
Kinetyka
Kinetyka
chemiczna
chemiczna

Kinetyka
chemiczna
zajmuje
się
badaniem przebiegu reakcji chemicznych
w czasie, pomiarem ich szybkości,
czynnikami wpływającymi na szybkość
reakcji. Ostatecznym celem kinetyki
chemicznej jest ustalenie mechanizmu
reakcji na poziomie molekularnym.

Reakcje mogą być :
Homogeniczne
– wszystkie reagenty są
w tej samej fazie np. mieszanina gazowa
lub roztwór
Heterogeniczne
– reagenty występują
w różnych fazach
Z punktu widzenia termodynamicznego
wszystkie reakcje są odwracalne i
zachodzą
aż
do
osiągnięcia
stanu
równowagi.
Reakcje kinetycznie nieodwracalne
to
takie, w których stan równowagi jest
bardzo silnie przesunięty w prawo (w
stronę produktów), a szybkość reakcji
odwrotnej jest znikomo mała
.
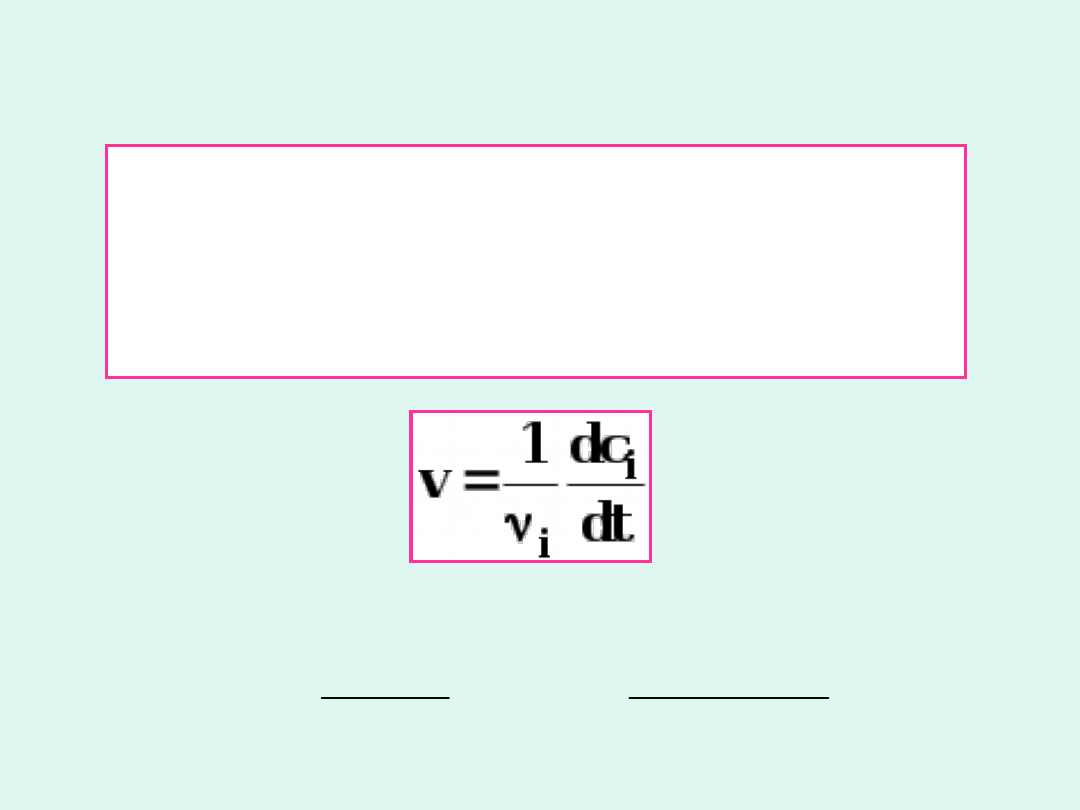
Szybkość reakcji
chemicznej
Szybkość reakcji chemicznej
to
ubytek stężenia molowego substratu
lub
przyrost
stężenia
molowego
produktu do czasu, w jakim nastąpiła
ta zmiana.
Jednostka szybkości reakcji to :
3
3
mol
mol
lub
dm s
dm min
�
�

Metody badania szybkości reakcji
chemicznych
Metody chemiczne
Metody fizykochemiczne
Ponadto istnieją specjalne metody badania
reakcji bardzo szybkich np. fotoliza
błyskowa czy technika „stopped flow”.
W
ustalaniu
mechanizmu
reakcji
chemicznych pomocne są różne techniki
identyfikacji produktów przejściowych.

Przykład
( )
( )
( )
katalizator
1
2
2
2
2 aq
l
2 g
H O
H O
O
�����
�
+
Szybkość tej reakcji można badać w
następujący sposób :
Z mieszaniny reakcyjnej pobierane są próbki, w
których stężenie H
2
O
2
oznaczane jest przez
miareczkowanie jodometryczne.
Z mieszaniny reakcyjnej pobierane są próbki, w
których stężenie H
2
O
2
oznaczane jest
kolorymetrycznie np. z odczynnikiem tytanowym.
Mierzona jest objętość tlenu tworzącego się w
reakcji.
Ponieważ H
2
O
2
silnie absorbuje światło w zakresie
UV, można w sposób ciągły mierzyć jego stężenie
metodą spektrofotometryczną.
Możliwy jest też pomiar elektrochemiczny stężenia
H
2
O
2
i tlenu przy użyciu odpowiednich elektrod
amperometrycznych.
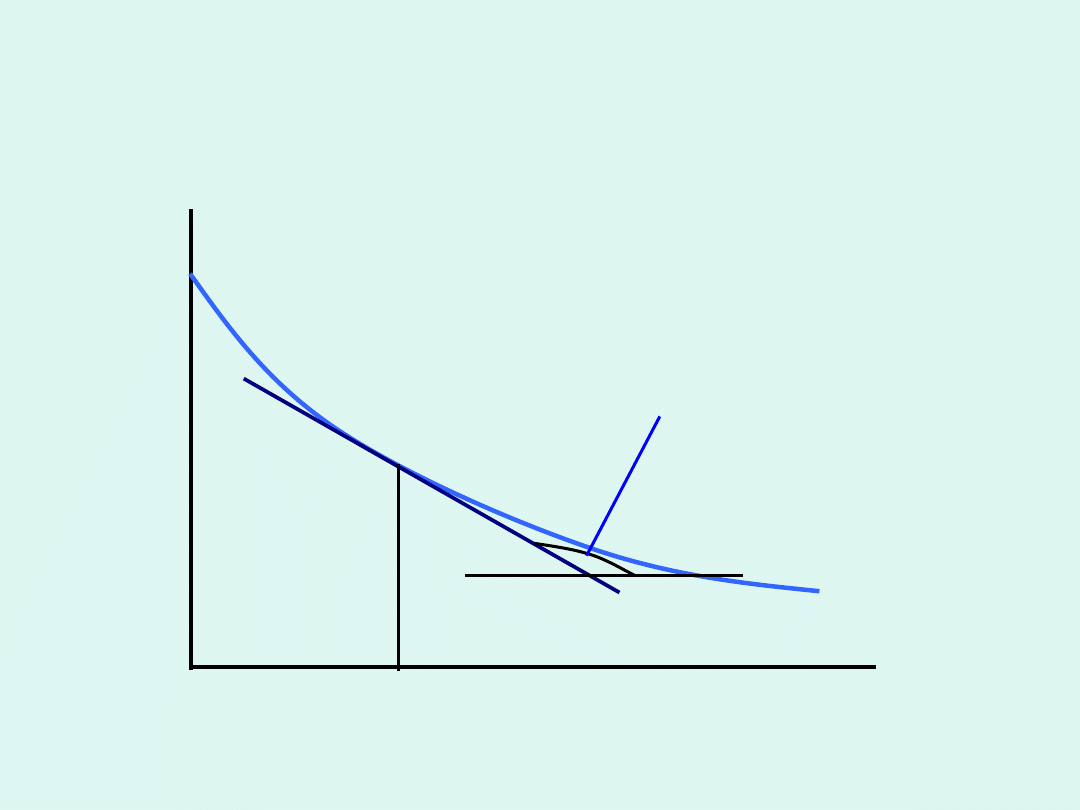
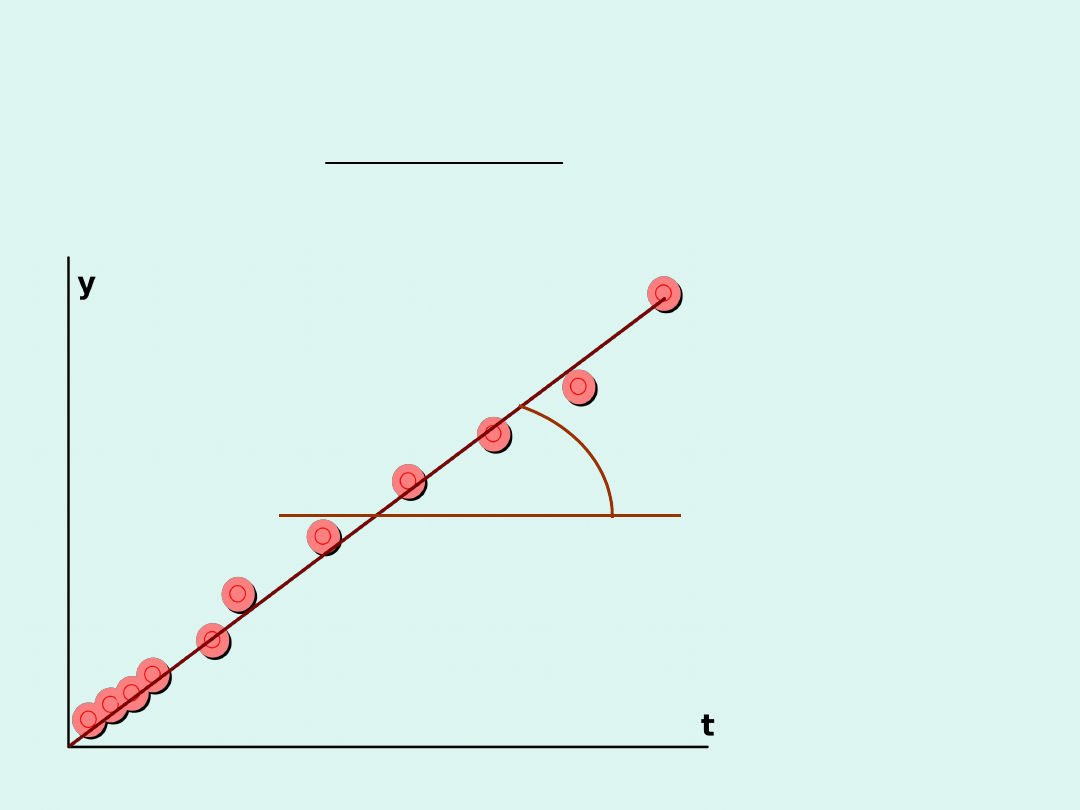
Szybkość reakcji chemicznej –
interpretacja graficzna
stężenie molowe
substratu
czas
t
s
s
v
tg
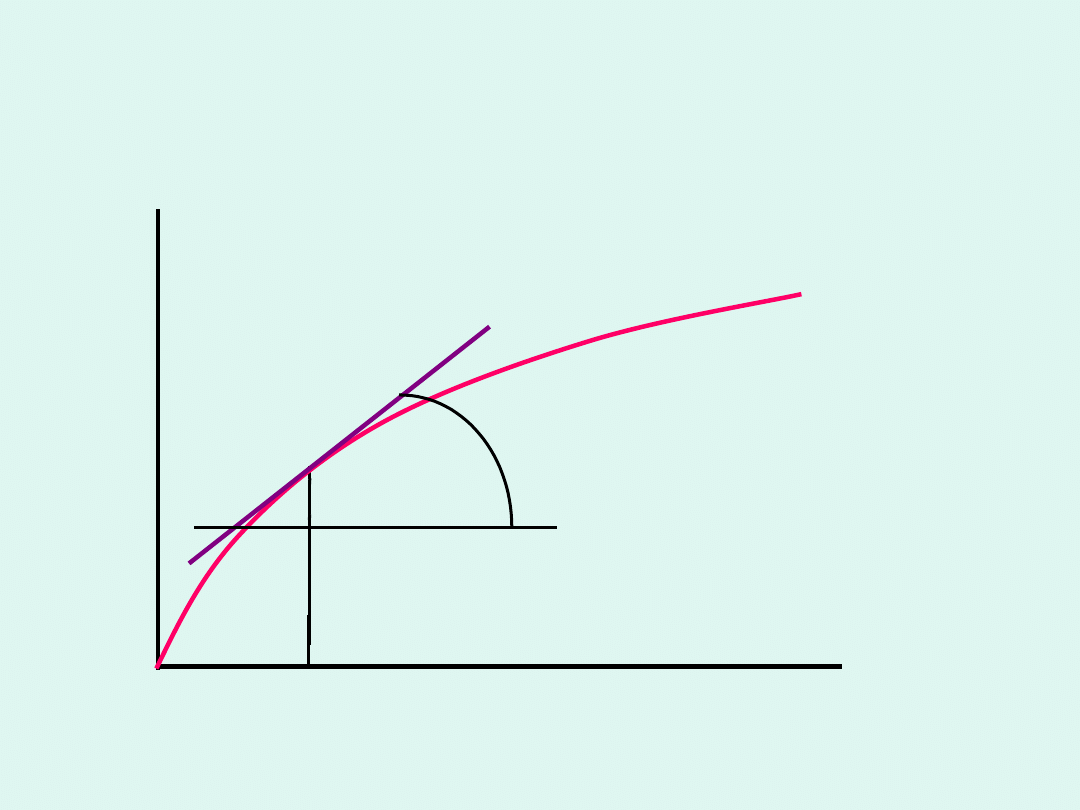
Szybkość reakcji chemicznej –
interpretacja graficzna
stężenie molowe
produktu
czas
t
p
p
v
tg

Czynniki wpływające na
szybkość reakcji
chemicznych
Stężenie substratów i
produktów
Temperatura
Rodzaj i stężenie katalizatora
Inne : rodzaj rozpuszczalnika,
siła jonowa, ciśnienie, światło,
kształt i rodzaj naczynia, w którym
przebiega reakcja i inne
Pierwsze trzy czynniki wpływają na szybkość każdej
reakcji chemicznej, natomiast czynniki z punktu
czwartego wpływają na szybkość tylko określonych
reakcji.
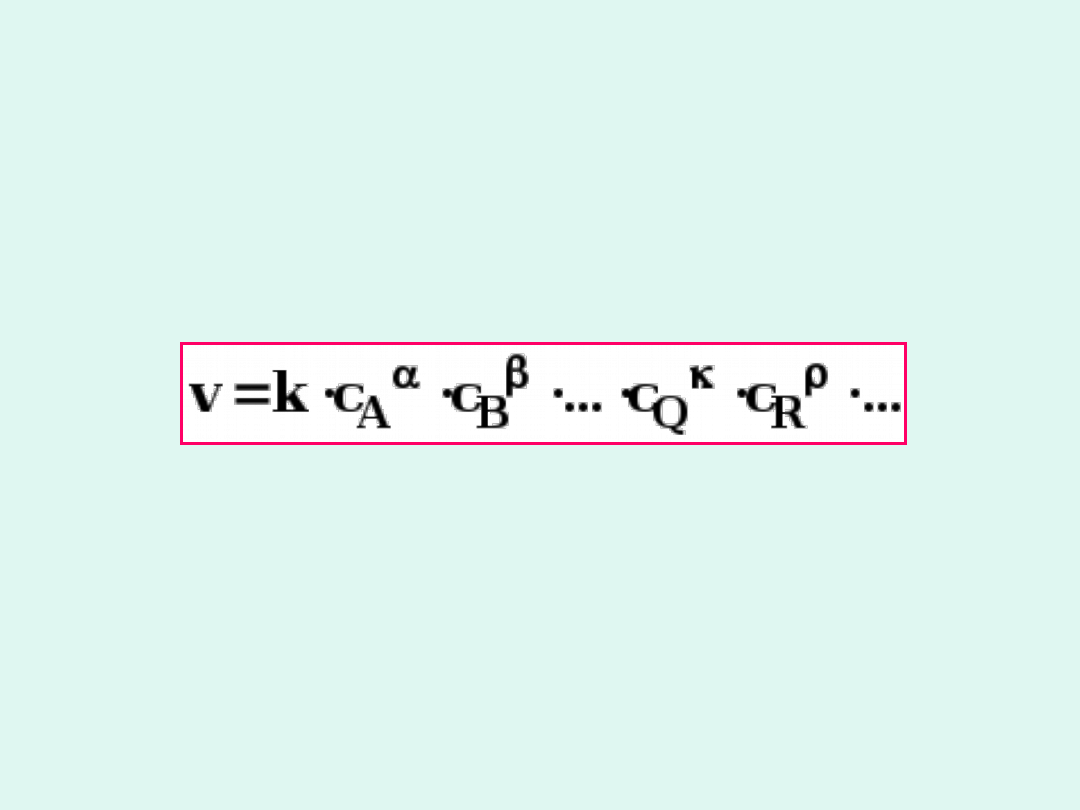
Wpływ stężenia reagentów
aA bB ....
qQ rR ...
+
+ �
+
+
W większości przypadków szybkość
reakcji zależy od stężeń reagentów w
następujący sposób :
Jest to
kinetyczne równanie reakcji
w
postaci różniczkowej.
k
– stała szybkości reakcji (ma stałą
wartość w określonych warunkach
prowadzenia reakcji)

- rząd reakcji względem
odpowiednio reagenta A, B, Q, R
(cząstkowe) (nie mają nic wspólnego ze
współczynnikami stechiometrycznymi a,
b, q, r)
Całkowity rząd reakcji chemicznej
to
suma wykładników potęgowych, w
jakich stoją stężenia reagentów w
kinetycznym równaniu reakcji.
r
...
...
=a +b+ +k+r +
Rząd reakcji może wyrażać się liczbami 0, 1, 2, 3. W
pewnych przypadkach mogą istnieć rzędy ułamkowe
np. ½. Istnieją reakcje, dla których nie można określić
rzędu, gdyż zależność szybkości reakcji od stężenia
reagentów jest bardziej złożona.

Cząsteczkowość
reakcji chemicznej to
liczba cząsteczek biorących udział w
elementarnym akcie reakcji, decydującym
o szybkości całej reakcji.
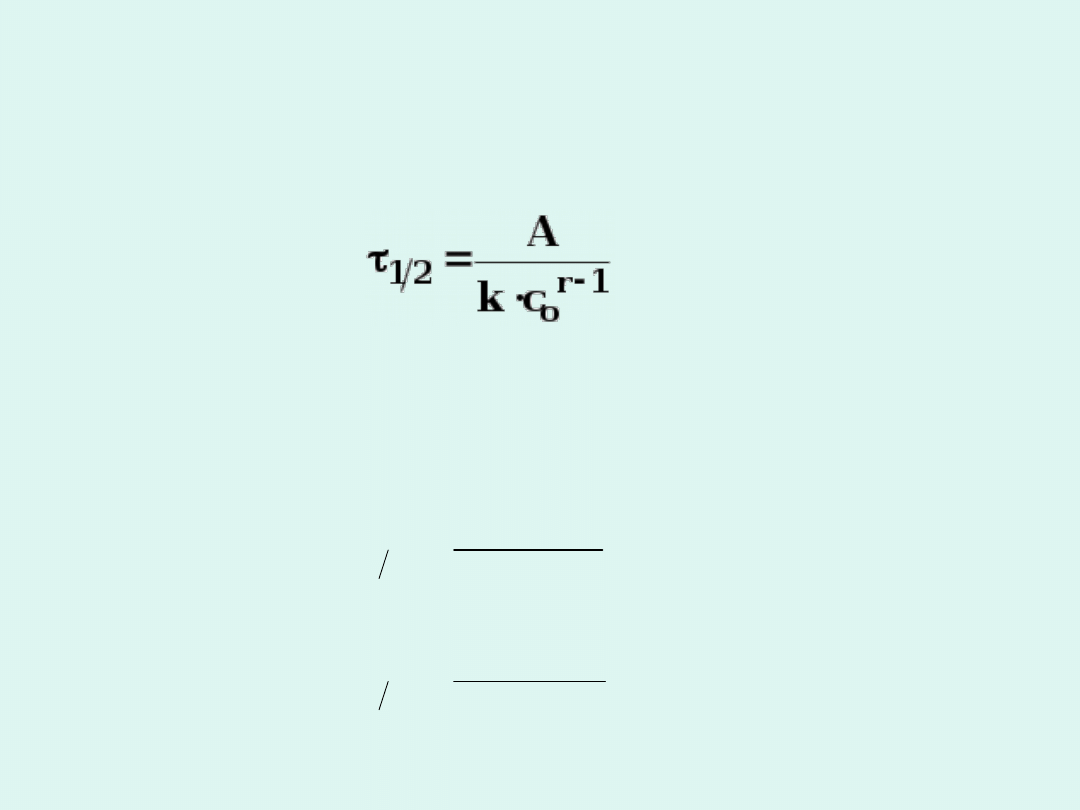
Czas połowicznej przemiany
jest to
czas, po którym stężenie substratu
maleje o połowę (osiąga połowę
stężenia początkowego).

Reakcja 0 rzędu
k
v
c
k
v
0
t
k
c
c
t
k
c
c
dt
k
dc
k
dt
dc
o
o
t
0
c
c
o
Szybkość reakcji zerowego rzędu jest stała.
Dla reakcji zerowego rzędu stężenie substratu maleje
liniowo w funkcji czasu.
Z reakcjami zerowego rzędu mamy do czynienia, gdy
szybkość reakcji zależy od jakiegoś innego czynnika, a
nie od stężenia reagentów np. od natężenia światła w
przypadku reakcji fotochemicznych.
k
2
c
k
c
o
2
1
2
1
2
c
o
o
min
dm
mol
k
s
dm
mol
k
t
c
c
k
3
3
o
Jednostka stałej szybkości :
Czas połowicznej przemiany :
Czas połowicznej przemiany w przypadku
reakcji zerowego rzędu jest wprost
proporcjonalny do stężenia początkowego
substratu.
c
t
tg =
-k
Reakcja 0 rzędu - wyznaczanie
stałej szybkości
t
k
c
c
o

Reakcja I rzędu
c
k
dt
dc
c
k
v
kt
o
o
o
t
0
c
c
e
c
c
t
k
c
c
t
k
c
c
dt
k
c
dc
o
ln
ln
t
k
c
c
o
ln
ln
W przypadku reakcji pierwszego rzędu logarytm
naturalny ze stężenia jest liniową funkcją czasu.
stężenie
substratu
stężenie
produktu
c
o
1/2
c
o
1/2
Reakcja I rzędu
(
)
k t
k t
A
Ao
P
Ao
A
S
c
c
e
c
c
1 e
- �
- �
�
=
�
=
-
min
1
k
s
1
k
t
k
c
c
o
ln
Jednostka stałej szybkości :
Czas połowicznej przemiany :
Czas połowicznej przemiany w przypadku
reakcji pierwszego rzędu nie zależy od
stężenia początkowego substratu.
k
2
k
2
c
c
2
1
2
1
o
o
ln
ln
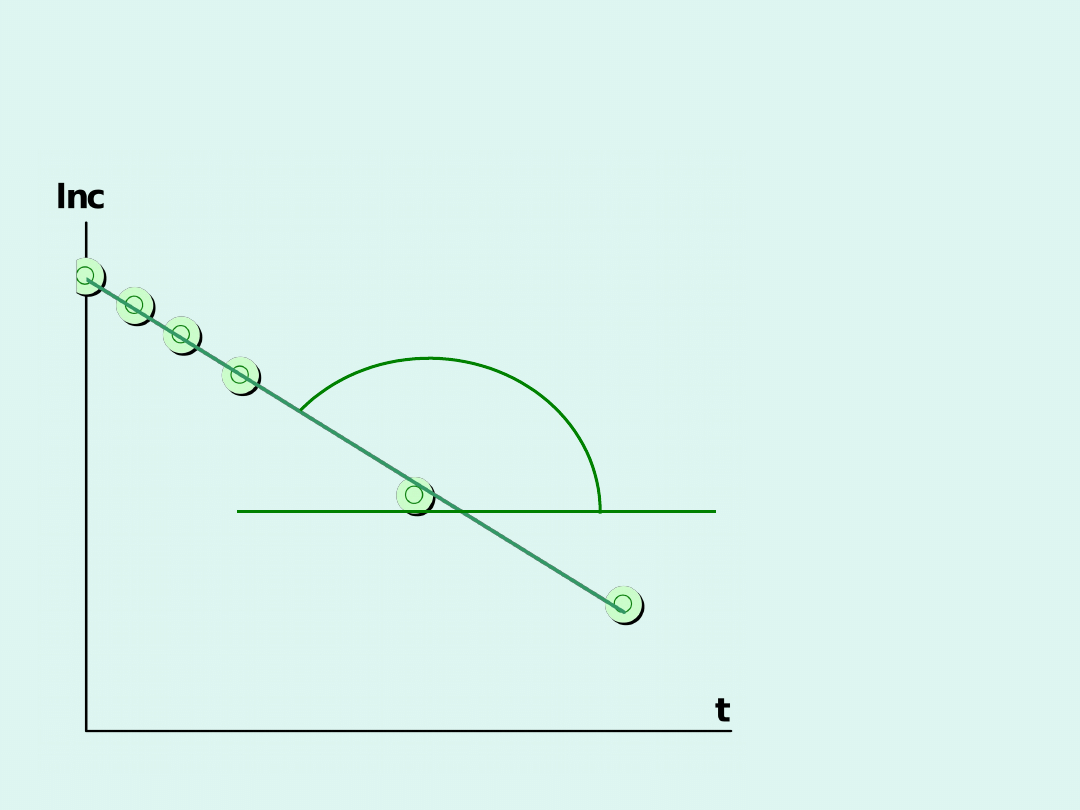
Wyznaczanie stałej szybkości dla
reakcji I rzędu
tg a = -k
t
k
c
c
o
ln
ln

Reakcje pierwszego rzędu są bardzo często
spotykane np. hydroliza estru w środowisku
kwaśnym.
Model kinetyki pierwszego rzędu stosuje się
bardzo często do opisu rozmaitych innych
zjawisk, na przykład :
rozpad izotopów promieniotwórczych
rozpad leku w organizmie po podaniu
śmiertelność mikroorganizmów w wyniku
działania temperatury (pasteryzacja, sterylizacja)
zanik substancji szkodliwych w środowisku

Reakcja II rzędu
2 A
P
A
1.
2.
B
P
�
+ �
2
dc
v
k c
dt
=-
= �
gdy
c
Ao
= c
Bo
gdy
c
Ao
≠ c
Bo
(
) (
)
Ao
Bo
dx
v
k c
x c
x
dt
=
=
-
-
o
o
2
c
t
2
c
0
c
c
o
o
dc
k c
dt
dc
k dt
c
1
k t
c
1 1
k t
c c
1
1
k t
c c
-
= �
-
=
� �
- -
= �
� �
� �
-
= �
=
+ �
�
�
W przypadku reakcji drugiego rzędu odwrotność
stężenia jest liniową funkcją czasu.
[ ]
[ ]
3
o
3
1 1 1
dm
k
k
t c c
s mol
dm
k
min mol
�
�
=
-
�
=
�
�
�
�
�
=
�
Jednostka stałej szybkości :
Czas połowicznej przemiany :
o
1 2
1 2
c
o
o
2
1
1
1
k
c
k c
-
= �
t
�
t
=
�
W przypadku reakcji drugiego rzędu czas
połowicznej przemiany jest odwrotnie
proporcjonalny do stężenia początkowego substratu.
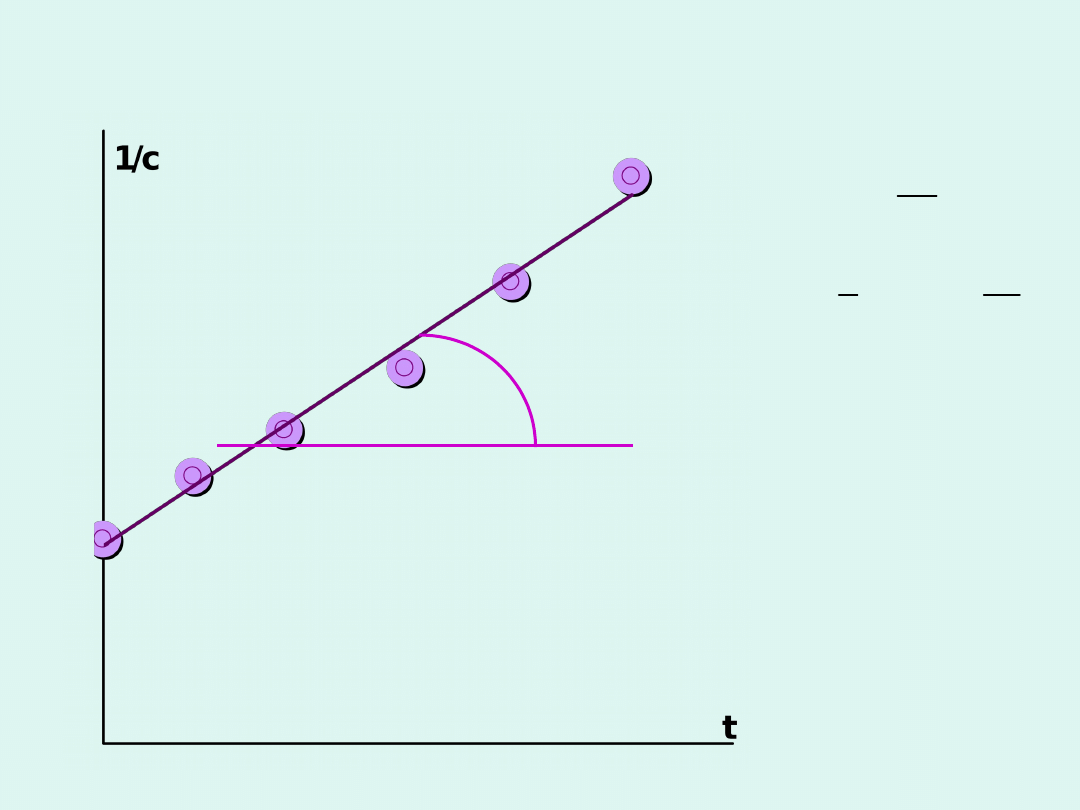
Wyznaczanie stałej szybkości
reakcji II rzędu
2
o
dc
v
k c
dt
1
1
k t
c
c
=-
= �
= �+
tg a = k
1/c
o

Bo
Ao
Bo
Ao
c
c
x
c
x
c
k
dt
dx
x
– ubytek stężenia substratu lub przyrost
stężenie produktu
Ao
A
Bo
B
A
Ao
B
Bo
x c
c
c
c
c
c
x c
c
x
=
-
=
-
=
-
=
-
t
0
x
0
Bo
Ao
Bo
Ao
dt
k
x
c
x
c
dx
kdt
x
c
x
c
dx
Aby wykonać powyższe całkowanie, musimy ułamek
pod całką po lewej stronie rozdzielić na ułamki
proste.

Bo
Ao
Bo
Ao
Bo
Ao
Ao
Bo
Ao
Bo
Bo
Ao
Ao
Bo
Bo
Ao
Bo
Ao
Ao
Bo
Bo
Ao
Bo
Ao
Bo
Ao
c
c
1
A
c
c
1
B
1
c
c
B
1
c
B
c
B
B
A
1
c
B
c
A
0
B
A
x
c
x
c
x
B
c
B
x
A
c
A
x
c
x
c
1
x
c
x
c
x
c
B
x
c
A
x
c
x
c
1
x
c
B
x
c
A
x
c
x
c
1
(
)
(
)
x
x
t
Ao
Bo
Ao
Bo
Ao
Bo
0
0
0
1
dx
1
dx
k dt
c
c
c
c
c
x
c
x
-
+
=
-
-
-
-
�
�
�
(
)
(
)
Ao
Bo
Ao
Bo
Ao
Ao
Bo
Bo
Ao
Bo
Ao
Bo
Ao
Bo
c
x
c
x
1
1
k t
c
c
c
c
c
c
c
x c
1
k t
c
c
c
c
x
ln
ln
ln
-
-
-
= �
-
-
-
�
= �
-
�
-
(
)
(
)
(
)
Ao
Bo
Ao
Bo
Ao
Bo
c
x c
c
c
k t
c
c
x
ln
-
�
=
-
��
�
-
W tym wypadku pojęcie czasu połowicznej przemiany
traci sens.
Wyznaczanie stałej szybkości
reakcji II rzędu
(
)
(
)
(
)
Ao
Bo
Ao
Bo
Ao
Bo
c
x c
y
c
c
k t
c
c
x
ln
-
�
=
=
-
��
�
-
0
tg = (c
Ao
- c
Bo
)·k

Reakcja III rzędu
Rozpatrzymy tylko jeden przypadek, gdy mamy
jeden substrat, lub kilka, ale ich stężenia
początkowe są jednakowe.
C
B
A
B
2
A
3
A
c
c
c
k
v
c
c
k
v
c
k
v
3
dc
k c
dt
-
= �
o
o
3
c
t
3
c
0
c
2
c
dc
k dt
c
dc
k dt
c
1 1
k t
2c
-
= �
-
=
�
�
- -
= �
�
�
�
�
�
�
t
k
c
1
2
1
c
1
2
1
2
o
2
t
k
2
c
1
c
1
2
o
2
Dla reakcji trzeciego rzędu odwrotność stężenia
podniesionego do kwadratu jest liniową funkcją
czasu.
[ ]
[ ]
6
2
2
2
o
6
2
1 1
1
dm
k
k
2t c
c
s mol
dm
k
min mol
�
�
�
�
=
-
�
=
�
�
�
�
�
=
�
Jednostka stałej szybkości :
Czas połowicznej przemiany :
o
1 2
1 2
2
2
2
c
o
o
2
1
1
3
2k
c
2k c
-
=
�
t
�
t
=
�
� �
� �
� �
W przypadku reakcji trzeciego rzędu czas
połowicznej przemiany jest odwrotnie
proporcjonalny do stężenia początkowego substratu
podniesionego do kwadratu.
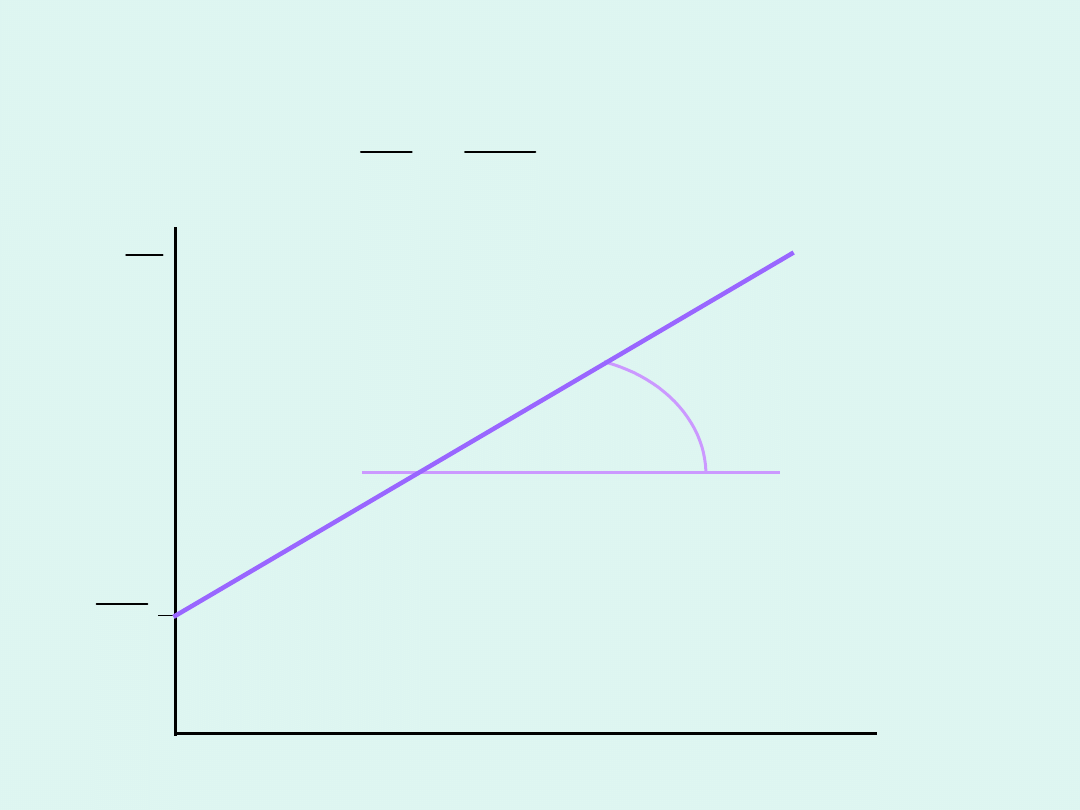
Wyznaczanie stałej szybkości
reakcji III rzędu
2
2
o
1
1
2 k t
c
c
=
+ ��
t
2
1
c
2
o
1
c
tg a = 2 k

Jednostka stałej szybkości reakcji zależy od rzędu
reakcji.

Metody wyznaczania rzędu
reakcji chemicznej
Metoda podstawienia do wzoru
Metoda czasów połowicznej
przemiany
Metoda różniczkowa (van’t
Hoffa)
Metoda izolacyjna Ostwalda –
pozwala na wyznaczenie
cząstkowych rzędów względem
poszczególnych reagentów

Wyznaczanie rzędu reakcji
metodą podstawiania do wzoru
Polega na podstawianiu do równań
scałkowanych danych doświadczalnych i
stwierdzeniu do którego z równań dane
pasują najlepiej. Można to robić w sposób
rachunkowy lub graficzny.
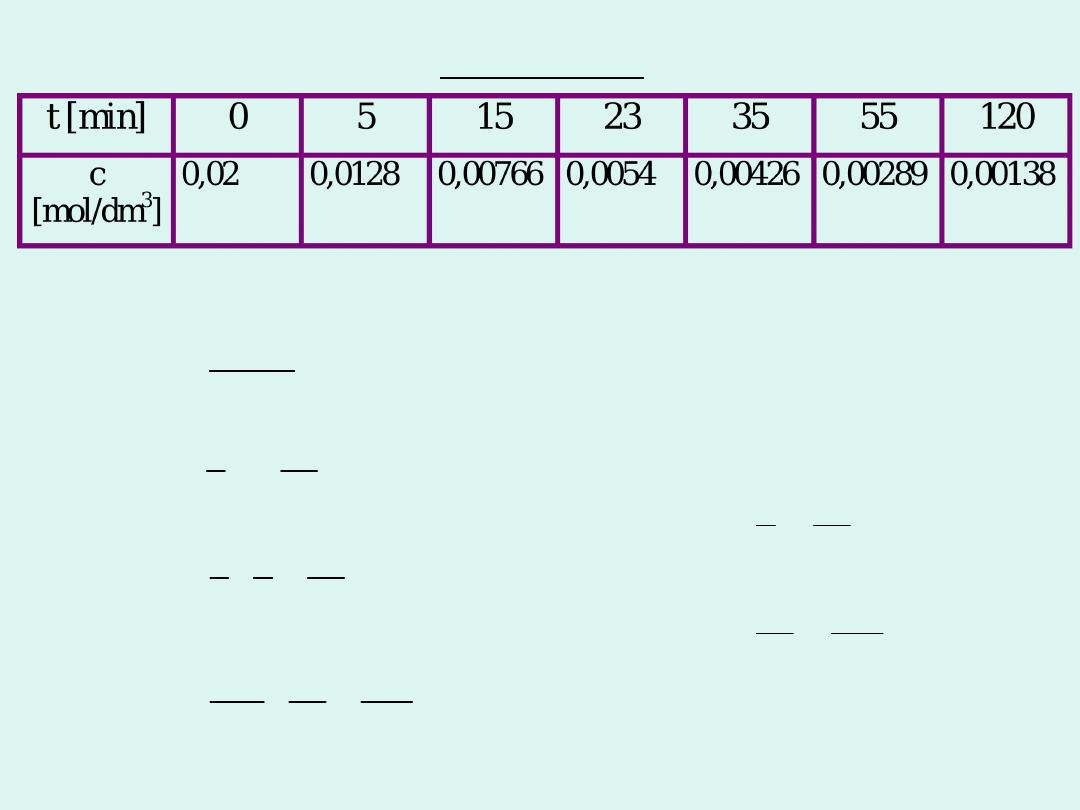
Przykład
o
o
o
2
2
o
0 c c
k t
1 lnc lnc
k t
1 1
2
k t
c c
1
1
3
2 k t
c
c
= - �
=
- �
=
+ �
=
+ ��
o
o
o
2
2
o
c
c
0 k
t
c
1
1 k
ln
t
c
1 1 1
2 k
t c c
1
1
1
3 k
2 t c
c
-
=
= �
�
�
=
-
�
�
�
�
�
�
=
-
�
�
�
�
��
�
sposób rachunkowy
sposób graficzny
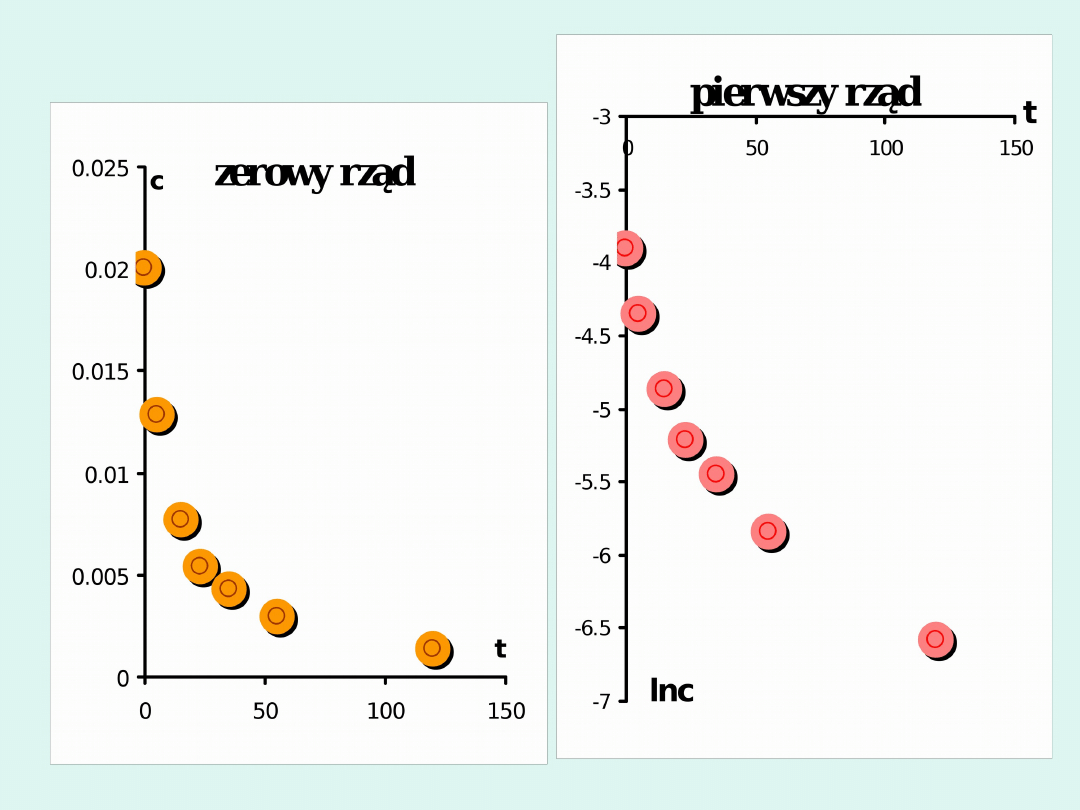
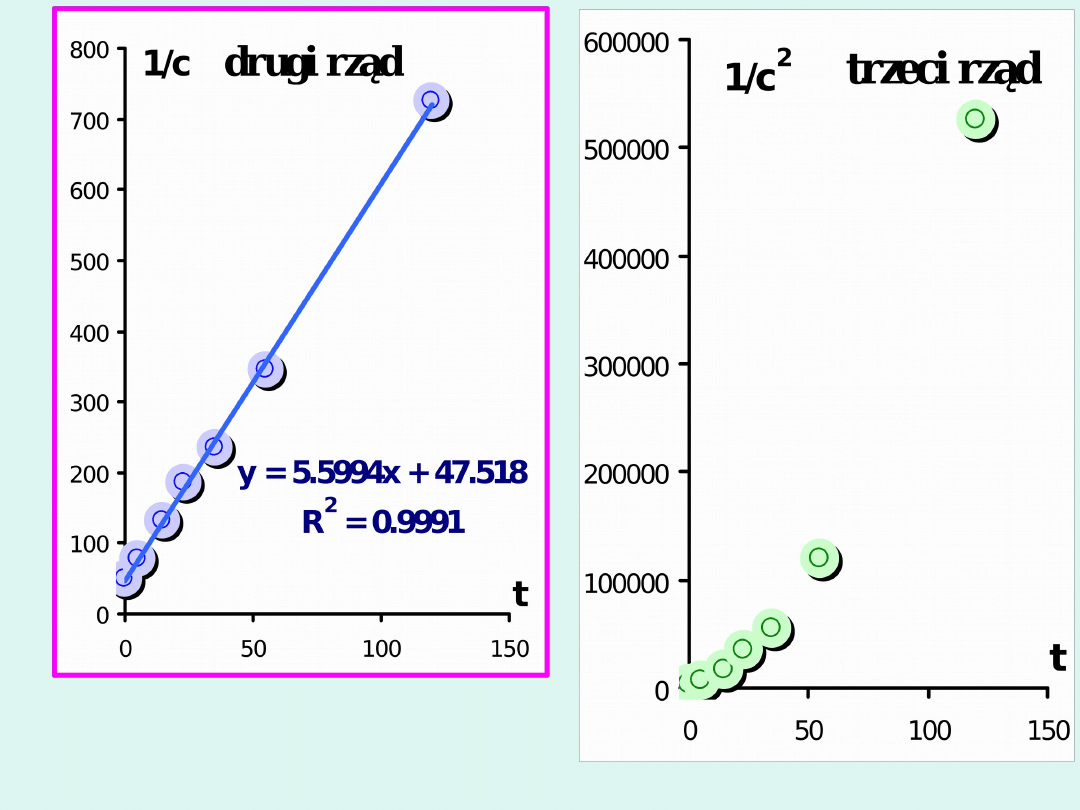
Reakcja jest II rzędu.
Wyznaczanie rzędu reakcji
metodą różniczkową
r
c
k
v
r
1
1
r
2
2
v
k c
v
k c
= �
= �
Dzielimy oba równania stronami i otrzymujemy :
r
r
1
1
1
r
2
2
2
v
k c
c
v
c
k c
�
� �
=
=� �
� �
�
( )
1
1
1
2
2
2
1
2
1
2
r
v
c
c
v
c
c
v
v
c
c
ln
ln
rln
ln
r
ln
=
=
=
Po zlogarytmowaniu stronami otrzymujemy :
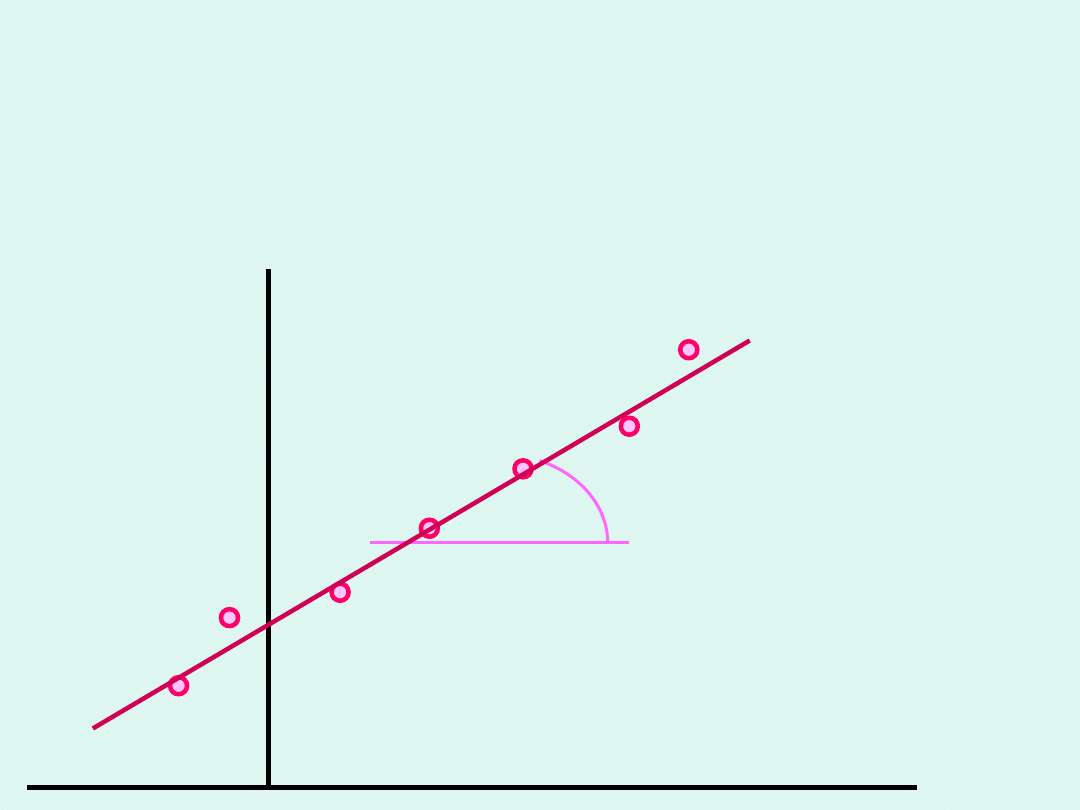
Wyznaczanie rzędu reakcji metodą
różniczkową
r
c
k
v
v
k r
c
ln
ln
ln
=
+ �
ln
v
ln c
tg = r
Po
zlogarytmowaniu
stronami :
Wyznaczanie rzędu reakcji
chemicznej na podstawie
czasów połowicznej przemiany
Wyznaczamy dwa czasy połowicznej przemiany dla
dwu różnych stężeń początkowych substratu w tej
samej temperaturze.
1
r
2
o
2
1
1
r
1
o
2
1
c
k
A
c
k
A
''
'
Dzielimy oba wyrażenia stronami :
1
r
1
o
2
o
1
r
1
o
1
r
2
o
c
k
A
c
k
A
2
1
2
1
c
c
c
c
1
r
2
o
1
r
1
o
''
'
Po zlogarytmowaniu stronami otrzymujemy :
''
'
''
'
ln
ln
''
'
ln
ln
ln
2
1
2
1
2
1
2
1
1
r
c
c
1
r
c
c
1
o
2
o
1
r
1
o
2
o
2
1
2
1
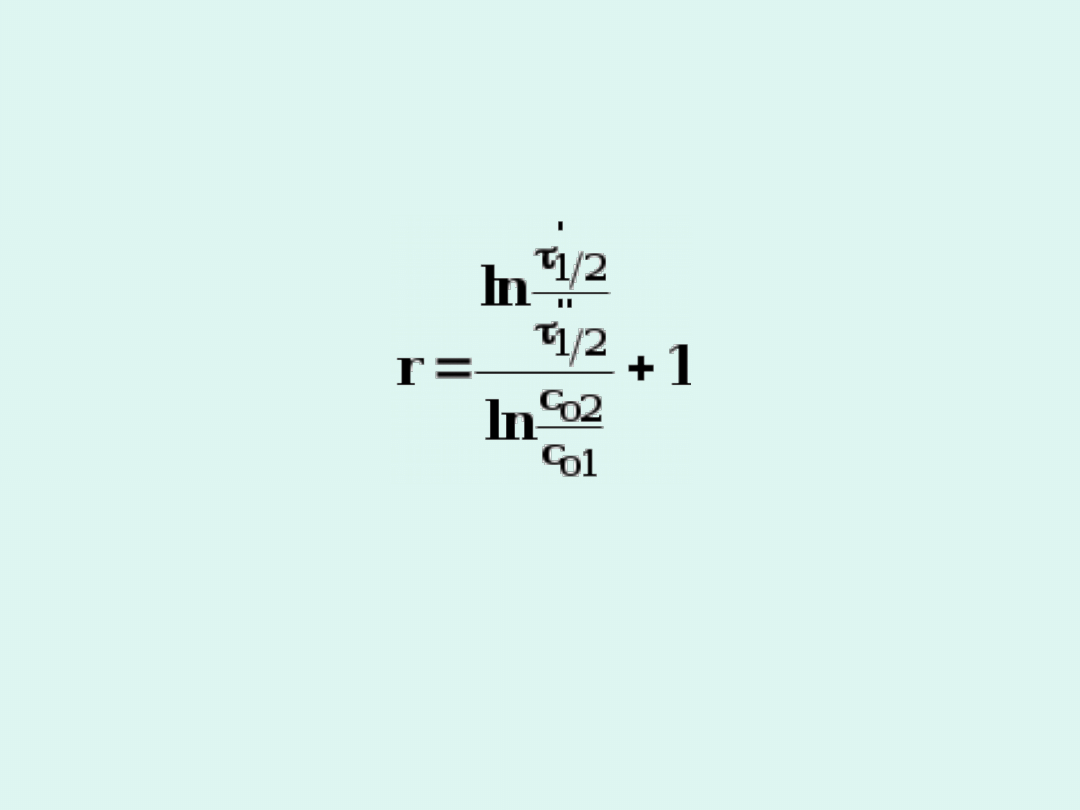
Ostatecznie rząd reakcji chemicznej obliczamy z
równania :
Wyznaczanie rzędu reakcji
chemicznej metodą izolacyjną
Ostwalda
A
B
v k c
c
a
b
= � �
Metoda
ta
pozwala
na
wyznaczenie
indywidualnych
rzędów
reakcji
dla
poszczególnych substratów.
Ustalamy warunki
reakcji tak, aby
c
Ao
>> c
Bo
.
Ustalamy warunki
reakcji tak, aby
c
Ao
<< c
Bo
.
Ao
'
B
'
Ao
A
A
c
k
k
c
k
v
c
c
const
c
Bo
''
A
''
Bo
B
B
c
k
k
c
k
v
c
c
const
c
r =
Rzędy
i
po „izolacji” wyznaczamy którąś z
poprzednio poznanych metod.
1.
Reakcje równoległe
2.
Reakcje odwracalne
3.
Reakcje następcze
Reakcje złożone
A
P
1
P
2
A
P
A
B
C
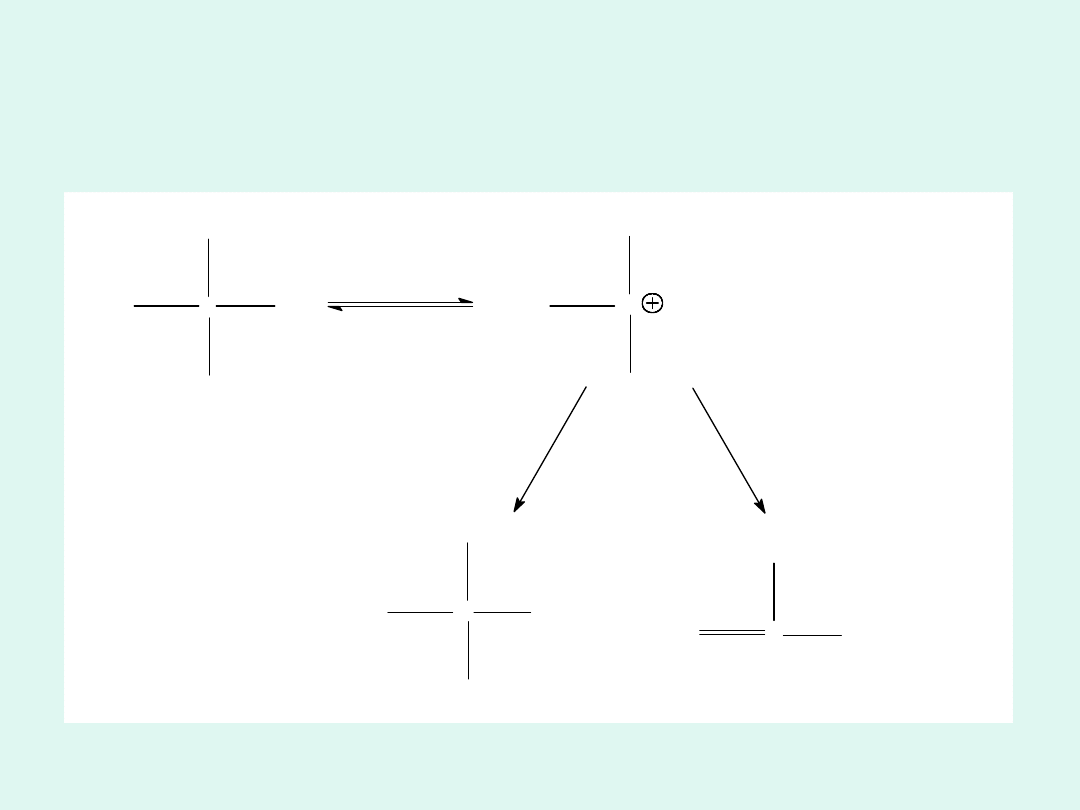
Rzeczywiste reakcje mogą być złożeniem
kilku różnych typów reakcji.
C
CH
3
CH
3
CH
3
Br
+ Br
-
C
CH
3
CH
3
CH
3
+ OH
-
+ OH
-
C
CH
3
CH
3
CH
3
OH
C
CH
3
CH
3
CH
2
+ H
2
O
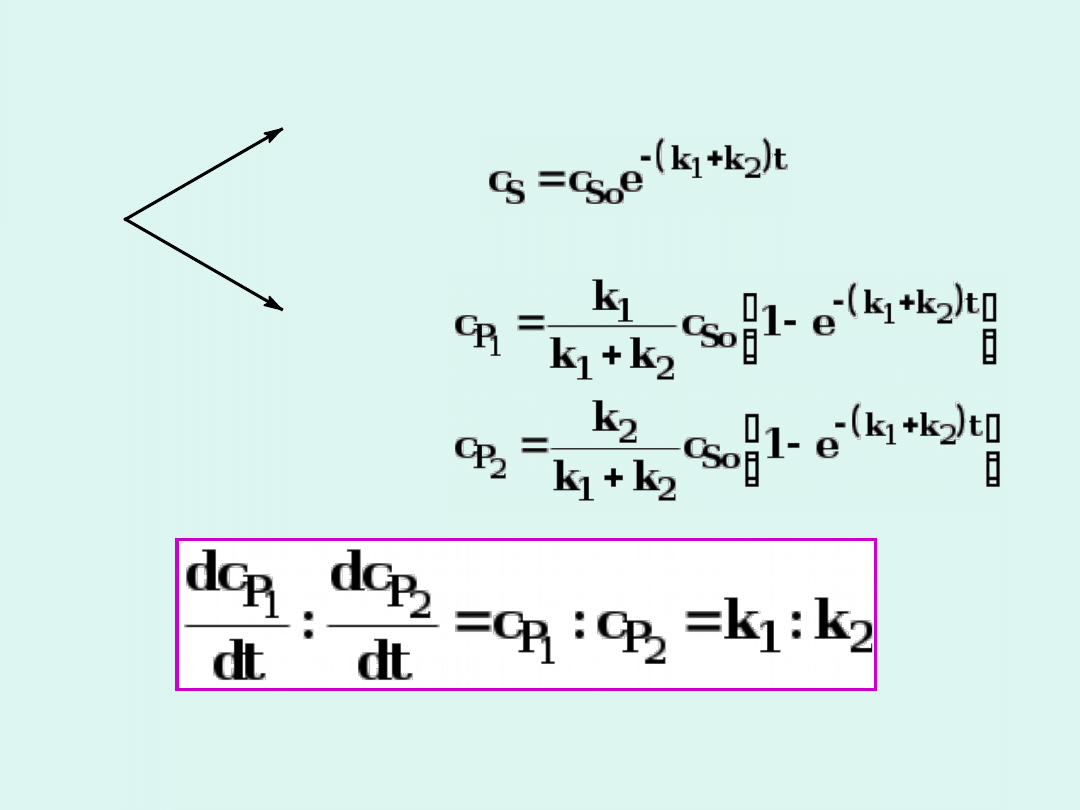
Reakcje równoległe
S
P
1
P
2
k
1
k
2
Stosunek stężeń produktów i szybkości ich tworzenia
mają się do siebie jak ich stałe szybkości.
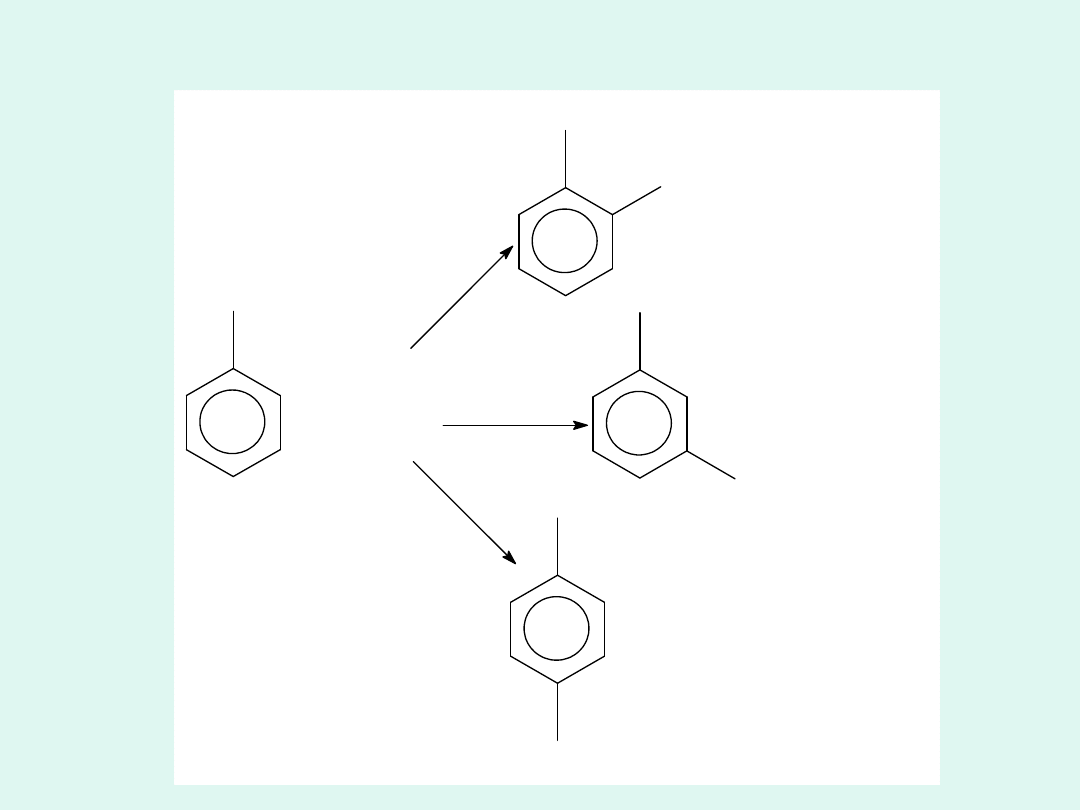
Reakcje równoległe - przykład
CH
3
+ HNO
3
CH
3
NO
2
CH
3
NO
2
CH
3
NO
2
+ H
2
O
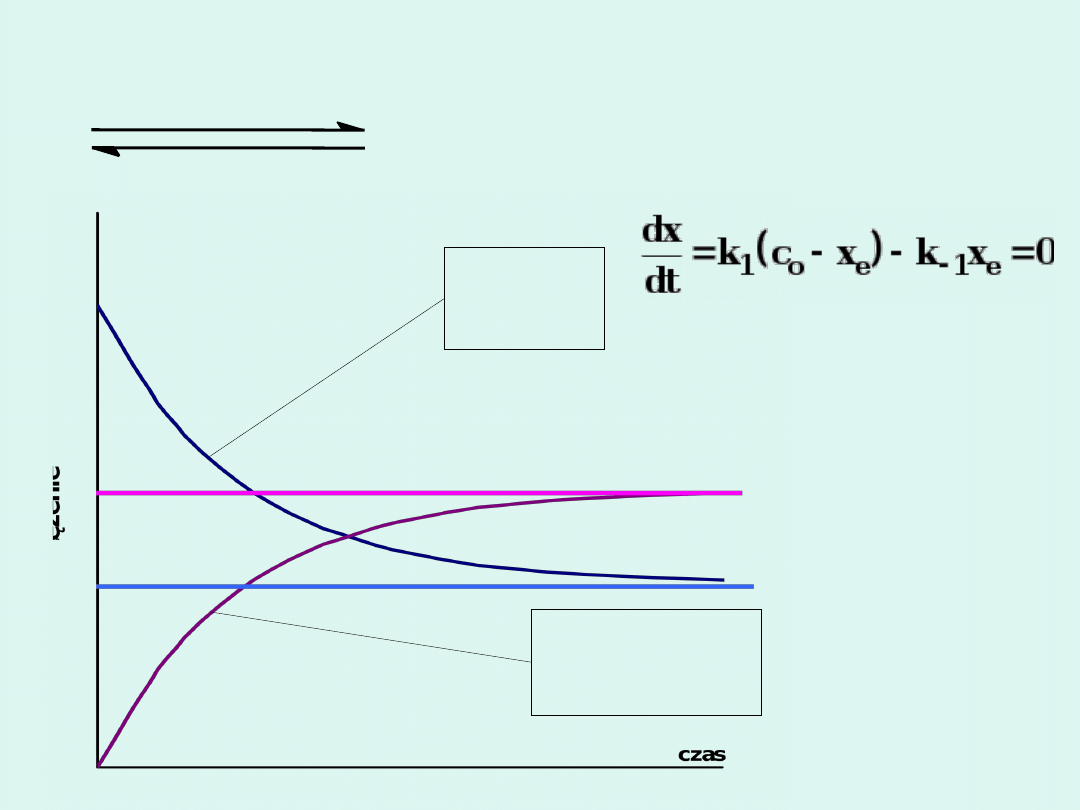
Reakcje odwracalne
A
P
k
1
k
-1
Zakładamy, że w obie strony
reakcja jest pierwszego
rzędu
.
stężeni
e
substra
tu
stężenie
produktu
c
o
- x
e
x
e
W stanie równowagi :
Wniosek ten jest słuszny dla reakcji
dowolnego rzędu.
1
1
c
k
k
K
Przykład reakcji odwracalnej
-
D
-
g
l
u
k
o
z
a
-
D
-
g
l
u
k
o
z
a
k
1
k
-
1
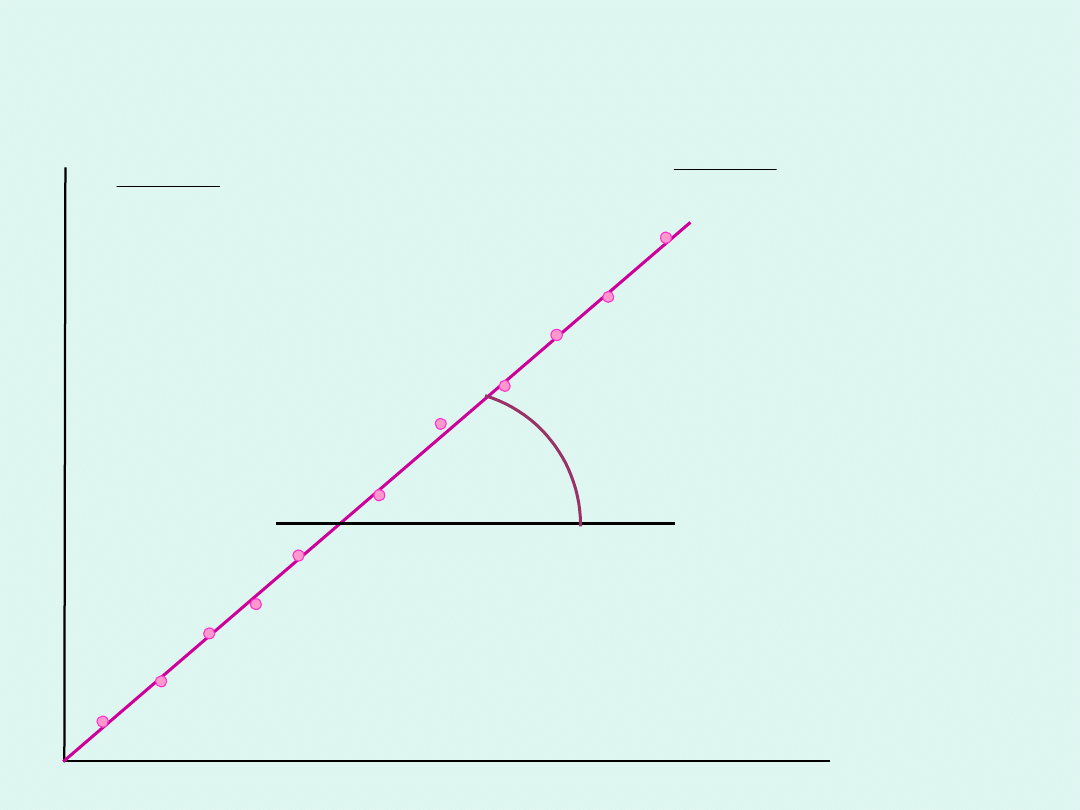
Scałkowane równanie kinetyczne dla
odwracalnej reakcji I rzędu
czas
t
k
k
x
x
x
ln
1
1
e
e
tg = k
1
+ k
-1
x
x
x
ln
e
e
Reakcje następcze
A
B
C
k
1
k
2
Zakładamy, że obie reakcje są pierwszego
rzędu.
Jeśli stałe szybkości obu etapów są różne,
rozwiązanie układu równań ma postać :
1
2
t
k
1
t
k
2
Ao
C
B
A
Ao
C
t
k
t
k
1
2
1
Ao
B
t
k
Ao
A
2
1
k
k
e
k
e
k
1
c
c
c
c
c
c
e
e
k
k
k
c
c
e
c
c
k
k
gdy
2
1
2
1
1

Przykład reakcji następczej
R
COOR'
COOR''
+ H
2
O
COOH
COOR'
R
+ R''OH
R
COOR'
COOH
+ H
2
O
COOH
COOH
R
+ R'OH
k
1
k
2
H
3
O
+
H
3
O
+
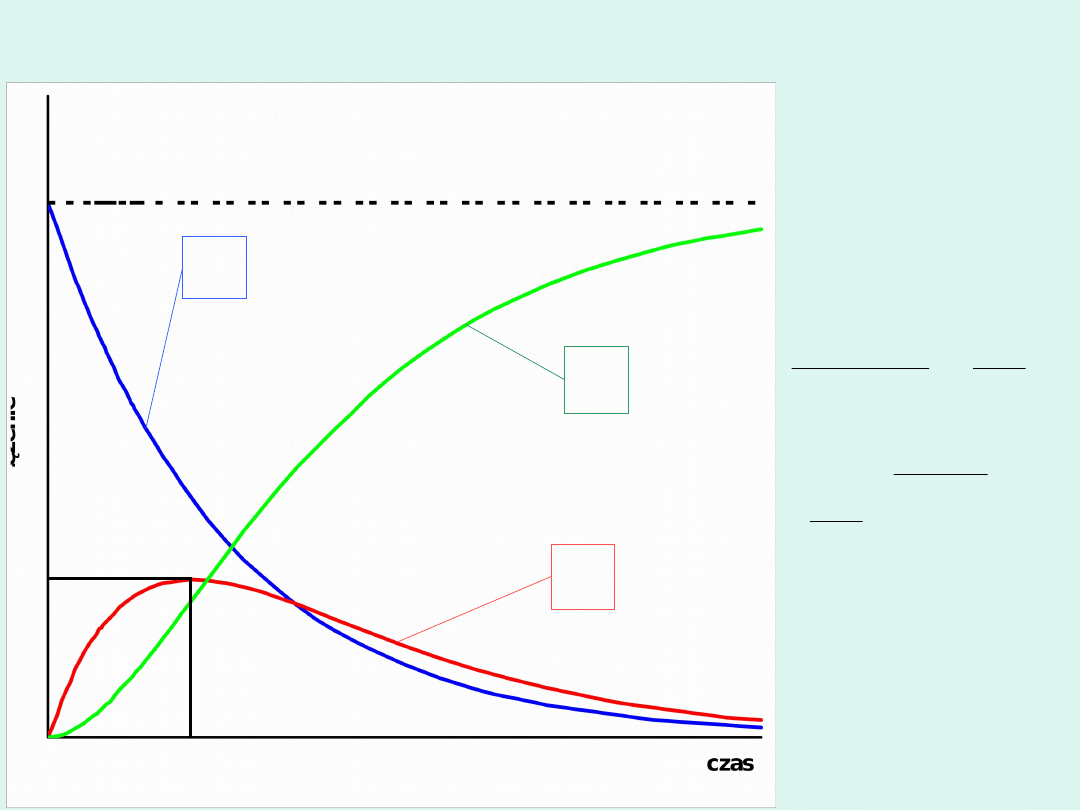
c
A
c
A
c
A
t
ma
x
c
B
max
c
Ao
Zmiany stężeń reagentów w typowej
reakcji następczej
Ao
k
k
k
2
1
max
B
1
2
1
2
max
c
k
k
c
k
k
ln
k
k
1
t
1
2
2

Przypadek graniczny w reakcji
następczej gdy :
k
1
>> k
2
t
k
t
k
2
1
e
e
1
k
1
t
t
k
Ao
C
t
k
Ao
B
2
2
e
1
c
c
e
c
c
Wówczas
i po czasie
równania sprowadzają się do
postaci
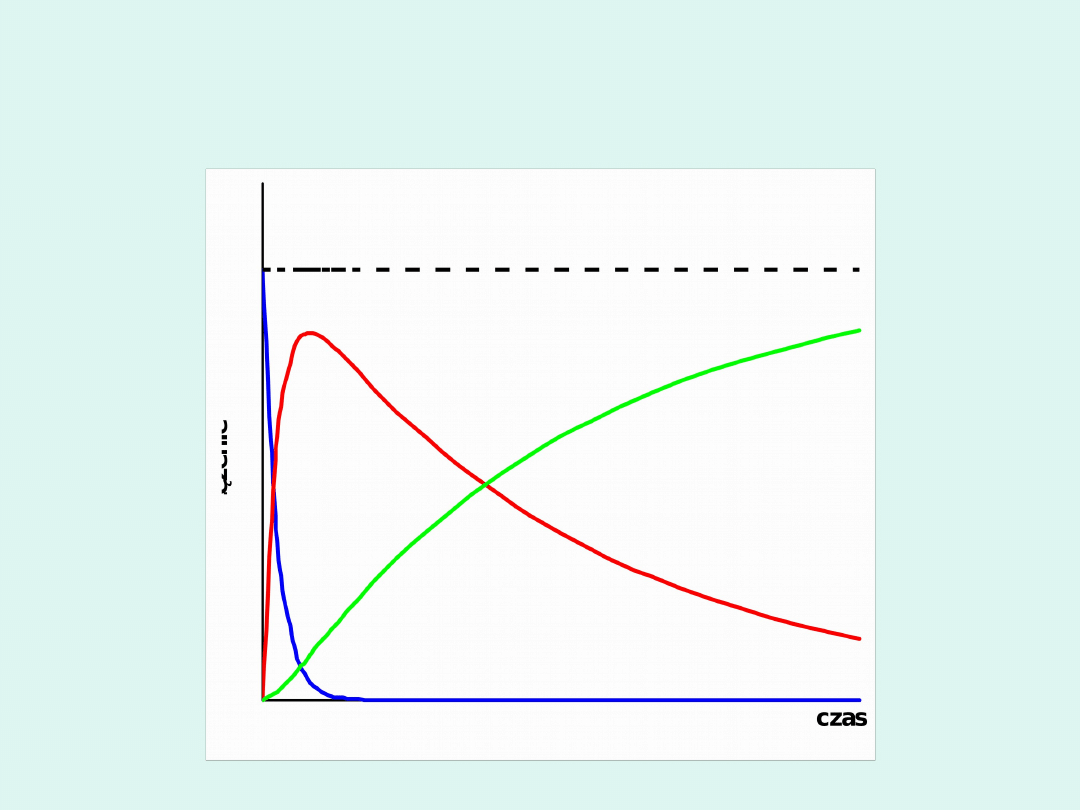
Przypadek graniczny w reakcji
następczej gdy :
k
1
>> k
2

Przypadek graniczny w reakcji
następczej gdy :
k
1
<< k
2
t
k
t
k
2
1
e
e
2
1
A
B
B
2
A
1
B
k
k
c
c
0
c
k
c
k
dt
dc
t
k
Ao
C
A
1
A
2
1
2
B
2
C
1
e
1
c
c
c
k
c
k
k
k
c
k
dt
dc
Wówczas
I po krótkim czasie zwanym
czasem
indukcji
równania przybierają postać :
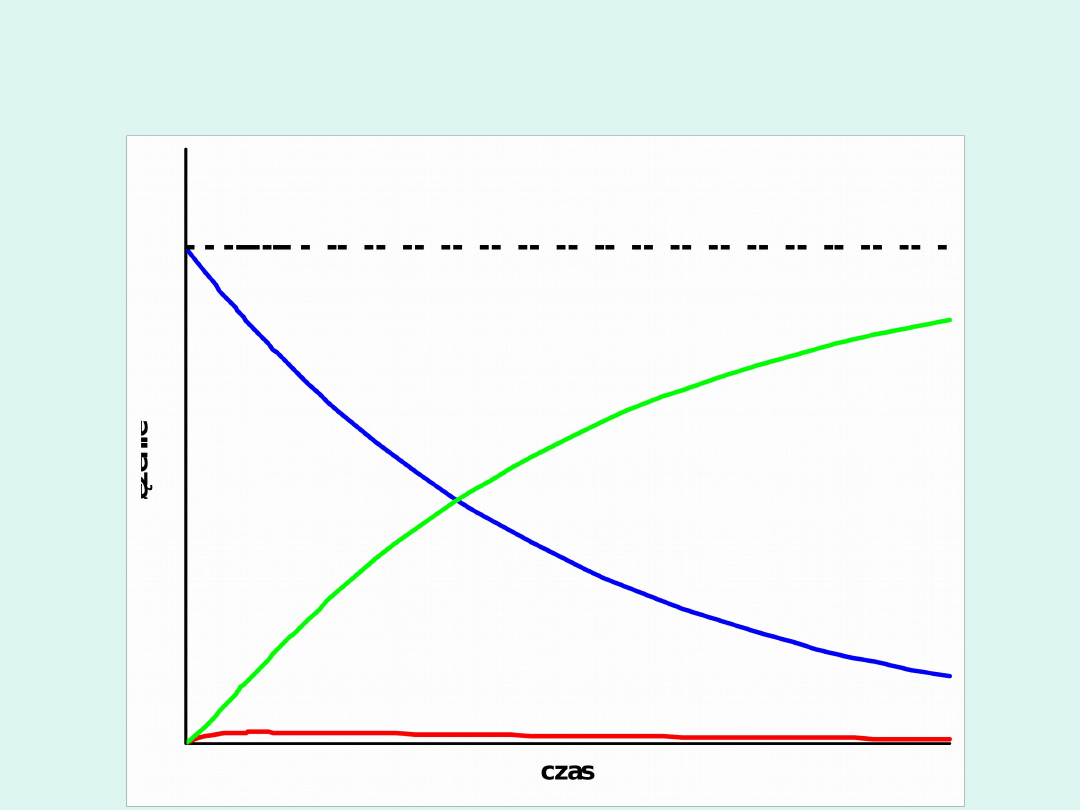
Przypadek graniczny w reakcji
następczej gdy :
k
1
« k
2
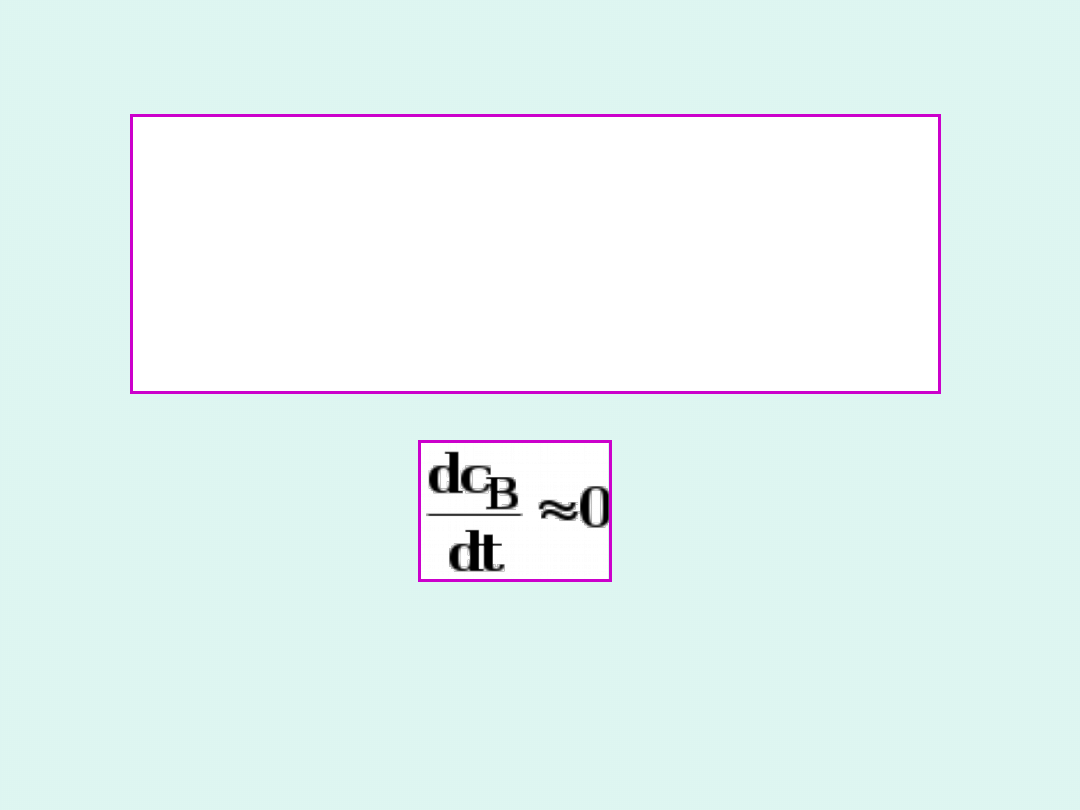
Przybliżenie stanu stacjonarnego
Bodensteina
W reakcji złożonej po pewnym
czasie indukcji stężenie substratu
pośredniego ustala się na niskim, w
przybliżeniu stałym poziomie, a
całkowita szybkość zmian jego
stężenia jest w przybliżeniu równa
zero.
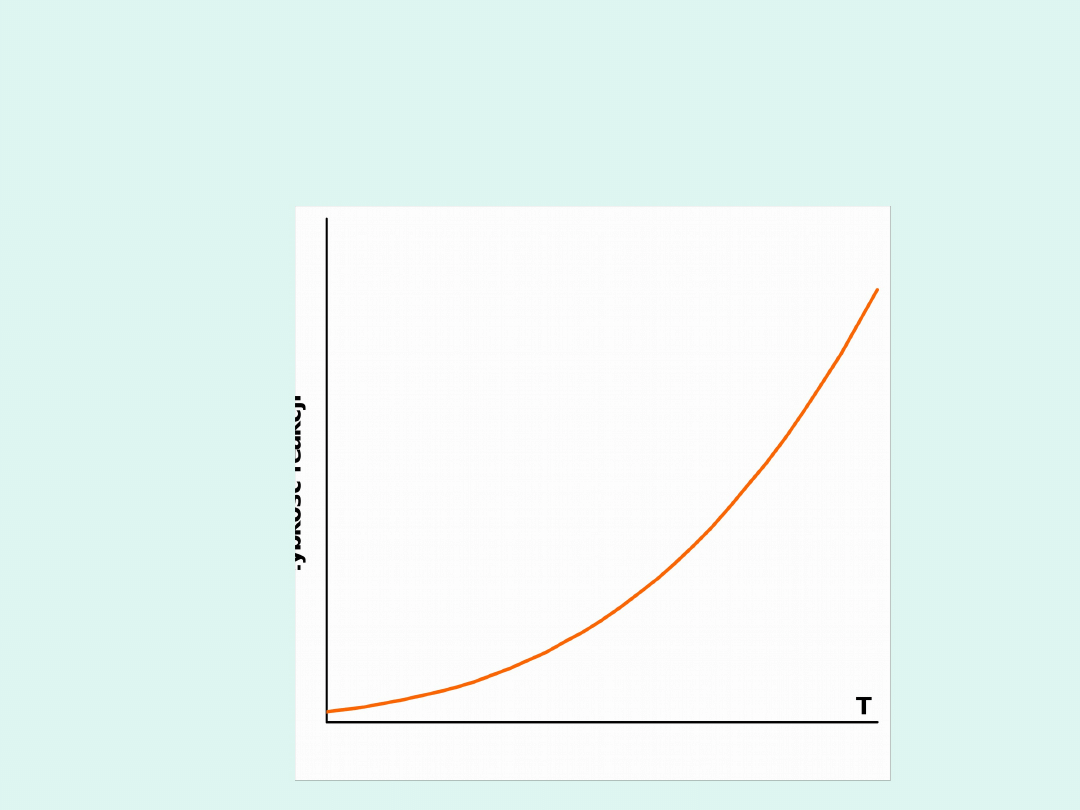
Wpływ temperatury na szybkość
reakcji chemicznych
Dla większości reakcji szybkość rośnie wraz
ze wzrostem temperatury.

Równanie Arrheniusa
Opisuje zależność szybkości reakcji od
temperatury.
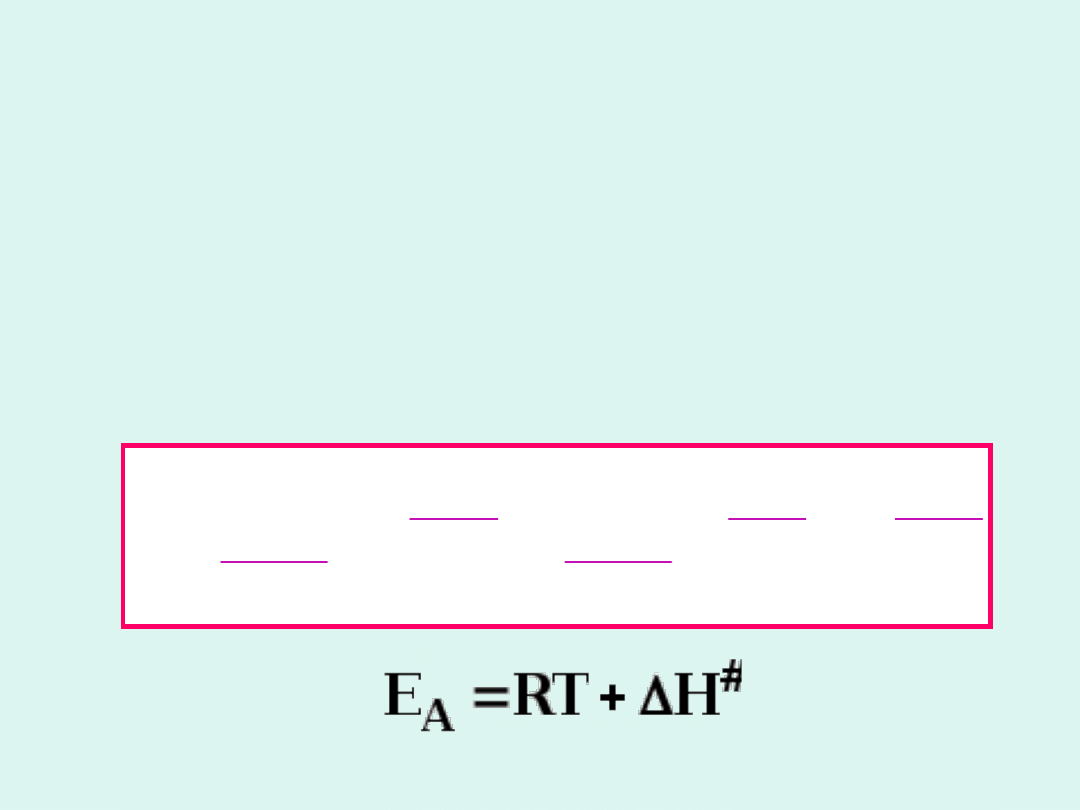
Energia aktywacji
to najmniejsza ilość
energii, jaką muszą posiadać substraty,
aby ulec przekształceniu w produkty,
liczona na 1 mol elementarnych aktów
reakcji.
E
A
– energia aktywacji
2
A
RT
E
dT
k
d
ln

o
A
A
2
A
2
A
2
A
k
T
1
R
E
C
T
1
R
E
k
T
dT
R
E
k
d
T
dT
R
E
k
d
RT
E
dT
k
d
ln
ln
ln
ln
ln
RT
E
o
A
e
k
k
k
o
– czynnik przedwykładniczy (czynnik częstości)
Oznacza szybkość reakcji w nieskończenie dużej
temperaturze, gdy wszystkie cząsteczki mają
wystarczającą energię do pokonania bariery
energetycznej na drodze reakcji.
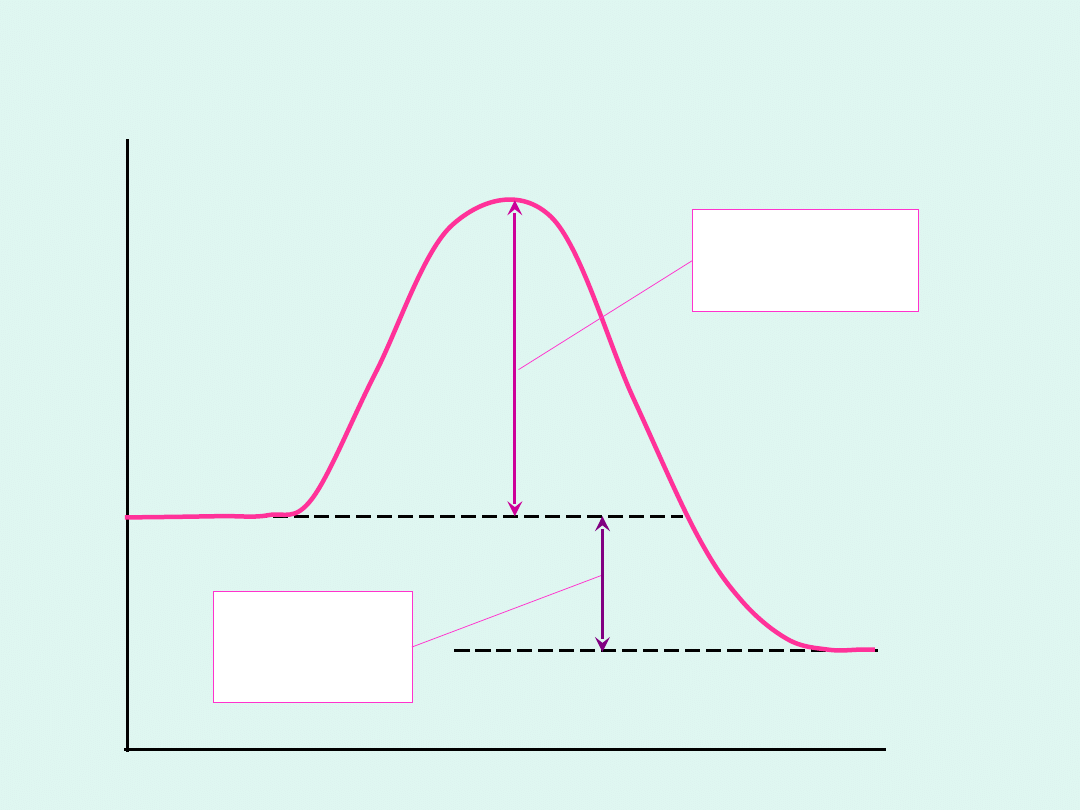
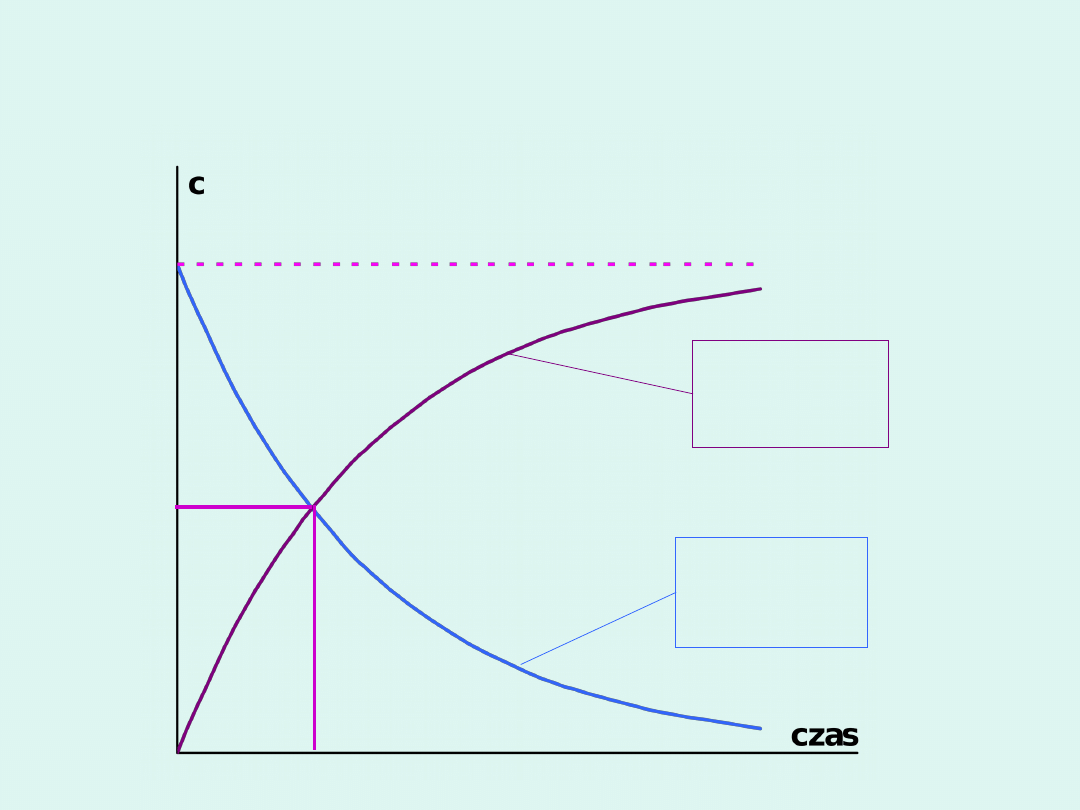
energia
aktywacji
E
A
ciepło
reakcji
Q
substr
aty
produk
ty
komlek
s
aktywn
y
droga
reakcji
energi
a
Energia aktywacji

Jak wyznaczamy energię
aktywacji ?
Trzeba wyznaczyć doświadczalnie stałe
szybkości w co najmniej dwu różnych
temperaturach. Następnie do obliczenia
energii aktywacji korzystamy z równania
Arrheniusa.
1
2
1
2
1
2
A
1
2
1
2
A
1
2
2
1
A
T
T
A
1
2
T
T
2
A
k
k
T
T
k
k
T
T
R
E
T
T
T
T
R
E
k
k
T
1
T
1
R
E
T
1
R
E
k
k
T
dT
R
E
k
d
2
1
2
1
2
1
ln
ln
ln
ln

W celu dokładniejszego wyznaczenia energii
aktywacji wyznaczamy stałe szybkości
reakcji w wielu różnych temperaturach, a
następnie wykreślamy zależność
lnk
w
funkcji
1/T
zgodnie z zależnością :
A
o
E
1
lnk lnk
R T
=
-
�
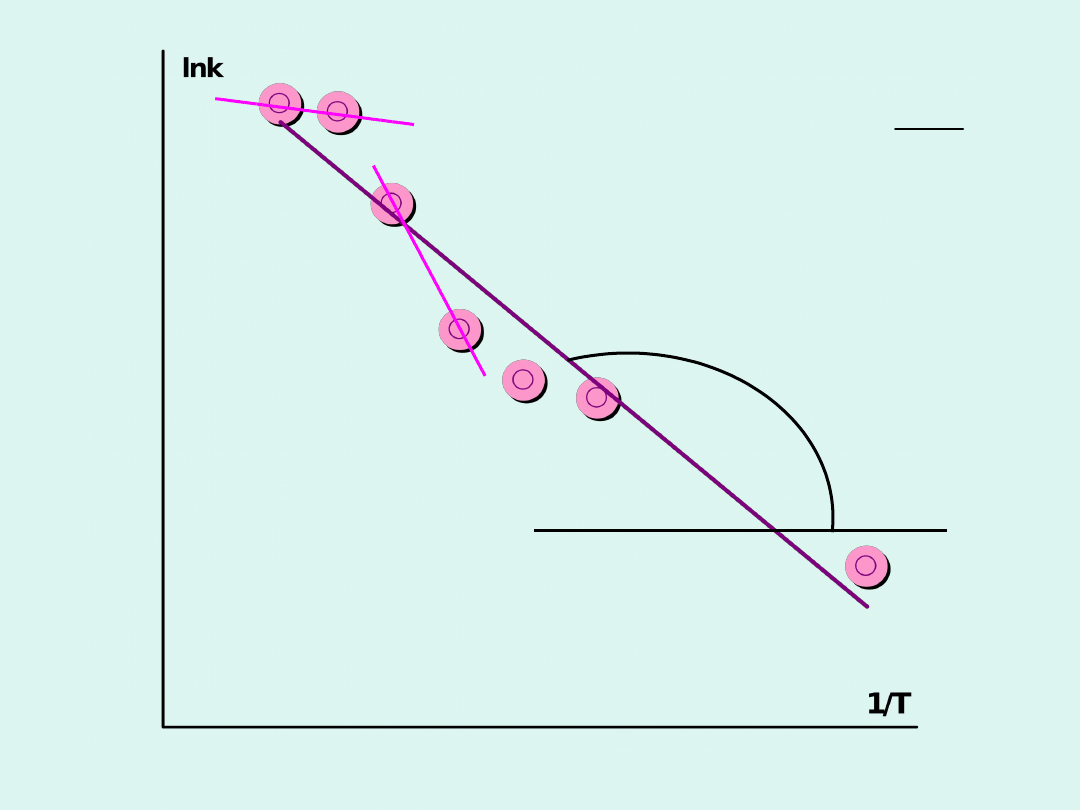
A
A
E
a tg
R
E
R tg
= a =-
=- �a

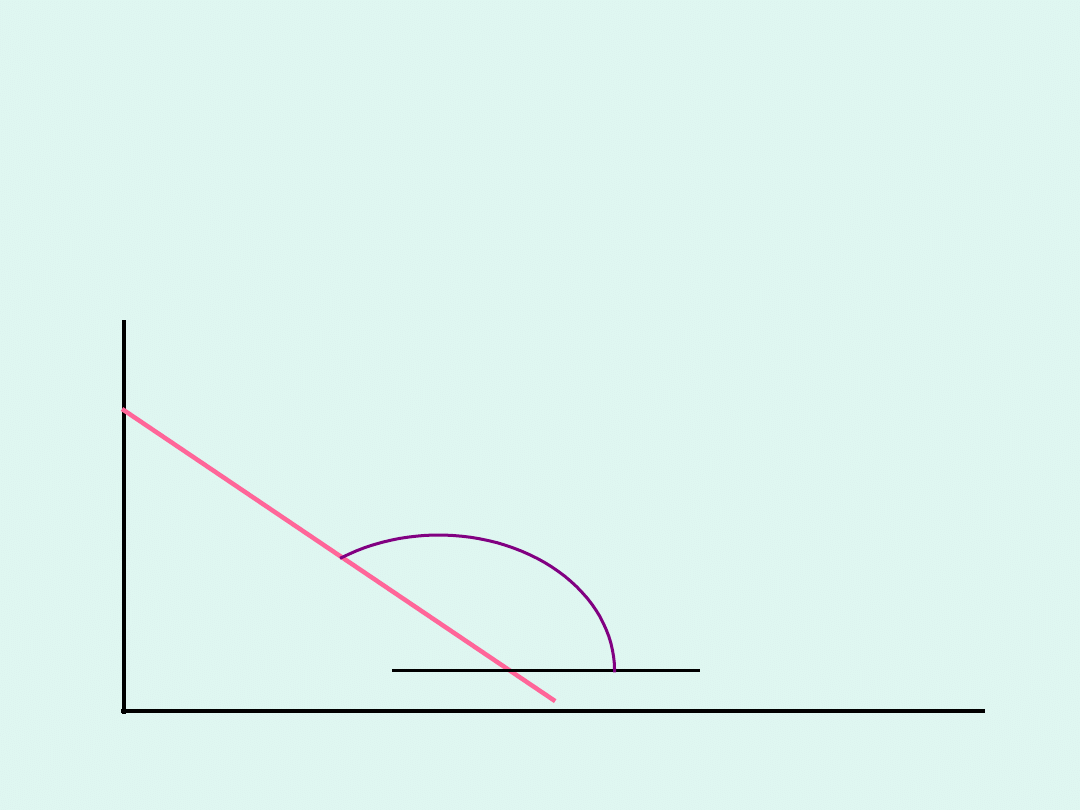
Teorie szybkości reakcji
chemicznych
Teoria zderzeń aktywnych (starsza)
Teoria kompleksu aktywnego zwana też
teorią absolutnych szybkości reakcji lub
teorią Eyringa-Polanyi’ego (nowsza)
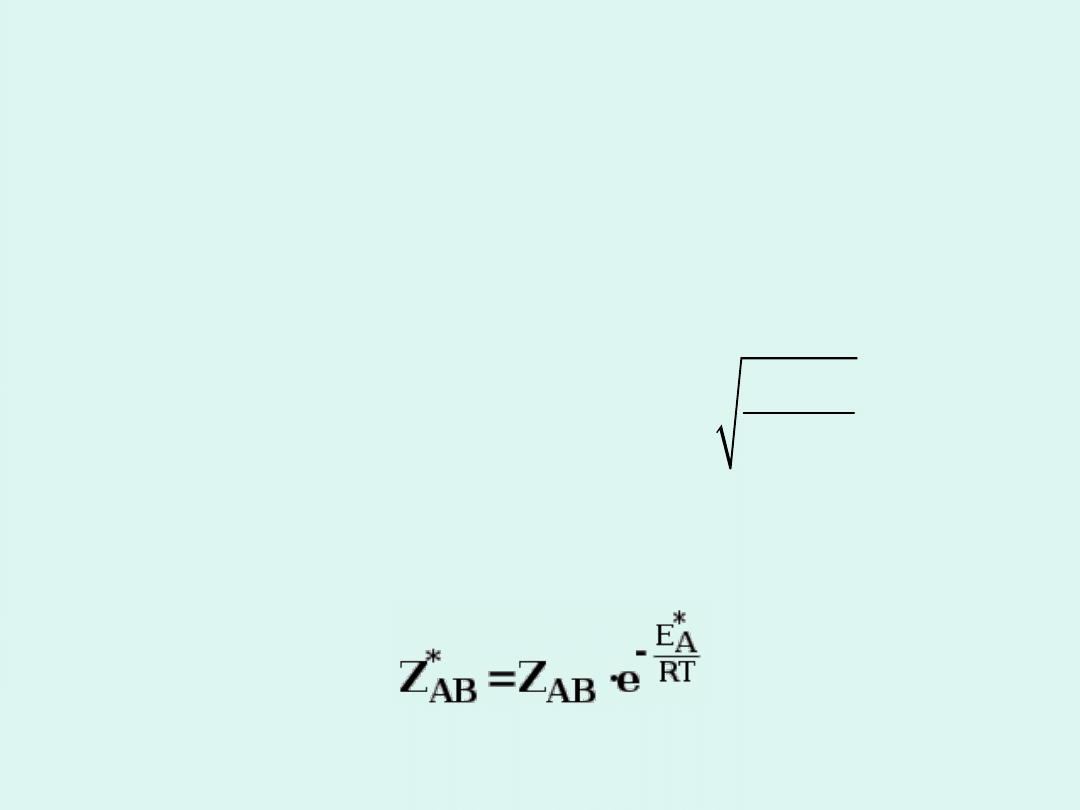
Teoria zderzeń
A B
P
+ �
Szybkość reakcji jest wprost
proporcjonalna do częstości zderzeń
cząsteczek
A
i
B.
(
)
2
AB
A
B
AB
AB
8RT
Z
=
� �
P s
Pm
N
N
Tylko zderzenia, których energia jest
większa od progowej
E
*
A
, prowadzą do
powstania produktu.

Energia progowa
E
*
A
jest wyznaczana
doświadczalnie
i
nie
można
jej
przewidzieć na podstawie teorii zderzeń.
Jest
ona
związana
z
wyznaczaną
doświadczalnie energią aktywacji :
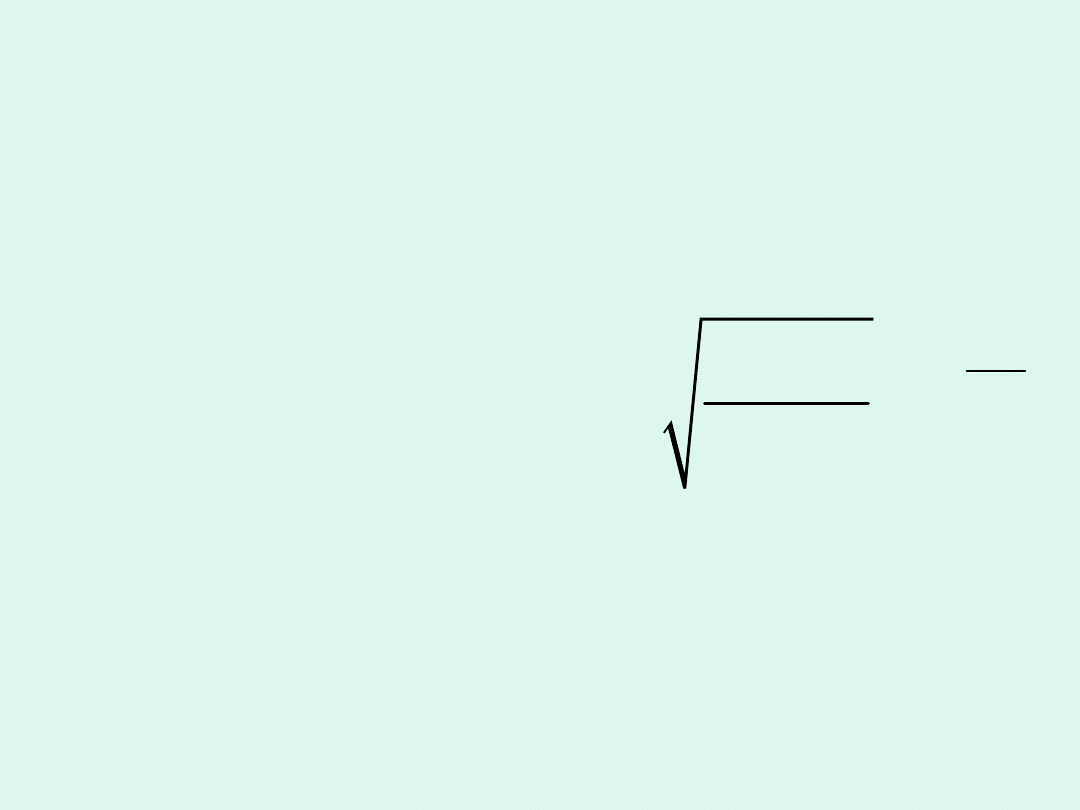
Współczynnik steryczny
Zwykle stała szybkości obliczona z teorii
zderzeń jest większa niż wyznaczona
doświadczalnie.
k
doświadczalna
< k
teoretyczna
(
)
*
EA
RT
2
A
AB
AB
8RT
k
N
e
P
-
= � �
P s
�
Pm
P – współczynnik steryczny
. Określa
prawdopodobieństwo zderzenia się cząsteczek
w taki sposób, aby mogła zajść pomiędzy nimi
reakcja.
P =
k
doświadczalna
/k
teoretyczna
P < 1
(zwykle)
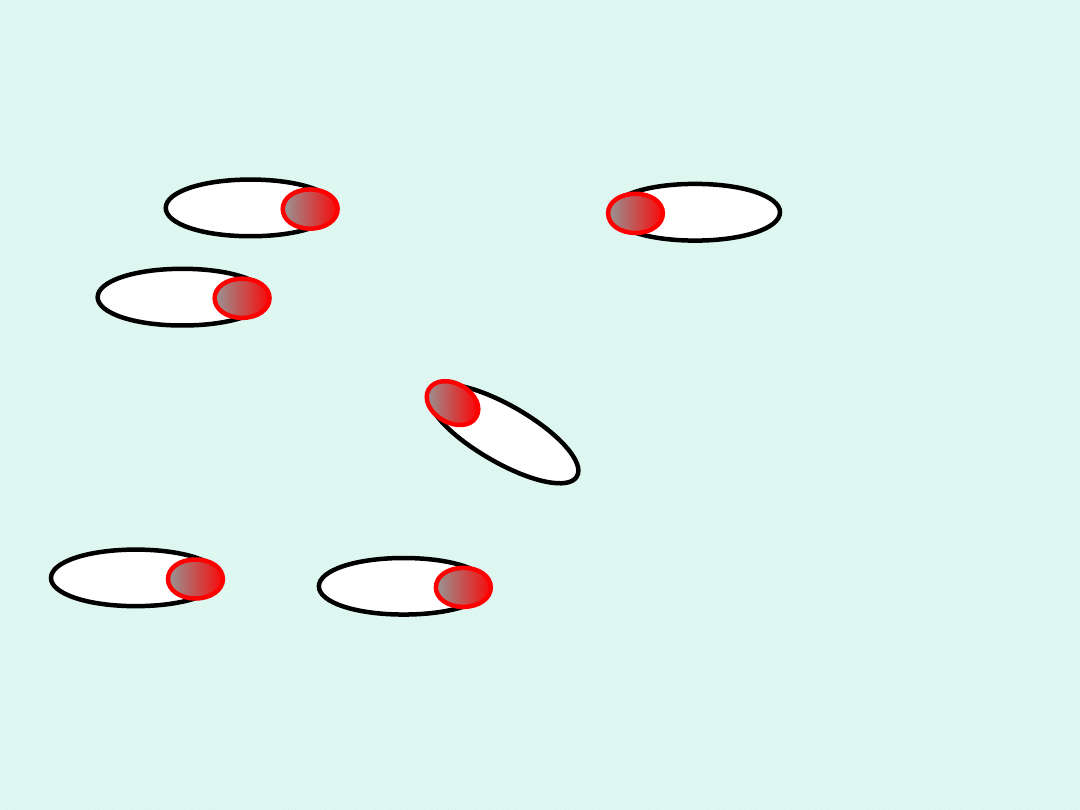
Interpretacja współczynnika
sterycznego
To
zderzenie
prowadzi
do reakcji.
Te zderzenia
są
nieefektywn
e
Im bardziej
złożone są
cząsteczki tym
mniejsza jest
wartość P.

Istnieją reakcje, dla których
P >1
.
Przyczyną jest wymiana elektronu pomiędzy
atomem K i Br
2
. W wyniku tego powstają jony
różnoimienne, które nawzajem się przyciągają i
zwiększają prawdopodobieństwo spotkania się
cząsteczek. Elektron działa jak „harpun” łączący
obie cząsteczki
Zwykle wartość
P > 1
wskazuje na mechanizm
łańcuchowy reakcji.
Wartości współczynnika sterycznego nie można w
zasadzie przewidzieć teoretycznie.
8
,
4
P
Br
KBr
Br
K
g
g
g
2
g
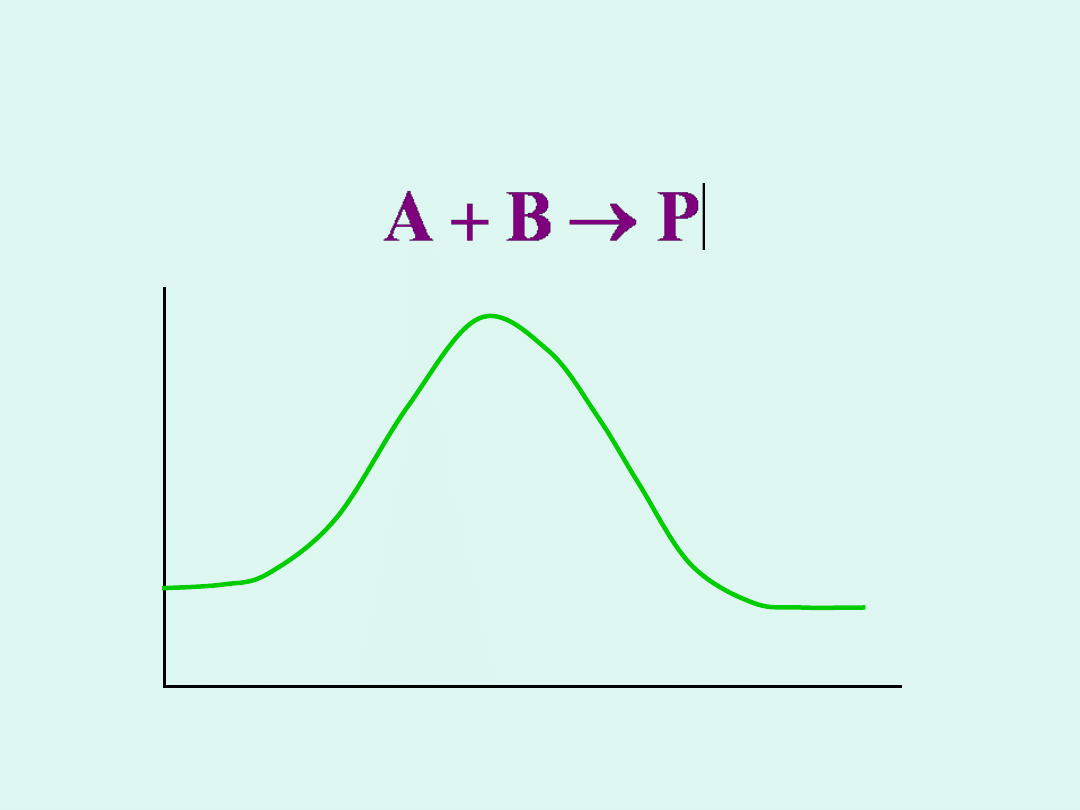
Teoria kompleksu aktywnego
(absolutnej szybkości reakcji,
Eyringa-Polanyi'ego)
A+
B
P
[A
....
B]
- kompleks
aktywny
współrzędna
reakcji
E

Formalnie możemy napisać, że reakcja zachodzi w
dwu etapach :
1.
Odwracalne tworzenie kompleksu aktywnego.
A + B [A
....
B]
#
2.
Rozpad kompleksu aktywnego do produktów.
[A
....
B]
#
P
X
X
X
Y
Y
Z
Z
Y
Z
Co się dzieje w trakcie
reakcji ?
komplek
s
aktywny
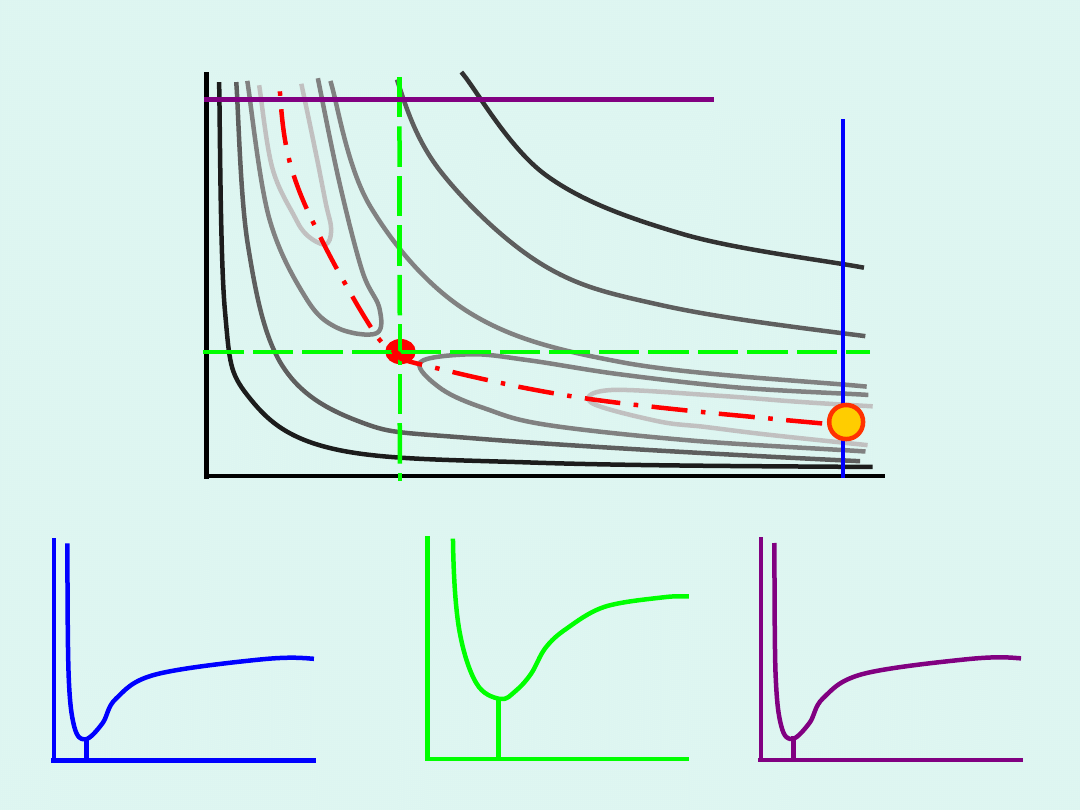
r
XY
r
YZ
D
C
B
#
A
r
XY
E
r
e
r
YZ
E
r
e
r
e
#
r
XY
r
XZ
E
Mapa energetyczna reakcji
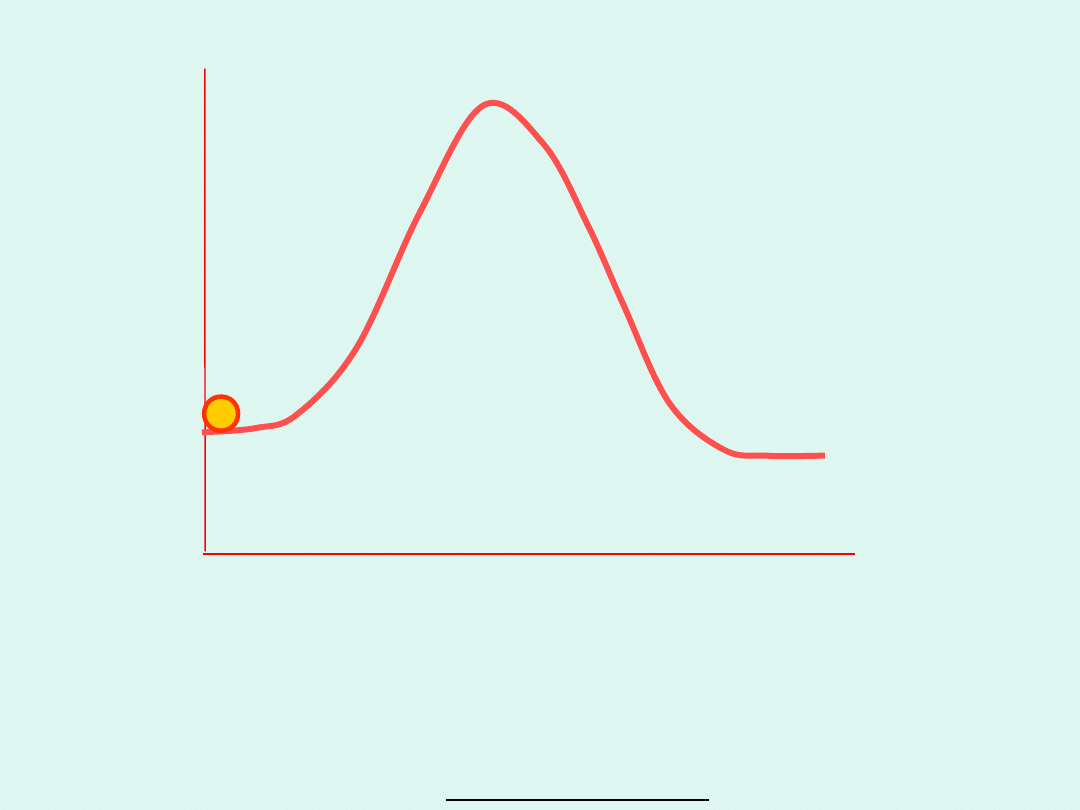
E
XY + Z
#
droga
reakcji
X + YZ
Cała mapę energetyczną reakcji a więc i
zależność energii od drogi reakcji oraz
ostatecznie energię kompleksu aktywnego
można obliczyć teoretycznie na gruncie
mechaniki kwantowej (w sposób przybliżony).
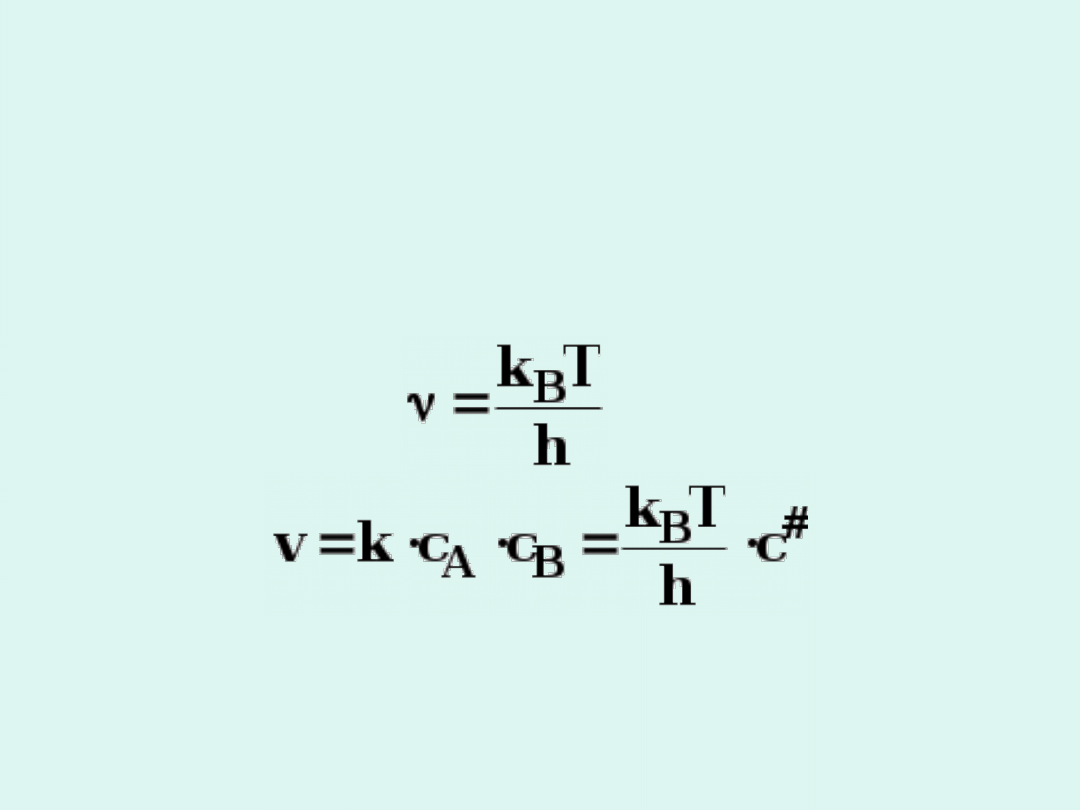
Szybkość reakcji jest wprost proporcjonalna
do stężenia kompleksu aktywnego :
#
v
c
i częstości drgań wiązania decydującego o
rozpadzie kompleksu aktywnego w kierunku
produktu

#
#
#
B
c
c
A
B
gdzi
k T
c
k
K
K
h
c c
e
=
=
�
Wielkość stałej równowagi tworzenia
kompleksu
aktywnego
K
c
#
można
wyznaczyć
teoretycznie
metodami
termodynamiki statystycznej i mechaniki
kwantowej.
Równanie Eyringa
Wielkość stałej równowagi tworzenia
kompleksu
aktywnego
K
c
#
można
powiązać z odpowiednimi funkcjami
termodynamicznymi tworzenia : entalpią
swobodną, entalpią i entropią.
#
#
c
#
#
#
G
RTlnK
G
H
T S
D
=-
D
=D
- D
#
#
#
G
S
H
B
B
RT
R
RT
k T
k T
k
e
e
e
h
h
D
D
D
-
-
=
�
=
�
�

Kataliza i katalizatory
Katalizatory
to substancje, które zwiększają
szybkość reakcji chemicznej, pozornie nie
wchodząc w nią. Po zajściu reakcji pozostają
niezmienione.
Katalizatory ujemne (zmniejszające szybkość
reakcji) to
inhibitory
.
Autokaliza
to przyspieszenie reakcji przez
jej produkty.

Kataliza homogeniczna
– katalizator i
reagenty są w tej samej fazie.
Kataliza heterogeniczna
– katalizator
jest w innej fazie niż reagenty. Zwykle
katalizator jest w tym wypadku ciałem
stałym. Taki typ katalizy nazywamy
katalizą kontaktową
, a katalizator
kontaktem
.
Ponadto
wyróżniamy
katalizę
enzymatyczną
i
fotokatalizę
.

Katalizator powoduje przyspieszenie
reakcji.
Katalizator zwiększa stałą szybkości
reakcji w danej temperaturze.
Obecność katalizatora obniża energię
aktywacji reakcji.
W obecności katalizatora mechanizm
reakcji jest inny niż bez niego.
Katalizator nie ma wpływu na stan
równowagi reakcji ani na jej efekt
cieplny.
Katalizator nie zmienia stałej
równowagi reakcji.
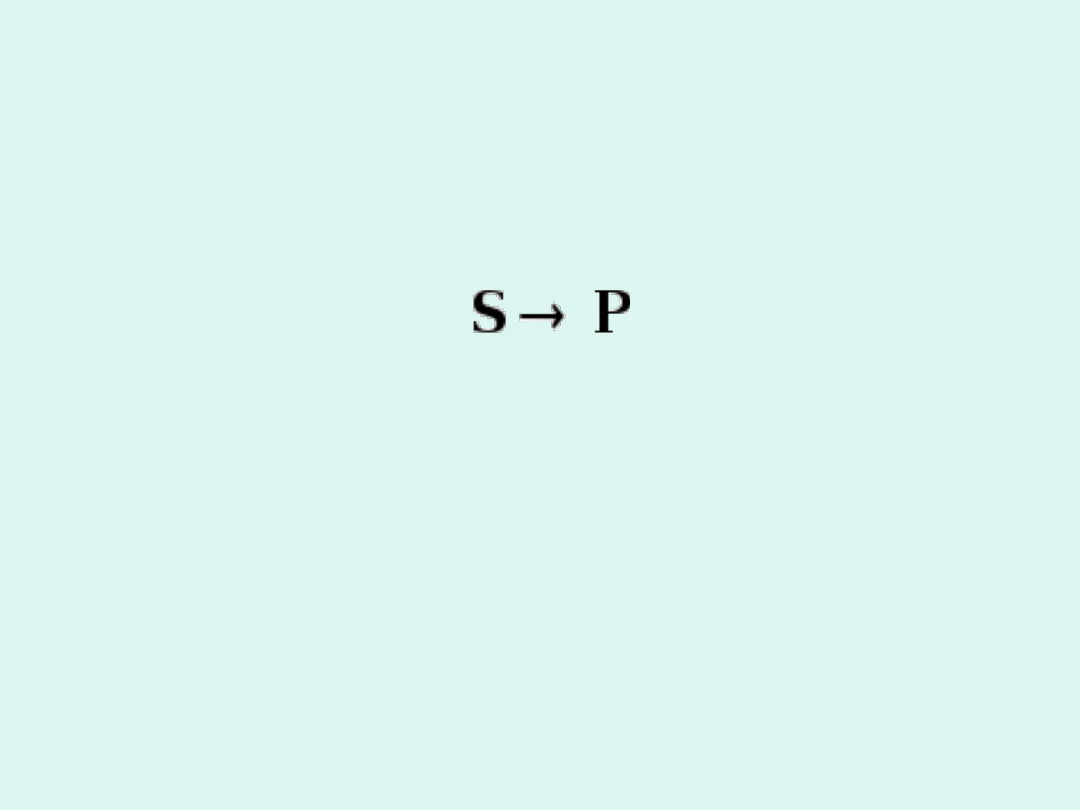
Jak działa katalizator ?
S K
SK
SK
P K
+ �
� +
Reakcja bez katalizatora :
W obecności katalizatora reakcja zachodzi
co najmniej w dwu etapach:
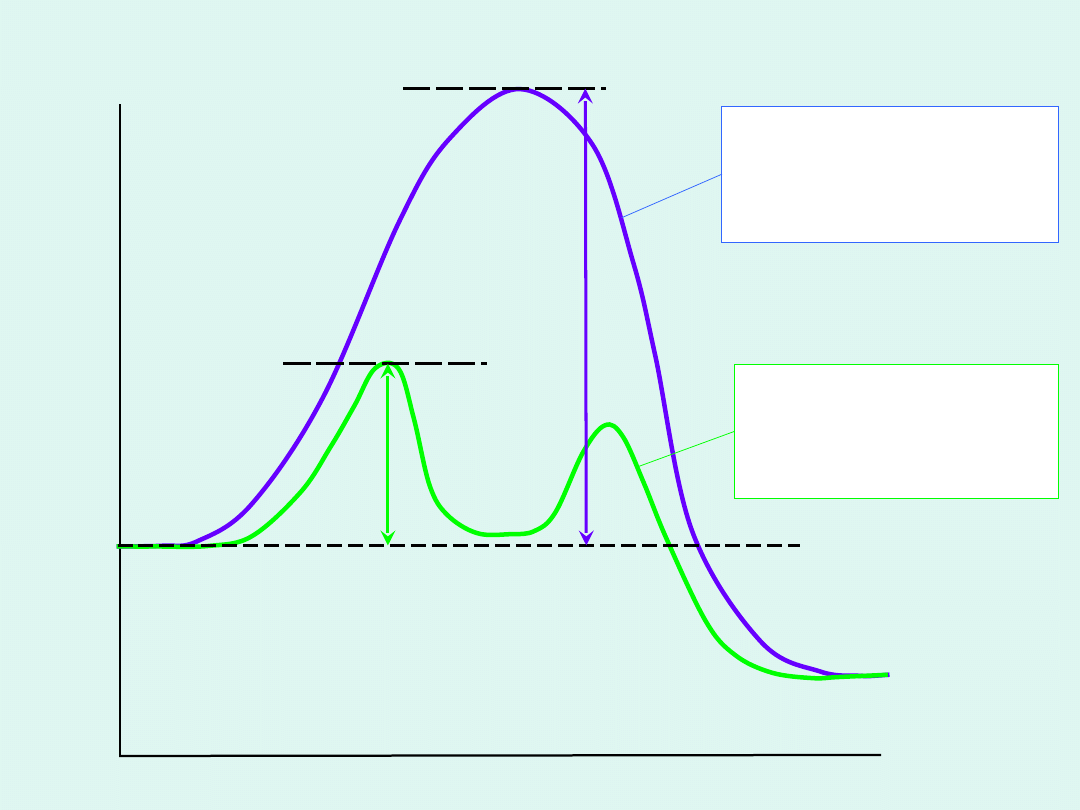
S
P
SK
E
współrzędna reakcji
E
A
'
E
A
Profil
energetyczny
reakcji bez
udziału
katalizatora
Profil
energetyczny
reakcji z
udziałem
katalizatora
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
Wyszukiwarka
Podobne podstrony:
15 Kinetyka reakcji 1 rzedu
15 Rejestryid 16210 ppt
15 autoprezentacjaid 16114 ppt
15 Odtrutkiid 16280 ppt
15 Kinetyka
15 NOWOTWORYid 16039 ppt
15 kinetyka
03 i 04 BIOCHEMIA kataliza kinetykaid 4367 ppt
15 inflacjaid 16275 ppt
15 osnowaid 16179 ppt
15 przysadkaid 16049 ppt
15 Rejestryid 16210 ppt
15 autoprezentacjaid 16114 ppt
więcej podobnych podstron