
1
CHOROBY
DZIEDZICZONE
AUTOSOMALNIE
RECESYWNIE
Dr med. Tomasz Byrski
Zakład Genetyki i Patomorfologii
PAM

2
CECHY DZIEDZICZENIA
AUTOSOMALNEGO RECESYWNEGO
1. Choroba występuje u homozygot
2. Małe ryzyko dla potomstwa
3. Jednakowa częstość i ciężkość
objawów
u obu płci
4. Stała ekspresja w rodzinie
5. Poziomy wzór rodowodu
6. Ważne pokrewieństwo rodziców

3
Choroby recesywne
autosomalne
CHOROBA
Częstość na 1000 urodzeń
Mukowiscydoza
0,5
Upośledzenie umysłowe rec.
0,5
Głuchota wrodzona
0,2
Fenyloketonuria
0,1
Rdzeniowy zanik mięśni
0,1
Ślepota recesywna
0,1
Zespół nadnerczowo-płciowy
0,1
Mukopolisacharydozy
0,1
Inne
0,3
Ogółem
2/1000

4
CYSTIC FIBROSIS –
CF
MUKOWISCYDOZA
CZĘSTOŚĆ NOSICIELSTWA:
1:20 – 1:25
CZĘSTOŚĆ CHOROBY:
1:2300

5
Mucoviscidosis
Objawy ze strony układu oddechowego
(występują u ponad 90% chorych)
gęsty i lepki śluz, który zalega w oskrzelach i jest
podłożem dla rozwoju bakterii
występuje uciążliwy kaszel, niekiedy duszność (pierwsze
objawy mogą wystąpić już w wieku niemowlęcym)
nawracające zapalenia oskrzeli i płuc, trudno poddające
się typowemu leczeniu, prowadzą do rozstrzeni oskrzeli i
włóknienia płuc
przewlekłe zapalenie zatok bocznych nosa z polipami
(głównie u starszych dzieci i dorosłych)

6
Mucoviscidosis
Objawy ze strony przewodu pokarmowego
(występują u około 75% chorych)
gęsty i lepki śluz blokuje przewody trzustkowe i
przyjmowane pokarmy nie są odpowiednio trawione i
wchłaniane, co prowadzi do zaburzeń odżywiania
występują obfite, nie uformowane, cuchnące,
tłuszczowate stolce (najczęściej od wczesnego
dzieciństwa)
powiększenie objętości brzucha, niekiedy wypadanie
odbytnicy
niedrożność smółkowa jelit w okresie noworodkowym
(często pierwszy objaw mukowiscydozy)

7
Mucoviscidosis
Niektóre inne objawy:
opóźnienie rozwoju fizycznego (niedobór masy i
wysokości ciała)
upośledzenie rozwoju mięśni (np. brak pośladków)
nadmierna męczliwość (szybkie męczenie się przy
wysiłku)

8
Mucoviscidosis
Szczególnego charakteru chorobie
nadaje fakt, że rozpoznanie
mukowiscydozy
u dziecka jest jednoznaczne z
rozpoznaniem rodziny wysokiego
ryzyka genetycznego!

9
Mucoviscidosis
Częstość występowania mukowiscydozy w Polsce
oceniana jest na 1:2300 żywo urodzonych.
Chorobą tą dotknięte są zarówno noworodki,
niemowlęta, dzieci starsze, jak i dorośli.
Długość przeżycia, jak i jakość życia pacjentów w
dużej mierze zależą od wczesnego rozpoznania i
prawidłowego leczenia.
Mimo znacznego postępu konwencjonalnych
metod terapii, średnia długość życia chorych nie
przekracza 30 roku życia.

10
Mucoviscidosis
Przy częstości urodzeń 400 000, w Polsce rocznie
rodzi się około 200 dzieci dotkniętych tą chorobą.
Około dwóch milionów Polaków, w równym
stopniu mężczyzn i kobiet, jest nosicielami
zmutowanego genu CFTR.
Polski Rejestr Mukowiscydozy, prowadzony w
Instytucie Gruźlicy i Chorób Płuc (Zespół
Pediatryczny im Jana i Ireny Rudników w Rabce), w
1998 zawierał informacje o 899 żyjących chorych z
mukowiscydozą.

11
Mucoviscidosis
Struktura genu
Gen CFTR (ang. cystic fibrosis transmembrane
regulator)
został sklonowany w 1989 roku. Stało
się to w niespełna cztery lata od chwili określenia
jego lokalizacji w długim ramieniu
chromosomu 7
(prążek q31.3).
Pozycja mapowa została ustalona
dzięki wykazaniu sprzężenia genu CFTR ze
znanymi markerami chromosomalnymi. Gen CFTR
zajmuje obszar
około 250 kb i składa się z 27
eksonów
. Jest jednym z większych znanych genów
człowieka. Wielkość eksonów jest zróżnicowana:
od 38 nukleotydów (ekson 14b) do 724
nukleotydów (ekson 13). Wielkości intronów
wahają się od 1,1 kb do 40 kb.

12
Mucoviscidosis
Funkcja białka
(1)
Sekwencja aminokwasów białka kodowanego
przez gen CFTR została określona na podstawie
analizy sekwencji nukleotydów. Kodowany w tym
genie polipeptyd, o masie cząsteczkowej około 170
kD, złożony jest z 1480 aminokwasów. Wyróżnia
się w nim szereg domen. Pozycję centralną białka
zajmuje domena R (regulacyjna). Po obu jej
stronach znajdują się domeny określane jako:
domena wewnątrzbłonowa i domena wiążąca
nukleotydy (ang. nucleotide binding folds, NBF).

13
Mucoviscidosis
Funkcje białka (2)
Białko CFTR ze względu na swoją strukturę i
funkcje zostało zaklasyfikowane do dużej grupy
białek transportowych tzw. "ABC family" (ang.
ATP-binding cassette) występujących zarówno u
bakterii, drożdży, muszki owocowej oraz u ssaków
Jest ono błonową glikoproteiną.
U człowieka pełni funkcje kanału chlorkowego w
komórkach nabłonkowych. Przypisuje mu się
również rolę w transporcie białek przez błonę
komórkową. Uszkodzenie tych funkcji, w wyniku
mutacji, prowadzi do zmiany kwasowości w
organellach komórkowych, powstawania lepkiego
(zahamowanie przepływu, wraz z jonami Cl-, wody)
o odmiennym składzie białek śluzu

14
Mucoviscidosis
Mutacje
Większość z opisanych w genie CFTR mutacji to
mutacje punktowe.
Mutacje missens - około 45%
Mutacje nonsens - około 18%
Mutacje o charakterze małych wstawek/delecji
(około 23% wszystkich znanych mutacji)
szereg mutacji na złączach intron/ekson/intron
oraz mutacji wewnątrzintronowych
odpowiedzialnych za błędną obróbkę transkryptu -
około 14%
wielkie delecje występują rzadko. Największa
znana delecja, około 40 tysięcy nukleotydów
obejmuje eksony od 11 do 18.

15
Mucoviscidosis
Heterogenność mutacji w genie CFTR
Do końca 2005 roku w genie CFTR opisano około
800 różnych mutacji odpowiedzialnych za
modyfikację lub brak jego funkcji. Trudno obecnie
wyjaśnić przyczynę tak wielkiej różnorodności
mutacji w tym genie. Nawet w populacjach
określanych jako wysoce homogenne
heterogenność mutacji jest znaczna.
Do wyjątków należy kanadyjska populacja
Hutteire, w której badając częstość występowania
dwóch mutacji (∆F08 i M1101K) identyfikuje się
wszystkie zmutowane geny CFTR.
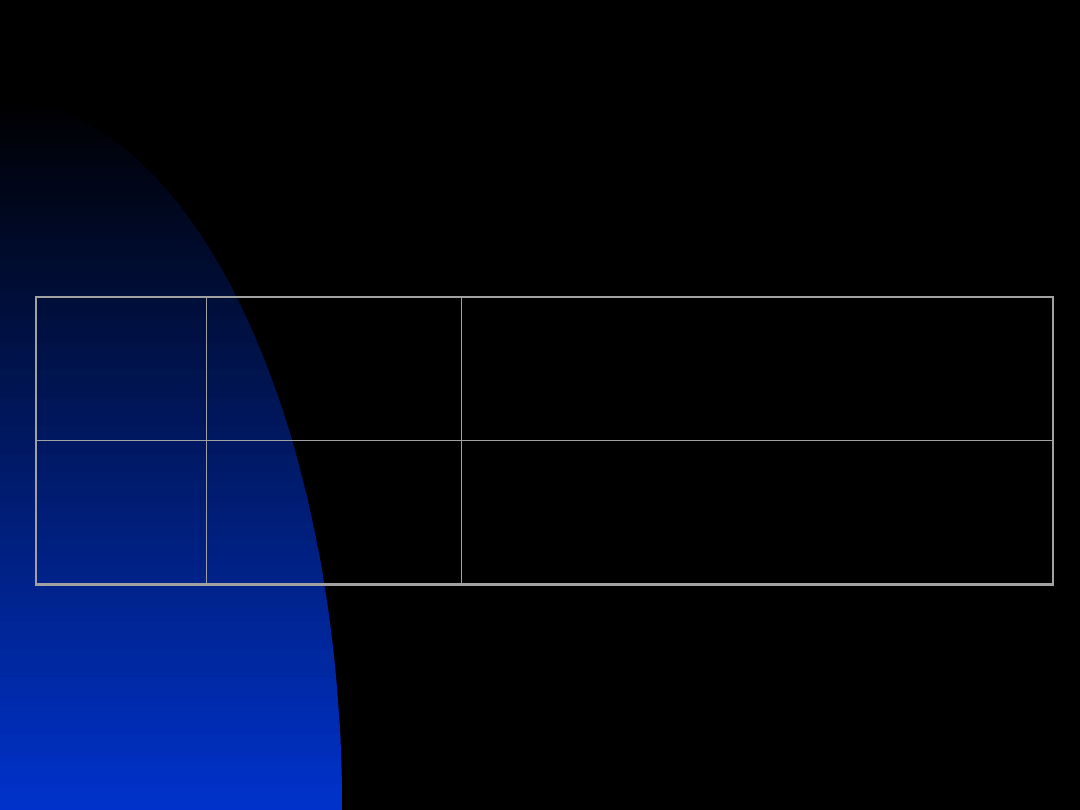
16
Molecular Genetics of CFTR-Related
Disorders
Gene
Symbol
Chromoso
mal Locus
Protein Name
CFTR
7q31.2
Cystic fibrosis transmembrane
conductance regulator

17
Mucoviscidosis
Mutacje najczęściej odpowiedzialne za defekt genu CFTR
Nazwa mutacji
Lokalizacja
Badana populacja (%)
Świat*
(%)
∆F508
ex 10
54
66
del21kb
del2,3
2,6
G542X
ex 11
2,5
2,4
3849+10kb >T
in 19
2,3
0,2
1717-1G>A
in 10
2,2
0,6
N1303K
ex 21
1,9
1,3
R553X
ex 11
1,2
0,7
2184insA
ex4
0,9
0,1
W1282X
ex 20
0,6
1,2
mutacje sporadyczne
5,0
allel CF (nieokreślony)
26,8
*
dane Cystic Fibrosis Genetic Analysis Consortium
Zacieniowano mutacje najczęściej występujące w populacji polskiej
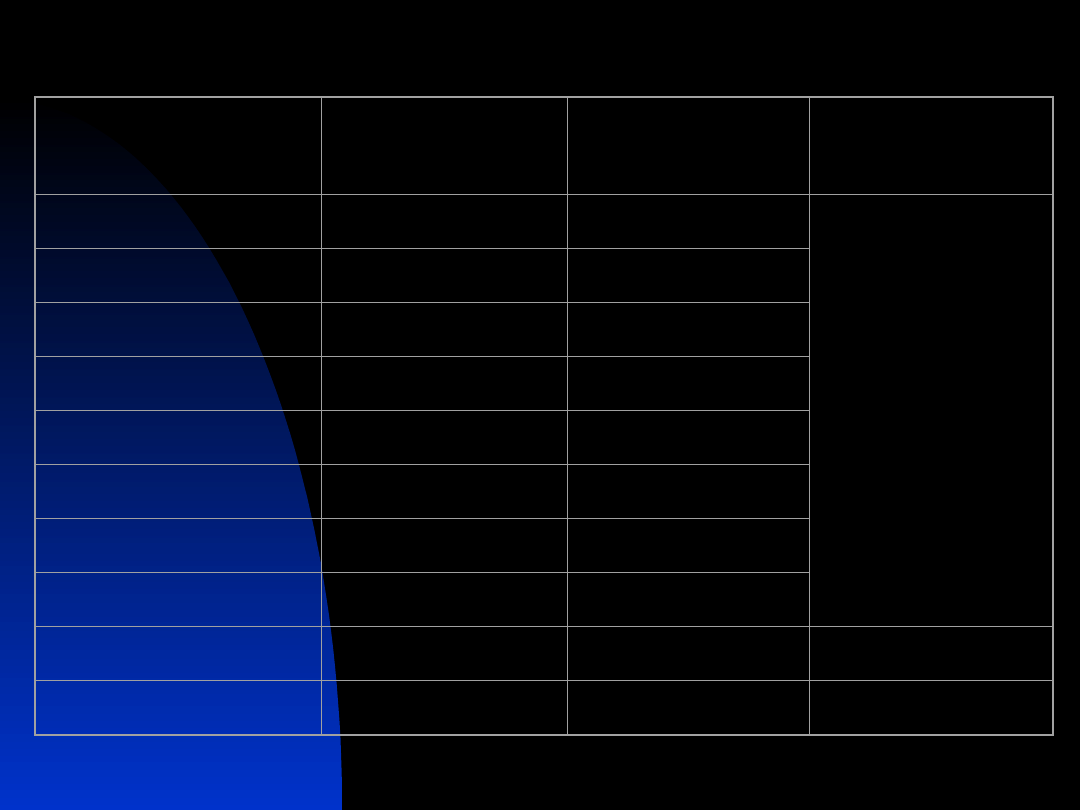
18
Ten Most Common CFTR
Relative
Frequency
Phenotype
F508
66.0%
II
Classic
G542X
2.4%
I
G551D
1.6%
III
N1303K
1.3%
II
W1282X
1.2%
I
R553X
0.7%
I
621+1G->T
0.7%
I
1717-1G->A
0.6%
I
R117H
0.3%
IV
Nonclassic
R1162X
0.3%
Not clear
2
Classic

19
Mucoviscidosis
Diagnostyka prenatalna
Diagnostyka prenatalna mukowiscydozy
oferowana jest w I i II trymestrze ciąży.
Źródłem
DNA do analizy molekularnej są komórki
trofoblastu lub amniocyty
.
Decyzje o poddaniu się badaniom prenatalnym
podejmowane są także przez te rodziny, które nie
decydują się na przerwanie ciąży po rozpoznaniu
prenatalnym choroby.

20
Mucoviscidosis
KOSZTY BADAŃ
W praktyce, w badaniach rutynowych
identyfikowane są te mutacje, które włączono w
handlowo dostępne zestawy diagnostyczne. W
chwili obecnej poza del21kb identyfikacja
wszystkich pozostałych mutacji jest możliwa za
pomocą takich testów.
Wykorzystywane do tej pory testy firmy
Inogenetics umożliwiały identyfikację 8 mutacji
(∆F508, ∆I507, G542X, 1717-1G>A, N1303K,
R553X, W1282X, G551D).
Mutacje del21kb i
3849+10kb wymagały wykonania dodatkowo
niezależnej analizy.
Koszty materiałowe testu Inogenetics dla jednego
pacjenta kształtowały się w granicach 250 zł.
Nowe zestawy umożliwiają identyfikację 27
mutacji za cenę dwukrotnie wyższą.

21
Mucoviscidosis
BADANIA MOLEKULARNE MUKOWISCYDOZY W
POLSCE
Analizę DNA w mukowiscydozie w Polsce
rozpoczęto w 1986 roku w Zakładzie Genetyki
Instytutu Matki i Dziecka.
Od kilku lat badania molekularne prowadzone są
również w Zakładzie Genetyki Człowieka PAN w
Poznaniu.
W obu tych ośrodkach rutynowo identyfikuje się
najczęściej występujące mutacje w genie CFTR.
Diagnostyka prenatalna mukowiscydozy
wykonywana jest jedynie w Zakładzie Genetyki
Medycznej Instytutu Matki i Dziecka.

22
Mucoviscidosis
Podstawowym celem molekularnych badań
diagnostycznych w mukowiscydozie jest
weryfikacja rozpoznania klinicznego oraz
zapewnienie rodzinom ryzyka
genetycznego odpowiedniej opieki
lekarskiej
.
Wydaje się jednak, że badania molekularne
mukowiscydozy, tak jak i innych chorób
dziedzicznych mają dodatkowy wymiar
jakim jest
profilaktyka. Jednym z
elementów profilaktyki wtórnej są badania
przesiewowe

23
Mucoviscidosis
The diagnosis of cystic fibrosis (CF) is established in
individuals with the following:
One or more characteristic phenotypic features of CF
AND
Evidence of an abnormality in cystic fibrosis
transmembrane conductance regulator (CFTR) function
based upon ONE of the following:
Presence of two
in the CFTR
OR
Two abnormal quantitative pilocarpine iontophoresis
sweat chloride values (>60 mEq/L)
OR
Transepithelial nasal potential difference (NPD)
measurments characteristic of CF

24
Mucoviscidosis
Sweat chloride.
A chloride concentration of greater than 60 mEq/L in
sweat on two separate occasions is diagnostic. This
test is positive in over 90% of individuals with CF.
Sweat chloride levels higher than 160 mEq/L are not
physiologically possible and should be attributed to
technical error.
False positive sweat chloride results may be
associated with other conditions, most notably
(Hurler syndrome).
False negative sweat chloride results may be obtained
in the setting of acute CF-related salt losses.

25
Mucoviscidosis
Transepithelial nasal potential difference (NPD).
Respiratory epithelia regulate ion transport and alter
content of the airway surface fluid by active transport
mechanisms. The absence of functional CFTR at the
apical surface with resultant alterations in chloride efflux
and sodium transport produces an abnormal electrical
potential difference across epithelial surfaces.
The protocol for NPD measurements in individuals over
six years of age is well described, standardized, and
safely performed in many specialized CF centers
worldwide.

26
Mucoviscidosis
Newborn screening has been implemented using
immunoreactive trypsinogen (IRT) assays performed on
blood spots.
IRT levels are elevated in cystic fibrosis.
Abnormal results are therefore further evaluated with
molecular genetic testing of the CFTR gene OR sweat
testing after the child is at least two months of age (if
two disease-causing mutations are not identified)

27
Mucoviscidosis
CFTR molecular genetic testing is the initial diagnostic
test:
Prenatal testing, in a high-risk fetus
Prenatal diagnosis in low-risk fetus with echogenic
bowel
Newborn screening
Symptomatic infants (i.e., those with
meconium ileus) who are too young to produce
adequate volumes of sweat.
Testing of symptomatic sibs of an affected
individual in whom both CFTR
have
been identified

28
Mucoviscidosis
Prenatal Testing
High-risk pregnancies - molecular genetic testing of
DNA extracted from fetal cells obtained by amniocentesis
at 16-18 weeks' gestation or chorionic villus sampling
(CVS) at about 10-12 weeks' gestation. The disease-
causing mutations of the CFTR gene must be identified in
both parents before prenatal testing can be performed.
Indeterminate-risk pregnancies. In cases in which
one parent is known to be a CFTR mutation carrier and
the other parent has tested negative for a panel of CFTR
alleles.
Low-risk pregnancies. The finding of fetal echogenic
bowel and/or dilated bowel on USG is associated with an
increased risk for CF in a pregnancy previously not known
to be at increased risk for CF.

29
Mucoviscidosis
Respiratory:
-
- lower airway inflammation and chronic endobronchial
infection (Staphylococcus aureus and Pseudomonas
aeruginosa)
-
- airway obstruction and intense neutrophilic
inflammation
-
(chronic cough, intermittent sputum production and
dyspnea)
-
- chronic endobronchitis, structural injury to the airways
-
(bronchiectasi)
-
- end-stage lung disease is characterized by extensive
damage to the airways (cysts/abscesses) and
accompanying fibrosis of lung parenchyma adjacent to
airways

30
Mucoviscidosis
Gastrointestinal:
Meconium ileus occurs (15-20%)
Pancreatic insufficiency with malabsorption (92% of
individuals - enzyme supplements)
Cystic fibrosis-related diabetes mellitus (CFRDM) - 3.1%
of children under 18 years of age require insulin. The
etiology is a combination of reduced insulin secretion
(secondary to fibrosis of the pancreas and reduced
number of islet cells) and peripheral insulin resistance
Hepatobiliary disease, with elevation of serum
concentration of liver enzymes in school-age children,
As liver disease progresses, individuals develop portal
hypertension and varices.

31
Mucoviscidosis
Fertility:
More than 95% of males with CF are infertile as a result
of azoospermia caused by altered vas deferens, which
may be absent, atrophic or fibrotic.
The body and tail of the epididymis and seminal vesicles
may be abnormally dilated or absent.
Women with CF are fertile, although a few females have
abnormal cervical mucus that may contribute to infertility.

32
Mucoviscidosis
Nowe kierunki leczenia
inhalacje liposomów - pozwalające na upłynnienie
śluzu i ułatwiające jego ewakuację;
inhalacje rekombimowanych antypro-teaz,
hamujące uszkodzenia oskrzeli przez enzymy
proteolityczne;
inhalacje (UTP) zwiększające uwodnienie płynu
okołorzęskowego poprzez zwiększenie wydzielania
Cl do światła oskrzeli;
stosowanie cytokin, ograniczające aktywność
stanu zapalnego;
immunoglobuliny hiperimmunonizowane
przeciwko najbardziej szkodliwej bakterii,
Pseudomonas aeuroginosa;
terapię genową, polegającą na wprowadzeniu
prawidłowego genu CFTR do komórek nabłonka
oddechowego; od trzech lat trwają próby
kliniczne.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
Wyszukiwarka
Podobne podstrony:
choroby dziedziczone autosomalnie recesywnie
DZIEDZICZENIE AUTOSOMALNE RECESYWNE
dziedziczenie autosomalne recesywne, genetyka, kolokwia pytania i odp
Dziedziczenie autosomalne recesywne
Mutacje genowe autosomalne dominujące i recesywne
choroby naczyn i serca(1)
ŻYWIENIE A CHOROBY 4b
Choroby układu nerwowego ppt
Produkty przeciwwskazane w chorobach jelit II
Choroba niedokrwienna serca
CZLOWIEK I CHOROBA – PODSTAWOWE REAKCJE NA
Choroby nerwów czaszkowych
Choroby Cywilizacyjne 5
więcej podobnych podstron