
Farmakologia Ogólna
prof. dr hab. med. Ivan KOCIĆ
KATEDRA I ZAKŁAD FARMAKOLOGII
GUMed

Podstawowe pojęcia
• Lek
• Farmakokinetyka
• Farmakodynamika
• Farmakologia kliniczna
• Toksykologia
• Stosowanie leków u dzieci, ludzi
starszych i kobiet w ciąży
• Niepożądane działania i interakcje
leków

LEK
Surowiec farmaceutyczny
Pochodzenia roślinnego
• Pochodzenia zwierzęcego
• Leki BIOLOGICZNE (insulina, EPO,
MONOKLON.P.)
• NAZWY LEKÓW
• Międzynarodowa
• Handlowa
• Oryginał i generik

Definicje
• Substancje lecznicze: związki czynne mogące
służyć zapobieganiu, łagodzeniu, leczeniu czy
rozpoznaniu chorób
• Środek leczniczy: substancja lecznicza w
odpowiedniej postaci przeznaczonej do stosowania u
ludzi lub zwierząt
• Trucizna (substancja szkodliwa) jest substancją
czynną wywołującą działanie szkodliwe nawet w
najmniejszych dawkach
• Siła działania leku: zależy od dawki lub stężenia
potrzebnego do wywołania efektu klinicznego
• Skuteczność leku: odnosi się do efektu klinicznego

• Farmakokinetyka: zajmuje się wpływem
organizmu na lek, to znaczy losem leku w
ustroju w zależności od czasu
• Farmakodynamika: to nauka o
działaniu leku na organizm, zajmuje się
mechanizmami działania leków
• Farmakologia kliniczna zajmuje się
klinicznymi aspektami stosowania leków,
od badan klinicznych, poprzez
farmakogenetykę i farmakoepidemiologię
do farmakoekonomiki.

Dawki
• Okno terapeutyczne
• Rząd wielkości dawki
• Wielkość cząsteczki-MW od 7 (Lit) do
50000 (streptokinaza)

Badania na zwierzętach
• LD50
• ED50
• IT=LD50/ED50

Badania na ludziach
• Faza1-wstępna ocena
• Faza 2-kontrolowana ocena
kliniczna:placebo,nocebo,
maskowane próby
• Faza3-rozszerzona badania kliniczne,
wieolośrodkowe
• Faza 4-od wprowadzenia leku do
lecznictwa do jego wycofania

Zmienność reakcji na lek
• Idiosynkrazja
• Alergia
• Wpływ choroby
Interakcje leków
• Synergizm
• Antagonizm
• Fizjologiczny i farmakologiczny

Interakcje w farmakologii
klinicznej
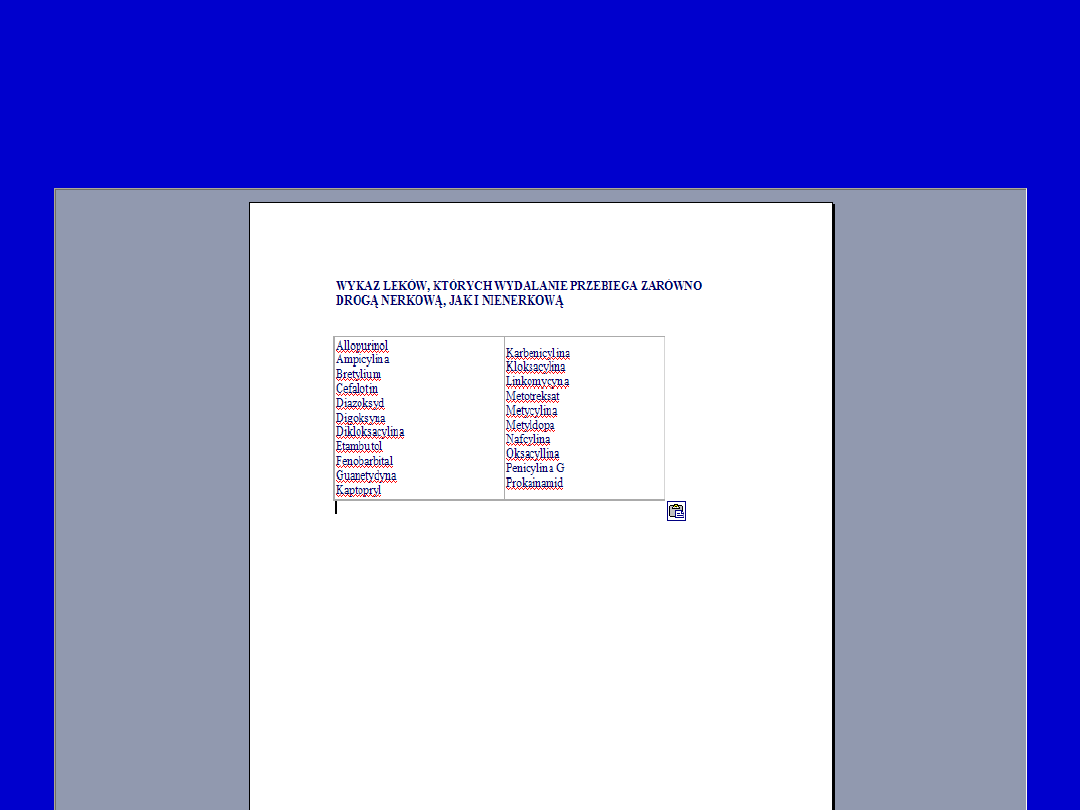
• Wpływ na wchłanianie
• Wpływ na wiązanie się z białkami
• Wpływ na wydalanie
• Hamowanie metabolizmu
• Aktywacja enzymów

Kumulacja, tolerancja,
tachyfilaksja
• KUMULACJA: digoksyna (lek gromadzi się w
niektórych tkankach)
• TOLERANCJA-azotany, opioidy (kolejna
taka sama dawka leku po kilkudniowym
leczeniu powoduje coraz mniejszy efekt)
• TACHYFILAKSJA-efedryna (każda kolejna
dawka leku daje coraz słabszy efekt,
działanie leku zależne od iłości substancji
endogennej zmagazynowanej w
odpowiednich strukturach biologicznych)

FARMAKOKINETYKA
• L-liberation (uwolnienie)
• A-absorption (wchłanianie)
• D-distribution (rozmieszczenie)
• M-metabolism (metabolizm)
• E-elimination (usuwanie leku z
organizmu)
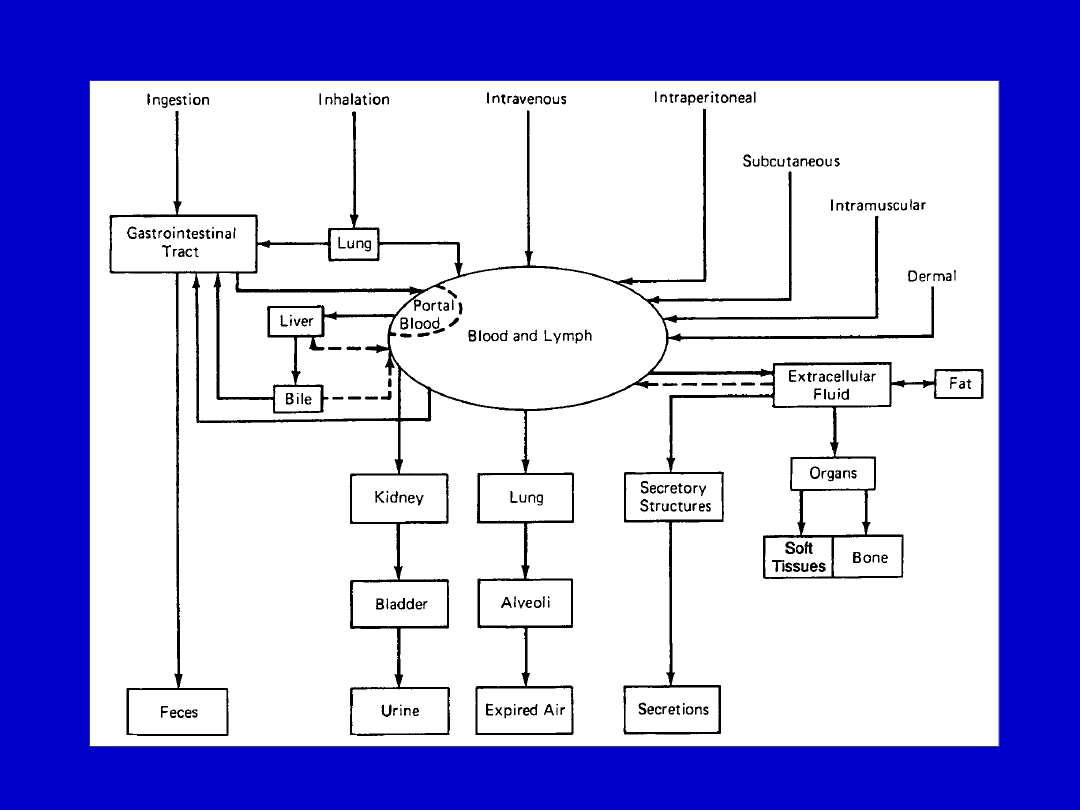
Rozmieszczenie leków w organizmie
The disposition of chemicals entering the body (from C.D. Klaassen, Casarett and Doull’s
Toxicology, 5th ed., New York: McGraw-Hill, 1996).
związa
ny
wolny
woln
y
związany
Miejsce działania
“RECEPTORY
”
Magazyn w tkance
Układ krążenia
Wolny lek
związany
WCHŁANIANIE
WYDALANIE
BIOTRANSFORMAC
JA

Reguła FICK-a
przepływ cząsteczek leku=
(C1-C2)x powierzchnia x PK/grubość
błony
Równanie HENDERSON-HASSELBALCH-a
log protonated/unprotonated=pKa-pH
Transport przez błony
•
Dyfuzja pasywna:
-
Przepuszczalność błony
- Różnica stężeń
- Powierzchnia wchłaniania
Przepuszczalność:
błona komórkowa:
- rozpuszczalność w tłuszczach
- pH środowiska
- pK leków
śródbłonek
wielkość
,
kształt i ładunek
Przejście przez błony
Pasywnie
ułatwione
Aktywnie

Wchłanianie w przewodzie
pokarmowym
• Rozpuszczalność leku
• Stabilność chemiczna leku w soku
żołądkowym
• perystaltyka
• Obecność i rodzaj pokarmu
• Rytm opróżniania
EFEKT PIERWSZEGO PRZEJŚCIA

Nasopharyn
gealRegion
5-30 µm
Trachea
Bronchi
Bronchiol
es
1-5 µm
Alveolar Region
1 µm
DEPOSITION OF
PARTICLES IN THE
RESPIRATORY SYSTEM

Skóra-wchłanianie
• Kilka warstw (stratum corneum,
epidermis, dermis) to reach blood
vessels.
• Ważne czynniki:
rozpuszczalność w tłuszczach
hydratacja skóry
miejsce (e.g. stopy vs. worek
mosznowy)

Inne drogi
• Dootrzewnowo
large surface area, vascularized, first
pass effect.
• Domięśniowo, podskórnie,
wśródskórnie
:
absorption through
endothelial pores into the circulation;
blood flow is most important + other
factors
• dożylnie

Biodostępność
Definicja: Ilość leku która dostaje się do
krążenia po podaniu innym niż i.v.
i.v.: 100%
nie i.v.: od 0 do 100%
e.g. lidocaine bioavailability 35% due to
destruction in gastric acid and liver metabolism
Efekt pierwszego przejścia
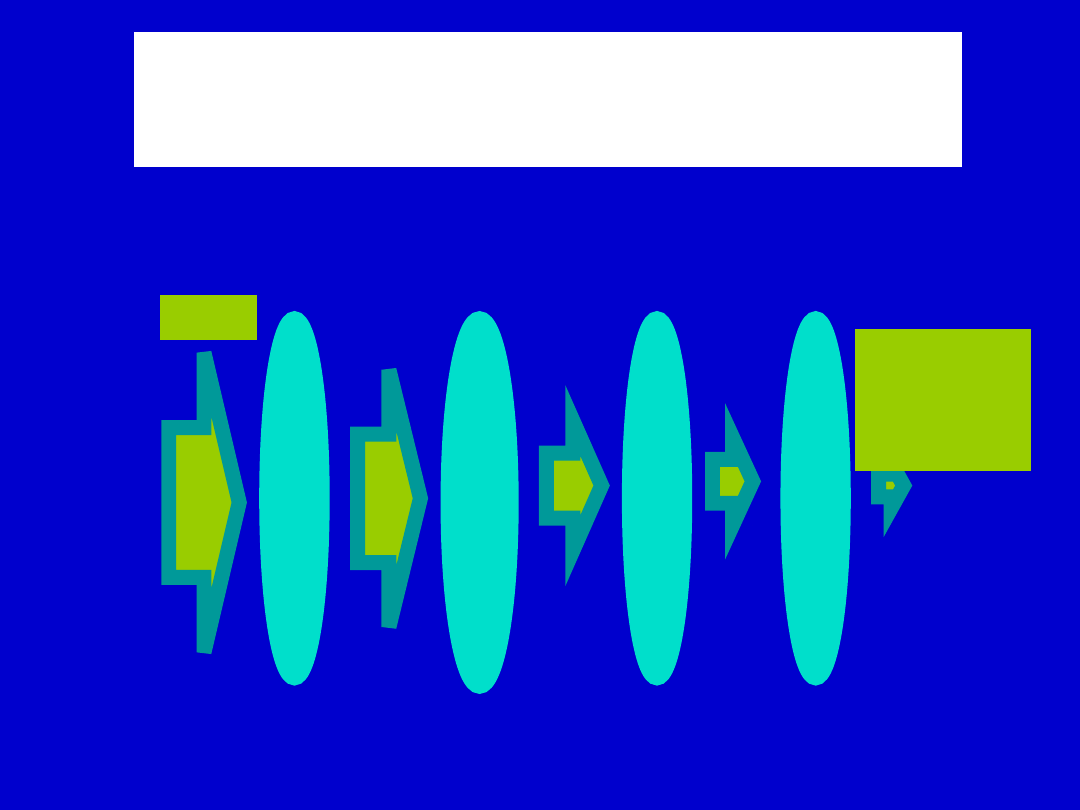
Bioavailability
Dose
Destroyed
in gut
Not
absorbe
d
Destroyed
by gut wall
Destroyed
by liver
to
systemic
circulatio
n
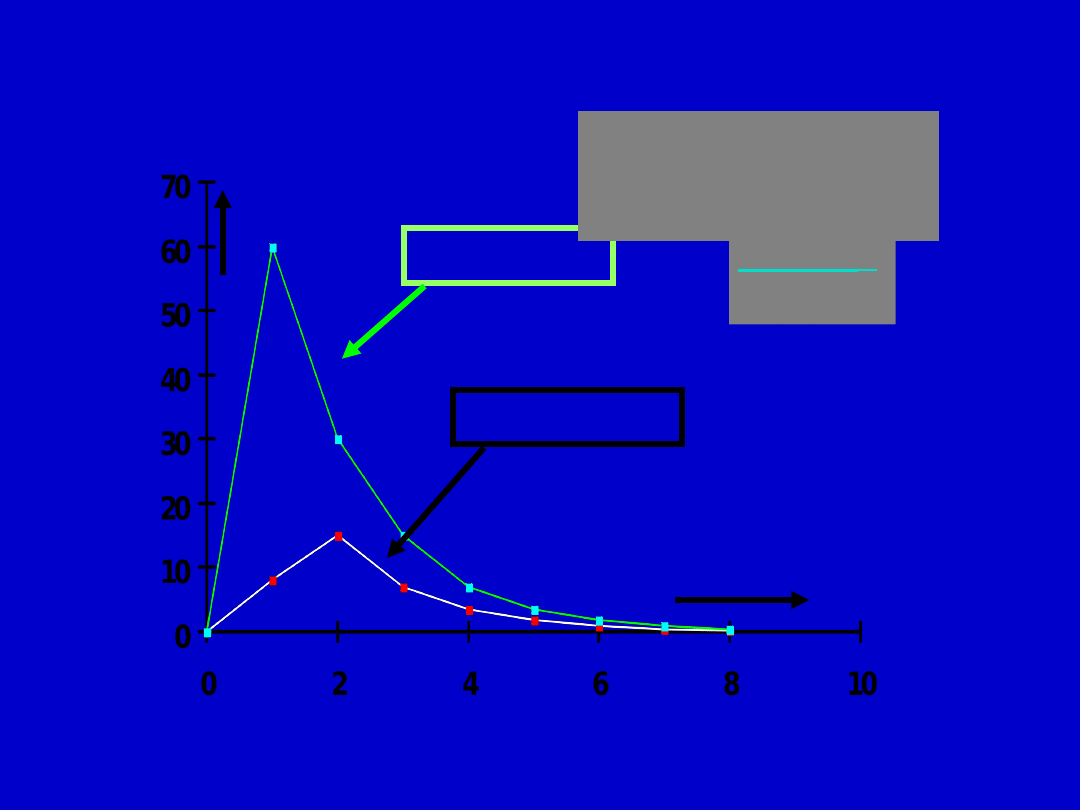
Plasma concentration
Time (hours
)
i.v. route
oral route
Bioavailabili
ty
(AUC)
o
(AUC)
iv

•Osocze
3.5 lit
.
(
heparin, plasma
expanders)
•Pozakomórkowe płynni
14 lit.
(tubocurarine, charged polar
compounds)
•Całkowita woda w ustroju
40
lit.
(ethanol)
Dystrybucja:
kompartmenty

Objętość dystrybucji
To objętość potrzebna do
rozmieszczenia całej podanej
ilości leku w stężeniu jakie
występuje w osoczu
Vd=D/Co

Metabolizm leku
• Wątroba
• Reakcje I fazy (acetylacja, oksydacja)
» aktywne metabolity
»Reakcje II fazy: sprzęganie z
kwasem glukuronowim czy
siarkowym-przygotowywanie
do eliminacji

Wiązanie leku z białkami
• Albuminy
• Alfa-glikoproteina
• Nieaktywny magazyn leku
• Pojęcie „bomby farmakologicznej”
Eliminacja leku
płuca
nerki
Inne drogi
Filtracja klębuszkowa
Aktywne wydzielanie kanalikowe
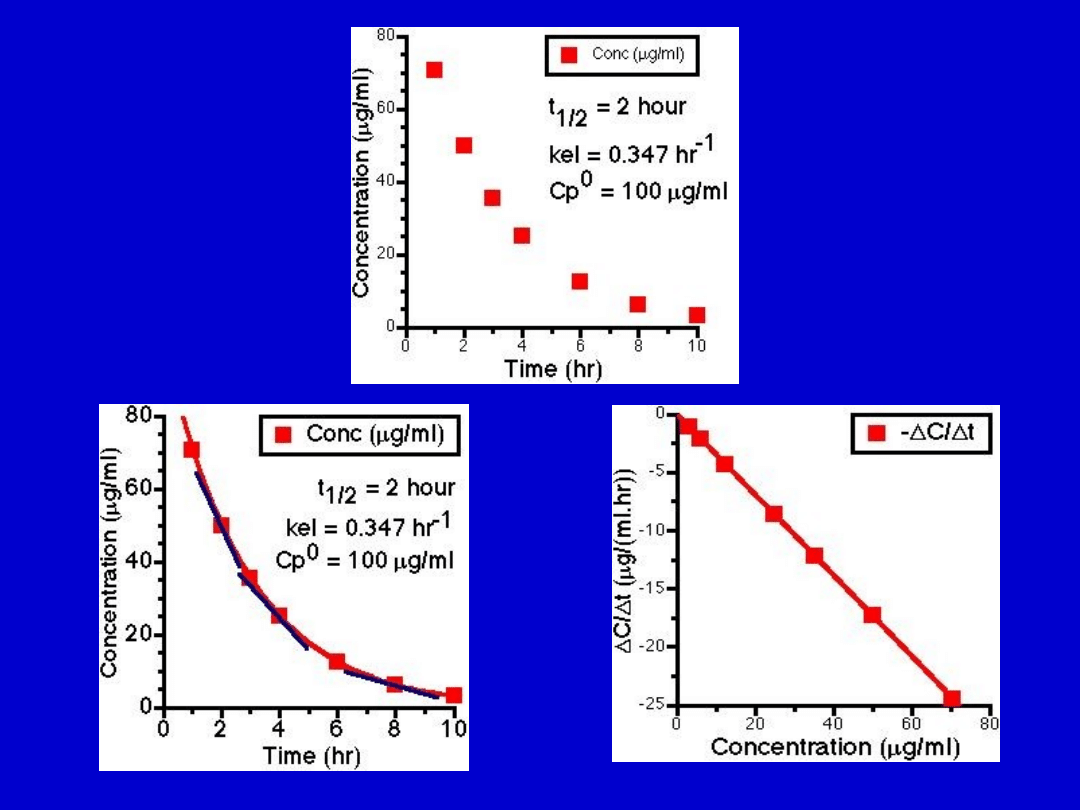
0-prosta linia
I-prosta w semi-log
II-prosta w 1/C
Kinetyka
Eliminacji
leków

Elyminacja c.d.
• Kinetyka zerowego rzędu:
stała iłość
leku eliminowana niezależnie od stężenia leku w
osoczu
• I- rzędu:
ilość eliminowanego leku
proporcjonalny do stężenia leku w osoczu.
Stała frakcja leku eliminowana w jednostce
czasu
• II-rzędu: eliminacja zależna od określonego enzymu
czy mechanizmu, gwałtownie się może zmniejszyć
Szybkość eliminacji = constant (CL) x
Conc.

T1/2, Cl
• t1/2-czas póltrwania leku jest funkcją
Vd i klirensu (Cl). Jest to czas po
którym stężenie leku w surowycy
zmniejszy się o połowę
• t1/2=0,693x Vd/Cl; stan stacjonarny
leku w organizmie po około 4-5 t1/2
• Cl= objętość krwi lub osocza z której
lek jest nieodwracalnie usuwany

Pojęcia podstawowe
• AUC (area under the curve) - POLE POD
KRZYWĄ STĘŻEŃ - miara ilości leku jaka dociera
do krążenia ogólnego w postaci nie zmienionej.
• BIOLOGICZNY OKRES PÓŁTRWANIA LEKU
(t0.5) - czas, po upływie którego stężenie leku
obserwowane we krwi zmaleje o połowę w wyniku
eliminacji po zakończeniu procesu wchłaniania i
dystrybucj
i
.
• Cmax - największe stężenie leku obserwowane
we krwi natychmiast po dożylnym podaniu lub po
pewnym czasie od chwili pozanaczyniowego
podania leku.

DOSTĘPNOŚĆ BIOLOGICZNA
(biodostępność, F)
• -
ułamek (procent) dawki leku, jaki
przechodzi do krążenia ogólnego po
pozanaczyniowym podaniu leku. Przyjmuje
się, że po dożylnym podaniu leku wartość
tego ułamka wynosi 1 (100%), natomiast po
pozanaczyniowym podaniu jest mniejsza od
jedności. Spowodowane jest to m.in.
niecałkowitym uwalnianiem leku z danej
postaci, niecałkowitym jego wchłanianiem z
miejsca pobrania lub efektem pierwszego
przejścia przez wątrobę. Jest to
biodostępność bezwzględna. Parametry
określające biodostępność: AUC, Cmax,
tmax

• DYSTRYBUCJA - proces przechodzenia leku z
przestrzeni naczyniowej do pozanaczyniowej oraz
rozmieszczania w płynach i tkankach organizmu.
• EFEKT PIERWSZEGO PRZEJŚCIA - leki podane
doustnie dostają się do krwiobiegu niemal wyłącznie
przez układ krążenia wrotnego, przez co cała
wchłonięta dawka leku przechodzi najpierw przez
wątrobę. W wątrobie leki poddane działaniu enzymów
ulegają biotransformacji. Jeżeli lek jest intensywnie
metabolizowany, to znaczny ułamek dawki zostaje
"wyekstrahowany" z krwi i ulega przemianom, zanim
dostanie się do krążenia ogólnego, co zmniejsza
ewidentnie jego dostępność biologiczną.

• FARMAKOKINETYKA - nauka o szybkości procesów
absorpcji (wchłaniania), dystrybucji, biotransformacji i
wydalania leków z organizmu. Jej praktycznym
zastosowaniem jest badanie przebiegu zmian
stężenia (ilości) leku i jego metabolitów w płynach
ustrojowych, tkankach i wydalinach z równoczesnym
ustalaniem zależności matematycznych opisujących i
tłumaczących znalezione dane analityczne.
• FARMAKOKINETYKA LINIOWA(I°) - opisuje procesy
farmakokinetyczne, których szybkości są liniową
funkcją stężenia, a wielkości parametrów
farmakokinetycznych nie zależą od podanej dawki
leku.
• FARMAKOKlNETYKA NIELINIOWA (0°) - opisuje
procesy farmakokinetyczne, których szybkości nie są
liniową funkcją stężenia leku, lecz zmieniają się w
sposób opisany przez równanie Michaelisa-Menten.
Konsekwencją nieliniowości tych procesów jest
zmienność parametrów farmakokinetycznych w
zależności od podanej dawki leku.

• FARMAKOKINETYKA POPULACYJNA -
dział farmakokinetyki zajmujący się
określaniem parametrów
farmakokinetycznych, przewidywaniem
stężeń oraz ustalaniem schematu
dawkowania u indywidualnego chorego na
podstawie jednego pomiaru stężenia leku
we krwi i na podstawie populacyjnych
wartości parametrów farmakokinetycznych,
ich odchyleń standardowych oraz danych
fizjologicznych, takich jak: wiek, płeć,
wzrost, masa ciała, klirens kreatyniny itp.
Metoda wymaga korzystania z programów
komputerowych.

• KLIRENS LEKU (CL) - objętość krwi
oczyszczana z leku w jednostce czasu. Jest
miarą wydajności eliminacji leku.
• KOMPARTMENT - obszar kinetycznie
jednorodny, tzn. taki, w którym lek po
rozmieszczeniu ma jednakowe stężenie w
danym momencie czasowym. Pojęcie
kompartmentu ma charakter czysto
funkcjonalny, przeważnie nie związany z
określonym obszarem anatomicznym.

• OBJĘTOŚĆ DYSTRYBUCJI (Vd) - jest to
taka objętość płynów ustrojowych, w której
należałoby rozpuścić ilość leku obecnego w
ustroju, aby otrzymać jego stężenie
stwierdzane we krwi. Im większe jest
stężenie leku w czasie t = 0, tym Vd jest
mniejsza. Jeśli stężenie jakiegoś leku w
czasie t = 0 jest bardzo małe, to objętość
dystrybucji jest duża, w przypadku
niektórych leków (np. benzodiazepiny
tiopental) przekraczająca wielokrotnie
objętość ciała. Objętość dystrybucji z reguły
jest obliczana po podaniu dożylnym.

• Vd = 10-20 l (4,3% masy ciała)lek
ulega dystrybucji w płynie
wewnątrzkomórkowym
• Vd = 25-30 l (20-30% masy ciała)lek
ulega dystrybucji w płynie
zewnątrzkomórkowym
• Vd = 40 l (35-42% masy ciała) lek
ulega dystrybucji we wszystkich
płynach ustrojowych
• Vd>100% masy ciała lek wiąże się w
bardzo znacznym stopniu z
materiałem biologicznym

• RÓWNOWAŻNOŚĆ BIOLOGICZNA
(BIOEKWIWALENCJA). Jeśli różnica
parametrów dostępności biologicznej dwóch
preparatów tego samego leku nie różni się
istotnie [nie przekracza 20% (80-120%)], to
preparaty te są równoważne biologicznie.
Jeden z badanych preparatów przeważnie
jest traktowany jako standardowy.
• STAŁA SZYBKOŚCI ELIMINACJI (K) - stała
szybkości pierwszego rzędu dla procesu
eliminacji leku z organizmu na drodze
wydalania i biotransformacji. Określa
ułamek dawki leku eliminowany z
organizmu w jednostce czasu.

• STAN STACJONARNY - stan, w którym szybkość
wprowadzenia leku do organizmu jest równa
szybkości jego eliminacji. Osiągany jest podczas
wlewu dożylnego prowadzonego przez czas równy
co najmniej 4-5 biologicznym okresom półtrwania
leku lub w wyniku wielokrotnego podawania leku
(dożylnie, pozanaczyniowo) przez ten sam okres.
• STĘŻENIE STACJONARNE - stężenie leku lub
metabolitu występujące we krwi i w tkankach po
osiągnięciu stanu stacjonarnego. W przedziale
dawkowania oscyluje ono pomiędzy wartością
maksymalną oraz minimalną.
• tmax - czas stężenia maksymalnego - czas, po
którym stężenie leku we krwi osiąga wartość
maksymalną Cmax (mierzony od chwili
pozanaczyniowego podania leku).


FARMAKODYNAMYKA
Relacje stężenie -efekt
• Pojęcie receptora
• Jonowy
• Metabotropowy
• Jądrowy
• Efekt maksymalny
• ED (EC)50
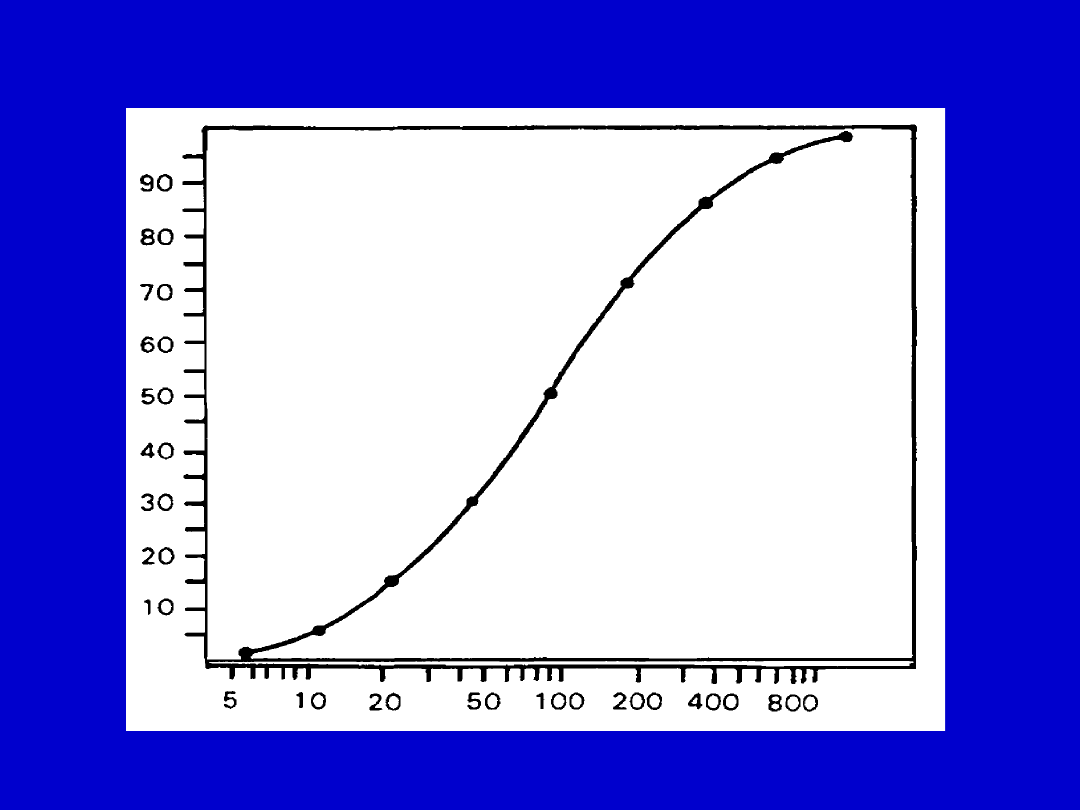
THE DOSE-RESPONSE RELATIONSHIP
The dose-response relationship (from C.D. Klaassen, Casarett and Doull’s
Toxicology, 5th ed., New York: McGraw-Hill, 1996).
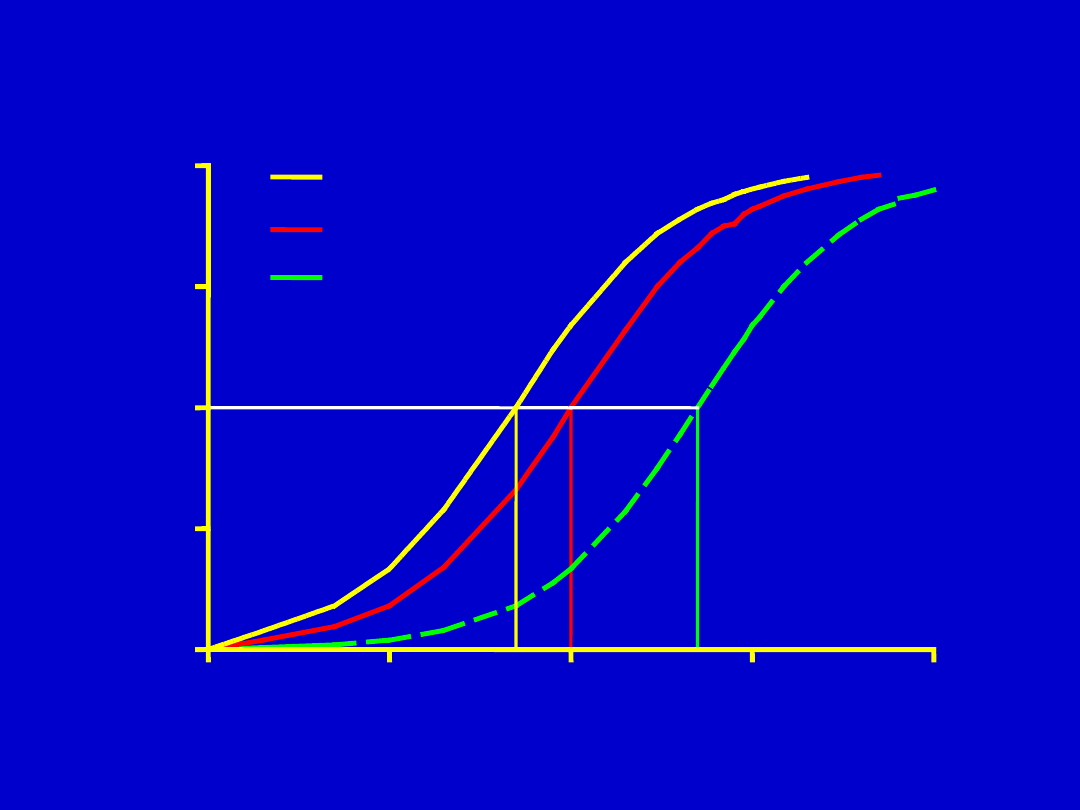
[D]
(concentration units)
[D
R
]/
R
T
0.01
0.10
1.00
10.00
100.00
0.00
0.25
0.50
0.75
1.00
Kd=1
kd=5
Kd=0.5
Compounds Have Different Affinities for the Same Receptor

Agonysta i Antagonysta
• Agonysta: substancja która wiąże się z
receptorem i powoduje efekt
• Antagonysta: substancja, która ma
powinowactwo do receptora, wiąże się z
nim, ale bez efektów komórkowych
• Częściowy agonysta: wiąże się z
receptorem , powoduje efekt ale istotnie
mniejszy niż efekt maksymalny
uzyskany pełnym agonystą
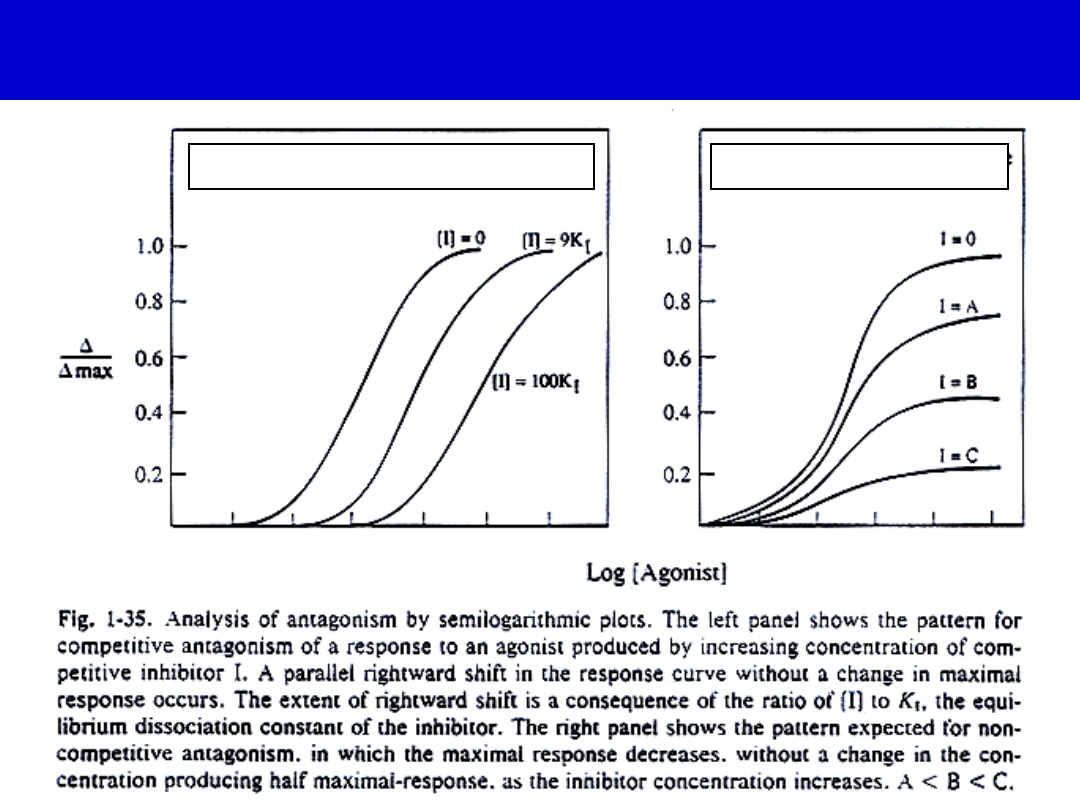
Competitive
Noncompetitive
Types of Receptor Antagonists
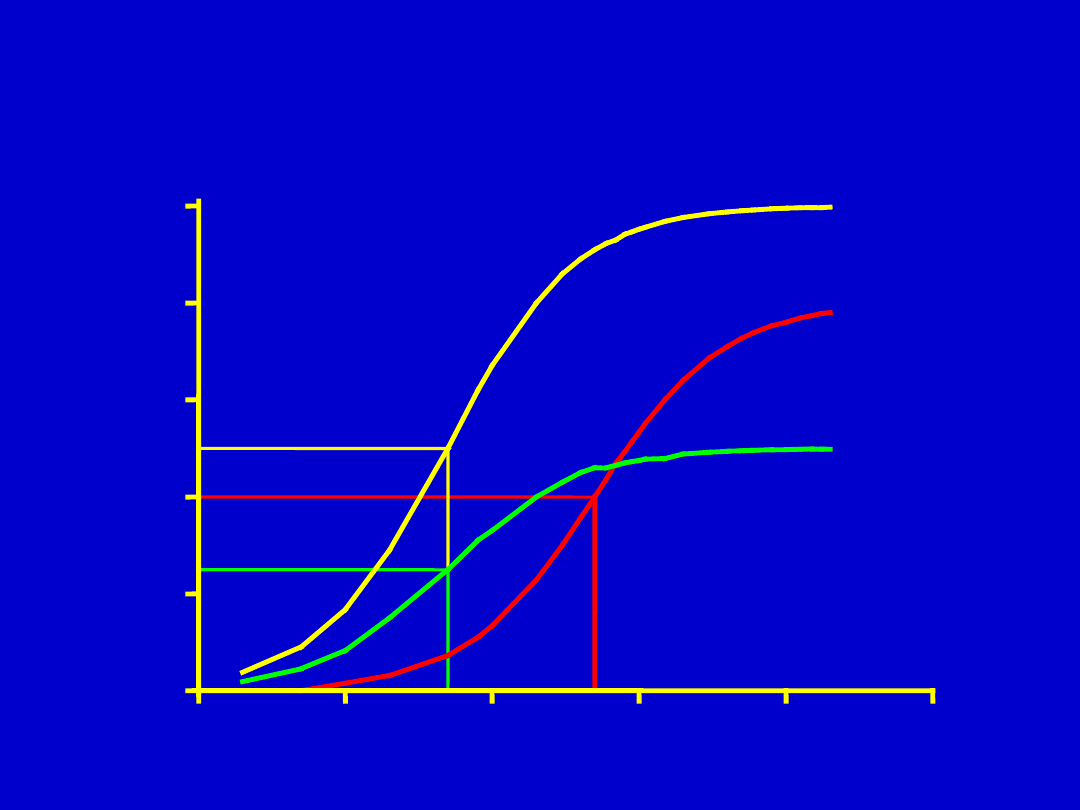
[D]
(concentration units)
%
M
a
x
im
a
l
E
ff
e
c
t
0.01
0.10
1.00
10.00
100.00
1000.00
0.0
0.2
0.4
0.6
0.8
1.0
Partial agonist
Full Agonist
Partial agonist
PARTIAL AGONISTS - EFFICACY
Even though drugs may occupy the same # of
receptors, the magnitude of their effects may
differ.

•Antagonizm
konkurencyjny
•Antagonizm
niekonkurencyjny
•Antagonizm funkcjonalny

Siła leku i jego skuteczność
• Siła zależy od dawki (stężenia)
• Skuteczność-od efektu wewnętrznego
• Z dwóch leków powodujących taki sam
efekt maksymalny silniejszy jest ten,
który osiąga ten efekt przy mniejszej
dawce
• Fentanyl 100 razy silniejszy od
morfiny!!!

Działania niepożądane
leków
• Specyficzne, zależne od rodzaju
leków i od dawki
• Reakcje alergiczne

Interakcje leków
• Im większa liczba naraz stosowanych
leków tym większe
prawdopodobieństwo poważnych
interakcji
• Interakcje farmakokinetyczne lub
farmakodynamiczne
• POLIPRAGMAZJA!!

FARMAKOGENETYKA
• Nauka o genetycznych
podstawach różnic w
odpowiedziach na leki
• Obejmuje farmakogenomikę-
wykorzystuje metodykę badan
całego genomu do wykrycia
wielogenowych uwarunkowań
odpowiedzi na lek!

Źródła danych o
skuteczności leków
.

Pub Med
Cochrain Institute
Podstawowymi bazami do poszukiwania badań pierwotnych są:
• Medline
• EMBASE®
• BioMed Central
Zalecane jest przeszukanie także innych baz informacji
medycznych, takich jak:
• BIOSIS Previews®
• CINAHL® Database
• PsycINFO®
• European Public Assessment Report (EPAR)
• Health Canada
• Netherlands Pharmacovigilance Centre Lareb
• The Uppsala Monitoring Centre
• Thompson Micromedex®
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
Wyszukiwarka
Podobne podstrony:
farmakologia ogólna
embriologia ogólna wykład III
w1.FARMAKOLOGIA OGÓLNA, wykłady PMWSZ w Opolu - Pielęgniarstwo, Farmakologia
Farmakologia Ogólna
Wykład Farmakologia ogólna 2, FARMAKOLOGIA
Farmakoterapia ogólna i miejscowa
FARMAKOLOGIA OGÓLNA ANALITYKA
WYKŁAD Mechanika Ogólna Część III
farmakologia ogólna cz2
Farmakologia ogólna 7
FARMAKOLOGIA OGÓLNA LEKÓW P
więcej podobnych podstron