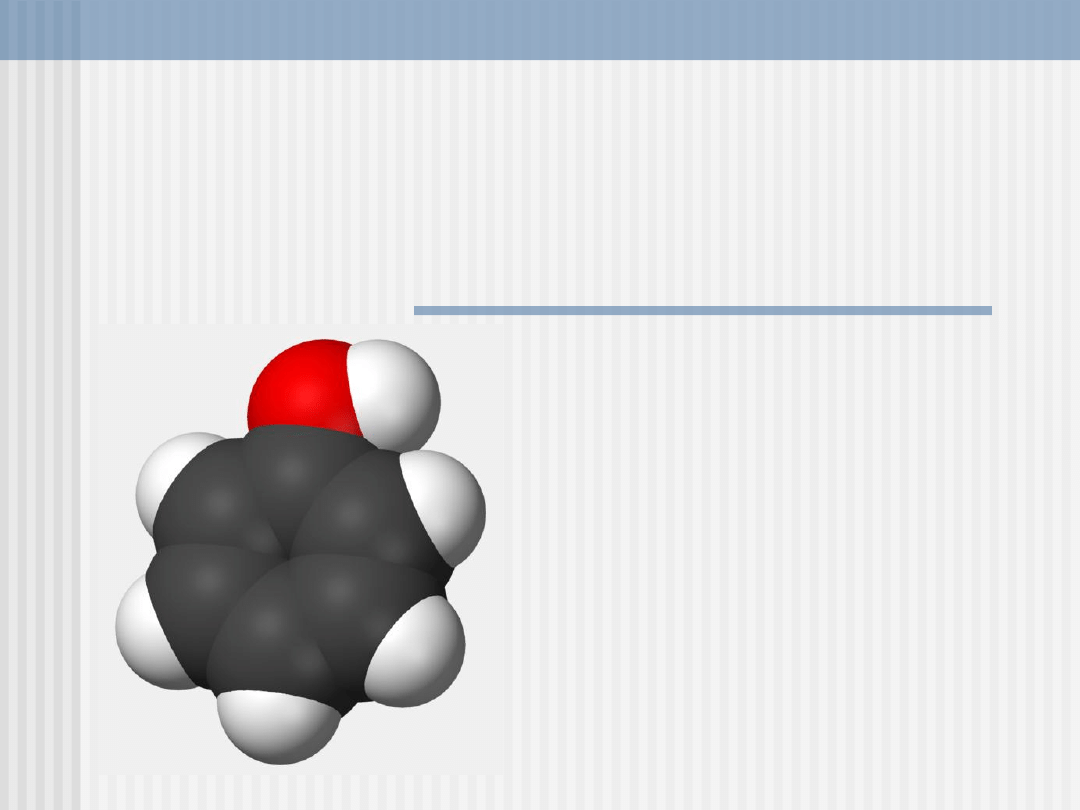
FENOL

Fenol (C
6
H
5
OH) inaczej hydroksybenzen (inne
nazwy: benzenol, kwas karbolowy, karbol)
Najprostszy związek z grupy fenoli. Od alkoholi
odróżnia go fakt, że grupa hydroksylowa
połączona jest bezpośrednio z pierścieniem
aromatycznym, co wpływa na właściwości
związku - m. in. na wzrost właściwości
kwasowych.
Po raz pierwszy został wydzielony ze smoły
węglowej w 1832 r. przez chemika niemieckiego
Podczas II wojny światowej więźniów niektórych
obozów koncentracyjnych (np. Auschwitz-
Birkenau) zabijano przez wstrzyknięcie fenolu.

Właściwości
Stan skupienia, barwa: w temperaturze pokojowej
czysty fenol jest bezbarwnym krystalicznym ciałem
stałym (pod wpływem światła następuje częściowe
utlenienie fenolu, w wyniku którego zmienia barwę na
różową, brunatną lub czarną)
Zapach: ostry, słodkawy
Gęstość: 1,07 g/cm³
Rozpuszczalność: niezbyt dobrze rozpuszcza się w
wodzie (w temp. 20°C 8,2 g na 100 cm³ H
2
O), lepiej w
rozpuszczalnikach organicznych np.: alkohol etylowy,
eter etylowy, gliceryna, chloroform, benzen
Temperatura topnienia: 41°C
Temperatura wrzenia: 182°C

Należy unikać źródeł zapłonu,
wysokiej temperatury, ponieważ
fenol jest substanją palną (w
środowisku pożaru wydzielają się
tlenki węgla)
Fenol ma słabe właściwości kwasowe
(stała dysocjacji K
a
= 1,3 · 10
-10
).
Tworzy sole i estry - fenolany.

Otrzymywanie
Najważniejszą metodą otrzymywania fenolu jest
obecnie metoda kumenowa.
Inne metody, obecnie głównie o znaczeniu
historycznym, to np.:
ekstrakcja ze smoły węglowej,
katalizowana hydroliza chlorobenzenu w
podwyższonej temperaturze,
stapianie kwasu benzenosulfonowego lub jego soli z
wodorotlenkiem sodu lub potasu i zakwaszanie
powstałych fenolanów.
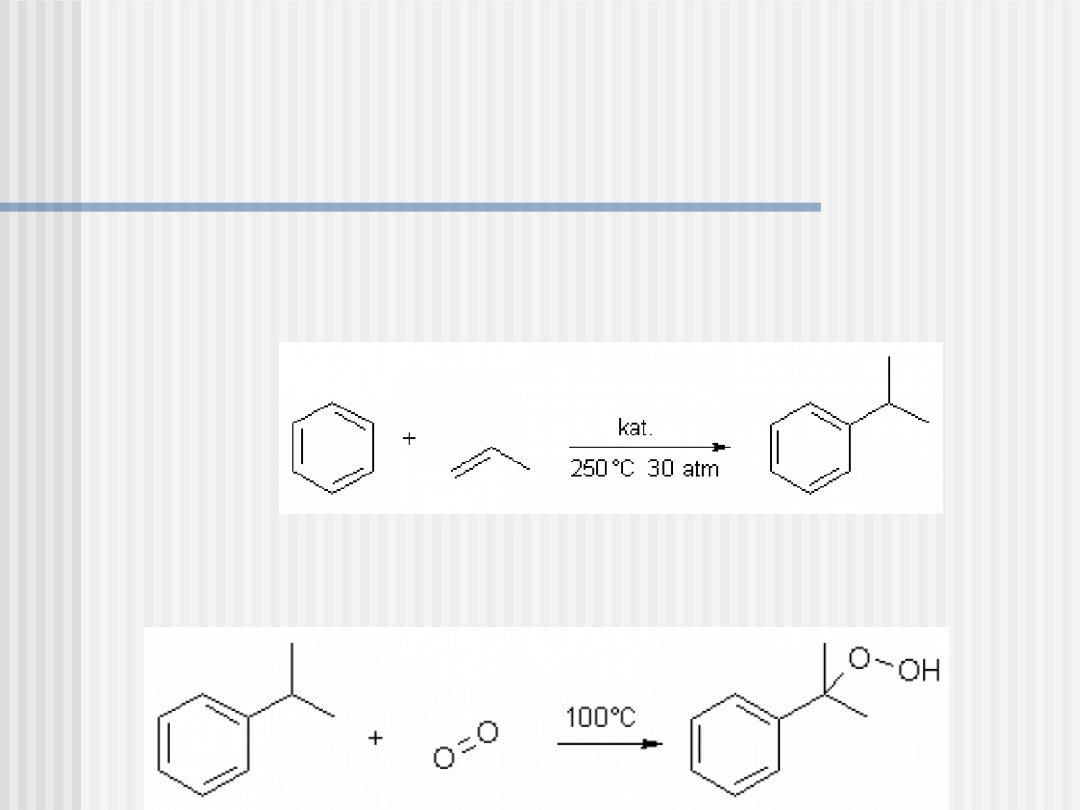
Otrzymywanie – metoda kumenowa
To trójetapowa metoda otrzymywania fenolu i acetonu z benzenu i
propenu. Nazwa tej metody pochodzi od kumenu
(izopropylobenzenu), który jest produktem pośrednim.
etap I - alkilacja benzenu propenem w temp. 250°C pod zwiększonym
ciśnieniem w obecności kwasu Lewisa (np. chlorku glinu) jako
katalizatora:
etap II – utlenianie kumenu tlenem w temp. 100°C do wodoronadtlenku
kumenu:
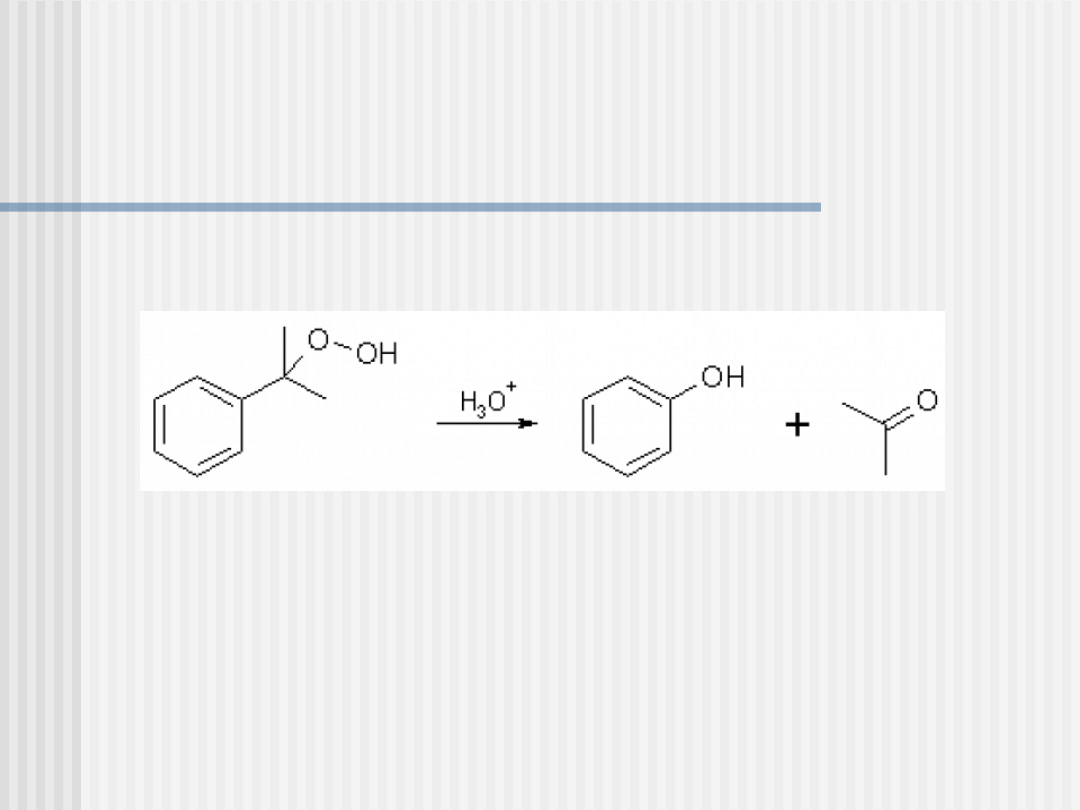
Otrzymywanie – metoda kumenowa
etap III - kwasowa hydroliza otrzymanego hydronadtlenku
kumenu:
Produkty są następnie rozdzielane przez destylację.
Zdecydowana większość światowej produkcji fenolu
i acetonu oparta jest na tej metodzie.

Zastosowanie
produkcja żywic fenolowo-
formaldehydowych (np. bakelitu),
leków (np. kwasu acetylosalicylowego),
detergentów, herbicydów, fungicydów i
barwników,
sam fenol był używany w roztworze
wodnym jako środek bakteriobójczy. Wodny
roztwór fenolu - karbol, używany był do
dezynfekcji pomieszczeń. Nazwa karbol
dawniej była niekiedy stosowana również na
określenie samego fenolu.

Zastosowanie
był jednym z najwcześniej stosowanych środków
przeciwbakteryjnych. W stężeniu 0,2% działa
bakteriostatycznie, 1,3% grzybobójczo, powyżej 2%
bakteriobójczo. Dzisiaj nie stosowany, ze względu na
dużą toksyczność; jedynie w stomatologii - pomocniczo
jako antyseptyk w leczeniu endodontycznym
(kanałowym) miazgi zębowej
stosowany w kosmetyce do tzw. peelingu
chirurgicznego. Fenol to najsilniejszy wśród związków
chemicznych używanych do złuszczania skóry. Zabieg
może być wykonywany tylko przez chirurga plastyka lub
specjalnie przeszkolonego lekarza.

Toksyczność
Fenol jest sklasyfikowany jako
substancja toksyczna oraz żrąca,
powoduje oparzenia
Drogi wchłaniania:
drogi oddechowe,
skóra,
z przewodu pokarmowego

Toksyczność
Pary fenolu dobrze wchłaniają się zarówno
przez układ oddechowy jak i przez skórę
(wchłanianie przez skórę może być nawet
przyczyną zatruć śmiertelnych).
Fenol stały lub w roztworach wchłania się
przez skórę, jednak denaturacja białek
naskórka powoduje opóźnienie wchłaniania.

Objawy zatrucia ostrego:
pary i mgła powodują podrażnienie spojówek, błon
śluzowych nosa, gardła, uczucie suchości w nosie,
gardle, kaszel. W wysokich stężeniach: zawroty i ból
głowy, mdłości, wymioty, duszność, przyspieszenie i
pogłębienie oddechów, zaburzenia oddychania,
zaburzenia orientacji, zapaść, utratę przytomności.
wchłanianie przez skórę mgły, par i roztworu
wywołuje zawroty, ból głowy, dezorientację,
zaburzenia oddechowe, zapaść, utratę przytomności.
skażenie skóry substancją stałą lub ciekłą wywołuje
miejscowe zbielenie i oparzenia, które początkowo
nie są bolesne, oraz pęcherze, martwicę.
skażenie oczu powoduje ostry stan zapalny,
uszkodzenie rogówki.

bezpośrednim następstwem zatrucia jest
uszkodzenie wątroby z żółtaczką, uszkodzenie nerek
z ich ostrą niewydolnością, zapalenie płuc.
połknięcie roztworu wywołuje rozległe oparzenia błon
śluzowych jamy ustnej, gardła i dalszych części
przewodu pokarmowego, bóle i krwawienie,
perforację ścian przewodu pokarmowego,
uszkodzenie wątroby i nerek.
Objawy zatrucia przewlekłego:
zaburzenia ze strony układu pokarmowego: wymioty,
bóle gardła, utrata łaknienia, ślinotok, biegunka i
jadłowstręt.
występuje ochronoza (ciemne zabarwienie skóry i
moczu) i wykwity skórne.
może wystąpić uszkodzenie wątroby i nerek.

Narażenie zawodowe na fenol występuje
głównie w trakcie stosowania żywic
fenolowych oraz ich obróbki cieplnej. Są one
wykorzystywane jako materiał wiążący w
materiałach izolacyjnych, płytach wiórowych,
farbach i jako składnik mas formierskich.
Narażenie na fenol może także występować
podczas produkcji koksu, produkcji fenolu i
jego pochodnych oraz kaprolaktamu.
Źródłem narażenia mogą być wędzone
produkty spożywcze oraz woda pitna.

Losy w organizmie
Biotransformacja fenolu polega głównie
na sprzęganiu z kwasem siarkowym i
glukuronowym głównie w wątrobie, jednak
płuca, jelita i nerki również odgrywają
istotną rolę w metabolizmie tego związku
Główną droga wydalania fenolu są nerki,
w ciągu 24 h wydala się ok. 90% dawki
fenolu głównie w postaci siarczanu

Mechanizm działania
toksycznego
Fenol i jego metabolity wiążą się kowalencyjnie z
białkami tkanek, głównie wątroby, z białkami
osocza a także z DNA
Uszkadza wątrobę i nerki, wywołuje kwasicę
metaboliczną
Pary fenolu mogą powodować podrażnienie dróg
oddechowych
Działa neurotoksycznie, powodując
demielenizację włókien nerwowych
Przeprowadzone badania wykazują, że fenol może
mieć działanie genotoksyczne oraz hamujące
erytropoezę

Oznaczanie fenolu w wodzie i
moczu:
Polega na wydzieleniu go z matrycy – najczęściej
poprzez destylację z parą wodną z kwaśnego roztworu i
następnie reakcji z 4-aminoantypiryną w środowisku
alkalicznym przy pH = 9,8 +/- 0,2 w obecności
heksacyjanożelazianu (III) potasu jako substancji
utleniającej
W wyniku tej reakcji powstaje barwny związek
(indofenol) mający zabarwienia w zależności od
stężenia fenolu od zielonożółtego do
czerwonowiśniowego
Następnie określa się stężenie za pomocą
spektrofotometru UV-Vis lub kolorymetru
fotoelektrycznego
Metodę stosuje się w zakresie stężeń 0,5 – 10,00
mg/dm
3
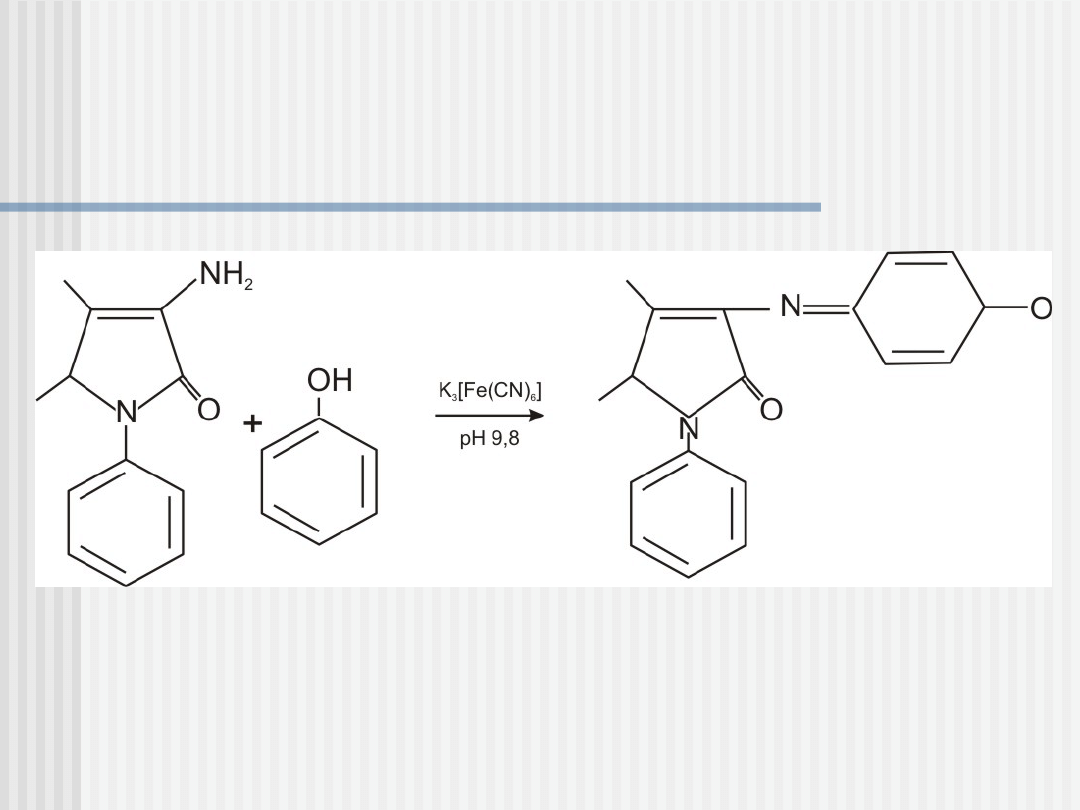
4-
aminoantypiryna
fenol
indofenol
Spektrofotometria
spektroskopia
Dział nauki zajmujący się
oddziaływaniem promieniowania
elektromagnetycznego z materią
spektroskopi
a
molekularna
Zajmuje się oddziaływaniem
promieniowania elektromagnetycznego
z cząsteczkami
W wyniku absorpcji promieniowania
elektromagnetycznego przez cząstki następuje
wzbudzenie odpowiednich poziomów energii,
zjawisko to obserwuje się w postaci widma
absorpcyjnego
spektrofotomet
ria UV-Vis
Bada elektronowe widma absorpcyjne, które
powstają wskutek wzbudzenia elektronów
promieniowaniem elektromagnetycznym z
zakresu UV-Vis, czyli o długościach fali od 100
do 780 nm
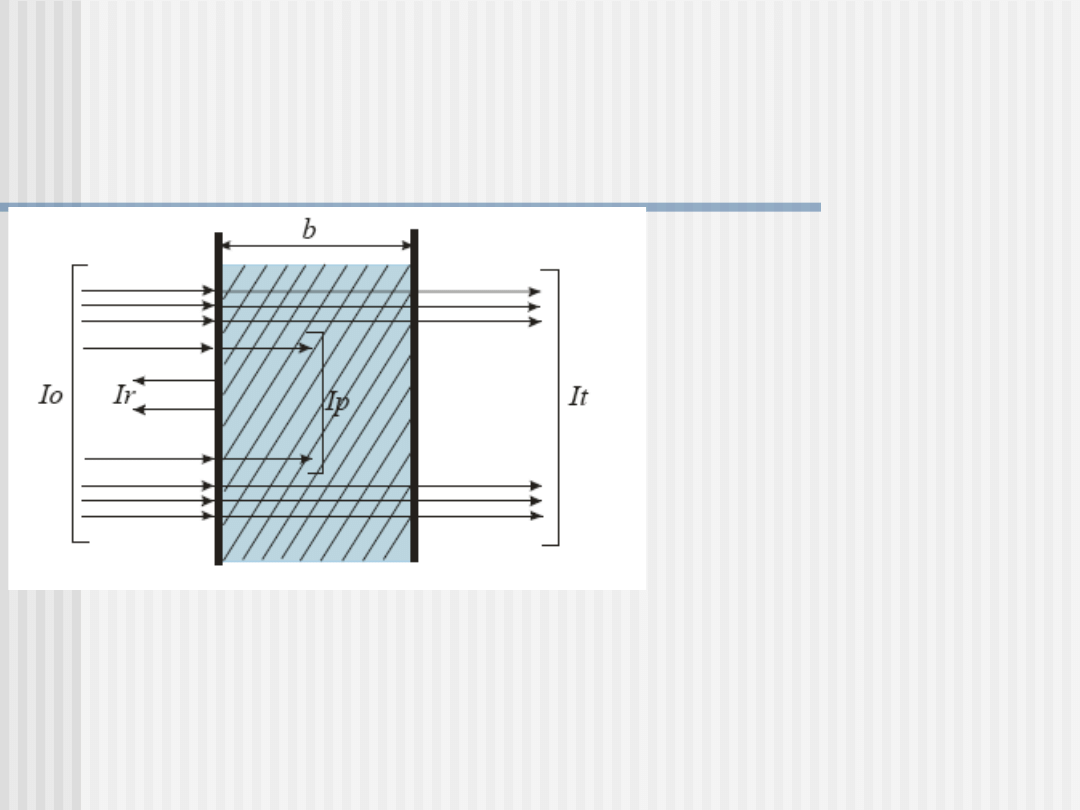
Wiązka promieniowania monochromatycznego przechodząca przez
warstwę roztworu jest osłabiona w stosunku do padającego.
Promieniowanie o natężeniu ulega częściowo odbiciu lub
rozproszeniu, częściowo pochłonięciu, a tylko część przechodzi
przez roztwór.
I
0
- natężenie wiązki
promieniowania
monochromatycznego
I
r
– natężenie promieniowania
rozproszonego i obitego
I
p
– natężenie promieniowania
pochłoniętego
I
t
– natężenie promieniowania
przechodzącego przez roztwór.
b – grubość warstwy
absorbującej
W przypadku roztworów, w których nie ma zawiesin, wartość I
r
można praktycznie pominąć
Powyższą obserwację można przedstawić w postaci równania
I
0
= I
p
+I
t

Do absorpcji promieniowania zdolne są
cząsteczki organiczne posiadające grupy
chromoforowe (sprzężone wiązania
podwójne lub potrójne, pierścienie
aromatyczne).
Nagromadzenie tych wiązań powoduje
wzrost absorpcji i przesunięcie jej w
kierunku podczerwieni.

I prawo absorpcji (prawo Lamberta)
Absorbancja jest proporcjonalna do grubości warstwy
absorbującej, jeżeli wiązka promieniowania
monochromatycznego przechodzi przez jednorodny ośrodek
absorbujący
I = I
o
* e
–kb
A = log I
o
/ I = a*b
I – natężenie promieniowania po przejściu przez ośrodek absorbujący
I
0
– natężenie wiązki promieniowania padającego na ośrodek absorbujący
b – grubość warstwy absorbującej
k - współczynnik absorpcji
a – 0,4343 k
e - podstawa logarytmów naturalnych
A - zdolność pochłaniania promieniowania zwana absorbancją

II Prawo absorpcji
(Lamberta-Beera)
Jeżeli współczynnik absorpcji rozpuszczalnika jest
równy zeru, to wiązka promieniowania
monochromatycznego, po przejściu przez jednorodny
roztwór substancji absorbującej o stężeniu c, ulega
osłabieniu według równania:
I = I
o
* e
–kbc
A = log I
o
/ I = abc
Prawo to dotyczy absorpcji promieniowania przez roztwory.
a - właściwy współczynnik absorpcji (gdy stężenie w
g/cm
3)
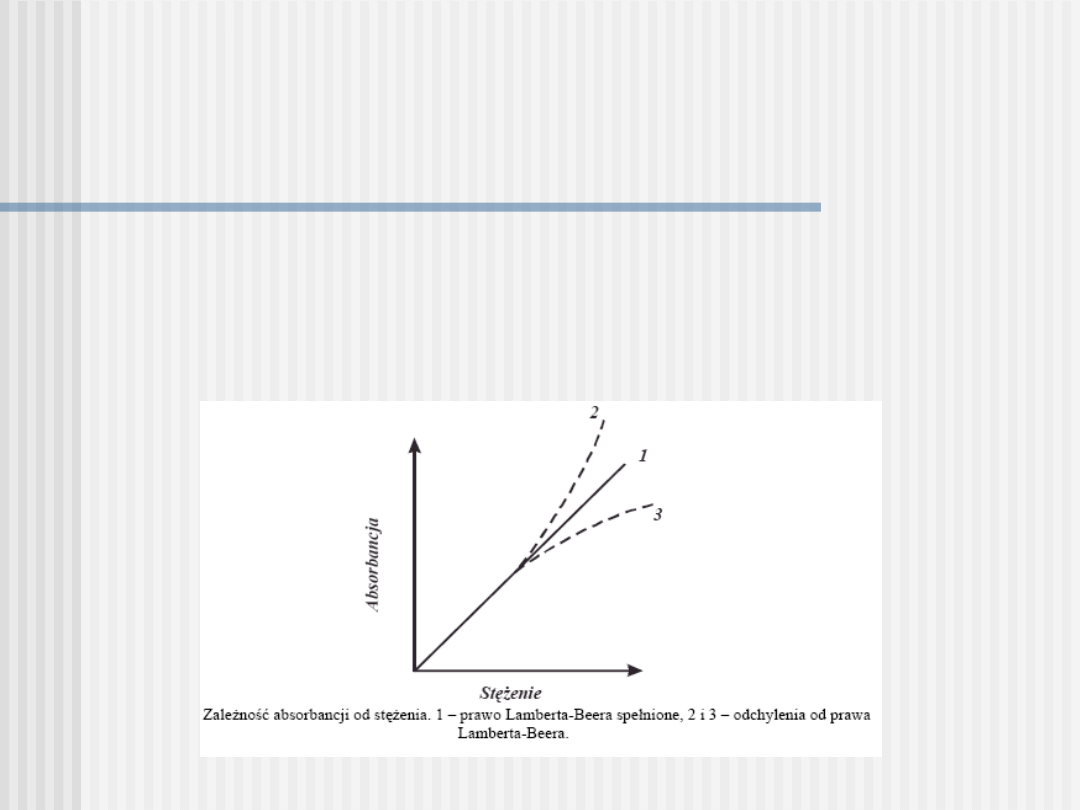
W przypadku gdy zależność
A = log Io / I = abc
jest liniowa, wówczas
dany roztwór podlega prawu Lamberta - Beera. Jednak w praktyce
spotyka się odchylenia od tego prawa głównie za sprawą reakcjami
chemicznymi zachodzącymi wraz ze zmianą stężenia lub błędami
aparaturowymi, np. niską monochromatycznością promieniowania.
Spełnienie prawa Lamberta-Beera nie jest jednak warunkiem
koniecznym wykonania analizy spektrofotometrycznej. Odchylenia
od prawa Lamberta-Beera powodują zwiększenie błędów
oznaczania.
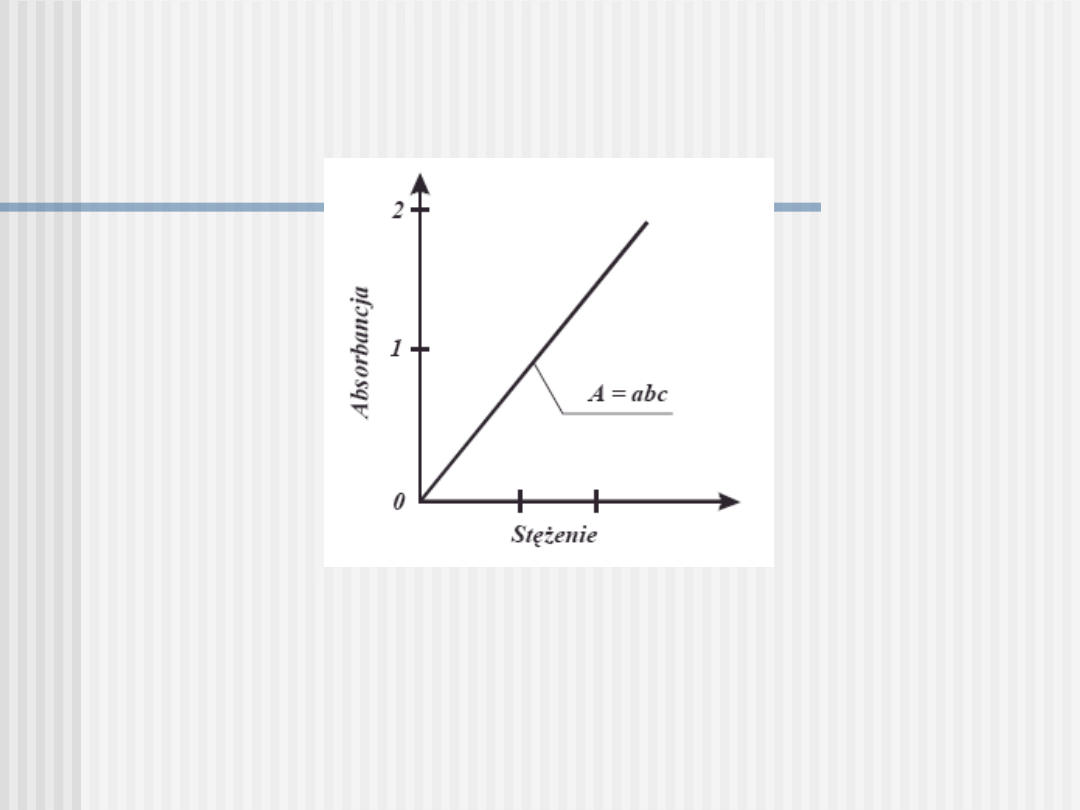
Zależność absorbancji od stężenia ma
charakter prostoliniowy:
Pomiar absorbancji należy wykonywać
przy długości fali, dla której uzyskuje się
maksymalną absorpcję, czyli dla
max
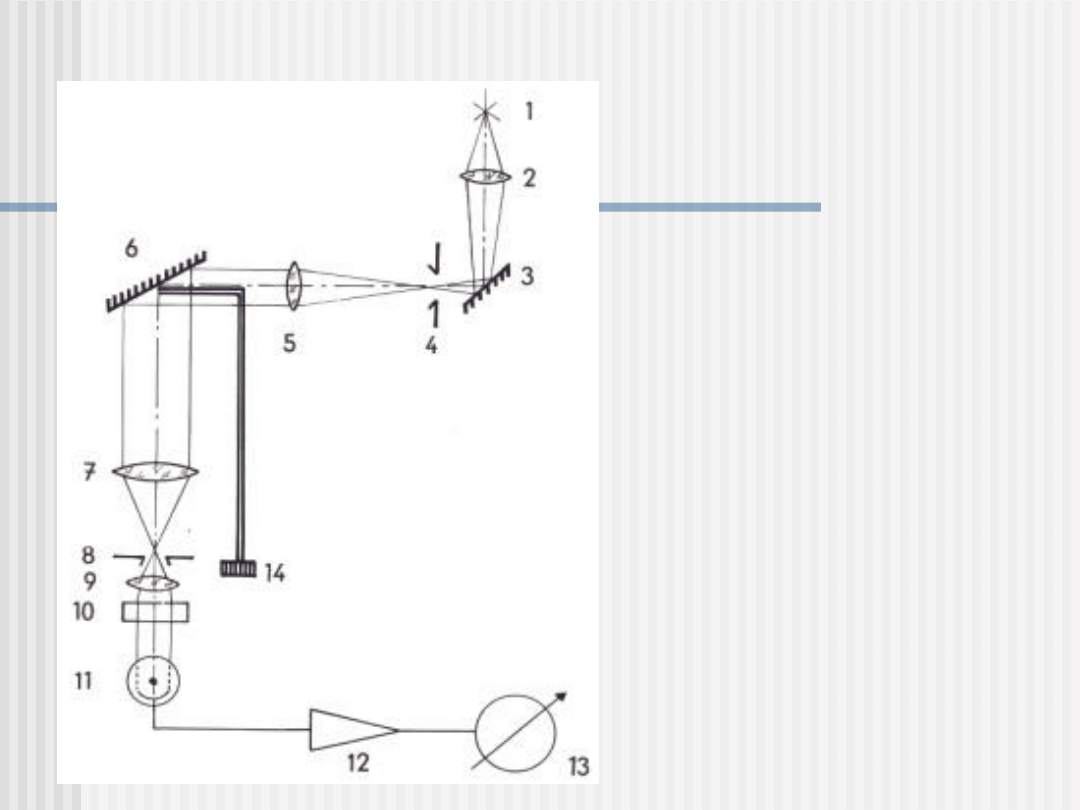
Budowa i zasada działania spektrofotometru
1-lampa wolframowa
2-kondensor
3-zwierciadło
4-szczelina wejściowa
5-układ achromatyczny
6-siatka dyfrakcyjna
7-soczewka achromatyczna
8-szczelina wyjściowa
9-kuweta
10-filtr
11-detektor
12-wzmacniacz tranzystora
13-urządzenie pomiarowe
14-bęben (pokrętło)
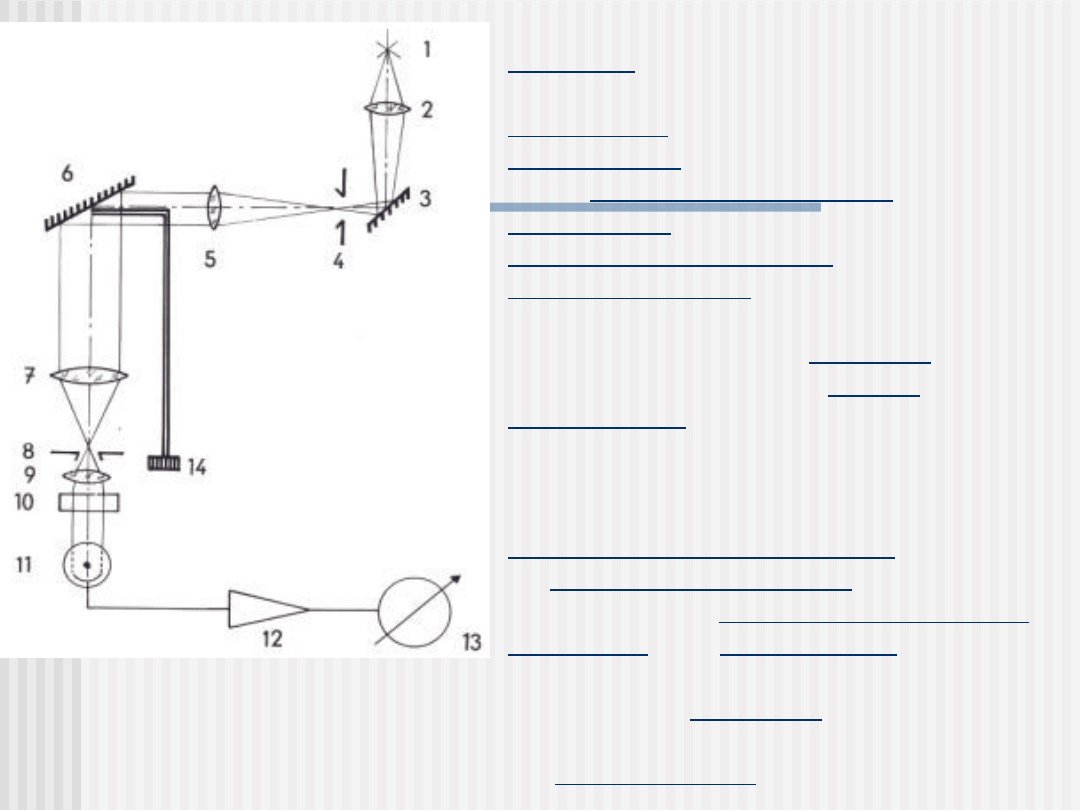
Źródłem (1) emitowanego światła jest
lampa. Światło przechodząc przez
kondensor (2) ulega odbiciu od
zwierciadła (3) następnie przechodzi
przez szczelinę wejściową (4) do
kolimatora, gdzie po przejściu przez
układ achromatyczny (5) pada na
monochromator (6), którym jest siatka
dyfrakcyjna. Przy pomocy układu
dźwigni, poruszanych bębnem (14),
możliwe jest obracanie siatką
dyfrakcyjną i dzięki temu w szczelinie
wyjściowej monochromatora uzyskuje się
wiązkę o żądanej długości fali. Wiązka o
wybranej długość, po przejściu przez
soczewkę achromatyczną (7), trafia
na szczelinę wyjściową (8) i po
przejściu przez kuwetę z roztworem
badanym (9) i filtr barwny służący do
pochłaniania promieniowania cieplnego
(10) pada na detektor (11). Powstały w
fotoogniwie fotoprąd ulega wzmocnieniu
we wzmacniaczu (12) a następnie
dostaje się do urządzenia
pomiarowego (13).
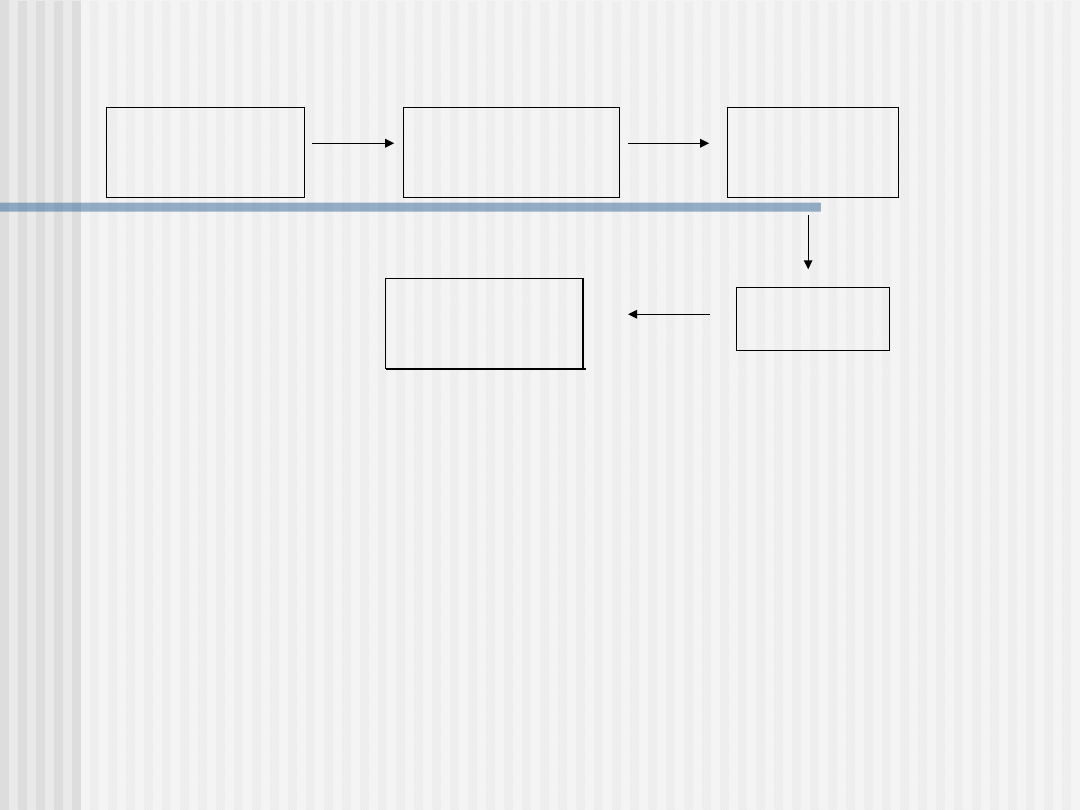
Schemat blokowy spektrofotometru UV-Vis
Kuweta
pomiarowa
monochromat
or
detektor
Wskaźnik i
rejestrator
źródło
promieniowani
a
• Źródło promieniowania musi pokryć cały zakres
UV-Vis
stosowane są lampy deuterowe, lampy
wolframowo halogenowe lub
wysokociśnieniowe łukowe lampy ksenonowe

Monochromator:
Ma za zadanie wybrać z emitowanego przez źródło ciągłego
promieniowania wąskie pasmo o żądanej długości fali i
przepuścić je przez komórkę z badaną substancją
Składa się ze szczeliny wejściowej, kolimatora (np.
zwierciadło), elementu rozszczepiającego promieniowanie
(np.siatka dyfrakcyjna) i szczeliny wyjściowej
Komórka pomiarowa:
Musi zapewnić dokładnie znaną grubość warstwy
absorbującej cieczy lub gazu
Wykazywać odporność na działanie analizowanych substancji
chemicznych
Zapewnić w max. stopniu transmisję promieniowania
Wykonane są na zakres UV z kwarcu lub stopionej
krzemionki a na zakres Vis ze szkła optycznego
Detektory:
Powinny charakteryzować się dobrą czułością i
proporcjonalnością przetwarzania sygnałów optycznych na
elektryczne
Najczęściej są to: fotokomórki, fotopowielacze lub fotodiody


Zastosowanie spektrofotometrii UV-Vis:
Jest to najczęściej wykorzystywana metoda instrumentalna w
analizie ilościowej
Główne jej zalety to : dobra czułość, precyzja i selektywność
oznaczeń
Stosowana jest w :
Analizie ilościowej kationów metali: najczęściej w postaci
barwnych kompleksów chelatowych z odczynnikami
organicznymi, barwnych kompleksów par jonowych a także
barwnych kompleksów z prostymi ligandami nieorganicznymi
W analizie ilościowej anionów nieorganicznych: najczęściej
azotany (V) i (III), fosforany, fluorki i krzemiany – w rolnictwie,
badaniach środków spożywczych i ochrony środowiska
W analizie ilościowej związków organicznych: jednak jest to
metoda nieselektywna i dlatego połączenie HPLC z detektorem
spektrofotometrycznym UV umożliwia analizę ilościową
olbrzymiej grupy związków organicznych
Do badania równowag reakcji chemicznych: zwłaszcza
wyznaczanie stałych dysocjacji kwasów i zasad, ustalanie
składu i stałych trwałości związków kompleksowych

Dziękuję za uwagę
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
Wyszukiwarka
Podobne podstrony:
FENOL ppt
fenol(1) ppt
13 Spektrometria masid 14798 ppt
03 Sejsmika04 plytkieid 4624 ppt
Choroby układu nerwowego ppt
10 Metody otrzymywania zwierzat transgenicznychid 10950 ppt
10 dźwigniaid 10541 ppt
03 Odświeżanie pamięci DRAMid 4244 ppt
Prelekcja2 ppt
2008 XIIbid 26568 ppt
WYC4 PPT
rysunek rodziny ppt
więcej podobnych podstron