
Zespoły
strukturalnych
aberracji
chromosomowych

Mutacje:
• genowe
•chromosomowe
strukturalne
•chromosomowe liczbowe
•genomowe

Mutacje (aberracje) chromosomowe –
to wszelkie zmiany w liczbie i
struktrurze chromosomów
Powstają w komórkach somatycznych
lub w gametach.
Mogą się dziedziczyć lub powstawać
de novo.

Aberracje chromosomowe strukturalne:
• Inwersja
• Translokacja
• Duplikacja
• Delecja (deficjencja)
• Chromosom kolisty
• Izochromosom
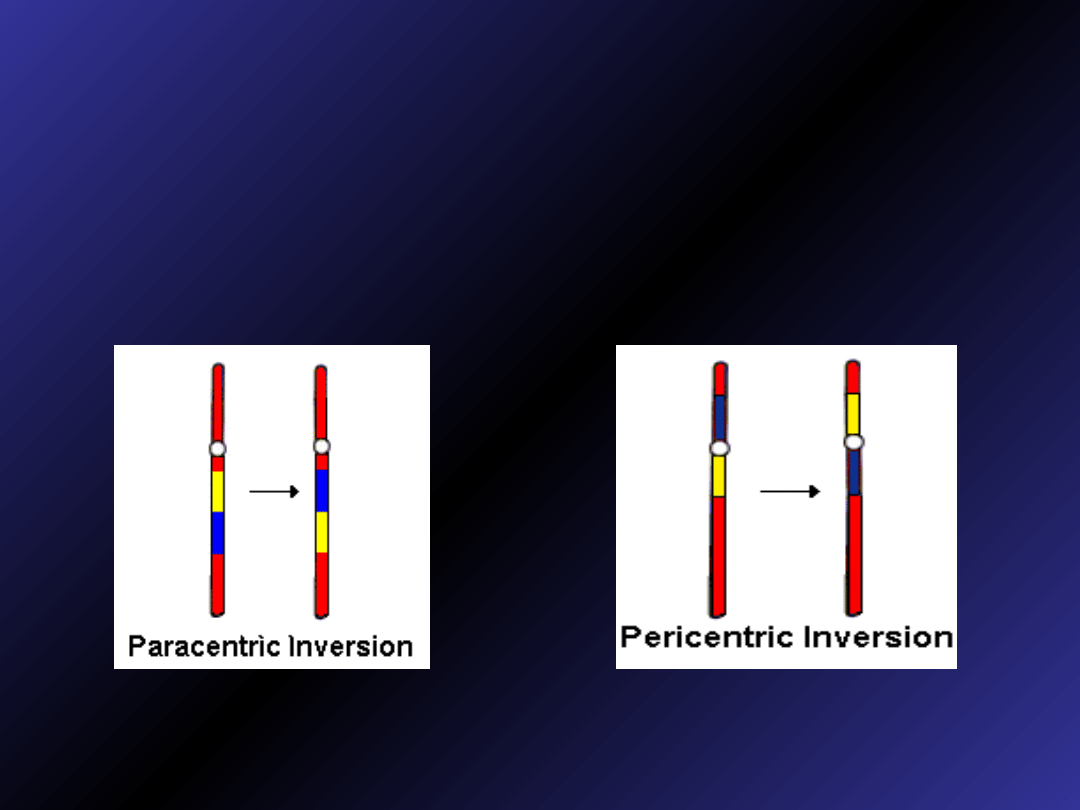
Inwersje
• Paracentryczne
•Perycentryczne
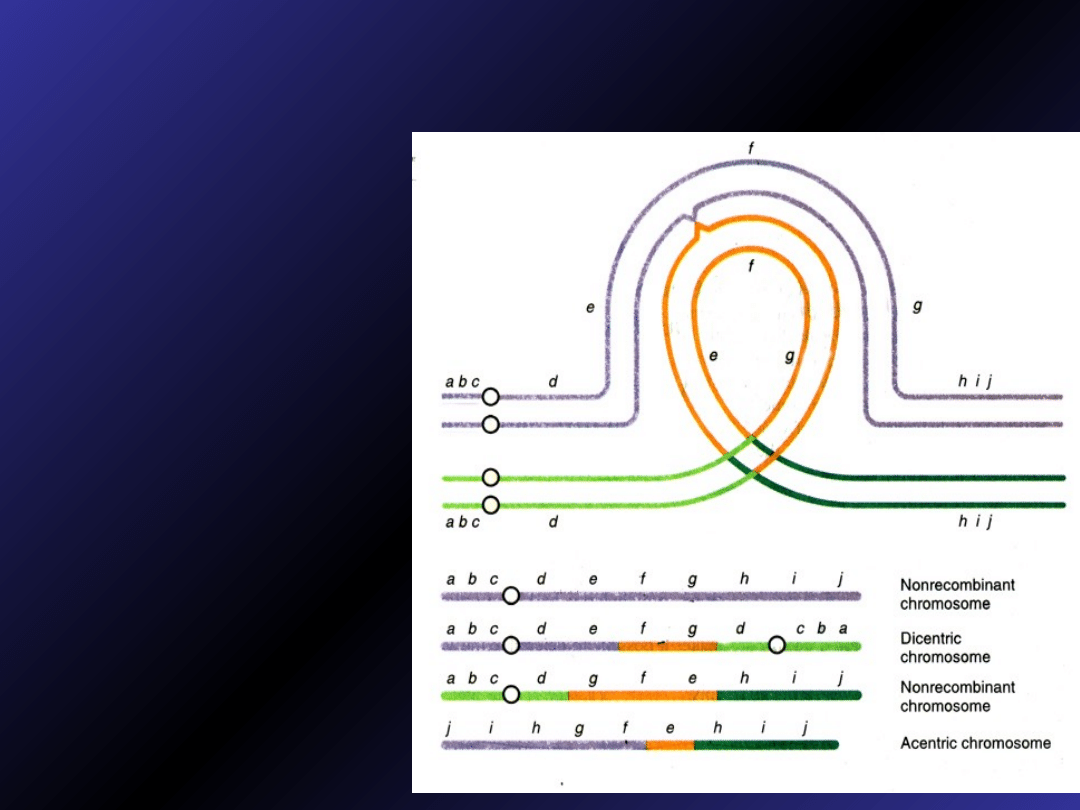
Inwersje
•
inwersje mogą
wpływać na proces
mejozy, wywołując
aberracje
chromosomowe u
potomstwa nosicieli
•
aby chromosom z
inwersją ustawił się w
idealnym porządku ze
swoim prawidłowym
homologiem podczas
profazy I, musi
uformować pętlę
•crossing over w ramach tej pętli może prowadzić do
wystąpienia duplikacji i delecji w chromosomach
komórek potomnych
•crossing over w ramach
tej pętli może prowadzić
do wystąpienia duplikacji
i delecji w chromosomach
komórek potomnych
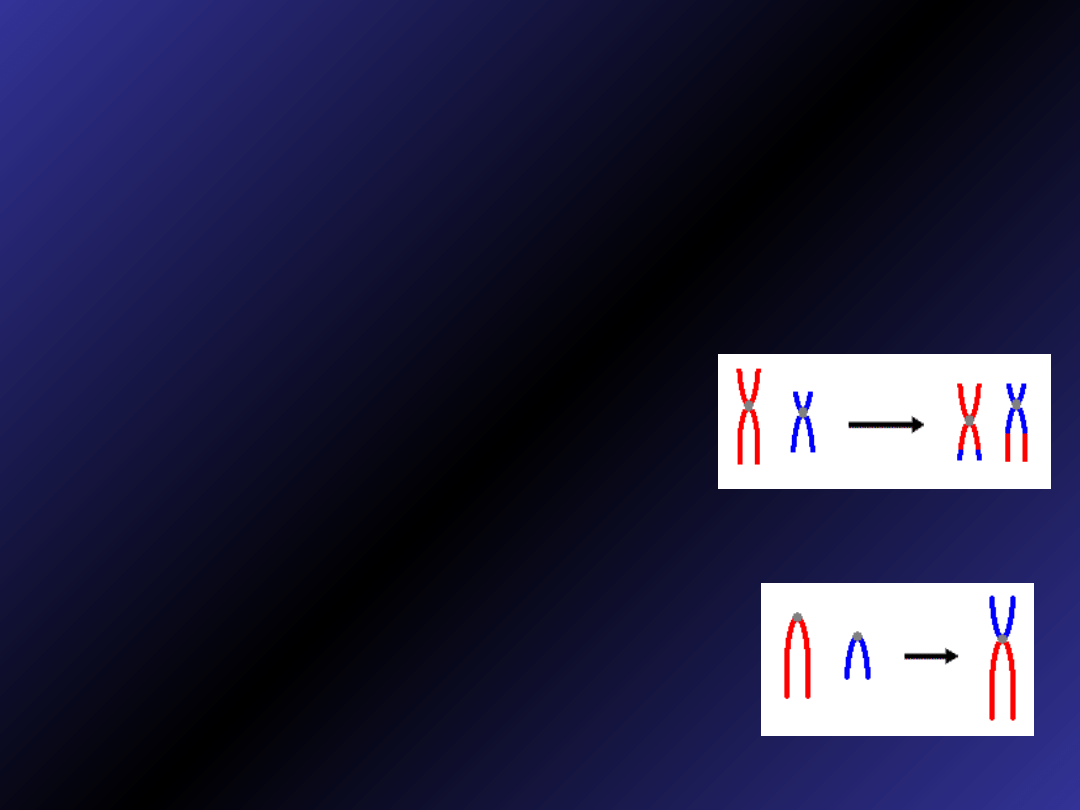
Translokacje
• Translokacja – wymiana materiału
genetycznego pomiędzy
niehomologicznymi chromosomami
• Translokacja wzajemna: dwa
chromosomy wymieniają między sobą
odcinki, co powoduje, że mają
nieprawidłowe kształty, całkowita liczba
chromosomów pozostaje niezmieniona
• Translokacja robertsonowska: łącza
się całe lub prawie całe ramiona długie
chromosomów. Miejscem połączenia
jest rejon centromeru. Dochodzi do
utraty ramion krótkich. W kariotypie
stwierdza się brak chromosomu.

Translokacje
• Zrównoważone - zasadniczo nie zmienia się ilość
materiału genetycznego, ale następuje zmiana jego
rozmieszczenia w genomie. Liczba chromosomów
może być prawidłowa lub zmieniona. Aberracja ta
może nie przejawiać się fenotypowo, ale może być
przekazywana potomswtu.
• Niezrównoważona - zmianie ulega ogólny skład
genowy. Ilość materiału jest większa, a liczba
chromosomów jest prawidłowa. W tym przypadku
zawsze dochodzi do ujawnienia fenotypowego
choroby

Translokacje
• t(2;5)(p23;q35)Anaplastyczny chłoniak
olbrzymiokomórkowy
• (ALCL)t(8;14)Chłoniak Burkitta
• t(9;22)(q34;q11)Przewlekła i ostra białaczka
szpikowa
• t(11;14)Chłoniak z komórek płaszcza
• t(11;22)(q24;q11.2-12)Mięsak Ewinga
• t(14;18)(q32;q21)Chłoniak pęcherzykowy
• t(17;22)Dermatofibrosarcoma protuberans
• t(15;17)Ostra białaczka promielocytowa
• t(1;12)(q21;p13)Ostra białaczka mielocytowa
• t(9;12)(p24;p13)Przewlekła i ostra białaczka
szpikowa
• t(X;18)(p11.2;q11.2)Mięsak błony maziowej

Duplikacja
•
podwojenie fragmentu chromosomu na skutek
niesymetrycznej wymiany odcinków chromatyd i
czasem błędnego crossing-over.
• mogą występować u potomstwa osób będących
nosicielami translokacji wzajemnej
• mogą zachodzić podczas rekombinacji
homologicznej w mejozie, w wyniku
nierównomiernego procesu crossing over
• duplikacja chromosomu 17p11.2 (choroba Charcota,
Mariego i Tootha typu 1A
• neuropatia, zaburzenia mielinizacji i regeneracji
aksonu, zanik włókien nerwowych, zanik mięśni

Delecja (deficjencja)
• Delecje powstają na skutek pęknięcia chromosomu i
następującej utraty materiału genetycznego; dotyczą
zwykle dość dużej liczby genów i wywołują
charakterystyczne zespoły chorobowe; są możliwe
do zobaczenia pod mikroskopem
• Delecja terminalna powstaje, kiedy dochodzi do
pojedynczego pęknięcia chromosomu i utraty
materiału z końca chromosomu
• Delecja interstycjalna jest efektem dwóch pęknięć
chromosomu i utraty materiału pomiędzy nimi

Delecja (deficjencja)
Choroby:
• Zespół Wolfa-Hirschhorna
• Zespół cri du chat
• Delecja długich ramion chromosomu 13

Mikrodelecja
• Delecja niewielkich fragmentów
chromosomów, niewidocznych w
mikroskopie świetlnym
• Wrodzone wady rozwojowe i/lub
cechy dysmorfii, upośledzenie
umysłowe

Mikrodelecja
Zespół
Cechy kliniczne
Delecje
chromosomo
we
Pradera-Willego
upośledzenie umysłowe, niski wzrost,
otyłość, hipotonia, charakterystyczne
rysy twarzy, małe stopy
15q11-13
Langera-
Giediona
charakterystyczne rysy twarzy, rzadkie
włosy, egzostoza, zmienne upośledzenie
umysłowe
8q24
Millera-Diekera
brak zakrętów mózgowych,
charakterystyczne rysy twarzy
17p13.3
DiGeorge’a
charakterystyczne rysy twarzy
rozszczep podniebienia, wada serca
22q11
Smitha-
Magenisa
upośledzenie umysłowe,
hiperaktywność, cechy dysmorficzne,
autoagresje
17p11.2
Williamsa
zaburzenia rozwojowe,
charakterystyczne rysy twarzy,
nadzastawkowe zwężenie aorty
7q1
Brak tęczówki/
guz Wilmsa
upośledzenie umysłowe, brak tęczówki,
predyspozycja do wystąpienia guza
Wilmsa, defekty narządów płciowych
11p13
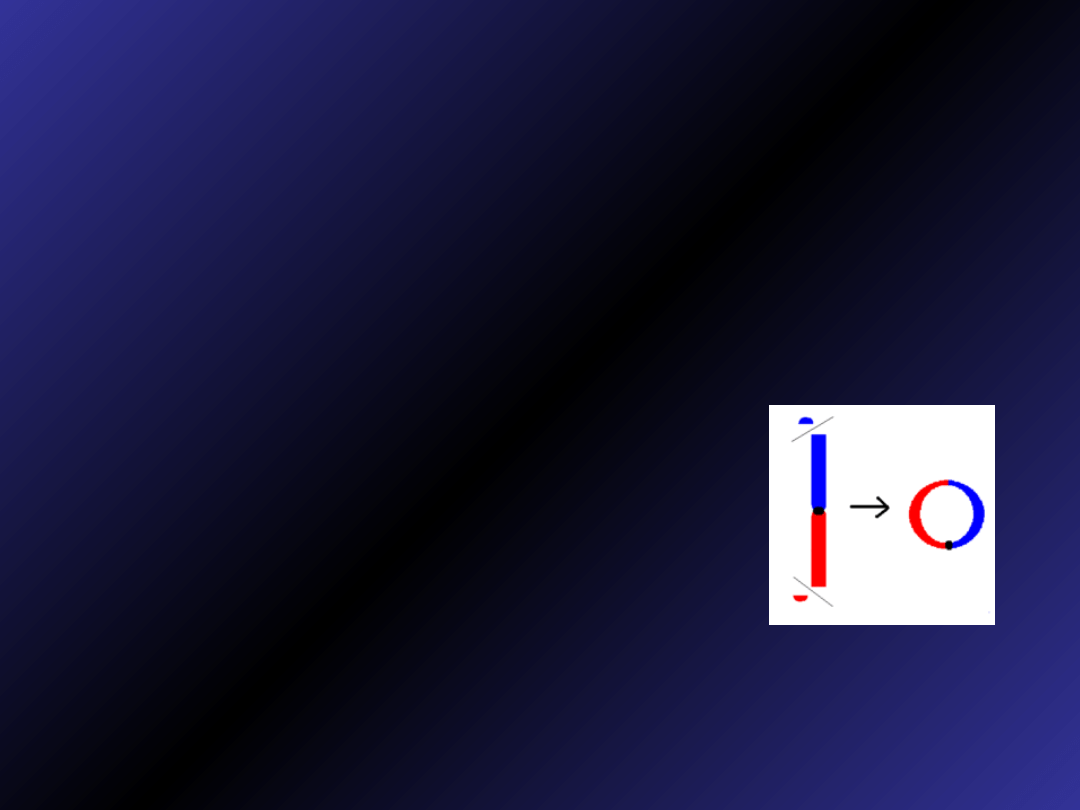
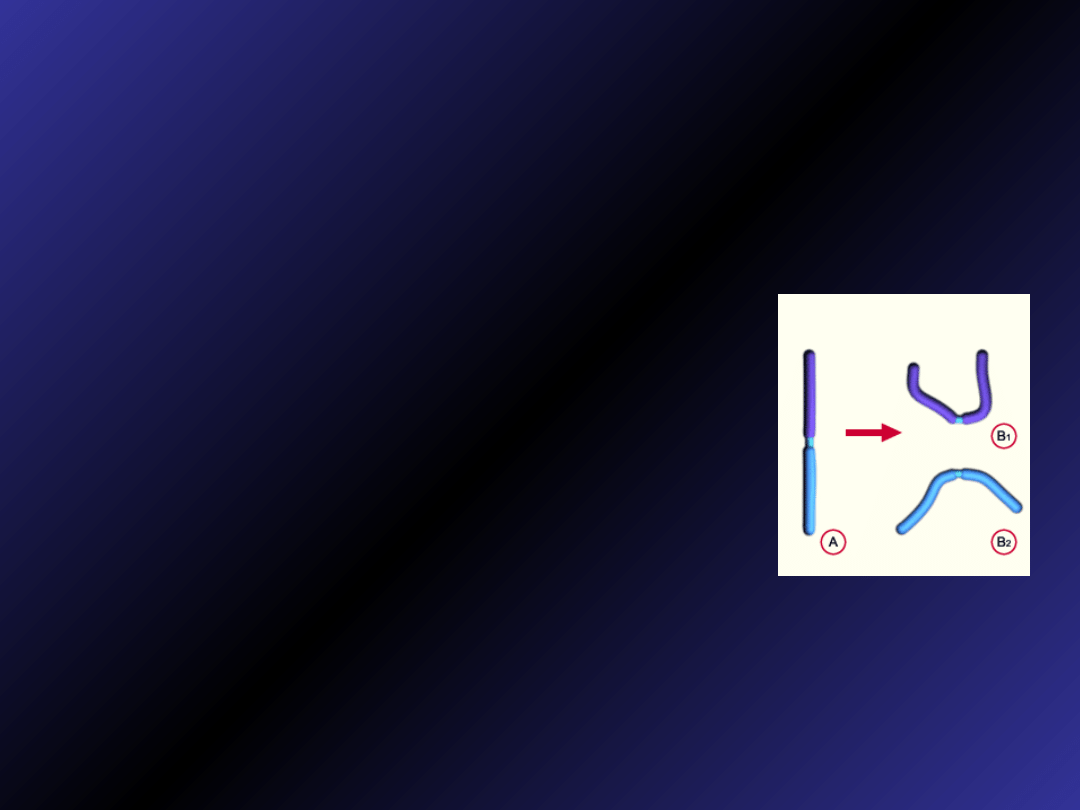
Chromosom kolisty
• powstają gdy na obu końcach
chromosomów dochodzi do delecji, a
następnie końce te łączą się ze sobą
• często ulegają utracie, co powoduje
monosomię chromosomową w
niektórych komórkach
• zostały opisane przynajmniej w
jednym przypadku dla każdego
autosomu człowieka
• chromosomy pierścieniowe 13 i 14
wywołują zespół związany z
upośledzeniem umysłowym
• zespół pierścieniowego chromosomu
20 powodujący padaczkę i
upośledzenie umysłowe
Izochromosomy
• powstają w rezultacie podziału
wzdłuż osi prostopadłej do zwykłej
osi podziału chromosomu
• posiadają dwie kopie jednego
ramienia i żadnej kopii drugiego
ramienia
• Izochromosomy większości
chromosomów są śmiertelne
• większość izochromosomów
obserwowanych u żywych
noworodków dotyczy chromosomu
X, a dzieci z chromosomem Xq
(46,X,i[Xq]) zwykle wykazują
cechy zespołu Turnera
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
Wyszukiwarka
Podobne podstrony:
2012.11.05 Zespoły aberracji chromosomowych, Lekarski I rok ŚUM, biologia
cw2 ZESPOŁY ABERRACJI CHROMOSOMÓW PŁCIOWYCH
aberracje strukturalne i genomowe chromosomów
aberracje strukturalne i genomowe chromosomów
Skrypt Budowa ciała i jego poszczególnych fragmentów w ujęciu antropometrycznym a wybrane zespoły ab
Rodzaje aberracji chromosomowych pop
Aberracje chromosomowe i mutacje
ABERRACJE CHROMOSOMOWE, BIOLOGIA MEDYCZNA
aberracje chromosomowe prezentacja
Aberracja chromosomowa referat, biologia, biologia medyczna
131 Aberracje chromosomow po napromieniowaniu
PRZYKLADY ZAPISU KARIOTYPOW Z ABERRACJAMI CHROMOSOMOWY MI
Slajd z wykładu (aberracje chromosomowe)
ZAPIS KARIOTYPÓW Z ABERRACJAMI CHROMOSOMOWYMI WWL
genetyka, RODZAJE ABERRACJI CHROMOSOMOWYCH WWL
2. Aberracje chromosomowe, VI rok, Genetyka, Genetyka, Egzamin
więcej podobnych podstron