
Genetyka molekularna chorób serca i
naczyń
W ostatnich dziesięcioleciach dokonał się
znaczny postęp w rozumieniu, jaką rolę
odgrywają czynniki dziedziczne w procesach
patologii człowieka, w tym również w chorobach
układu sercowo-naczyniowego.

Genetyka molekularna chorób serca i
naczyń
Choroby układu krążenia rozwijają się na skutek:
-
czynników wrodzonych tzn wskutek nieprawidłowości
genetycznych (=choroba genetyczna serca = choroba
dziedziczna, obecna od urodzenia)
lub
-
czynników nabytych, zależnych głównie od naszego
trybu życia.

Genetyka molekularna chorób serca i
naczyń
Choroby genetyczne serca
mogą się ujawniać już przy urodzeniu, w czasie
dorastania lub dopiero w wieku dorosłym. W niektórych
przypadkach nie dochodzi do ujawnienia się choroby
-zależne jest to od typu defektu, ale także od wpływu
dodatkowych czynników genetycznych i środowiskowych.
niosą ze sobą ryzyko nagłej śmierci sercowej w przebiegu
groźnej arytmii lub pęknięcia aorty.
w wielu przypadkach choroba nasila się z wiekiem i może
prowadzić do rozwoju niewydolności serca i/lub
przedwczesnej śmierci.

Genetyka molekularna chorób serca i
naczyń
Po rozpoznaniu choroby uwarunkowanej genetycznie
bardzo ważna jest ocena ryzyka wystąpienia poważnych
powikłań (nagły zgon, niewydolność krążenia).
Chorym w dobrym stanie z wysokim ryzykiem można
zaproponować zabiegowe postępowanie zapobiegawcze,
np wszczepienie kardiowertera-defibrylatora w przypadku
arytmii czy profilaktyczną operację aorty w zespole
Marfana.

Genetyka molekularna chorób serca i
naczyń
Choroby genetyczne serca mogą być dziedziczone:
•
jednogenowo lub wielogenowo
•
autosomalnie lub w sprzężeniu z chromosomem X
•
dominująco lub recesywnie
.

Genetyka molekularna chorób serca i
naczyń
Jednogenowe choroby genetyczne serca
to m.in.
kardiomiopatie (przerostowa, rozstrzeniowa)
kanałopatie (zespół wydłużonego lub skróconego
odcinka QT, zespół Brugadów)
choroby spichrzeniowe (choroba Pompego, Fabry’ego)
zespół Marfana
choroba węzła zatokowego
hipercholesterolemia rodzinna

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia przerostowa
Kardiomiopatia przerostowa:
wtórna do przeciążenia skurczowego mięśnia lewej
komory
(wskutek nadciśnienia tętniczego, wady zastawkowej)
idiopatyczna
dziedziczna

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia przerostowa
Typowe objawy kliniczne kardiomiopatii przerostowej:
duszność (głownie przy wysiłku)
postępujący spadek wydolności wysiłkowej
bóle wieńcowe
zaburzenia rytmu serca
(nagły zgon)
W badaniu fizykalnym: szmer serca, arytmia, objawy
niewydolności krążenia

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia przerostowa
W badaniach obrazowych (UKG) stwierdza się znaczny
przerost lewej komory serca.
Pogrubienie mięśnia może dotyczyć dowolnego fragmentu
komory, najczęściej jednak występuje asymetryczny
przerost przegrody komorowej.
Jama lewej komory jest zwykle mała.
Anatomopatologicznie stwierdza się przerost i
dezorganizację miocytów oraz włóknienie śródmiąższowe,
które występują w całym mięśniu sercowym.

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia przerostowa
Kardiomiopatii przerostowa jest jedną z najczęstszych
chorób genetycznych w kardiologii i najczęstszą
przyczyną nagłego zgonu wśród dzieci i młodych
dorosłych.

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia przerostowa
Kardiomiopatia przerostowa dziedziczy się
najczęściej jako cecha autosomalna dominująca,
rzadziej - jako cecha autosomalna recesywna sprzężona z
chromosomem X
lub choroba wynikająca z zaburzeń DNA
mitochondrialnego.
Schorzenie to spowodowane jest przez mutacje genów
kodujących białka aparatu kurczliwego mięśnia
sercowego.

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia przerostowa
Znanych jest ponad 100 mutacji odpowiedzialnych za rozwój
kardiomiopatii przerostowej.
Odkryto liczne mutacje genów przynajmniej 10 różnych
elementów białek sarkomeru, tj: łańcuch ciężki sercowej
beta-miozyny, sercowe białko wiążące miozynę, sercowa
troponina T, sercowa troponina I, alfa-tropomiozyna, sercowe
białko C, niezbędne i regulatorowe łańcuchy lekkie oraz
sercowa aktyna.
W rzadkich przypadkach kardiomiopatia występuje wskutek
mutacji genów nie związanych z białkami sarkomeru. Wtedy,
poza typowymi zmianami w mięśniu sercowym, obserwuje
się współistnienie dodatkowych objawów tj zespół WPW,
głuchotę czuciowo-nerwową, zaburzenia napięcia mięśni,
encefalopatia.

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia przerostowa
Ta sama mutacja może powodować różnorodny przebieg
kliniczny w danej rodzinie.
Za zmienność obrazu klinicznego mogą być odpowiedzialne:
Czynniki środowiskowe
Płeć
Czynniki nabyte (np. choroba wieńcowa, wady
zastawkowe serca…)

Geny, których mutacje są najczęściej związane z
kardiomiopatią przerostową
Gen /
kodowane białko
Częstość mutacji (%)
Obraz kliniczny i
rokowanie
MYH7
łańcuch ciężki beta-
miozyny
35-50
Mutacja R403Q (często
łącznie z R719W i
R453C) –
niekorzystne
rokowanie
Mutacje: V1606M, L908V,
G256Q, P513C –
rokowanie dobre
MYBPC3
myosin binding
protein C
30-47
Późne objawy kliniczne,
duże ryzyko zgonu w
wyniku arytmii
TNNT2
troponina T2
6,5-15
Łagodny przerost,
duże
ryzyko nagłego zgonu
w wyniku arytmii
TMP1
alfa-tropomiozyna
Współwystępowanie
przerostu
serca
z
zespołem WPW, rzadko
występowanie rodzinne

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia rostrzeniowa
Znane
przyczyny
„nabytej”
kardiomiopatii
rozstrzeniowej:
• choroba wieńcowa,
• przebyty zawał serca,
• wady zastawkowe,
• nadciśnienie tętnicze,
• zapalenie mięśnia sercowego
• choroby układowe.
W przypadkach gdy nie stwierdzamy czynnika
sprawczego mówimy o
kardiomiopatii idiopatycznej.
W części przypadków kardiomiopatia roztrzeniowa ma
podłoże genetyczne.

Jednogenowe schorzenia układu sercowo-naczyniowego
Kardiomiopatia rostrzeniowa
Kardiomiopatia rozstrzeniowa dziedziczy się najczęściej
w sposób autosomalny dominujący,
ale
występują
też
przypadki
dziedziczenia
autosomalnego
recesywnego,
związanego
z
mitochondrialnym DNA i sprzężonego z chromosomem
X.

Geny, których mutacje są najczęściej związane z
kardiomiopatią rozstrzeniową
Geny, których mutacje są najczęściej związane z
kardiomiopatią rozstrzeniową to geny kodujące:
białka szkieletu komórkowego
białka połączeń międzykomórkowych
białka otoczki jądrowej i blaszki jądrowej
białka sarkomeru

Geny, których mutacje są najczęściej związane z
kardiomiopatią rozstrzeniową
1. Białka szkieletu komórkowego
DES
(Desmina)
Kardiomiopatii mogą towarzyszyć:
miopatia szkieletowa, bloki
przewodzenia w sercu, zaburzenia
rytmu serca
SGCD, SGCB
(--sarkoglikan)
„Czysta” postać kardiomiopatii lub w
przypadku SGCB dziedziczona
autosomalnie recesywnie ciężka
odmiana dystrofii mięśniowej obręczy
kończyn
DMD
(dystrofina)
Postać sprzężona z chromosomem X,
małe stężenie dystrofiny sercowej przy
prawidłowych poziomach dystrofiny
mięśni szkieletowych
2. Białka połączeń międzykomórkowych
VCL
(winkulina),
DSP, CSRP3
Kardiomiopatii jest skutkiem
zaburzenia interakcji metawinkuliny z
aktyną i nieprawidłowego
zakotwiczenia cytoszkieletu do
sarkolemy miocytu
Geny, których mutacje są najczęściej związane z
kardiomiopatią rozstrzeniową
3. Białka otoczki jądrowej i blaszki jądrowej
LMNA (
laminina A i C
)
Autosomalna dominująca postać
kardiomiopatii z towarzyszącymi
zaburzeniami przewodzenia i bez
zajęcia mięśni szkieletowych lub
autosolana dominująca dystrofdia
mięśniowa Emery’ego-Dreifussa
Emeryna
Dystrofia mięśniowa Emery’ego-
Dreifussa z poszerzeniem jam serca i
zaburzeniami przewodzenia
4. Białka sarkomeru
ACTC (
aktyna)
MYH7 (
łańcuch ciężki miozyny
sercowej
)
TINT2 (
troponina sercowa T)
Alleliczne formy kardiomiopatii
przerostowej lub końcowe stadium tej
kardiomiopatii

Kardiomiopatia rozstrzeniowa typu
left ventricular non-compaction
Odmianą genetycznie uwarunkowanej kardiomiopatii
rozstrzeniowej jest
izolowana kardiomiopatia
gąbczasta
(left ventricular non-compaction)
Charakteryzuje się ona przerostem lewej komory z
głębokim beleczkowaniem i upośledzeniem czynności
skurczowej ze współistniejącą często rozstrzenia.
Podobne zmiany mogą dotyczyć również prawej komory.

Kardiomiopatia rozstrzeniowa typu
left ventricular non-compaction
Kardiomiopatii typu left ventricular non-compaction
towarzyszą często różnego rodzaju wady wrodzone serca,
np. ubytki w przegrodzie między przedsionkowej lub
międzykomorowej oraz zwężenie zastawki pnia płucnego.
Choroba może ujawniać się w niemowlęctwie lub w
wieku późniejszym.
Jej przebieg jest zwykle niekorzystny z szybko
postępującą dysfunkcją lewej komory i jawną klinicznie
niewydolnością serca.

Zaburzenia rytmu serca
Nagłe zgony związane z groźnymi zaburzeniami rytmu
serca ze względu na ich wielką liczbę stanowią bardzo
istotny problem kliniczny w kardiologii.
Czynniki genetyczne mogą modyfikować ryzyko arytmii
związane z typowym podłożem patologicznym.
Opisano geny sprzyjające zaburzeniom rytmu, co
pozwoliło na pogłębienie wiedzy na temat molekularnego
podłoża różnych typów tych zaburzeń.

Zespół wydłużonego odstępu QT
Zespół wydłużonego odstępu QT (
zespół
LQT
) jest
wrodzoną chorobą arytmogenną
Częstość występowania: 1-2 przypadki na 10 tys.osób
Zidentyfikowano 8 genów odpowiedzialnych za
wystepowanie róznych form zespołu LOD. Kodują one
podjednostki sercowych kanałów jonowych.
Choroba
występuje w strukturalnie prawidłowym
sercu !
Jest związana z dużym ryzykiem nagłego zgonu.

Zespół wydłużonego odstępu QT
Podstawy rozpoznania: w EKG nieprawidłowe wydłużenie
okresu repolaryzacji (= odstępu QT), nieprawidłowy
kształt załamka T oraz groźne dla życia zaburzenia rytmu
serca.
Zaburzenia rytmu serca wyzwalane są wysiłkiem
fizycznym lub stresem emocjonalnym.
Pierwsze objawy choroby (omdlenia lub nagły zgon)
występują średnio w wieku 12 lat.
Postępowanie terapeutyczne: stosowanie beta-blokerów,
profilaktyczne wszczepienie kardiowertera-defibrylatora
(ICD).

Zespół wydłużonego odstępu QT
Opisano
dwie główne formy fenotypowe
zespołu LQT:
zespół Romano-Warda
dziedziczony w sposób
autosomalny dominujący
oraz rzadziej występujący
zespół Jervella-Langego-Nielsena
dziedziczony w
sposób autosomalny recesywny i współwystępujący z
głuchotą czuciowo-nerwową.

Geny, których mutacje są związane z
występowaniem
różnych postaci zespołu LQT
Kana
ł
Gen (białko)
Postacie
choroby
Częstość
(%)
Obraz kliniczny
I
Ks
KNCQ1
(KvLQT1)
KCNE1 (MinK)
LQT1 lub JLN1
LQT5 lub JLN2
50
2-3
LQT1 – zmniejszona
penetracja, łagodniejszy
przebieg, objawy
wywołane bezpośrednio
bodźcem
adrenergicznym zwykle
występują w czasie
wysiłku
I
Kr
KCNH2 (HERG)
KCNE2 (MiRP)
LQT1
LQT6
35-40
<1
LQT2-większa penetracja i
ciężkość niż LQT1,
zwłaszcza u kobiet,
zaburzenia rytmu
wyzwalane bodźcem
dźwiękowym
LQT6-niepełna penetracja,
łagodny przebieg

Geny, których mutacje są związane z
występowaniem
różnych postaci zespołu LQT
Kana
ł
Gen (białko)
Postacie
choroby
Częstość
(%)
Obraz kliniczny
I
Na
SCN5A (Nav
1.5)
LQT3
10-15
Gorsze rokowanie,
zwłaszcza u mężczyzn,
zaburzenia rytmu
występują w spoczynku
-
ANK2
(ankrynina
B)
LQT4
Towarzyszy bradykardia,
napadowe migotanie
przedsionków,
wielofazowe załamki T
I
Ca
CACNA1c
(CaV1.2)
LQT8
Zespół
Timothy’ego
Znaczne wydłużenie QT,
towarzyszy syndaktylia
skórna, blok AV 2:1
wrodzone wady serca,
opóźnienie umysłowe,
autyzm, zaburzenia
metaboliczne, duża
śmiertelnosć

Zespół skróconego odstępu QT
Zespół ten został po raz pierwszy opisany w 2000r.
Charakteryzuje się on w zapisie EKG nieprawidłowo
krótkim okresem repolaryzacji (=odstęp QT < 300ms)
oraz wąskimi
i spiczastymi załamkami T.
Klinicznie zespół ten objawia się napadowym migotaniem
przedsionków, omdleniami i nagłym zgonem.

Zespół skróconego odstępu QT
Pierwszym genem, którego mutacja została wskazana
jako leżąca u podłoża tego zespołu, był KCNH2 kodujący
białko HERG.
Opisana mutacja powoduje wzrost aktywności
odśrodkowego prądu potasowego I
kr
.
Do tej pory opisano również mutacje w innych genach
KCNQ1, SCN5A, KCNE2 i KCNJ2.

Zespół Brugadów
Jest to wrodzona choroba arytmogenna dziedziczona w
sposób autosomalny dominujący.
Wyraźnie częściej jest stwierdzana wśród mężczyzn niż u
kobiet (8:1).
Występuje w prawidłowym strukturalnie sercu !
Podstawy rozpoznania w EKG: zupełny lub niezupełny
blok prawej odnogi pęczka Hisa oraz uniesienie odcinka
ST w odprowadzeniach V1-V2 (V3); występowanie
groźnych dla życia tachyarytmii komorowych.

Zespół Brugadów
Objawy kliniczne w postaci omdleń lub nagłego
zatrzymania krążenia ujawniają się zwykle w 3 – 4
dekadzie życia i występują zwykle w spoczynku lub
podczas snu.
W 20% przypadków podłożem molekularnym zespołu
Brugardów jest mutacja w genie SCN5A kodującym białko
(Nav1.5) będące składową kanału sodowego.
W 80% udało się jedynie określić locus na ramieniu
krótkim chromosomu 3 (3p22-25) ale bez określenia
konkretnego genu. Czynnościowo mutacje te również
upośledzają prąd sodowy.

Zespół Brugadów
W 80% przypadków udało się jedynie ustalić locus
związane z zespołem Brugadów zlokalizowane na
ramieniu krótkim 3 chromosomu (3p22-25), bez
określenia genu odpowiedzialnego za chorobę.
Czynnościowo mutacje leżące u podłoża tego zespołu
prowadzą do upośledzenia prądu sodowego.

Zespół Brugadów
Rozpoznanie stawia się na podstawie: wystąpienie
nagłego
zatrzymania
krążenia
(skutecznie
reanimowanego) wywiadu rodzinnego (nagłe zgony w
młodym wieku) oraz badania EKG.
Leczenie
zespołu
polega
na
profilaktycznym
zabezpieczeniu chorego przed nagłym zatrzymaniem
krążenia. Wszczepia się kardiowerter-defibrylator

Polimorficzny katecholaminergiczny
częstoskurcz komorowy
Obraz kliniczny charakteryzuje się występowaniem w
trakcie wysiłku fizycznego, bądź pod wpływem bodźca
emocjonalnego polimorficznych komorowych zaburzeń
rytmu prowadzących do omdlenia lub zatrzymania
krążenia,
przy braku strukturalnej choroby serca.
Spoczynkowe EKG jest prawidłowe lub stwierdza się
bradykardie zatokowa z wyraźnymi załamkami U.
Pierwsze objawy (w tym nagły zgon) występują zwykle w
dzieciństwie lub w młodości.

Polimorficzny katecholaminergiczny
częstoskurcz komorowy
Choroba dziedziczona autosomalnie dominująco.
Przyczyną choroby jest mutacja genu RyR2, kodującego
sercowy receptor rianodynowy uwalniający wapń
zjonizowany w mięśniu sercowym, który odgrywa rolę w
regulacji wewnątrzkomórkowego prądu wapniowego co
warunkuje prawidłowe sprzężenie elektromechaniczne.
Mutacja genu RyR2 prowadzi do niekontrolowanego
uwalniania Ca
2+
z siateczki sarkoplazmatycznej podczas
stymulacji adrenergicznej.

Wieloczynnikowe schorzenia
układu sercowo-naczyniowego
Powszechnie występujące schorzenia układu sercowo-
naczyniowego ujawniają się wskutek współdziałania
czynników genetycznych i środowiskowych.
„wrodzona podatność” + czynniki środowiskowe
CHOROBA

Wieloczynnikowe schorzenia
układu sercowo-naczyniowego
„Wrodzona podatność”
= mutacje i polimorfizmy licznych
genów
Czynniki ryzyka
chorób sercowo-naczyniowych:
•
Otyłość
•
nadciśnienie tętnicze
•
zaburzenia lipidowe
•
cukrzyca
•
mała aktywność fizyczna
•
wiek
•
płeć męska
•
palenie tytoniu
•
nadmierne spożycie alkoholu
•
dodatni wywiad rodzinny
•
czynniki psychologiczne

Grupy genów kandydatów potencjalnie sprzyjających
ujawnianiu się miażdżycy i choroby niedokrwiennej serca
GENY KODUJĄCE białka
Metabolizm lipidów (apolipoproteiny, enzymy lipolityczne, receptory
lipoprotein itp.)
Układ krzepnięcia i trombolizyny (fibrynogen, czynniki krzepnięcia,
inhibitor aktywatora plazminogenu 1 PAI-1 itp.)
Glikoproteiny płytkowe (GPIIb/IIIa, GPIa/IIa)
Układ renina-angiotensyna-aldosteron (angiotensynogen, ACE, receptor
AT1, syntetaza aldosteronu)
Czynniki wazoaktywne (ANP, BNP, CNP)
Czynniki adhezyjne i migracyjne dla monocytów i makrofagów (VCAM i
ICAM)
Czynniki zapalenia (cytokiny, TNF-)
Czynniki proliferacji komórek mięśni gładkich naczyń

Ryzyko zawału serca związane z polimorfizmem insercyjno-
delecyjnym genu enzymu konwertującego angiotensynę (I/D
ACE)
Badanie
Iloraz szans wystąpienia zawału
serca dla DD vs. ID + II
Cambien i wsp. 1992, ECTIM Study
1,34, p < 0,007
Lindpaintner
I
wsp.
1995,
Physicians’ Health Study
1,09 (95% CI 0,85-1,38, p=0,31)
Samani I wsp. 1996, metaanaliza
1,26 (95% CI 1,15-1,39, p<0,0001)
Staessen I wsp. 1997, metaanaliza
1,43 (95% CI 1,28-1,59, p<0,001)
Agerhol, - Larsen I wsp. 2000,
metaanaliza
1,21 (95% CI 1,11-1,32)
Keabney I wsp. 2000, ISIS Study
1,10 (95% CI 1,00-1,21, p<0,001)
Ciećwierz I wsp. 2000, populacja
Polski Północnej
1,02 (95% CI 0,70-1,49, p=0,92)
Genetyka cukrzycy

Genetyka cukrzycy typu 1
Przyczyną cukrzycy typu 1 jest uszkodzenie komórek beta
trzustki w przebiegu procesu autoimmunologicznego.
Niszczenie komórek beta trzustki wiąże się z istnieniem
genetycznej predyspozycji oraz współdziałaniem
czynników środowiskowych.
Skłonność do rozwoju cukrzycy typu 1 jest uwarunkowana
dziedziczeniem poligenowym
Potwierdzono znaczenie licznych regionów w genomie
mających związek z DM typu1; są one zlokalizowane na
chromosomach:
1, 2, 3, 5, 6, 10, 11, 14, 15,18, 20.

Genetyka cukrzycy typu 1
Ryzyko zachorowania na cukrzycę typu 1 wynosi:
w populacji ogólnej 0.2-0.4%
u dzieci matek z cukrzycą 2-3%
u dzieci ojców z cukrzycą 5-9%
Badanie wykazują częstsze zachorowanie na cukrzycę
typu 1
u krewnych pierwszego stopnia oraz bliźniąt
jednojajowych.

Jednogenowe postaci cukrzycy typu 1
Zespół autoimmunologicznej poliendokrynopatii
typu 1
Zespół XPID (X-linked poliendocrinopathy, immune
dysfunction, diarrhoea) związany z chromosomem X.
Mutacje występujące w tych zespołach dotyczą genów
kodujących czynniki transkrypcyjne, odpowiednio:
AIRE (autoimmune regulator) na chromosomie 23
Fox P3 na chromosomie X

Genetyka cukrzycy typu 2
Biorąc pod uwagę znaczenie czynników genetycznych w
rozwoju
cukrzycy typu 2 można dokonać podziału na 2 formy:
Formy monogenowe cukrzycy typu 2. Są one konsekwencją
rzadkich mutacji w pojedynczych genach, które
skutkują defektem w zakresie wydzielania insuliny bądź
też głębokim upośledzeniem wrażliwości na insulinę.
Formy poligenowe cukrzycy typu 2. Ich obraz kliniczny
wynika z interakcji czynników środowiskowych i
genetycznych (wpływ wielu genów).

Jednogenowe postaci cukrzycy typu 2
Najczęstsza postać DM2 charakteryzuje się zaburzeniami
wydzielania insuliny
Inne postaci DM2 mogą wynikać z mutacji genu
kodującego insulinę co prowadzi do zmiany struktury
insuliny i osłabienia lub zniesienia jej czynności.
U osób z mutacją genu insuliny pojawia się nietolerancja
glukozy, której towarzyszy hiperinsulinemia, prawidłowa
insulino- wrażliwość, bez współwystępujących zaburzeń
immunolo-gicznych.
Postać DM2 tzw MIDD (Maternal inheredited diabetes
with deafness) wiąże się z mutacjami mitochondrialnego
DNA. Towarzyszą jej upośledzenie słuchu i głuchota.

Jednogenowe postaci cukrzycy typu 2
z dominującą insulinoopornością
Insulinooporność jest spowodowana mutacją w jednym
lub dwóch allelach genu receptora insuliny.
Mutacje w genie receptora insuliny są bardzo rzadkie.
krasnoludkowatość
(leprechaunism). Krasnoludkowatość
charakteryzuje się nasiloną insulinoopornością, cukrzycą, niższą
masą urodzeniową, powolnym wzrostem. Zespół ten źle rokuje.
Zespół Robsona – Mendeholla
. Ujawnia się klinicznie w okresie
dzieciństwa. Charakteryzuje się szybkim wzrostem, hipertrofią
paznokci i zębów, przedwczesnym dojrzewaniem oraz wzrostem
szyszynki.
Zespół insulinooporności typu A
. Zespół ten dotyczy młodych
kobiet. Charakteryzuje się hiperandrogenizmem, zespołem
policystycznych jajników oraz zmianami skórnymi o typie
acanthosis nigricaus.

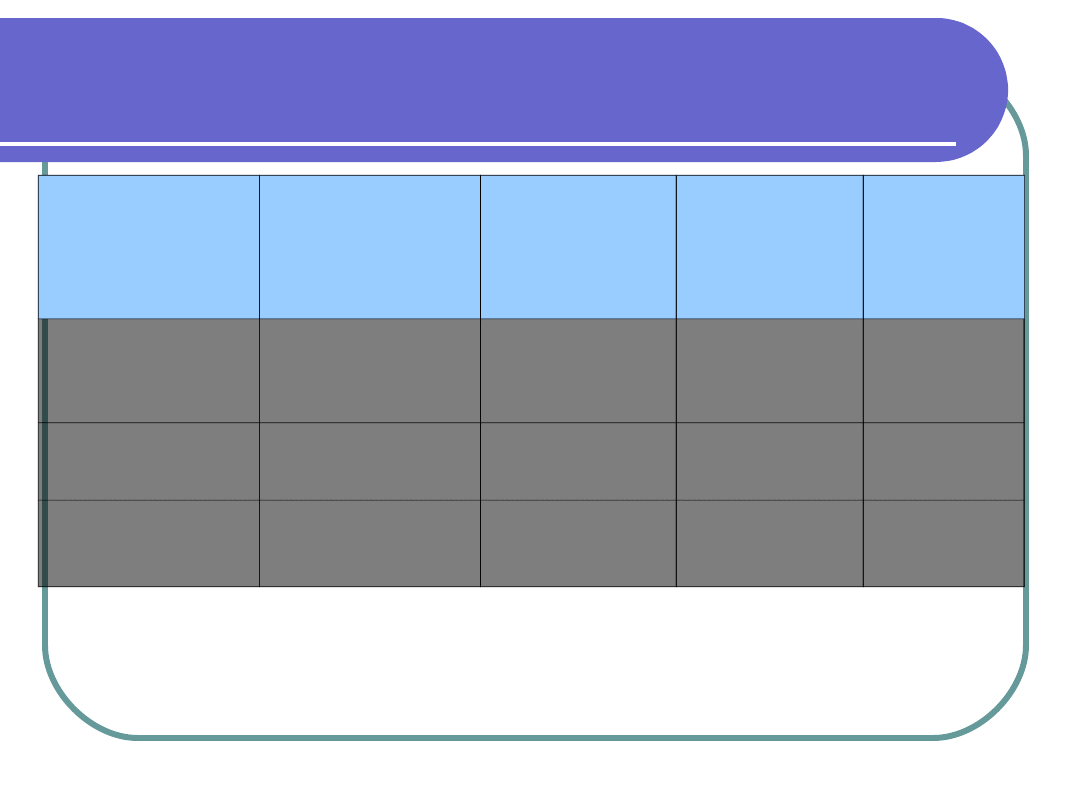
TYP
MODY
LOKALIZACJA
ZMIANY
NAZWA
GENU
CZĘSTOŚĆ
WYSTĘPOWANI
A
MODY1
20q
HNF-4α
(TCF4)
Rzadko 1
– 2%
MODY2
7q
GCK
10 – 65%
MODY3
12q
HNF-1α
(TCF1)
20 – 65%
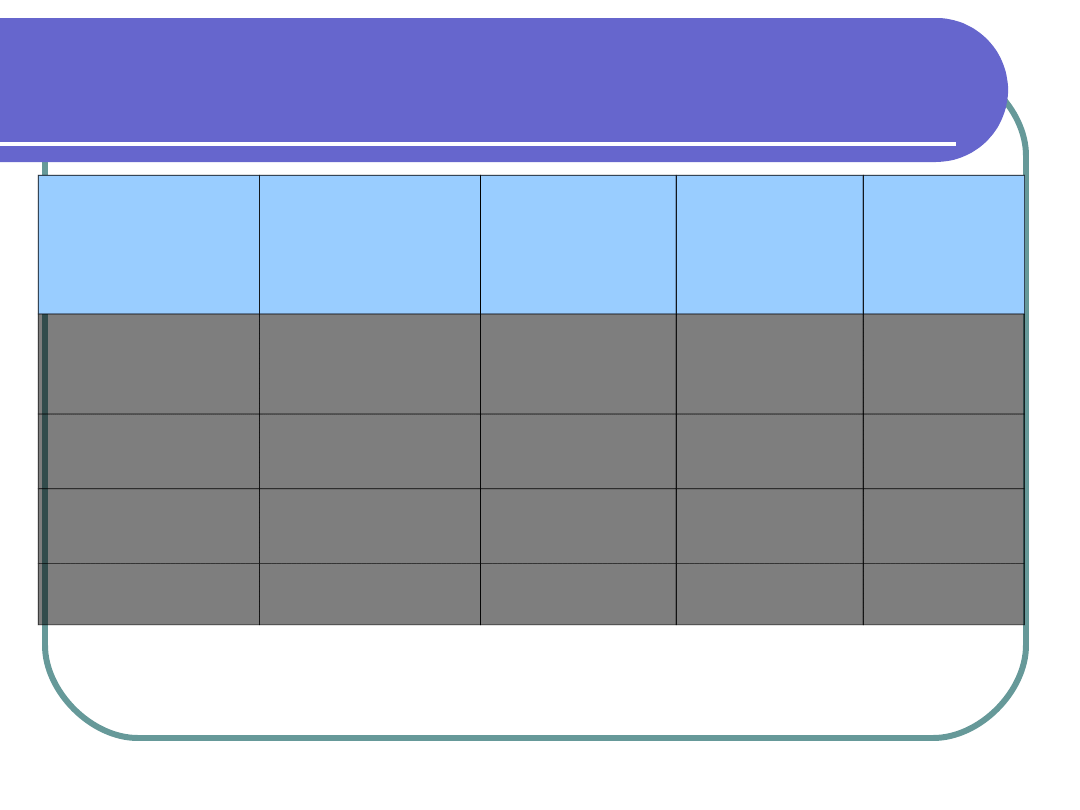
Genetyka cukrzycy typu 2

TYP
MODY
LOKALIZACJA
ZMIANY
NAZWA
GENU
CZĘSTOŚĆ
WYSTĘPOWANI
A
MODY4
13q
IPF-1
Rzadko
MODY5
17q
HNF-1β
(TCF2)
Rzadko
MODY6
2q32
Neurod1
Rzadko
MODY7
11q15.1
SUR1
E1506K
Rzadko
Genetyka cukrzycy typu 2
TYP
MODY
WIEK
UJAWNIENIA
CUKRZYCY
ZMIANA
PIERWOTNA
CIĘŻKOŚĆ
CUKRZYC
Y
POWI
KŁANIA
MODY1
młodzieniec,
młody,
dorosły.
Trzustka/
inne
Ciężka
Często
MODY2
po urodzeniu
Trzustka/
inne
Przebieg
łagodny
Rzadko
MODY3
młodzieniec,
młody
dorosły.
Trzustka/
inne
Ciężki?
Często
Genetyka cukrzycy typu 2
TYP
MODY
WIEK
UJAWNIENIA
CUKRZYCY
ZMIANA
PIERWOTNA
CIĘŻKOŚĆ
CUKRZYC
Y
POWI
KŁANIA
MODY4
młody
dorosły
Trzustka/
inne
Ciężki
Niezna
ne
MODY5
?
Trzustka/
inne
Ciężki
Chorob
y nerek
MODY6
młody
dorosły
Trzustka
Ciężki
Niezna
ne
MODY7
dorośli
Trzustka
Łagodny
Niezna
ne
Genetyka cukrzycy typu 2

Poligenowe formy cukrzycy typu 2
Stwierdzono związek z cukrzycą typu 2 następujących genów:
Gen kodujący białko
KALPAINĘ
Kalpaina zmniejsza uwalnianie insuliny pobudzane glukozą.
Kalpainy zmniejszają pobudzaną insuliną syntezę glikogenu
w mięśniach. Niski poziom RNA dla kalpainy 10 w
mięśniach prowadzi do narastania insulinooporności.
Gen Kapaliny 10 jest zlokalizowany na chromosomie 2q.
Ryzyko zachorowania na DM2 nie wiąże się z wariantem
jednego polimorfizmu genu kalpainy, ale wynika raczej z
obecności kilku niekorzystnych haplotypów tworzonych
przez allele trzech SNP – ów o numerach: 19, 43, 63.
Wszystkie te SNP – y zlokalizowane są w intronach, a więc
nie wpływają na strukturę aminokwasową białka.
Znaczenie kapaliny 10 w patogenezie cukrzycy typu 2 jest
różne w różnych populacjach.

Poligenowe formy cukrzycy typu 2
Gen kodujący
PPAR γ czyli peroksysomalny aktywowany
proliferacyjnie receptor γ
(peroximose proliferator
activated receptor)
Ostatnio opublikowane badanie zarówno osób
niespokrewnionych, jak i rodzin bezspornie udowodniło
znaczenie polimorfizmu Pro12Ala w patogenezie cukrzycy
typu 2.
Ryzyko rozwoju cukrzycy u osób homozygot Pro12Pro
PPARγ zwiększa się o 25%.
Polimorfizm Gly482Ser koaktywatora PPAR γ zwiększa
ryzyko rozwoju cukrzycy o 34%.

Poligenowe formy cukrzycy typu 2
Gen
KCJN11.
Gen KCJN11 koduje podjednostkę Kir6.2 ATP – zależnego
kanału potasowego komórek . Polimorfizm GLU23Lys w tym
genie zwiększa ryzyko rozwoju cukrzycy typu 2. Wiąże się to
prawdopodobnie z wpływem tej mutacji na czynność kanału
potasowego.
Gen
receptora insulinowego
Gen ten jest zlokalizowany na chromosomie 19q. Opisano
około 50 mutacji w genie receptora insulinowego. Są one
jednak zjawiskiem rzadkim. Opisano do tej pory kilkadziesiąt
przypadków cukrzycy z leżącą u jej podłoża bardzo nasiloną
insulinoopornością związaną z mutacjami jednego lub obu
alleli receptora insulinowego.

Jednogenowe postaci
nadciśnienia tętniczego
Choroba
Gen lub locus
Dziedziczenie
Niedobór 11-hydroklsylazy steroidowej
Niedobór 17-hydroksylazy steroidowej
Zespół Liddle’a
Pozorny nadmiar mineralokortykosteroidów
(AME)
Mutacja S810L genu receptora
mineralokortykosterodiów
Rodzinny hiperaldosteronizm Typu I (FH-I, GRA)
Rodzinny hiperaldosteronizm typu II (FH-II)
Zespół Gordona (PAHII)
Nadciśnienie z brachydaktylią
Nadciśnienie z hipercholesterolemią i
hipomagnezemią
CYP11B1
CYP17
SCNN1B
SCNN1G
HSD11B2
NR3C2
CYP11B1/B2
7p22
1q31-q42
PRKWNK1
PRKWNK4
12p
mtDNA
Autosomalne recesywne
Autosomalne recesywne
Autosomalne dominujące
Autosomalne recesywne
Autosomalne dominujące
Autosomalne dominujące
Autosomalne dominujące
(?)
Autosomalne dominujące
Autosomalne dominujące
Mitochondrialne
Genetyka
chorób nerek

Choroby nerek wywołane mutacją
pojedynczego genu:
Zespół Alporta
Zespół Alporta jest wrodzoną, dziedziczną glomerulopatią
spowodowaną genetycznie uwarunkowanym
zaburzeniem syntezy jednego z łańcuchów kolagenu IV.
Zespołowi temu towarzyszy zwykle upośledzenie słuchu.
Rzadko występują zmiany w narządzie wzroku

Choroby nerek wywołane mutacją
pojedynczego genu:
Zespół Alporta
Przyczyną choroby jest zaburzenie syntezy błony
podstawnej spowodowane genetycznie uwarunkowanym
brakiem łańcucha kolagenu IV.
W 80 – 85% przypadków choroba jest dziedziczona w
sposób dominujący związany z płcią.
Defekt dotyczy wówczas łańcucha 5 kolagenu IV, a
zmutowany gen jest położony na dystalnym ramieniu
chromosomu X (COL4A5, Xq22).
W genie COL4A5 zidentyfikowano ponad 300 różnych
mutacji. Są to mutacje bezsensu, mutacje procesu
składania RNA lub delecje mniej niż 10 par zasad.

Choroby nerek wywołane mutacją
pojedynczego genu:
Zespół Alporta
Mężczyźni chorzy na zespół Alporta (związany z
chromosomem X) nie przenoszą tej choroby na synów,
lecz tylko na córki.
Kobiety
chore
na
zespół
Alporta
(związany
chromosomem X) przenoszą chorobę w 1/3 do ½
przypadków.
U kobiet zwykle jedynym objawem jest mikrohematuria.
U mężczyzn z delecjami w genie COL4A5 stwierdzono
połączoną
z
głuchotą
progresję
do
krańcowej
niewydolności nerek w 2-3 dekadzie życia.

Choroby nerek wywołane mutacją
pojedynczego genu:
Zespół Alporta
Choroba przebiega łagodniej u kobiet, natomiast w
sposób umiarkowany lub ciężki u mężczyzn.
W postaci dziedziczonej w sposób autosomalny
recesywny defekt dotyczy łańcuchów 3 i 4 kolagenu
IV, a geny kodujące COL4A3 i COL4A4 znajdują się na
chromosomie 2 (2q36 – 37).
Prawdopodobnie defekty genetyczne mogą występować
również na innych chromosomach (chromosom 13?).
Błona podstawne z omówionym defektem genetycznym
ma unikatowe właściwości antygenowe: nie wiąże ona
przeciwciał przeciwko błonie podstawnej.
Wymienione zmiany w błonie podstawnej występują
również w naskórku, w śliniankach i soczewkach.

Choroby nerek wywołane mutacją
pojedynczego genu:
Zespół Alporta
Rozpoznanie zespołu Alporta stawia się na postawie
następujących
charakterystycznych
objawów
oraz
wyników badań dodatkowych:
rodzinne
występowanie
objawów
klinicznych
wymienionych powyżej
wynik badania biopsyjnego nerki, a zwłaszcza wygląd
błony podstawnej w mikroskopie elektronowym
wykrycie mutacji genowej po przeprowadzeniu badań
genetycznych
wzmożone wydalanie z moczem produktów degradacji
kolagenu z moczem.

Choroby nerek wywołane mutacją
pojedynczego genu:
Zespół Alporta
W rozpoznaniu różnicowym należy wziąć pod uwagę stany
chorobowe przebiegające z krwiomoczem o charakterze
rodzinnym.
Wśród takich chorób wymienić należy:
łagodny rodzinny krwiomocz
zespół cienkich błon podstawnych
stany zagrożenia kamicą.
Nie należy zapominać o tym, że aż do 20% przypadków
zespołu Alporta rozwija się w wyniku nowych mutacji.

Choroby nerek wywołane mutacją
pojedynczego genu:
Zespół Alporta
Nawroty krwiomoczu mogą utrzymywać się przez wiele
lat przed rozwojem niewydolności nerek.
Rozwój niewydolności nerek postępuje powoli.
U mężczyzn terminalna niewydolność nerek rozwija się w
4 – 5 dekadzie życia.
U kobiet terminalna niewydolność nerek rozwija się
rzadziej.
Brak zadowalających metod terapeutycznych.
Choroba nie przenosi się zwykle na przeszczepioną
nerkę).
Poradnictwo genetyczne prenatalne może zapoznać
rodziców z ryzykiem rozwoju choroby u dziecka.

Choroby nerek wywołane mutacją
pojedynczego genu:
Zwyrodnienie torbielowate (wielotorbielowatość ) nerek
Zwyrodnienie torbielowate nerek jest częstą przyczyną
krańcowej niewydolności nerek. Aż > 10% leczonych
nerkozastępczo to chorzy ze zwyrodnieniem
torbielowatym nerek.
Wielotorbielowatość nerek jest schorzeniem
charakteryzującym się obecnością niezliczonych ilości
torbieli różnej wielkości, rozmieszczonych zarówno w
korze, jak i w rdzeniu obu nerek.
Torbiele te powodują powiększenie i zniekształcenie
narządów.

Molekularna i kliniczna
charakterystyka
AD
PKD i
AR
PKD
ADPKD
ARPKD
Charakterystyka molekularna
-
Sposób dziedziczenia
-
Gen
- Produkt genu – białko
- Struktura białka
- Lokalizacja tkankowa
- Lokalizacja komórkowa
- Funkcja
Charakterystyka kliniczna
-
częstość występowania
-
wiek wystąpienia ESRD
- umiejscowienie torbieli
nerkowych
- objawy pozanerkowe
Autosomalnie dominujący
PKD1 (16p13.3)
PKD2 (4q21-q22)
Policystyna 1 (PC-1) – 4302 reszty
aminokwasowi
Policystyna 2 (PC2) – 968 reszt
aminokwasowych
PC-1L integralne białko błonowe
PC-2: integralne białko błonowe,
podobne do kanału TRPC
PC-1, PC-2: heterogeniczne
PC-1: błona cytoplazmatyczna,
rzęski pierwotne
PC-2: retikulum endoplazmatyczne,
rzęski pierwotne
PC-1: receptor?. Wspólne z PC-2
towarzyszy kanał jonowy
PC-2: kanał kationowy aktywowany
przez Ca
2+
1:400 do 1:1000
6. lub 7. dekada
Wszystkie segmenty nefronu
Torbiele wątrobiwe, trzustce,
śledzionie, nadciśnienie
tętnicze, tętniaki
śródczaszkowe
Autosomalnie recesywny
PKHD1 (16p21.1 – p12)
Fibrycystyna/poliducyna – 4074 reszty
aminokwasowi
Fibrycystyna/poliducyna – integralne
białko błonowe
Fibrycystyna/poliducyna – nerki, trzustka,
wątroba
Fibrocystyna/poliducyna – błonka
cytoplazmatyczna, rzęski pierwotne,
ciałko podstawowe
Funkcja nieznana
1:6000 do 1:40 000
Wczesne dzieciństwo
Kanaliki zbiorcze
Zaburzenia rozwoju dróg żółciowych,
zwłóknienie wątroby, nadciśnienie
tętnicze, nadciśnienie wtórne.

Główne jednogenowe choroby
kłębuszków nerkowych
Typ
glomer
ulopatii
Choroba
(zespół)
Początek
choroby
Dziedziczenie
Fen
Locus
Produkt genu
Zaburzenia
budowy
błony
podstaw
nej
kłębuszk
a
Zespół Alporta
Późny
AR
AD
COL4A3
lub
COL4A4
2q35-
3
7
Łańcuch 3 lub 4
kolagenu
typu IV
Zespół cienkich
błon
AD
Zespół Alporta
Sprzężone z
płcią
COL4A5
Xq22.3
Łańcuch 5
kolagenu
typu IV
Zespół Alporta
z rozsianą
mięśniako
watością
COL4A5
i
CO
L4
A6
Łańcuch 5 i 6
kolagenu
typu IV
Zespół Alporta
z
opóźnienie
m
umysłowy
m
COL4A5
i
AC
SL
4
(F
AC
L4
)
Łańcuch 5
kolagenu
typu IV i
synteza
acylo-CoA
swoista dla
długołańcuc
howych
kwasów
tłuszczowych
Zespół
paznokcio
wo-
rzepkowy
(NPS)
AD
LMXIB
9q34
Czynnik
transkypcyjn
y LMX1B
Zespół Piersona
Okres niemowlęcy
AR
LAMB2
3p14-
2
2
Łańcuch
2
lamininy

Główne jednogenowe choroby kłębuszków
nerkowych
Zaburzenia
budowy
cytoszkieletu
podocytów
OSSK
Późny
AD
ACTN4
19q13
Alfa-aktynina 4
Zespół Ebsteina
Późny
AD
MYH9
22q11.
2
Ciężki łańcuch IIA
miozyny
niemięśniowej
Zespół
Fechnera
Anomalia
Maya-Hegglina
Zespół
Sebastiana
Zaburzenia
czynności
organelli
komórkowych
Cytopatia
mitochondrialn
a
Dzieciństwo/młod
zi dorośli
Mitochondrialni
e
MTTL1
modna
tRNA dla leucyny
(tRNA
Leu
)
Zespół
Fabry’ego
Dorośli
Sprzężone z
płcią
GAL
Xq22
Alfa-
galaktozydaza
Zaburzenia
budowy
filtracyjnej
błony
szczelinowatej
ZN typu
fińskiego
Okres niemowlęcy
AR
NPHS1
19q13.
1
Nefruna
ZN
steroidooporny
Różny
AR
NPHS2
1q25-
31
Podoczna
Zespół Denysa-
Drasha
Okres niemowlęcy
AD
WT1
11p13
Białko WT1
Zespół Frasiera
OSSK
Dorośli
AD
CD1AP
6p12
CD2AP
OSSK
Późny
AD
TRPC6
11q21-
22
TRPC6
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
Wyszukiwarka
Podobne podstrony:
Niebisz A B Karnafel W Prewencja pierwotna chorób układu sercowo naczyniowego w cukrzycy
Rola diety bogatoresztkowej w profilaktyce i leczeniu zaparc, otylosci, cukrzycy i chorob ukladu ser
Choroby układu sercowo naczyniowego 1
Choroby układu sercowo naczyniowego
Fizjoterapia w chorobach układu sercowo naczyniowego I Demczyszak
choroby układu sercowo - naczyniowego, farmacja, układ krążenia
flawonoidy w zwalczaniu chorób układu sercowo naczynioweg
Medyczne czynności ratunkowe w chorobach układu sercowo naczyniowego
Choroby ukladu sercowo naczyniowego(3)
CHOROBY UKŁADU SERCOWO NACZYNIOWEGO W CIĄŻY
FARMAKOTERAPIA CHORÓB UKŁADU SERCOWO NACZYNIOWEGO
Choroby układu sercowo naczyniowego
Badania biochemiczne w chorobach układu sercowo naczyniowego
Fizjoterapia w chorobach układu sercowo naczyniowego I Demczyszak
CHOROBY UKŁADU SERCOWO NACZYNIOWEGO 2
W13 Patofizjologia układu sercowo naczyniowego
więcej podobnych podstron