
UKŁADY KOLOIDALNE
• Koloidy – układy dyspersyjne, o
wyglądzie układów fizycznie
jednorodnych, chociaż składniki nie są
ze sobą wymieszane na poziomie
cząsteczkowym.
• Faza ciągła układu – ośrodek
dyspersyjny, rozpraszający
• Faza rozproszona lub składnik
rozproszony
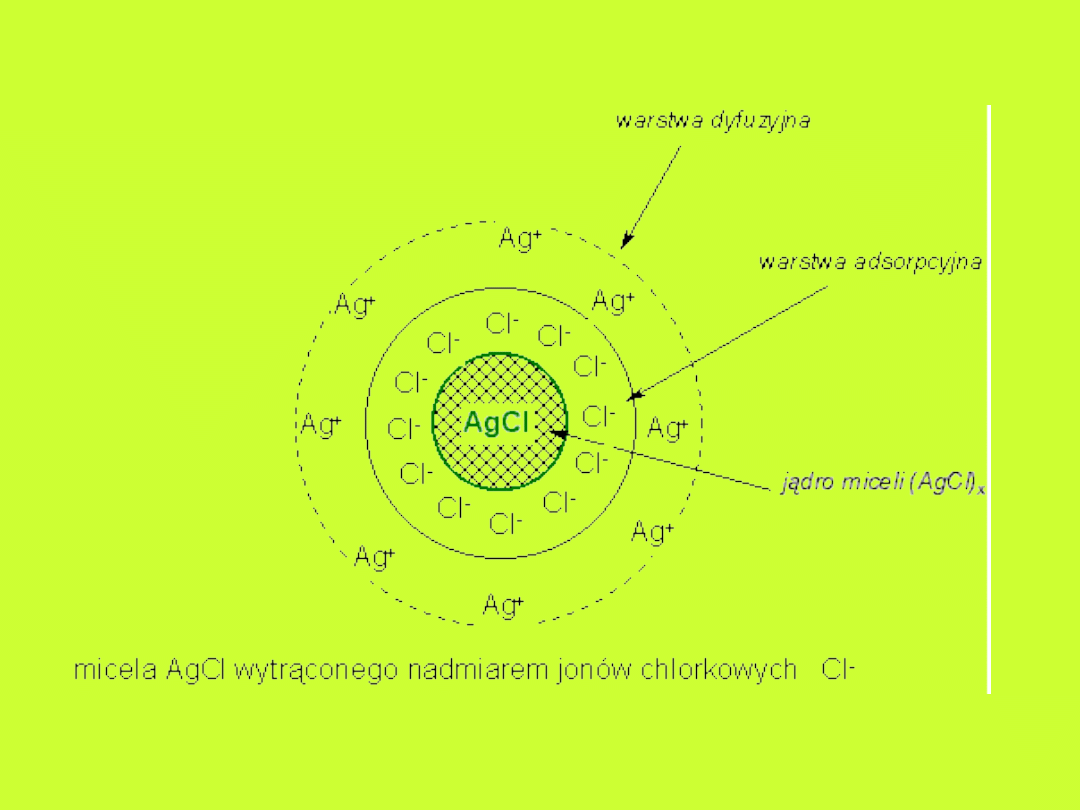

Faza rozproszona:
Wielkość cząstek: 1 – 500nm
Układy z cząstkami:
• trójwymiarowymi
• blaszkowatymi
• nitkowymi
Układ izodyspersyjny – cząstki o
jednakowej wielkości
Układ polidyspersyjny – cząstki o różnej
wielkości

• Ciekły ośrodek dyspersyjny: koloidy,
liozole, zole
woda – hydrozole
alkohol – alkozole
benzen – benzenozole
ciecz organiczna – organozole
gaz – gazozole
powietrze - aerozole

Koloidy liofilowe i liofobowe
Liofilowe (emulsoidy) - z cząstkami
solwatowanymi przez cząstki fazy
rozpraszającej (duże powinowactwo
do cząstek rozpuszczalnika)
Liofobowe (suspensoidy)– z cząstkami
niesolwatowanymi

WŁAŚCIWOŚCI UKŁADÓW
KOLOIDALNYCH
• ruchy Browna: ciągły,
nieuporządkowany ruch
• nie przenikają przez błony
przepuszczalne przez które
przechodzi rozpuszczalnik
• ruch pod wpływem pola
elektrycznego (elektroforeza)

• KOAGULACJA (żelowanie) –
zmniejszanie stopnia dyspersji
układów koloidalnych; wydzielenie
cząstek z roztworów (flokulacja)
ZOL
ŻEL
KOAGULACJA (ŻELOWANIE)
PEPTYZACJA (DYSPERGOWANIE)

Czynniki wywołujące koagulację:
• Dodatek elektrolitu
• Dodatek koloidu o ładunku przeciwnym
• Naświetlanie radiochemiczne
• Działanie mechaniczne (mieszanie,
wytrząsanie)
• Ogrzewanie
• Desolwatacja przez dodanie środków
odwadniających
• Odparowanie lub wymrażanie ośrodka
dyspergującego

Wysalanie – koagulacja koloidów liofilowych,
polega na wydzielaniu fazy rozproszonej
pod wpływem większych stężeń elektrolitu
Synereza – polega na wydzielaniu się żeli z
ośrodka dyspersyjnego po dłuższym
odstaniu i jest związana z procesami
koagulacji (wydzielanie serwatki z
kwaśnego mleka)
Tiksotropia – pod wpływem bodźców
mechanicznych (wytrząsanie, mieszanie)
żele przechodzą w zole, po pewnym czasie
zastygając w żel

Stechiometria
• Ustalenie równania reakcji
• Ustalenie bilansu materiałowego –
równania mas
• Ułożenie proporcji

Równanie
chemiczne
węglan + kwas → chlorek + dwutlenek
+woda
wapnia solny wapnia węgla
wzór
ilość
subst.
(mol)
masa, g
CaCO
3
+2HCl → CaCl
2
+CO
2
+H
2
O
1 + 2 → 1 + 1 +
1
100 +73 → 111 + 44 +
18
Ile wapna palonego można otrzymać z 500kg wapienia,
przyjmując zawartość CaCO
3
równą 100%?

Równowaga chemiczna
• Układ znajduje się w stanie
równowagi jeśli nie zachodzi w nim
zmiana stężeń jego składników.
aA+bB↔cC+dD, T=cons.,
p=const.
K=
c
C
c
c
D
d
c
A
a
c
B
b

Stan równowagi zależy od:
- temperatury
- ciśnienia
- dodatku reagentów

Termodynamika procesów
chemicznych
Służy do:
• określania kierunków i możliwości
przemian chemicznych
• obliczania bilansów energetycznych
• kolejności przebiegu przemian
• określania trwałości układów
• określania warunków procesów

Termodynamika procesów
chemicznych
FUNKCJE TERMODYNAMICZNE
U = ENERGIA WEWNĘTRZNA
U - jest sumą energii ruchu cząsteczek, drgań
atomów, stanu
jąder i elektronów
U - nie może być wyznaczona, ale można określić jej zmianę (U)
Zmiana energii wewnętrznej jest równa pracy
wymienianej przez układ z otoczeniem w
przemianie adiabatycznej (Q = 0)

ENTROPIA
S - opisuje sposób dystrybucji energii w
układzie
S - jest miarą uporządkowania układu


Procesy prowadzone są zwykle w specyficznych
warunkach:
adiabatycznie Q = 0
izotermiczne T = const
izobarycznie P = const
izochorycznie V = const
izentalpowe H = const
termoelastyczne T, V, P
zmienne
Rodzaje pracy: objętościowa,
mechaniczna, chemiczna,
powierzchniowa, elektryczna, itp

I zasada termodynamiki
W układzie izolowanym (bez wymiany
ciepła i masy) ilość energii pozostaje stała
∆U=Q+W
∆U – zmiana energii wewnętrznej układu
Q – ilość energii przekazana na sposób
ciepła
W – ilość energii przekazana na sposób
pracy

II zasada termodynamiki
• Podstawą tej zasady jest nieodwracalność
samorzutnych procesów przyrody.
Zachodzą one od stanu o wyższym
potencjale do stanów o potencjale niższym,
dążąc do osiągnięcia stanu równowagi
dQ
T
dS=
S- entropia
Q – ciepło przemiany
odwracalnej
T=const

III zasada termodynamiki
Układowi można odebrać pewną
skończoną ilość energii na sposób
ciepła, wówczas temperatura zmierza
do zera bezwzględnego a entropia do
pewnej minimalnej wartości.
W temp. zera bezwzględnego entropia
dla materiałów krystalicznych wynosi 0

Kinetyka reakcji
chemicznych
• opisuje przebieg reakcji w czasie,
• w czasie trwania reakcji zmienia się
stężenie reagentów
• chwilowa szybkość reakcji
chemicznej:
v=-dc
s
/dt lub v=dc
p
/dt
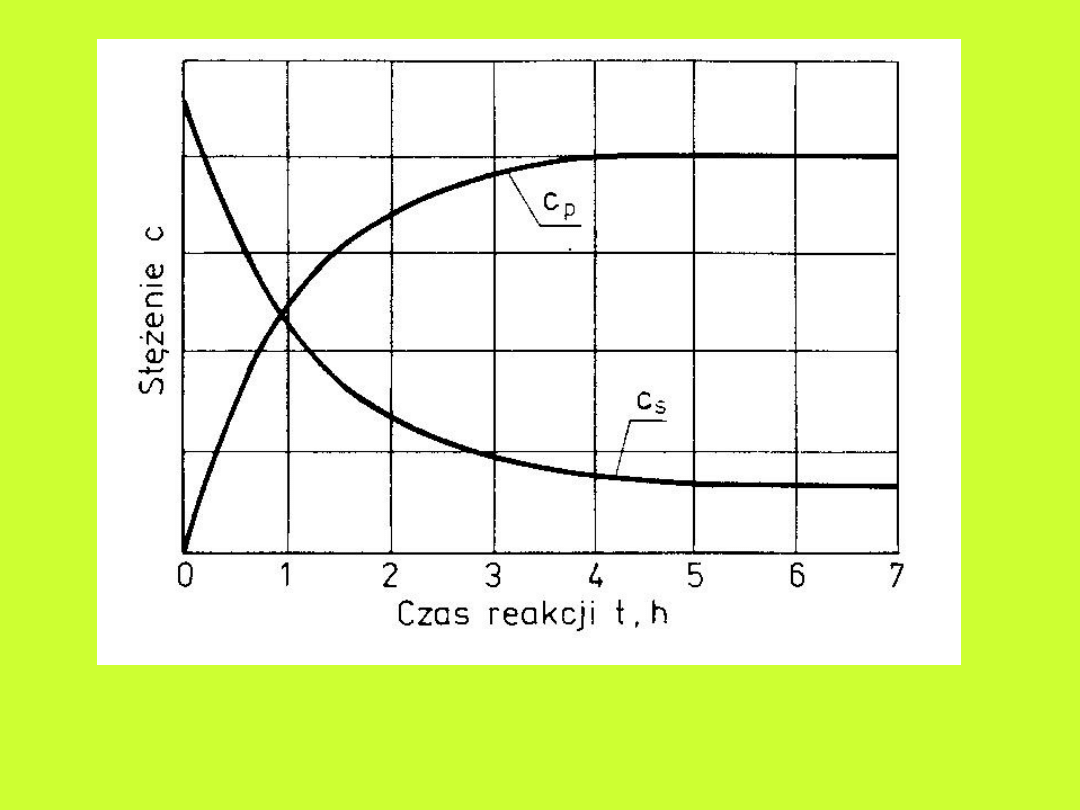
Zmiana stężenia reagentów w czasie

Szybkość reakcji chemicznej zależy od:
- stężenia substratów
- stanu skupienia reagentów
- stopnia kontaktu:
stopnia rozdrobnienia (ciała stałe)
intensywności mieszania (ciecze)
ciśnienia (gazy)
- temperatury
- katalizatorów

Równanie opisujące szybkość reakcji
v
=
-
dc
A
dt
=kc
n
k – stała szybkości reakcji,
n – rząd reakcji
c
A
– stężenie substratu

Zależność szybkości reakcji od
temperatury
wzór Arrheniusa
k=k
o
e
-Ea/RT
lnk=lnk
o
-(E
a
/RT)
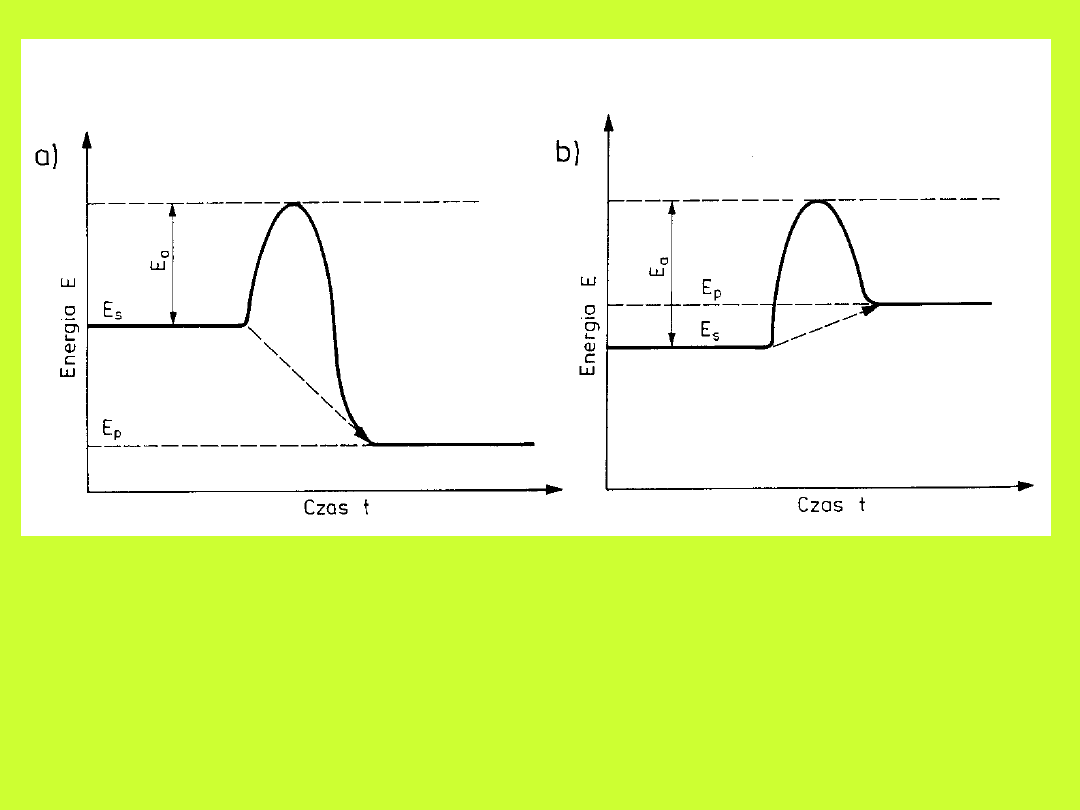
Zmiany energii podczas przebiegu reakcji
a) egzotermicznej, b) endotermicznej
E
s
- energia substratów, E
a
– energia aktywacji, E
p
–
energia produktów

ELEKTROCHEMIA
• przemiana energii elektrycznej w
chemiczną (elektroliza)
• przemiana energii chemicznej w
elektryczną (ogniwa galwaniczne)

I prawo Faraday’a
Masa substancji wydzielonej na elektrodzie
jest proporcjonalna do wielkości ładunku
elektrycznego, który przepłynął przez
elektrolit
m = kIt [g]
k=M/(z*F)
M – masa molowa [g/mol],
F – stała Faradaya=96500[C/mol],
z – liczba elektronów wymienionych w reakcji
I – natężenie prądu elektrycznego [A]
t – czas przepływu prądu [s]

II prawo Faraday’a
Masy różnych substancji wydzielonych
na elektrodach podczas przepływu
jednakowego ładunku elektrycznego
są proporcjonalne do ich
równoważników elektrochemicznych
m
1
/m
2
=k
1
/k
2

OGNIWA ELEKTRYCZNE
• proces odwrotny do elektrolizy –
reakcji chemicznej towarzyszy
przepływ prądu elektrycznego,
• Ogniwo – dwa półogniwa (elektrody)
zanurzone w elektrolicie (roztworze
swoich jonów)
• SEM – różnica potencjałów półogniw
SEM=E
1
– E
2

• Potencjał normalny elektrody (E
0
):
płytka metalu zanurzona w 1M
roztworze elektrolitu, potencjał
mierzony w stosunku odniesienia do
elektrody wodorowej


• Im bardziej ujemny potencjał normalny, tym
większa skłonność metalu do przejścia w
stan jonowy, czyli oddania elektronów,
• Potencjały normalne metali szlachetnych –
dodatnie, nieszlachetnych – ujemne
• Metale nieszlachetne wypierają metale
szlachetne z ich roztworów, np.
Zn+Cu
2+
→Zn
2+
+Cu
• Metale o ujemnym potencjale normalnym
przeprowadzają wodór ze stanu jonowego w
cząsteczkowy, rozpuszczają się w kwasach
• Skłonność niemetali do przechodzenia w
stan jonowy jest tym większa im bardziej
dodatni E
0
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
Wyszukiwarka
Podobne podstrony:
Wyklad 2 Zaoczni
PrawoUpadłościoweINaprawcze Wykład zaoczne całość 2012
pp program wykladu zaoczne 03, wisisz, wydzial informatyki, studia zaoczne inzynierskie, podstawy pr
Wyklad 6 Zaoczni
Wyklad 4 5 Zaoczni
wyklady 1-3 zaoczne, Administracja UŁ, Administracja I rok, Zasady tworzenia i stosowania prawa
wyklad 6 zaoczne, Administracja UŁ, Administracja I rok, Zasady tworzenia i stosowania prawa
wyklady 4-5 zaoczne, Administracja UŁ, Administracja I rok, Zasady tworzenia i stosowania prawa
wyklady 7-8 zaoczne, Administracja UŁ, Administracja I rok, Zasady tworzenia i stosowania prawa
wyklad 9 zaoczne, Administracja UŁ, Administracja I rok, Zasady tworzenia i stosowania prawa
Wyklad 3 Zaoczni
Ekonomia społeczna Wyklady zaoczne
controlling wyklad zaoczne
kryminologia wykladydadak zaoczni
Wyklad 4 5 Zaoczni
więcej podobnych podstron