
Źródła informacji naukowej o
leku
Elżbieta Wojtasik
WUM
Zakład Opieki
farmaceutycznej
1
Kategorie prawne produktów
leczniczych
Produkty lecznicze referencyjne
Procedura centralna - podlegają jej produkty lecznicze wymienione w załączniku
do Rozporządzenia (WE) 726/2004 Parlamentu Europejskiego i Rady z dnia
31.03.2004 r. oraz leki sieroce. Wydana w wyniku procedury centralnej decyzja
Komisji Europejskiej (KE) o pozwoleniu na dopuszczenie do obrotu obowiązuje
we wszystkich państwach członkowskich Unii Europejskiej oraz państwie
członkowskim EFTA – stronie umowy o Europejskim Obszarze Gospodarczym.
W państwach EFTA – stronie umowy o EOG – Islandii, Norwegii, Liechtensteinie
pozwolenie dla produktu zarejestrowanego centralnie obowiązuje po wydaniu
przez te państwa stosownych decyzji na podstawie decyzji KE.
2
EPAR/SPC
Kategorie prawne produktów
leczniczych
• Odpowiedniki czyli produkty lecznicze
odtwórcze do leku referencyjnego -
generyczne
• Produkty lecznicze o ugruntowanym
zastosowaniu medycznym
• Produkty lecznicze roślinne
EPAR/SPC
SPC
SPC/Assessme
nt Reports

Informacja naukowa o leku
• Źródła informacji naukowej o leku
– Dla pacjenta /ulotka/materiały informacyjne
• Materiały informacyjne ?
• Reklama?
– Dla fachowego personelu medycznego
EPAR/SPC/literatura
fachowa /medyczne bazy danych
– Internet/ literatura fachowa /terapia i leki-kwartalniki
4

Informacja naukowa o leku
• Informacja naukowa o działaniach niepożądanych
• Informacja naukowa o bezpieczeństwie stosowania leku
• Informacja o interakcjach
• Informacja na temat farmakokinetyki
– Różnice pomiędzy lekiem referencyjnym i jego odpowiednikiem
5

Informacja naukowa o leku
• Informacja na temat tolerancji miejscowej leków
stosowanych zewnętrznie / miejscowo
– Zamierzone i niezamierzone stosowanie leku.
- Tolerancja miejscowa ze względu na drogę podania
- na skórę
- do oka
- do ucha
- dopochwowo
- doodbytniczo
- dożylnie, domięśniowo, podskórnie, dotętniczo
.
• Informacja naukowa na temat toksykologii leku
6

Informacja naukowa o leku
•
Toksykologia leków generycznych
– Produkty lecznicze referencyjne i generyczne
Dyrektywa 2001/83/EC
• “generic medicinal product” shall mean a medicinal product which
has the same qualitative and quantitative composition in active
substances and the same pharmaceutical form as the reference
medicinal product, and whose
• bioequivalence with the reference medicinal product has been
demonstrated by appropriate bioavailability studies.
• The different salts, esters, ethers, isomers, mixtures of isomers,
complexes or derivatives of an active substance shall be considered
to be the same active substance, unless they differ significantly in
properties with regard to safety and/or efficacy.
7

Różnice :
Skład substancji pomocniczych,
Jakość substancji czynnej i substancji pomocniczych
Postać farmaceutyczna (w pewnych granicach)
Dostępność biologiczna
Chemiczne i fizyczne modyfikacje substancji czynnej
8

Informacja naukowa o leku
EPAR/SPC
• Skutek zmiany lek referencyjny – generyk
– Skuteczność/
odmienna farmakokinetyka, brak
biorównoważności
– Bezpieczeństwo
/zanieczyszczenia substancji
czynnej, jakość substancji pomocniczych
9

Toksykologia zanieczyszczeń
• Kwalifikacja zanieczyszczeń w badaniach na
zwierzętach – limit kwalifikacji
• Badania porównawcze z produktem
referencyjnym
– badania jakościowe
• Ocena in silico
– Źródła naukowe danych na temat toksykologii
zanieczyszczeń
10

Informacja naukowa o leku
Nie zawsze prosta, pozornie nieistotna modyfikacja
chemiczna, pozwala spodziewać się na tyle małych różnic
w bezpieczeństwie i skuteczności, by substancja spełniała
kryteria zasadniczego podobieństwa do substancji
wyjściowej.
Przykładem pozornie błahych, lecz istotnych z
farmakologiczno-toksykologicznego punktu widzenia
różnic, są
różne estry kortykosteroidów,
które nie ulegają
łatwej hydrolizie uwalniając „aktywną” część cząsteczki,
lecz pozostając z nią w trwałym połączeniu, decydują o
powstaniu nowej substancji czynnej.
11

Informacja naukowa o leku
EPAR/SPC
Przykład:
• propionian
flutikazonu i
pirośluzan
flutikazonu –
każdy z tych związków, chociaż rejestrowany w
podobnych wskazaniach, podlegał odrębnemu,
pełnemu programowi badań toksykologicznych,
takiemu jak dla nowej substancji leczniczej
• „….The different salts, esters, ethers, isomers, mixtures of
isomers, complexes or derivatives of an active substance
shall be considered to be the same active substance, unless
they differ significantly in properties with regard to safety
and/or efficacy”
12

Informacja naukowa o leku
• MZ
– Interakcje
– Komunikaty bezpieczeństwa
– Urzędowy Wykaz produktów leczniczych (BIP)
• GIF – wycofania z obrotu, wstrzymania
w obrocie, leki podrobione
• URPL WM PB
– ostrzeżenia o działaniach niepożądanych
– wycofania z obrotu ze względu na bezpieczeństwo
– SPC
– Publikuje wykaz produktów leczniczych (BIP)
• Komisja Eurpejska/EMA
– ograniczenia rejestru
– wycofania z obrotu
– EPAR
– SPC
13
Microsoft Office Word 2007.lnk
SPC, EPAR, Assessment
Repords, Komunikaty
bezpieczeństwa , „Dear
doctor letter”

Inne fachowe dane o leku
• Rzędowy wykaz produktów leczniczych
(BIP)
• Nomenklatura
• EDQM –
– European Pharmacopoea
– Standard Terms
• Kody ATC
– Collaborating Centre for Drug Statistics Methodology) w
Norwegii
(Centrum Współpracy nad Metodologią
Statystyczną Lekó
w)
14

Standard terms
EDQM
• The List of Standard Terms covers :
– dosage forms
,
– routes of administration
and
– containers
used for medicines for human and veterinary use.
• It gives the equivalents of several hundred terms in 31
world languages:
Albanian, Bulgarian,
Chinese,
Croatian, Czech, Danish, Dutch, English,
Estonian, Finnish, French, German, Greek, Hungarian, Icelandic, Italian,
Latvian, Lithuanian, Macedonian, Maltese, Norwegian,
Polish,
Portuguese,
Romanian,
Russian
, Serbian, Slovak, Slovenian, Spanish, Swedish and Turkish
.
15
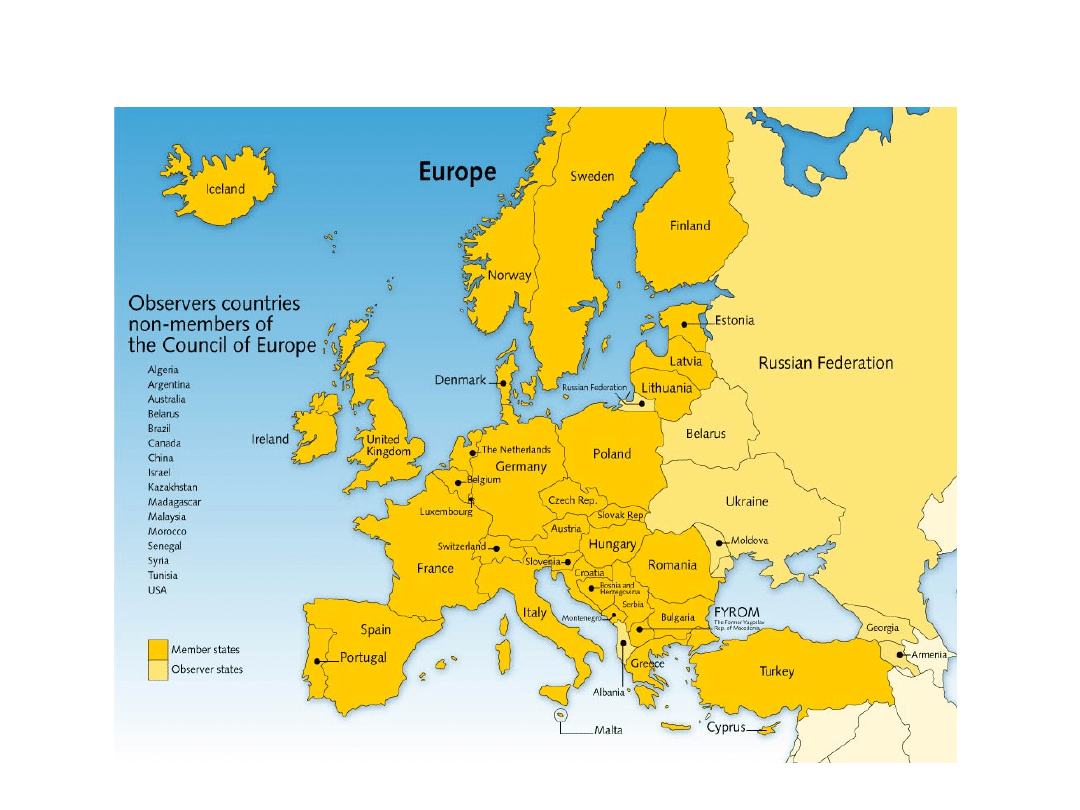
16

EDQM
• 36 Member States and the European Union are
signatory to the
Convention on the
Elaboration of a European Pharmacopoeia
.
• 8 European countries
• 14 non-european countries and the World Health
Organization
(WHO)
are observers.
17

EDQM
wydawnictwa
• Blood Transfusion Guides
Guide to the preparation, use and quality assurance of
blood components - 15th Edition (2009). This guide is
available in English and French.
• European Pharmacopoeia 7th Edition
• Pharmeuropa
18

EDQM –
OMCL
Control of Medicines
General European OMCL
Network
OMCL Network of the Council of
Europe
GENERAL DOCUMENT
PA/PH/OMCL (07) 89 8R
Annex 1: Definition of an OMCL
and OMCL Status within the
GEON
It is the right of the patient to receive
medicines of good quality and the duty of the
Competent Authorities (organisations,
administrations, agencies) to ensure that the
markete products comply with the
specifications laid down in the application file
and in any other
relevant compendial requirements, which these
same authorities had approved. The aim of
Official Laboratory Testing
is to support the
Competent Authorities in controlling the quality
of medicinal products on the market for the
benefit of the human patient and/or animals.
19
An Official Medicines Control
Laboratory
(OMCL) is a
public
institution
, which only
performs laboratory testing for a
Competent Authority, independently
from the manufacturer,
for medicinal products prior to and/or
after marketing for the general
surveillance of
medicines in relation to the
safety of
human patient and/or animals.

• OMCL Code OMCL Name
• NL_RIVM-B Centre for Biological Medicines and Medical Technology, BMT
• NL_RIVM-C Centre for Quality Control of Chemical-Pharmaceutical Products, KCF
• NO_NOMA Norwegian Medicines Agency
• PL_IL National Medicines Institute, NIL
• PL_PZH National Institute of Public Health - National Institute of Hygiene, DSVE
• PT_INFARMED INFARMED I.P. National Authority for Medicines and Health
Products
• RO_ANM National Medicines Agency
• SE_MPA Medical Products Agency, MPA
• SI_JAZMP Agency for Medicinal Products and Medical Devices, JAZMP
• SK_SUKL State Institute for Drug Control, SUKL
• SK_USKVBL Institute for State Control of Veterinary Biologicals and Medicines,
USKVBL
• SRB_ALIMS Agency for Medicines and Medical Devices of Serbia, ALIMS
• UK_MHRA_LGC MHRA Laboratory of the Government Chemist, LGC
• UK_NIBSC National Institute for Biological Standards and Control, NIBSC
20

Postać leku
Powder and solvent for
suspension for injection in
pre-filled
Concentrate and solvent
for cutaneous solution
Bee-hive gel (Vet.) –
Powder and solvent for
suspension for injection
in pre-filled syringe –
Proszek i rozpuszczalnik do
sporządzania zawiesiny do
wstrzykiwań w ampułko-
strzykawce
Koncentrat i rozpuszczalnik
do sporządzania roztworu na
skórę
Żel do stosowania w ulu
(Wet.)
Proszek i rozpuszczalnik do
sporządzania zawiesiny do
wstrzykiwań w ampułko-
strzykawce
Drogi podania
Intraosseous use
Posterior juxtascleral use
Buccal use
Retrobulbar
use
Periosseous use
Podanie śródkostne
Podanie tylne okołotwardówkowe
Podanie podpoliczkowe
Podanie pozagałkowe
Podanie okołokostne
21

Komisja Europejska
• Community Register
– EU Centralized Procedure
• Human medicinal products by ATC
• Adopted Commission Decisions of the last six
month
• General index on active ingredients
• General index on brand name
22
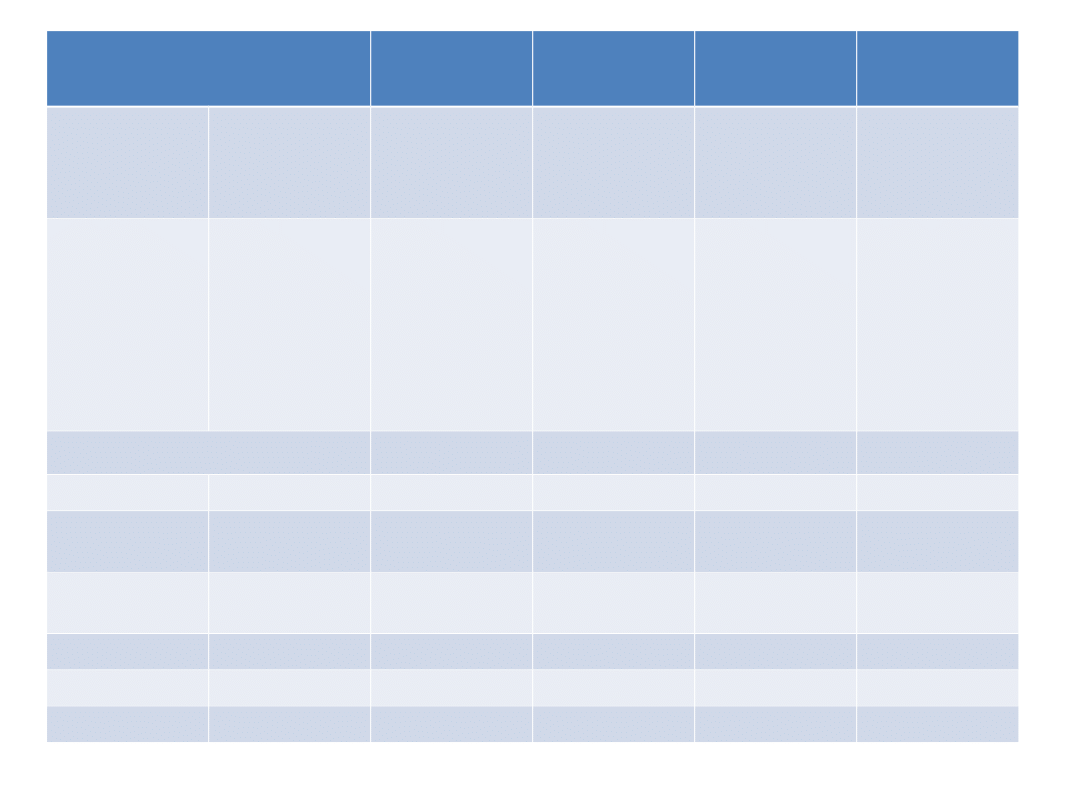
Community
Register
Brand
name
EU Number
INN
Human or
veterinary
medicinal
product
Procedure
type
details
4-[[[4-(4-
Chloropheno
xy)phenyl]su
lfonyl]methyl
]tetrahydro-
N-hydroxy-
2H-pyran-4-
carboxamide
Human
Refusal
A.A. Xylazine
Veterinary
Referral
A.M.
Piroxicam
Piroxicam
Human
Referral
Abalgin
dextropropox
yphene
Human
Referral
Abilify
EU/1/04/276/
aripiprazole
Human
Centralised
Abraxane
EU/1/07/428/
paclitaxel
Human
Centralised
Abseamed
EU/1/07/412/
epoetin alfa
Human
Centralised
23

Kod ATC
• Klasyfikację kontroluje
Centrum Współpracy
nad Metodologią Statystyczną Leków
(Collaborating Centre for Drug Statistics Methodology) w
Norwegii
• Podlega pod
(WHO).
• Pierwszy spis został opublikowany w
roku.
24

Kod ATC
• Kod leku w klasyfikacji ATC jest siedmiopozycyjny i ma
postać
(L – litera, C – cyfra): LCCLLCC
• Pierwszy poziom (jedna litera) - określa grupę anatomiczną.
• Drugi poziom (dwie cyfry) - określa podgrupę terapeutyczną,
• Trzeci poziom (jedna litera) - określa podgrupę farmakologiczną,
• Czwarty poziom (jedna litera)- podgrupę chemiczną
• Piąty poziom (dwie cyfry) - wskazuje na konkretną substancję
chemiczną.
25

Kod ATC
• In the Anatomical Therapeutic Chemical (ATC) classification system, the
active substances
are divided into different groups according to the
organ
or
system on which they act
and their
therapeutic,
pharmacological and chemical properties.
• Drugs are classified in groups at five different levels.
– The drugs are divided into fourteen main groups (1st level)
– Pharmacological/therapeutic subgroups (2nd level).
– The 3rd and 4th levels are chemical/pharmacological/therapeutic
subgroups and the 5th level is the chemical substance.
– The 2nd, 3rd and 4th levels are often used to identify pharmacological
subgroups when that is considered more appropriate than therapeutic or
chemical subgroups.
26

The complete classification of
metformin
illustrates the structure of the code:”
27
A
Alimentary tract and metabolism
(1st level, anatomical main
group)
A10
Drugs used in
diabetes
(2nd level, therapeutic
subgroup)
A10B
Blood glucose lowering drugs,
excl.
insulins
(3rd level, pharmacological
subgroup)
A10BA
Biguanides
(4th level, chemical subgroup)
A10BA02
metformin

• Lokalizację w ATC
, posiadającej alfanumeryczny kod
C01CA24:
• Epinefryna (adrenalina)
C01C A24
– Układ sercowo-naczyniowy
→ (anatomiczna grupa główna)
– Leki stosowane w chorobach serca
→ (podgrupa terapeutyczna)
C01C – Leki pobudzające układ sercowo-naczyniowy (bez glikozydów
nasercowych)
→ (podgrupa farmakologiczna)
C01C A – Leki wpływające na receptory adrenergiczne i
dopaminergiczne
→ (podgrupa chemiczna)
C01C A24 –
→ (substancja chemiczna)
28

All centralized human medicinal
product by ATC code
29

ATC code
30

ATC code
• B
- Blood and blood forming organs - krew i układ krwiotwórczy
• C
- Cardiovascular system - układ sercowo - naczyniowy
• D
- Dermatologicals - dermatologia
• G
- Genito urinary system and sex hormones - układ moczowo-płciowy i
hormony płciowe
• H
- Systemic hormonal prep, excl sex hormones – leki hormonalne
• J
- General antiinfectives for systemic use - leki stosowane w zakażeniach
• L
- Antineoplastic and immunomodulating agents - leki przeciwnowotworowe i
immunomodulujące
• M
- Musculo-skeletal system - układ mięśniwo-szkieletowy
• N
- Nervous system – Ośrodkowy Układ nerowy
• P
- Antiparasitic products – leki przeciwpasożytnicze , owodobójcze i repelenty
• R
- Respiratory system – układ oddechowy
• S
- Sensory organs - narządy wzroku i słuchu
• V
- Various - inne
–
V04 - Diagnostic agents
31

Nomenclature
• International non-proprietary names
(INN)
are preferred.
• If INN names are not assigned,
USAN
(United States Adopted
Name) or
• BAN
(British Approved Name) names are usually chosen.
32

Jakie są kryteria włączenia substancji do systemu
ATC ?
1.new
chemical
entities (active ingredients) or
biologicals
proposed
for licensing in a range of countries.
A new chemical entity is normally not included in the ATC system
before an application for marketing authorisation is submitted in
at least one country.
2. they are existing
well defined chemical entities
used in a variety
of countries.
3. An
INN should preferably be established
for the active ingredient.
4. Alternatively other official names, e.g.
USAN or BAN
names
should be available.
5. there are
herbal medicinal products
assessed and approved by
regulatory authorities based on
dossiers including efficacy,
safety, and quality data
(e.g. the
well-established use procedure
in EU).
33

Kod ATC
• Prednisolone
in single ingredient products is
given several ATC codes due to different
therapeutic use and different local application
formulations.
– A07EA01 Intestinal antiinflammatory agents
C05AA04 Antihemorrhoidals for topical use
D07AA03 Dermatological preparations
H02AB06 Corticosteroids for systemic use
R01AD02 Nasal decongestants
S01BA04 Ophthalmologicals
S02BA03 Otologicals
34

Classification of combination products
• Combination products containing two or more
active ingredients belonging to the same 4th level
are normally classified using the 5th level codes
20 or 30.
N01BB02 lidocaine
N01BB04 prilocaine
N01BB20 combinations (e.g.
lidocaine and prilocaine)
35

Human medicinal products by ATC
Community Register
•
ATC: N - Nervous system
N02 - Analgesics
N02A - Opioids
N02AB - Phenylpiperidine derivatives
N02AB03 - Fentanyl
Indication: PecFent is indicated for the management of breakthrough pain (BTP)
in adults who are already receiving maintenance opioid therapy for chronic cancer pain.
Breakthrough pain is a transitory exacerbation of pain that occurs on a background of otherwise
controlled persistent pain.
Patients receiving maintenance opioid therapy are those who are taking at least 60 mg of oral
morphine daily, at least 25 micrograms of transdermal fentanyl per hour, at least 30 mg of oxycodone
daily, at least 8 mg of oral hydromorphone daily or an equianalgesic dose of another opioid for a
week or longer. Marketing Authorisation Holder: Archimedes Development Ltd
• EPAR:
This
is a summary of the
European Public Assessment Report
(EPAR) for PecFent.
• „It explains how the Committee for Medicinal Products for Human Use
(CHMP) assessed the medicine to reach its opinion in favour of granting a
marketing authorisation and its recommendations on the conditions of use
for PecFent”
36

EPAR
• What is PecFent ?
• What is PecFent used for ?
• How is used ?
• How does PecFent work?
• How has PecFent been sudies?
• What benefit has PenFect shown during the studies?
• What is the risk associated with PecFent ?
• Why has PecFent been approved ?
• What measures are being taken to ensure the safe
use of PecFent?
• Other information about PecFent.
37

Patient safety
• This page lists major changes made to the authorisation of
medicines, which were recommended by the Committee for
Medicinal Products for Human Use (CHMP) to improve
safety for patients.
• The page lists patient safety information from the last two
years. For a full list of all changes made to centrally
authorised medicines, see the
• For information on referrals, see
.
38

Patient safety
• European Medicines Agency recommends
suspension of Octagam in all EU Member
States
• About
• What is Octagam ?
• Why was Octagam refused ?
• Which data has the CHMP reviewd?
• What are the conclusion of the CHMP?
• What are the recommendations for prescribers and
patients?
Octagam (human normal immunoglobulin 5% and 10%)
39

23 September 2010
EMA/588737/2010
EMEA/H/A-107/001278
• Questions and answers on the suspension of the
marketing authorisations for Octagam (human
normal immunoglobulin 5% and 10%)
• Outcome of a procedure under Article 107 of
Directive 2001/83/EC
•
„The European Medicines Agency has completed a review of
Octagam triggered by reports of serious
thromboembolic events
(incydenty zatorowo-zakrzepowe) The Agency’s Committee for
Medicinal Products for Human Use (CHMP) has recommended that
all marketing authorisations for the medicine be suspended
throughout the European Union (EU) and that Octagam currently on
the market be recalled”
40

EPAR
What is Octagam?
• Octagam is a solution for infusion (drip into a vein) that contains human normal
immunoglobulin extracted from blood as the active substance. Human normal
immunoglobulins are antibodies (types of protein) normally found in the blood
that help the body to fight infections and other diseases.
• Octagam is used in patients who are at risk of infection because they do not
have sufficient antibodies including people with
primary immunodeficiency
syndrome
, or children born with acquired immune deficiency syndrome
(AIDS).
It
is also used in people with certain immune disorders such as
idiopathic
thrombocytopenic purpura
( małopłytkowość)
and in patients who have had a
bone marrow transplant
(przeszczep szpiku kostnego)
• Octagam is made by Octopharma.
• It is authorised in Austria, Belgium, Bulgaria, Cyprus, Czech Republic, Denmark,
Estonia, Finland, France, Germany, Greece, Hungary, Iceland, Italy, Latvia,
Lithuania, Luxembourg, Malta, Netherlands, Norway,
Poland,
Portugal,
Romania, Slovakia, Slovenia, Spain, Sweden and United Kingdom
.
41

Why was Octagam reviewed
?
• In September 2010, the German and Swedish medicines regulatory
agencies
suspended marketing authorisations
for Octagam.
• This follows an unexpectedly high number of reports of serious
thromboembolic events
(problems due to the formation of blood
clots in the blood vessels –
problemy zakrzepowo-zatorowe
) in patients
taking the medicine.
• These events were thought to be related to problems with the
medicine’s manufacture, and included stroke, myocardial infarction
(heart attack) and pulmonary embolism (clot in a blood vessel
supplying the lungs).
u
dar mózgu, zawał mięśnia sercowego (atak serca) i zatorowość płucną (zakrzepy w naczyniach krwionośnych
zaopatrujących płuca).
42

• As required by Article 107, the German
and Swedish agencies
informed the CHMP
of their actions so that the
Committee
could prepare an opinion
on whether the
marketing authorisations for Octagam
should be maintained, changed,
suspended or withdrawn across the EU.
43
What are the conclusions of the CHMP?
• The CHMP noted that, based on the available
information, there was clear evidence of a recent
increase in thromboembolic events associated with
Octagam but that the exact cause of the problems
could not be identified with certainty.
• The CHMP therefore recommended that, because of
the safety concerns with Octagam, the marketing
authorisations for the medicine be suspended in the
EU.
• While the marketing authorisations are suspended,
Octagam will not be available. The suspension will
remain in place until the problem has been rectified.
44

What are the recommendations for
prescribers and patients?
• Doctors should stop using Octagam and
switch their patients to the most
appropriate alternative treatment.
• Patients who have any questions should
speak to their doctor or pharmacist.
• A European Commission decision on this
opinion will be issued in due course.
45

European Commission
• Withdrawn or suspended
•
•
• Official Journal
of the European Union
• ISSN 1725-2423
C 305
Volume 51
28 November 2008
•
46

Pending EC decisions
• This search allows you to find medicines that have been
evaluated by the
Committee on Human Medicinal Products
(CHMP) and are pending a decision by the European
Commission.
• The Committee gives a positive or negative recommendation
on whether to grant the product a Community marketing
authorisation.
• This opinion is replaced by a full European Public Assessment
Report
(EPAR)
once the European Commission has decided -
taking the European Medicines Agency's opinion into
consideration - to grant a marketing authorisation.
• Opinions are also given for medicines with an existing
marketing authorisation where changes to the medicine are
sought by the pharmaceutical company.
47

Withdrawn applications
• This search allows you to find information on the
decision by a pharmaceutical company to
withdraw an application made to the European
Medicines Agency for the evaluation of a medicine
or for changes to an existing authorised medicine.
• When an application is withdrawn after the first
stage of evaluation by the
Committee on Human Medicinal Products
(CHMP),
a
withdrawal assessment report is published.
48

Opinions and Decisions on Paediatric
Investigation Plans (PIPs)
• This search allows you to find information on opinions and
decisions on a Paediatric Investigation Plan (PIP)
including deferrals and waivers.
• A PIP is a development plan aimed at ensuring that the
necessary data is obtained through studies in children to
support the authorisation of the medicine for children
.
• The plan is submitted by a pharmaceutical company to the
(PDCO) at the European Medicines
Agency which is responsible for agreement or refusal of the
plan and publishes an opinion with its decision.
49
Rare disease (orphan) designations
• This search allows you to find information on rare
disease (orphan) designations.
• A designation from the European Medicines Agency's
Committee on Orphan Medicinal Products
(COMP)
permits a pharmaceutical company to benefit from
incentives from the European Union to develop a
medicine for a rare disease such as a genetic disorder
or a rare cancer.
• A large number of these diseases affect children and
newborn babies.
• Once orphan designation is granted a medicine may be
developed by the pharmaceutical company.
50

Opinions on medicines for use outside the
European Union
• Article 58 of Regulation (EC) No 726/2004 1 ("the
Regulation") allows the Agency's Committee for
Medicinal Products for Human Use (CHMP) to give
opinions, in cooperation with the World Health
Organization (WHO), on medicinal products for
human use that are intended exclusively for
markets outside of the EU.
• Medicines eligible for this new procedure are used
to prevent or treat diseases of major public health
interest.
51

• This includes vaccines used in the
WHO Expanded Programme on Immunization
or for
protection against a public health priority disease, as
well as medicines for WHO target diseases such as
HIV/AIDS, malaria, or tuberculosis.
• European Public Assessment Reports (EPARs)
•
• Lamivudine GSK, lamivudine
• Lamivudine / Zidovudine GSK, lamivudine / zidovudine
• Withdrawals of application
• Globorix
52

Veterinary medicines
• EPARS
• Veterinary Health Alerts
• Pending EC decisions
• Withrawn applications
• MRL Reports
53

MRL Reports
• The maximum residue limit, or MRL, is the maximum
concentration of residue accepted by the European Union (EU) in
a food product obtained from an animal that has received a
veterinary medicine.
• The assessment for the
safety of residues is carried out by the by
the Committee for Medicinal Products for Veterinary Use (CVMP).
• Any MRL opinion given by the CVMP is published in the first
instance as a Summary of Opinion as part of the Agency
strategy to improve transparency in the regulatory process.
• It is without prejudice to the final Commission Regulation.
• More detailed information is published later, following the
publication of the Commission Regulation, as a European Public
MRL Assessment Report (EPMAR, formerly calledSummary
Reports).
54

Herbal medicines for
human use
• This search allows you to find herbal
substances that
are designated for assessment by the European Medicines
Agency's Committee on Herbal Medicinal Products (HMPC). Each
substance will be at a different stage of assessment and various
documents will be associated with the substance depending on
where it is in the assessment process. The HMPC conclusions on
the herbal substance at the end of the assessment process can
be found in the final Community Herbal Monograph and may also
be found in Community list entry.
• Status type
55

EMA
European Medicines Agency
What are you looking for ?
• Human medicines
• Veterinary medicines
• Herbal medicin
es
56

European Public Assessment
Reports (EPARs)
• Once a medicine has been granted a Community marketing
authorisation by the European Commission, the European Medicines
Agency publishes a full scientific assessment report called a
European Public Assessment Report (EPAR).
• Using this search you will find
key information for a medicine
including a
Q&A
on the medicine and the patient leaflet.
• You will also find information on medicines which were refused a
marketing authorisation or have been suspended or withdrawn post-
approval.
• Please be aware that the Agency does not evaluate all medicines
currently in use in Europe. If you cannot find the medicine you need
through this search, please visit the website of your national health
authority. More information is available on the centrally-authorised
procedure managed by the Agency. More information on European
Public Assessment Reports.
57
Ministerstwo Zdrowia
Informator o lekach
Interakcje leków
Wyniki sprawdzenia interakcji
•
Sprawdzane leki: -
Lovastin - Acenocumarol WZF - Amlodipine 1A
Pharma - Bromocorn
•
Lek Nr 1 2 3 4: Acenocumarol WZF 1 X Bromocorn 2 X Lovastin 3 X
Amlodipine 1A Pharma 4 X
INTERPRETACJA WYNIKÓW - OPIS RODZAJU INTERAKCJI
•
W badanej grupie leków wystąpiły interakcje.
•
Lovastin Amlodipine (1A Pharma ) , interakcja nieistotna
Możliwy wzrost stężenia lowastatyny we krwi i zwiększenie ryzyka jej
działania niepożądanego ( miopatii i rabdomiolizy).
58
BRAK INFORMACJI
NTERAKCJA
NIEISTOTNA
INTERAKCJA
ISTOTNA
INTERAKCJA B.
ISTOTNA

Informacja dla farmaceuty
•
Amlodipine 1A Pharma + Lovastin = interakcja NIEISTOTNA
Możliwy wzrost stężenia lowastatyny we krwi i zwiększenie ryzyka jej działania
niepożądanego (
miopatii i rabdomiolizy).
•
Bromocorn + Amlodipine 1A Pharma = NIEISTOTNA
Możliwe nasilenie działania hipotensyjnego.
„Dane zamieszczone w serwisie mają charakter informacyjny, nie zastępują
przepisów prawa
i nie mogą być podstawą do jakichkolwiek roszczeń”
•
„W razie jakichkolwiek wątpliwości dotyczących zażywanych leków,
prosimy o skontaktowanie się z lekarzem lub farmaceutą!”
59
Informacja dla pacjenta
Lek 1
Lek 2
Moc
interakcji
Objawy
Amlodipine
1A Pharma
Bromocorn
NIEISTOTNA Możliwe
nasilenie
działania
hipotensyjn
ego
W wyniku
interakcji
działanie
leków nie
powinno
ulec zmianie
60

Urząd Rejestracji Produktów
Leczniczych, Wyrobów
Medycznych, Produktów
Biobójczych
• Biuletyny i wykazy : wykaz produktów leczniczych dopuszczonych
do obrotu
– Stosowanych u ludzi i weterynaryjnych
• SPC/ChPL – produktu leczniczego weterynaryjnego
• Aktualności :
– Komunikat Ministra Zdrowia w sprawie produktów leczniczych
zawierających rozyglitazon
Komunikaty bezpieczeństwa dla produktów leczniczych
61

Komunikat do pracowników ochrony zdrowia
dotyczący dostaw leku Cerezyme®
(imigluceraza)
Uaktualnienie informacji dotyczących dostaw i
zaleceń odnośnie leczenia pacjentów
od 1 października 2010 r. dostawy produktu leczniczego Cerezyme zostaną zwiększone do około 85%
ogólnoświatowego
zapotrzebowania. Przewidujemy, że taki poziom dostaw będzie utrzymany co najmniej do czerwca
•
Spodziewamy się, że będziemy mogli dostarczać wystarczającą ilość produktu leczniczego
Cerezyme, aby pacjenci obecnie leczeni tym produktem leczniczym w zmniejszonej dawce lub ze
zmniejszoną częstotliwością infuzji, otrzymywali lek w dawkach zgodnych z zatwierdzoną
Charakterystyką Produktu Leczniczego, pod kontrolą lekarza prowadzącego.
•
Zachęcamy lekarzy, aby z rozwagą rozpatrywali zmianę dawki i dali pierwszeństwo pacjentom z grup najbardziej
zagrożonych:
• Noworodki, dzieci i młodzież
• Dorośli pacjenci, u których występuje duże ryzyko ciężkiej, zagrażającej życiu progresji choroby lub
kobiety
w ciąży , z objawową chorobą Gauchera.
•
Pacjenci narażeni na duże ryzyko to osoby spełniające co najmniej jedno z następujących kryteriów: l
–
iczba płytek krwi poniżej 20.000/μl, trombocytopenia i krwawienia, niedokrwistość
–
objawowa, ciężka choroba współistniejąca (na przykład stan powodujący u pacjenta ryzyko krwawienia, m.in.
–
marskość wątroby, duży zabieg chirurgiczny), konieczność chemioterapii, choroba płuc wywołana naciekaniem przez
komórki Gauchera lub nowe ostre zdarzenie ze strony kości w ciągu minionych 12 miesięcy.
•
Nie zaleca się, aby nowi pacjenci rozpoczynali leczenie produktem leczniczym
Cerezyme oraz aby pacjenci, którzy obecnie są poddawani innym metodom leczenia,
powracali do leczenia produktem leczniczym Cerezyme.
62
23 WRZEŚNIA 2010

63
Informacje
dotyczące
bezpieczeństwa
URPL

64

•
Rozporządzenie Ministra Zdrowia z dnia 18 grudnia 2002 r. w sprawie dokonywania zmian
w pozwoleniu i dokumentacji dotyczącej wprowadzenia do obrotu produktu leczniczego
(Dziennik Ustaw nr 27) zawiera zapis:
•
„W przypadku nowych istotnych informacji dotyczących bezpieczeństwa stosowania
produktu leczniczego, podmiot odpowiedzialny powinien niezwłocznie wprowadzić
tymczasowe środki bezpieczeństwa
, powiadamiając Prezesa Urzędu Rejestracji Produktów
Leczniczych, Wyrobów Medycznych i Produktów Biobójczych o otrzymanych doniesieniach
i prowadzonych tymczasowych środkach bezpieczeństwa.
•
Jeżeli Prezes Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów
Biobójczych w
ciągu 24 godzin nie wniesie zastrzeżeń, podmiot odpowiedzialny
wprowadza tymczasowe zmiany w Charakterystyce Produktu Leczniczego i ulotce,
niezwłocznie występując z wnioskiem o dokonanie odpowiedniej zmiany typu II”.
•
Jednym ze sposobów przekazania fachowym pracownikom opieki zdrowotnej nowych
istotnych informacji jest rozesłanie komunikatu, znanego pod nazwą
„Dear Doctor Letter”.
•
Sytuacja, w której rozpowszechnienie informacji może wpływać na bezpieczeństwo
pacjentów nakłada na wszystkie zainteresowane strony konieczność szybkiego działania.
65

66

67

URPL – Kampanie
• Ogólnopolska Kampania „LEK BEZPIECZNY
”
2010-06-15
• „Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i
Produktów Biobójczych stojąc na straży interesów Pacjenta postanowił
zainicjować Kampanię Społeczną „Lek Bezpieczny”, mającą na celu:
– Uświadomienie społeczeństwu zasad bezpiecznego stosowania leków
– Przeciwdziałanie nieuzasadnionemu nadużywaniu leków
– Wyeliminowanie stosowania leków pochodzących z nielegalnych źródeł,
będących poza oficjalnym obiegiem farmaceutycznym, nie dopuszczonych do
obrotu przez Urząd
– Uwrażliwienie pacjentów na możliwość występowania działań niepożądanych
związanych z terapią
– Uświadomienie zagrożeń wynikających z interakcji leków, w tym także interakcji
leków i żywności oraz suplementów diety.
• Skuteczność naszej Kampanii niepomiernie wzrośnie, jeśli media,
obdarzone przecież ogromnym zaufaniem społecznym, zechcą się
aktywnie włączyć do jej upowszechniania.”
68

URPL
• W każdym numerze Almanachu znajdziecie Państwo
stałe działy:
•
O Urzędzie – informacje dotyczące bieżącej pracy Urzędu
Produkty lecznicze - publikacje o produktach leczniczych stosowanych u
ludzi
Produkty lecznicze weterynaryjne - publikacje o produktach leczniczych
stosowanych
w weterynarii
Wyroby medyczne - artykuły poświęcone tematyce wyrobów medycznych
Produkty biobójcze - informacje z zakresu produktów biobójczych.
69

MHRA
• Problemy bezpieczeństwa
• Adverse Drug Reactions
• Healthcare Professional Reporting
• Drug Analysis Prints (DAPs)
• Information for the Paharmaceuitical
Industry
• Defective Medicine
• Devices
• Blood
70
71
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
Wyszukiwarka
Podobne podstrony:
żródła inf bibliotekoznastwo specjalne, informacja naukowa i bibliotekoznawstwo 3 semestr
ŹRÓDŁA INFORMACJI, Studia INiB, informacja naukowa i bibliotekoznawstwo
Małgorzata Janiak Informacja naukowa w historii sztuki, czyli encyklopedie online jako podstawowe ź
JHP, Informacja naukowa i bibliotekoznastwo 2 semestr, Analiza i opracowaniw dokumentów, Analiza i o
- 2. Książka w Grecji, Informacja naukowa i bibliotekoznastwo 2 semestr
tiob2, Informacja Naukowa i Bibliotekoznawstwo, Teoria i organizacja bibliografii
SYSTEMY DOKUMENTACYJNE I SYSTEMY FAKTOGRAFICZNE(1), informacja naukowa i bibliotekoznawstwo 3 semest
Katalog Mediów Polskich, Informacja naukowa i bibliotekoznawstwo, Technologia informacyjna
biblioteki cyfrowe ocena, Informacja Naukowa i Bibliotekoznawstwo, Materiały
Polonica Zagraniczne, Informacja naukowa i bibliotekoznastwo 2 semestr
kristanioea, informacja naukowa i bibliotekoznawstwo 3 semestr
referat, informacja naukowa i bibliotekoznawstwo 3 semestr
Informacja Naukowa kolokwium
Statystyki RK opis, Informacja Naukowa i Bibliotekoznawstwo, Materiały
Metodyka sporządzania adnotacji i analiz dokumentacyjnych i ich rodzaje, Informacja naukowa i biblio
nauka o książce, Informacja Naukowa i Bibliotekoznawstwo, Materiały
Dziennki urzędowe, Informacja Naukowa i Bibliotekoznawstwo, Materiały
gramatyka opisowa, Informacja naukowa i bibliotekoznastwo 2 semestr
Bibliografia specjalna.BABiN, Informacja naukowa i bibliotekoznastwo 2 semestr
więcej podobnych podstron