córki
przekazują go swoim córkom. Zatem wszystkie jego córki
będą nosicielkami. Córki zwykle nie będą chore,
będą nosicielkami. Córki zwykle nie będą chore,
ale istnieje ryzyko, że będą miały dotkniętych chorobą
ale istnieje ryzyko, że będą miały dotkniętych chorobą
synów.
synów.
Jeśli mężczyzna dotknięty chorobą sprzężoną
Jeśli mężczyzna dotknięty chorobą sprzężoną
z chromosomem X ma syna
z chromosomem X ma syna
, jego syn
, jego syn
nigdy
nigdy
nie odziedziczy chromosomu X ze zmienionym
nie odziedziczy chromosomu X ze zmienionym
genem. Dzieje się tak, ponieważ mężczyzna zawsze
genem. Dzieje się tak, ponieważ mężczyzna zawsze
przekazuje swoim synom chromosom Y (jeśli przekazałby
przekazuje swoim synom chromosom Y (jeśli przekazałby
chromosom X to miałby córkę).
chromosom X to miałby córkę).

Zespół Retta
Zespół Retta

Zespół Retta
Zespół Retta
Jest to
Jest to choroba
genetyczna –
genetyczna –
wynik mutacji genu MECP2,
wynik mutacji genu MECP2,
zlokalizowanego w
zlokalizowanego w
chromosomie płci – X. Zespół
chromosomie płci – X. Zespół
ten dotyczy dziewczynek,
ten dotyczy dziewczynek,
ponieważ chłopcy obarczeni
ponieważ chłopcy obarczeni
tą wadą nie rodzą się lub
tą wadą nie rodzą się lub
w niedługim czasie
w niedługim czasie
po narodzinach umierają.
po narodzinach umierają.
Zespół Retta pojawia się raz
Zespół Retta pojawia się raz
na dziesięć tysięcy
na dziesięć tysięcy
urodzonych dzieci i może
urodzonych dzieci i może
dotyczyć każdej rodziny,
dotyczyć każdej rodziny,
także „nieobciążonej”.
także „nieobciążonej”.

Zespół Retta
Zespół Retta
Na obraz kliniczny składa się szereg zaburzeń
Na obraz kliniczny składa się szereg zaburzeń
neurorozwojowych, które w większości wypadków
neurorozwojowych, które w większości wypadków
prowadzą do znacznej i głębokiej niepełnosprawności
prowadzą do znacznej i głębokiej niepełnosprawności
ruchowej oraz znacząco ograniczają możliwość
ruchowej oraz znacząco ograniczają możliwość
komunikacji z otoczeniem. Charakterystyczna dla
komunikacji z otoczeniem. Charakterystyczna dla
Zespołu Retta jest nasilona dyspraksja (w wielu
Zespołu Retta jest nasilona dyspraksja (w wielu
wypadkach apraksja) połączona ze specyficznymi
wypadkach apraksja) połączona ze specyficznymi
ruchami stereotypowymi w obrębie
ruchami stereotypowymi w obrębie
kończyn górnych. Uważa się, że Zespół Retta stanowi
kończyn górnych. Uważa się, że Zespół Retta stanowi
jedną z najczęstszych przyczyn obniżenia poziomu
jedną z najczęstszych przyczyn obniżenia poziomu
rozwoju intelektualnego u dziewczynek.
rozwoju intelektualnego u dziewczynek.

Zespół Retta często jest mylnie rozpoznawany
Zespół Retta często jest mylnie rozpoznawany
jako autyzm, porażenie mózgowe czy ogólne
jako autyzm, porażenie mózgowe czy ogólne
zaburzenia rozwoju intelektualnego. Typowe
zaburzenia rozwoju intelektualnego. Typowe
objawy:
objawy:
normalny rozwój od urodzenia do 6-18
normalny rozwój od urodzenia do 6-18
miesiąca życia
miesiąca życia
utrata sprawności manualnej i zdolności
utrata sprawności manualnej i zdolności
mówienia
mówienia
ataksja
ataksja
niski wzrost, małe ręce i głowa (wtórna
niski wzrost, małe ręce i głowa (wtórna
mikrocefalia)
mikrocefalia)
stereotypowe ruchy rąk (klaskanie, stukanie,
stereotypowe ruchy rąk (klaskanie, stukanie,
wkładanie do ust), zgrzytanie zębami
wkładanie do ust), zgrzytanie zębami
problemy z kontaktami społecznymi, ataki
problemy z kontaktami społecznymi, ataki
paniki, unikanie kontaktu wzrokowego
paniki, unikanie kontaktu wzrokowego

napady padaczkowe w 81%
napady padaczkowe w 81%
problemy żołądkowo-jelitowe i oddechowe
problemy żołądkowo-jelitowe i oddechowe
boczne skrzywienie kręgosłupa
boczne skrzywienie kręgosłupa
przykurcze mięśniowe
przykurcze mięśniowe
Hiperamonemię (choroba cyklu moczniowego)
Hiperamonemię (choroba cyklu moczniowego)
ogólne stępienie afektu
ogólne stępienie afektu
atrofię kończyn dolnych
atrofię kończyn dolnych
objawy charakterystyczne dla autyzmu
objawy charakterystyczne dla autyzmu
dziecięcego.
dziecięcego.

Leczenie:
Leczenie:
Leczenie przyczynowe zespołu Retta nie istnieje.
Leczenie przyczynowe zespołu Retta nie istnieje.
Terapia skupia się wokół trudnej i długotrwałej
Terapia skupia się wokół trudnej i długotrwałej
rehabilitacji, dzięki której dziewczynki mogą w jakiś
rehabilitacji, dzięki której dziewczynki mogą w jakiś
sposób kontaktować się ze światem i funkcjonować
sposób kontaktować się ze światem i funkcjonować
fizycznie.
fizycznie.
Pomimo braku sposobu leczenia postępowanie
Pomimo braku sposobu leczenia postępowanie
powinno być interdyscyplinarne i prowadzone przez
powinno być interdyscyplinarne i prowadzone przez
rehabilitanta, ortopedę, neurologa, psychologa i
rehabilitanta, ortopedę, neurologa, psychologa i
genetyka. Opisane przez specjalistów zostało
genetyka. Opisane przez specjalistów zostało
znaczenie leków: nootropowych - piracetamu,
znaczenie leków: nootropowych - piracetamu,
L-karnityny, przeciwpadaczkowych, czynników
L-karnityny, przeciwpadaczkowych, czynników
neurotroficznych, witamin oraz melatoniny jako
neurotroficznych, witamin oraz melatoniny jako
znaczących w terapii zespołu Retta.
znaczących w terapii zespołu Retta.

Wrodzona
Wrodzona
hipoplazja skóry
hipoplazja skóry

WRODZONA HIPOPLAZJA SKÓRY jest
WRODZONA HIPOPLAZJA SKÓRY jest
przykładem dziedziczenia dominującego
przykładem dziedziczenia dominującego
sprzężonego z płcią, gdzie:
sprzężonego z płcią, gdzie:
chorują zarówno kobiety jak i mężczyźni
chorują zarówno kobiety jak i mężczyźni
chora kobieta ma 50 % szans przekazania
chora kobieta ma 50 % szans przekazania
zmutowanego genu swoim dzieciom niezależnie od
zmutowanego genu swoim dzieciom niezależnie od
ich płci
ich płci
chorzy mężczyźni maja wszystkie córki chore,
chorzy mężczyźni maja wszystkie córki chore,
synowie nie otrzymują patologicznego genu
synowie nie otrzymują patologicznego genu
w niektórych chorobach zarodki płci męskiej-letalne
w niektórych chorobach zarodki płci męskiej-letalne
kobiety w układzie homozygotycznym letalne
kobiety w układzie homozygotycznym letalne

Wrodzona hipoplazja
Wrodzona hipoplazja
skóry:
skóry:
Jest to rzadka choroba uwarunkowana
Jest to rzadka choroba uwarunkowana
genetycznie, spowodowana jest mutacją w
genetycznie, spowodowana jest mutacją w
genie
genie
IKBKG
IKBKG
w locus Xq28, kodującym
w locus Xq28, kodującym
aktywator jądrowego czynnika
aktywator jądrowego czynnika
transkrypcyjnego kappa B. U płci męskiej
transkrypcyjnego kappa B. U płci męskiej
wada jest zazwyczaj letalna. Istotą choroby
wada jest zazwyczaj letalna. Istotą choroby
jest niezdolność komórek podstawnych
jest niezdolność komórek podstawnych
skóry do gromadzenia melaniny, która
skóry do gromadzenia melaniny, która
gromadzi się w skórze właściwej i w
gromadzi się w skórze właściwej i w
makrofagach indukując stan zapalny.
makrofagach indukując stan zapalny.
Wrodzona hipoplazja
Wrodzona hipoplazja
skóry
skóry
(
(
Incontinentia
Incontinentia
pigmenti
pigmenti
czyli nietrzymanie
czyli nietrzymanie
barwnika
barwnika
– zespół Blocha i Sulzbergera)
– zespół Blocha i Sulzbergera)
Zaburzenie dotyczy
Zaburzenie dotyczy
tkanek ektodermalnych
tkanek ektodermalnych
i
i
mezodermalnych,
mezodermalnych,
Częstość 1: 75 000,
Częstość 1: 75 000,
Choroba wiąże się z
Choroba wiąże się z
mutacją Locus genu – Xq
mutacją Locus genu – Xq
27-q28,
27-q28,
Chorują głównie
Chorują głównie
dziewczęta,
dziewczęta,
Dla płodów męskich –
Dla płodów męskich –
zespół letalny.
zespół letalny.
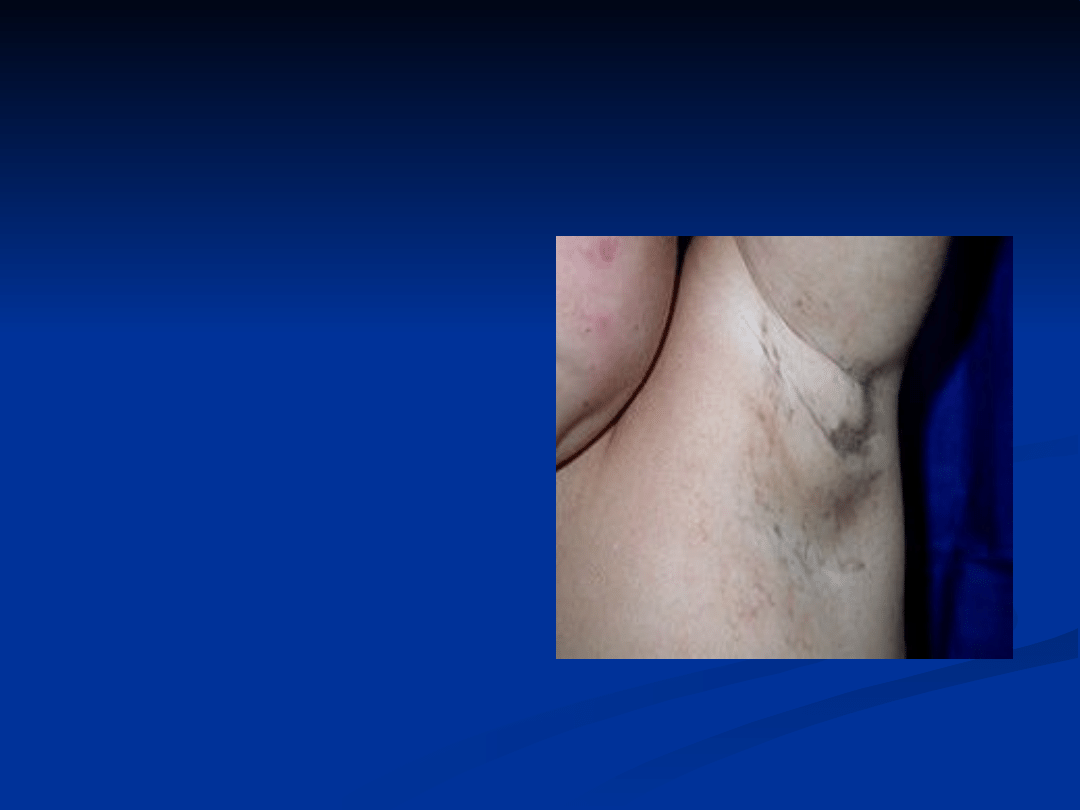
Zmiany skórne układające się wzdłuż linii
Blaschko u 3-letniej dziewczynki.

Objawy:
Objawy:
po urodzeniu na skórze dziecka pojawiają się
po urodzeniu na skórze dziecka pojawiają się
plamy rumieniowe, zawierające małe
plamy rumieniowe, zawierające małe
pęcherzyki,
pęcherzyki,
w dalszym etapie: rogowacenie paznokci,
w dalszym etapie: rogowacenie paznokci,
pękanie skóry stóp,
pękanie skóry stóp,
naturalny tatuaż skóry,
naturalny tatuaż skóry,
zez,
zez,
wady układu kostnego i serca,
wady układu kostnego i serca,
u 50% chorych – niedorozwój umysłowy,
u 50% chorych – niedorozwój umysłowy,
porażenia, napady drgawek,
porażenia, napady drgawek,

Przebieg objawów:
Przebieg objawów:
Zmiany skórne mają różny charakter, opisano
Zmiany skórne mają różny charakter, opisano
cztery następujące kolejno fazy choroby z różnymi
cztery następujące kolejno fazy choroby z różnymi
objawami skórnymi:
objawami skórnymi:
W pierwszej fazie powstają pęcherze, rumieniowe
W pierwszej fazie powstają pęcherze, rumieniowe
prążki, plamki i krosty. Typowo zmiany te
prążki, plamki i krosty. Typowo zmiany te
układają się w przebiegu linii Blaschko.
układają się w przebiegu linii Blaschko.
Faza druga (brodawkowa) opisywana jest u dzieci
Faza druga (brodawkowa) opisywana jest u dzieci
w 2-8 tygodniu życia i charakteryzuje się
w 2-8 tygodniu życia i charakteryzuje się
pęcherzami na odsiebnych częściach
pęcherzami na odsiebnych częściach
kończyn, ewoluującymi w
kończyn, ewoluującymi w
brodawkowate płytki.
brodawkowate płytki.

W fazie trzeciej (tygodnie-miesiące
W fazie trzeciej (tygodnie-miesiące
od urodzenia) pojawiają się
od urodzenia) pojawiają się
przebarwienia, również układające
przebarwienia, również układające
się wzdłuż linii Blaschko.
się wzdłuż linii Blaschko.
W fazie czwartej spotyka się
W fazie czwartej spotyka się
odbarwione plamki i prążki, głównie
odbarwione plamki i prążki, głównie
w okolicach zgięciowych.
w okolicach zgięciowych.
Choroba ta jest zespołem wad, gdzie
Choroba ta jest zespołem wad, gdzie
dodatkowymi nieprawidłowościami
dodatkowymi nieprawidłowościami
składającymi się na obraz kliniczny są:
składającymi się na obraz kliniczny są:
wady uzębienia
wady uzębienia
(hipodoncja)
(hipodoncja)
wady ośrodkowego
wady ośrodkowego
układu nerwowego
układu nerwowego
wady narządu wzroku
wady narządu wzroku
wady układu kostnego
wady układu kostnego
(półkręgi, skolioza,
(półkręgi, skolioza,
rozszczep kręgosłupa,
rozszczep kręgosłupa,
syndaktylia)
syndaktylia)
nowotwory (rzadko)
nowotwory (rzadko)

W diagnostyce różnicowej
W diagnostyce różnicowej
nietrzymania barwnika należy
nietrzymania barwnika należy
uwzględnić:
uwzględnić:
bielactwo Ito
bielactwo Ito
liszajec pęcherzowy
liszajec pęcherzowy
zakażenie HSV
zakażenie HSV
podłużna i wirowa
podłużna i wirowa
znamieniopodobna hipermelanoza
znamieniopodobna hipermelanoza
(LWNH)
(LWNH)

Leczenie:
Leczenie:
Leczenie objawowe:
Leczenie objawowe:
Stała opieka dermatologiczna – stosowane
Stała opieka dermatologiczna – stosowane
kremy z witaminami A, E oraz maści sterydowe,
kremy z witaminami A, E oraz maści sterydowe,
Opieka stomatologiczna (dochodzi do
Opieka stomatologiczna (dochodzi do
zniekształceń już w zawiązkach ząbków),
zniekształceń już w zawiązkach ząbków),
Opieka okulistyczna,
Opieka okulistyczna,
Często potrzebna opieka psychologiczna,
Często potrzebna opieka psychologiczna,
ponieważ widoczne zmiany na skórze powodują
ponieważ widoczne zmiany na skórze powodują
często brak zrozumienia ze strony
często brak zrozumienia ze strony
społeczeństwa.
społeczeństwa.

Zespół łamliwego
Zespół łamliwego
chromosomu
chromosomu
X
X

ZESPÓŁ ŁAMLIWEGO
ZESPÓŁ ŁAMLIWEGO
CHROMOSOMU X
CHROMOSOMU X
Choroba występuje u chłopców z
Choroba występuje u chłopców z
częstością 1:1200-3600, a u dziewczynek
częstością 1:1200-3600, a u dziewczynek
z częstością 1:4000-6000. Jest to
z częstością 1:4000-6000. Jest to
najczęstsza dziedziczna przyczyna
najczęstsza dziedziczna przyczyna
upośledzenia umysłowego
upośledzenia umysłowego
u chłopców i druga co do częstości
u chłopców i druga co do częstości
wśród przyczyn genetycznych (po
wśród przyczyn genetycznych (po
zespole Downa). W większości
zespole Downa). W większości
przypadków nie jest prawidłowo
przypadków nie jest prawidłowo
rozpoznawana, zwłaszcza u kobiet.
rozpoznawana, zwłaszcza u kobiet.

Zespół łamliwego chromosomu X
Zespół łamliwego chromosomu X
Jest to mutacja dynamiczna – polega na
Jest to mutacja dynamiczna – polega na
powieleniu segmentu genu o sekwencji
powieleniu segmentu genu o sekwencji
nukleotydów CGG. 65-200 powtórzeń to
nukleotydów CGG. 65-200 powtórzeń to
tzw. premutacja, najczęściej nie dająca
tzw. premutacja, najczęściej nie dająca
objawów chorobowych, ale mająca
objawów chorobowych, ale mająca
tendencję do "wydłużania się" w kolejnych
tendencję do "wydłużania się" w kolejnych
pokoleniach. Ponad 200 powtórzeń to pełna
pokoleniach. Ponad 200 powtórzeń to pełna
mutacja, która daje objawy u
mutacja, która daje objawy u
wszystkich obciążonych nią chłopców
wszystkich obciążonych nią chłopców
i u około połowy dziewczynek.
i u około połowy dziewczynek.

Objawy:
Objawy:
Kobiety z pełna mutacją – upośledzenie umysłowe w
Kobiety z pełna mutacją – upośledzenie umysłowe w
stopniu lekkim (20-30%) lub umiarkowanym (1-2%), u
stopniu lekkim (20-30%) lub umiarkowanym (1-2%), u
1/3 kobiet z pełną mutacją genu FMR1
1/3 kobiet z pełną mutacją genu FMR1
występują:
występują:
- zmiany w obrębie twarzoczaszki, upośledzenie
- zmiany w obrębie twarzoczaszki, upośledzenie
umysłowe średniego stopnia ( IQ 24-41),
umysłowe średniego stopnia ( IQ 24-41),
- spowolnienie ruchowe,
- spowolnienie ruchowe,
- trudności wymowy,
- trudności wymowy,
- problemy z koncentracją,
- problemy z koncentracją,
Mężczyźni z pełną mutacją ciężki stopień
Mężczyźni z pełną mutacją ciężki stopień
upośledzenia
upośledzenia
(max. IQ = 31)
(max. IQ = 31)

Objawy:
Objawy:
u noworodków i niemowląt płci męskiej
u noworodków i niemowląt płci męskiej
:
:
niska waga urodzeniowa,
niska waga urodzeniowa,
mały obwód głowy,
mały obwód głowy,
zwiększona objętość jąder,
zwiększona objętość jąder,
duże małżowiny uszne,
duże małżowiny uszne,
hipotonia mięśniowa,
hipotonia mięśniowa,
u dzieci
u dzieci
:
:
autyzm, zaburzenia mowy,
autyzm, zaburzenia mowy,
opóźnienie rozwoju psychoruchowego,
opóźnienie rozwoju psychoruchowego,
u dorosłego mężczyzny:
u dorosłego mężczyzny:
deformacje twarzoczaszki (wydatne guzy czołowe, duże i odstające małżowiny
deformacje twarzoczaszki (wydatne guzy czołowe, duże i odstające małżowiny
uszne, duża żuchwa, wysokie podniebienie, hiperteloryzm),
uszne, duża żuchwa, wysokie podniebienie, hiperteloryzm),
powiększenie jąder, bladoniebieskie tęczówki, zniekształcenia kręgosłupa,
powiększenie jąder, bladoniebieskie tęczówki, zniekształcenia kręgosłupa,
padaczka, encefalopatia,
padaczka, encefalopatia,

Objawy cd.
Objawy cd.
zaburzenia rozwoju
zaburzenia rozwoju
umysłowego – szerokie
umysłowego – szerokie
spektrum: od problemów z
spektrum: od problemów z
mówieniem w wieku
mówieniem w wieku
przedszkolnym i nauką szkolną
przedszkolnym i nauką szkolną
po głębokie upośledzenie
po głębokie upośledzenie
(85% pacjentów ma IQ w
(85% pacjentów ma IQ w
granicach 20-70); objawy są
granicach 20-70); objawy są
bardziej nasilone u mężczyzn
bardziej nasilone u mężczyzn
nieśmiałość, utrudniony
nieśmiałość, utrudniony
kontakt wzrokowy
kontakt wzrokowy
w części przypadków pojawiają
w części przypadków pojawiają
się też objawy zbliżone do
się też objawy zbliżone do
ADHD i autyzmu
ADHD i autyzmu
(autoagresja, trzepotanie
(autoagresja, trzepotanie
rękami)
rękami)
obniżone napięcie mięśniowe
obniżone napięcie mięśniowe
Typowy wygląd twarzy u mężczyzny
z zespołem łamliwego chromosomu X.

Objawy cd.
Objawy cd.
charakterystyczne cechy wyglądu
charakterystyczne cechy wyglądu
zewnętrznego występują głównie
zewnętrznego występują głównie
u mężczyzn w różnych
u mężczyzn w różnych
kombinacjach; są widoczne u
kombinacjach; są widoczne u
60% pacjentów, najczęściej
60% pacjentów, najczęściej
dopiero po osiągnięciu
dopiero po osiągnięciu
dojrzałości:
dojrzałości:
pociągła twarz, wypukłe czoło
pociągła twarz, wypukłe czoło
duży obwód czaszki
duży obwód czaszki
odstające uszy, zez, wystająca
odstające uszy, zez, wystająca
żuchwa (prognatyzm)
żuchwa (prognatyzm)
nadmierna ruchomość
nadmierna ruchomość
w stawach, płaskostopie,
w stawach, płaskostopie,
skolioza, klatka piersiowa
skolioza, klatka piersiowa
lejkowata
lejkowata
makrorchidyzm – duże jądra
makrorchidyzm – duże jądra
(powyżej 25 ml u dorosłych)
(powyżej 25 ml u dorosłych)

u pacjentów częściej pojawiają się
u pacjentów częściej pojawiają się
także dodatkowe schorzenia:
także dodatkowe schorzenia:
szmery sercowe, zespół wypadania
szmery sercowe, zespół wypadania
płatka zastawki mitralnej
płatka zastawki mitralnej
przewlekłe zapalenia zatok i ucha
przewlekłe zapalenia zatok i ucha
środkowego
środkowego
refluks żołądkowo-przełykowy
refluks żołądkowo-przełykowy
napady padaczkowe – u 25% chorych
napady padaczkowe – u 25% chorych
zaburzenia nastroju.
zaburzenia nastroju.

LECZENIE
LECZENIE
Aktualnie nie istnieje żadna
Aktualnie nie istnieje żadna
sprawdzona metoda
sprawdzona metoda
leczenia przyczynowego.
leczenia przyczynowego.
Wskazane jest leczenie
Wskazane jest leczenie
objawowe, m.in.
objawowe, m.in.
opieka logopedy
opieka logopedy
i psychologa dziecięcego,
i psychologa dziecięcego,
terapia integracji
terapia integracji
sensorycznej,
sensorycznej,
nadpobudliwości czy
nadpobudliwości czy
zachowań agresywnych.
zachowań agresywnych.
Do łagodzenia tych
Do łagodzenia tych
ostatnich w przypadku
ostatnich w przypadku
pacjentów przed okresem
pacjentów przed okresem
dojrzewania próbowano
dojrzewania próbowano
stosować kwas foliowy, ale
stosować kwas foliowy, ale
wyniki badań były
wyniki badań były
niejednoznaczne.
niejednoznaczne.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
Wyszukiwarka
Podobne podstrony:
Dziedziczenie jednogenowe autosomalne i sprzężone z płcią
Dziedziczenie jednogenowe i sprzężone z płcią
dziedziczenie cech sprzężonych z płcią i zw z nią
Biologia część III, Cechy sprzężone z płcią
Prelekcja 9 Chromosomy i?chy sprzężone z płcią poprawione
dziedziczenie sprzężone z chromosomem x u człowieka
Choroby sprzężone z płcią, Choroby sprzężone z płcią
sprzężone z płcią, genetyka, kolokwia pytania i odp
wyklad 7-genetyka czlowieka, far, genetyka, dziedziczenie związane z płcią 7 ćwiczenie
Cwiczenie-5-cechy sprzezone z plcia i rodowody
sprzężenia z płcią, Biologia II, Genetyka
Ćw 13 Dziedziczenie sprzężone z X
Ćwiczenie 7 - cechy sprzężone z płcią, podstawy genetyki
Cwiczenie 5 cechy sprzezone z plcia i rodowody
więcej podobnych podstron