
Wykorzystanie myszy
szczepu NOD w badaniach
nad cukrzycą
insulinozależną (typ 1)

• Cukrzyca typu I (insulinozależna, T1D) to przewlekła
autoimmunologiczna
choroba
spowodowana
stopniowym
niszczeniem komórek wysp trzustkowych,
występująca u osób podatnych genetycznie. Typową
jej
postać cechuje nagły początek z koniecznością
insulinoterapii, wskutek bezwzględnego niedoboru
insuliny, do którego doprowadził agresywny proces
autoimmunologiczny.

• Myszy szczepu NOD (nonobese diabetic) to zwierzęcy model
ludzkiej cukrzycy insulinozależnej, który spontanicznie
rozwija chorobę autoimmunologiczną za pośrednictwem
komórek T, charakteryzującą się intensywną infiltracją wysp
trzustkowych oraz innych organów (okołożuchwowych
ślinianek oraz tarczycy) przez komórki CD4+ oraz CD8+.
• Różnorodne linie myszy NOD różnią się częstością
występowania spontanicznej cukrzycy, np. 93% samic myszy
NOD/Lt w porównaniu do 43% samic NOD/Wehi rozwinęło
cukrzycę po 250 dniach.

Pochodzenie i opis myszy NOD
• Makino i wsp. stworzyli szczep myszy NOD w Japonii
podczas
selekcji
szczepu
podatnego
na
zaćmę
pochodzącego z hodowli niekrewniaczej myszy linii
JCL:ICR. Podczas tej selekcji zaobserwowali myszy bez
zaćmy, ale charakteryzujące się niską wagą, wielomoczem
(poliurią), polidypsją (nadmiernym spożywaniem płynów)
oraz cukromoczem (glikozurią). Szczep NOD ustalono
poprzez powtarzające się kojarzenie brat-siostra, jako linię,
która spontanicznie rozwija cukrzycę.
• Rozwój spontanicznej cukrzycy u myszy NOD waha się w
granicach od 60% do 80% u samic oraz od 20% do 30% u
samców.

• Samice NOD stają się trwale hiperglikemiczne
w młodszym wieku niż samce (początek
między 16 a 20 tygodniem, w porównaniu do
21 – 28 tygodnia) i charakteryzują się większą
częstością zachorowań.
• Steroidy płciowe są istotnym modulatorem
patogenezy: wycięcie jąder zwiększa, a
wycięcie
jajników
zmniejsza,
częstość
występowania choroby.

• Badania histologiczne wykazały obecność niewielkiej ilości
infiltratów
komórek
immunologicznych
w
wyspach
trzustkowych do ok. 3-4 tygodnia życia. W tym okresie
zarówno u samców, jak i samic myszy NOD zaczynają się
pojawiać infiltraty komórek jednojądrzastych, które otaczają
wyspy trzustkowe (peri-insulitis). Te infiltraty przemieszczają
się do i atakują wyspy trzustkowe (insulitis) przez kolejne
kilka tygodni, tak, że większość myszy wykazuje ostre
zapalenie ok. 10 tygodnia życia.
• Odkrycie, że samce chorują z mniejszą częstością niż samice,
mimo, że mają podobny poziom wczesnego zapalenia,
sugeruje, że późniejsze mechanizmy regulatorowe kontrolują
rozwój choroby.

• Infiltraty komórek jednojądrzastych wysp trzustkowych
mają złożony skład. Większość komórek stanowią
komórki T CD4+, ale występują też komórki T CD8+,
komórki NK, limfocyty B, komórki dendrytyczne i
makrofagi.
• T1D u myszy jest przede wszystkim zależna od komórek
CD4+ oraz CD8+.

• Wiele loci odpowiada za podatność genetyczną na cukrzycę
u myszy NOD. Myszy tego szczepu posiadają unikalny
haplotyp MHC, zwany H-2
g7
, który jest czynnikiem
genetycznym mającym największy wpływ na podatność na
rozwój cukrzycy.
• Wiele badań, mających na celu sprawdzenie roli MHC u
myszy NOD w rozwoju zapalenia i cukrzycy, wykazało, że
homozygotyczność haplotypu H-2
g7
może być niezbędna dla
rozwoju T1D.

• Myszy NOD są również podatne na rozwój innych
autoimmunologicznych
chorób,
takich
jak:
autoimmunologiczne
zapalenie
ślinianek,
autoimmunologiczne
zapalenie
tarczycy,
autoimmunologiczna
obwodowa
polineuropatia,
choroba podobna do tocznia rumieniowatego oraz
zapalenie gruczołu krokowego (u samców).

Patogeneza T1D u myszy NOD
• Etiologia cukrzycy typu 1 u myszy NOD jest zarówno
złożona, jak i wieloczynnikowa.
• Myszy NOD charakteryzują się obecnością licznych
defektów układu immunologicznego, które przyczyniają
się
do
wystąpienia
autoimmunizacji.
Układ
immunologiczny
posiada
defekty
w
różnorodnych
subpopulacjach leukocytów. U myszy NOD obserwuje się
niewłaściwe dojrzewanie i funkcjonowanie makrofagów,
niski poziom aktywności komórek NK, defekty w
komórkach NKT, niedobór ich regulatorowych komórek T
CD4+CD25+ oraz brak dopełniacza hemolitycznego i C5a.

• U myszy NOD, autoimmunologiczny atak na
komórki beta w wyspach trzustkowych jest
prawdopodobnie wynikiem nieprawidłowej selekcji
komórek T w grasicy.

• Wyniki wielu badań wskazują, że zarówno komórki T
CD4+, jak i CD8+ biorą udział w rozwoju i progresji
cukrzycy typu 1. Komórki T CD4+ są niezbędne we
wczesnej i późnej fazie rozwoju choroby. Komórki T
CD4+ są niezwykle istotne dla patogenezy T1D i mogą
bezpośrednio wpływać na destrukcję komórek wysp
trzustkowych.
• Komórki T CD8+ również promują rozwój choroby.
Komórki T CD8+ biorą udział we wczesnej fazie
choroby,
być
może
poprzez
powodowanie
wystarczającej destrukcji komórek wysp do aktywacji
silniejszej odpowiedzi komórek T CD4+. Komórki T
CD8+ mogą również odgrywać rolę w funkcji
efektorowej.

• Antygeny rozpoznawane przez CD4+ i CD8+ są to np.
insulina, GAD, IA-2 czy Hsp60. Wszystkie te antygeny
są wytwarzane przez wyspy trzustkowe, ale tylko
insulina jest produkowana wyłącznie przez te wyspy.

• Wyniki doświadczeń sugerują, że komórki B odgrywają
istotną rolę w rozwoju autoreaktywności w T1D myszy NOD.
• Mechanizm bądź mechanizmy, za pomocą których komórki B
wpływają na patogenezę T1D, są niejasne. Komórki B mogą
wpływać
na
chorobę
autoimmunologiczną
poprzez
produkcję
autoprzeciwciał
lub
jako
komórki
APC
zaangażowane w selekcję lub aktywację autoreaktywnych
komórek T.

• Pomimo, że wysokie miana autoprzeciwciał przeciwko
insulinie
lub
anty-GAD
są
wykrywane
u
przedklinicznych myszy NOD i u ludzi z cukrzycą,
transfer tych przeciwciał nie indukuje choroby u myszy
NOD.
• W odniesieniu do immunopatofizjologii, jasne jest, że
T1D jest chorobą związaną z komórkami T we
wszystkich modelach gryzoni, z patogennym wsparciem
ze strony limfocytów B, raczej jako komórek APC, niż
producentów przeciwciał.

• Zidentyfikowano genetyczne loci, które są związane z
wystąpieniem cukrzycy typu 1 u myszy, podczas gdy pełen
rozwój cukrzycy wymaga ekspresji unikalnego genu MHC I-
A
NOD
.
• Myszy NOD spontanicznie rozwijają cukrzycę, poprzedzoną
okresem zapalenia wysp trzustkowych podczas którego
wyspy Langerhansa są infiltrowane przez autoreaktywne
komórki T i komórki APC.
• Zapalenie stopniowo rozprzestrzenia się z obrzeży wysp
Langerhansa do ich wnętrza. Skutkuje to spadkiem liczby
komórek wysp, ilość komórek beta również się zmniejsza i
są one niszczone. W ten sposób, zapalenie stale postępuje.
• Wystąpienie zapalenia jest związane z nieprawidłowością w
genie MHC klasy II.
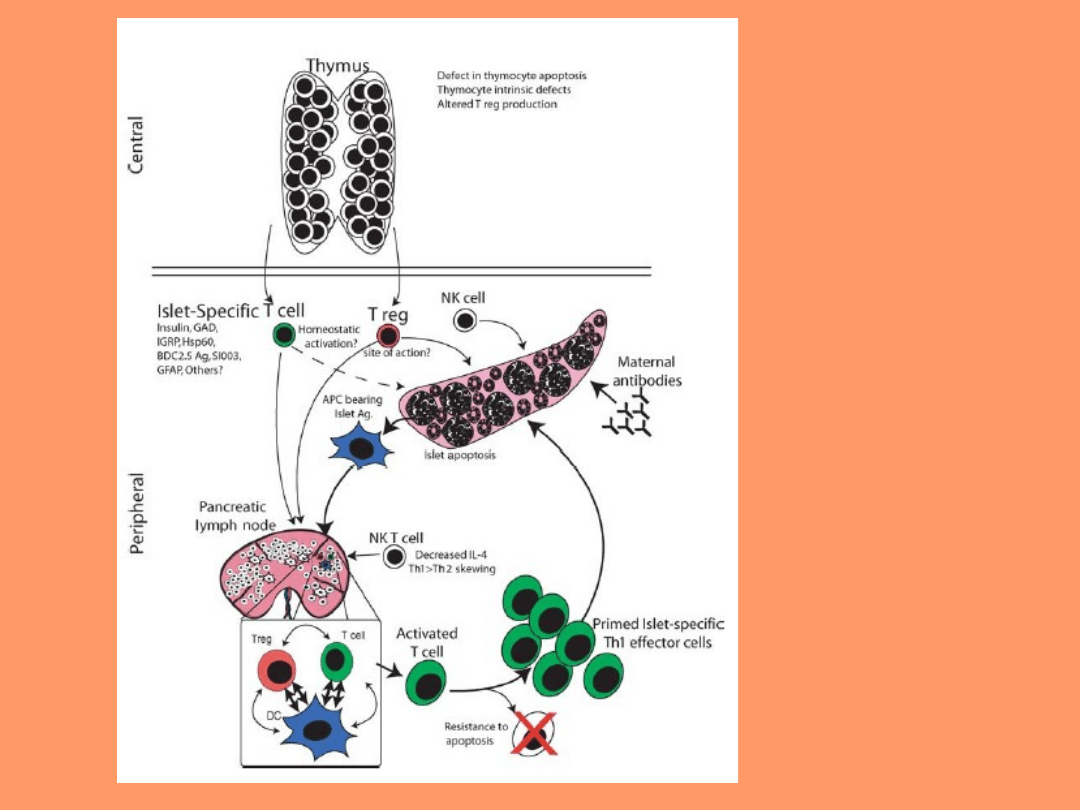
(„THE NOD MOUSE:
A Model of Immune
Dysregulation”
Mark S. Anderson, Jeffrey
A. Bluestone
)

• Rozwój repertuaru komórek T w grasicy prawdopodobnie
jest nieprawidłowy i pozwala komórkom T reaktywnym
przeciw wyspom trzustkowym na uniknięcie zniszczenia oraz
zapobiega właściwej generacji komórek Treg. Na obwodzie
komórki T reaktywne napotykają antygeny w węzłach
chłonnych trzustki i są niewłaściwie aktywowane do bycia
efektorami. Otoczenie cytokin, APC, komórek wrodzonej
odporności, interakcje z cząsteczkami kostymulatora, które
są obecne podczas tego etapu prawdopodobnie prowadzą do
początkowej aktywacji i ekspansji odpowiedzi przeciw
wyspom trzustkowym, czyli do insulitis.

• Następnie, odpowiedź przeciw wyspom trzustkowym
przekształca się na dłuższy okres czasu w skomplikowaną
immunoregulatorową „bitwę” angażującą komórki Treg,
NK oraz predysponującą do odpowiedzi typu Th1, która
kończy się niewłaściwą odpowiedzią efektorową wobec
wysp trzustkowych i rozwojem cukrzycy.

Podobieństwa i różnice między
cukrzycą typu 1 u myszy i u ludzi
• Podobieństwa:
- gwałtowny początek choroby pomiędzy 90-120 dniem
(co odpowiada wczesnemu okresowi dojrzewania u ludzi)
- hiperglikemia
- glikozuria
- hipercholesterolemia
- ketonuria
- polidypsja
- poliuria
- polifagia

- insulitis
- podwyższony w surowicy poziom Ab przeciw wyspom
trzustkowym
-
autoimmunologiczny
udział
innych
organów
endokrynnych
- udział komórek T w patogenezie
- makrofagi mają osłabioną zdolność aktywowania
regulatorowych komórek T (podobne osłabienie funkcji
występuje w komórkach dendrytycznych u ludzi z T1D)
- predyspozycja genetyczna (geny MHC klasy II)
- złożona kontrola poligenetyczna
- choroba przenoszona przez szpik kostny

• Różnice:
- brak uszkodzeń nerek
- brak neuropatii
- brak retinopatii
- homozygotyczność dla genów MHC niezbędna dla
rozwinięcia się choroby (u ludzi heterozygotyczność dla
genów MHC jest częsta)
- wpływ płci
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
Wyszukiwarka
Podobne podstrony:
12 Patogeneza cukrzycy
12 Ubóstwo i pomoc społecznaid 13305 ppt
12 Ćwiczenia wspomagane i samowspomaganeid 13428 ppt
(12) Cechy indyw uczniaid 831 ppt
1 12 Zaburzenia czynnościowe biegunka ostra ppt
14 Mysie modele nowotworow jelitid 15475 ppt
12 HORMONY TRZUSTKA, PRZYTARCZYCE, SZYSZYNKA ppt
17(45) Modele systemów informatycznychid 17383 ppt
12 Modele atomu
12 wartość pieniądza w czasieid 13309 ppt
cukrzyca choroba społeczna ppt
10 12 2010 Niezawodność państwaid 10742 ppt
12 Układ Endokrynny 3id 13634 ppt
10 Mięśnie klatki piersiowej 12 05 2007 komentarzid 10951 ppt
Etyka psychologiczna Teoplitz Wiśniewska wykład 12 Badanie naukowe Planowanie i publikowani ppt
2 Cukrzyca w ciązyid 19480 ppt
więcej podobnych podstron