
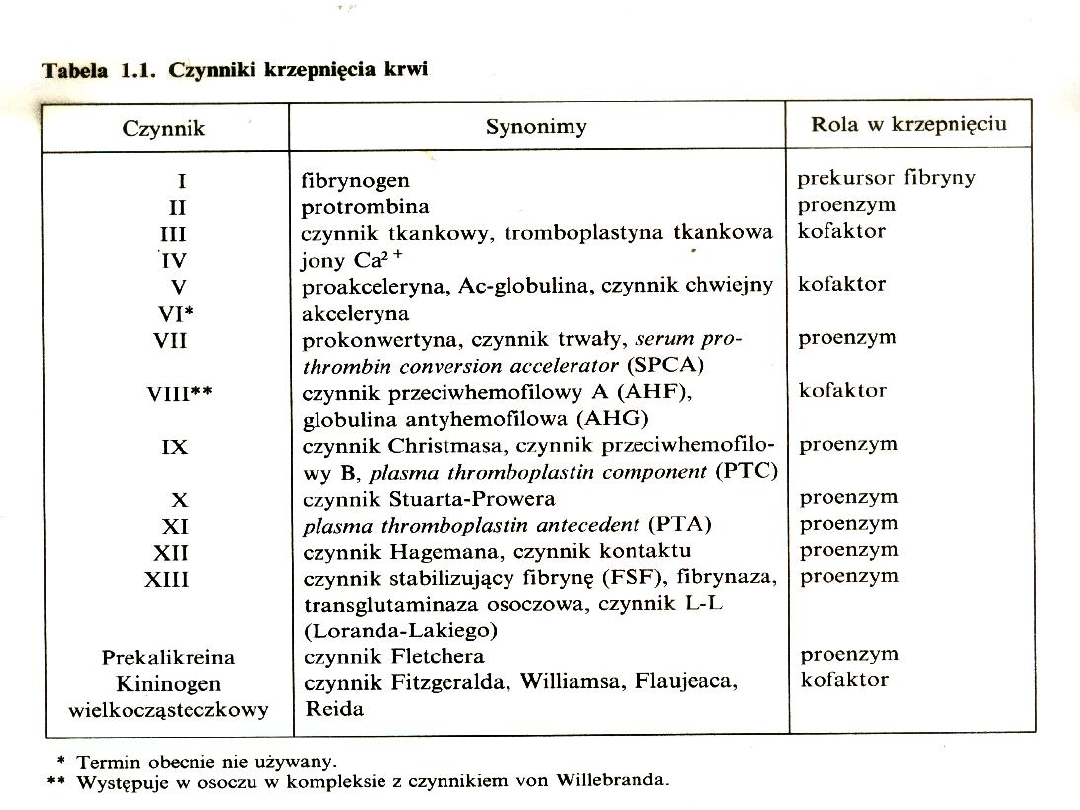
Skazy krwotoczne
osoczowe
Krzysztof Chojnowski
Klinika Hematologii UM w
Łodzi

Skaza krwotoczna
Definicja
Występowanie krwawień
samoistnych lub nadmiernie
obfitych i długotrwałych w
stosunku do urazu, który je
wywołał, w następstwie
niewydolności mechanizmów
hemostazy

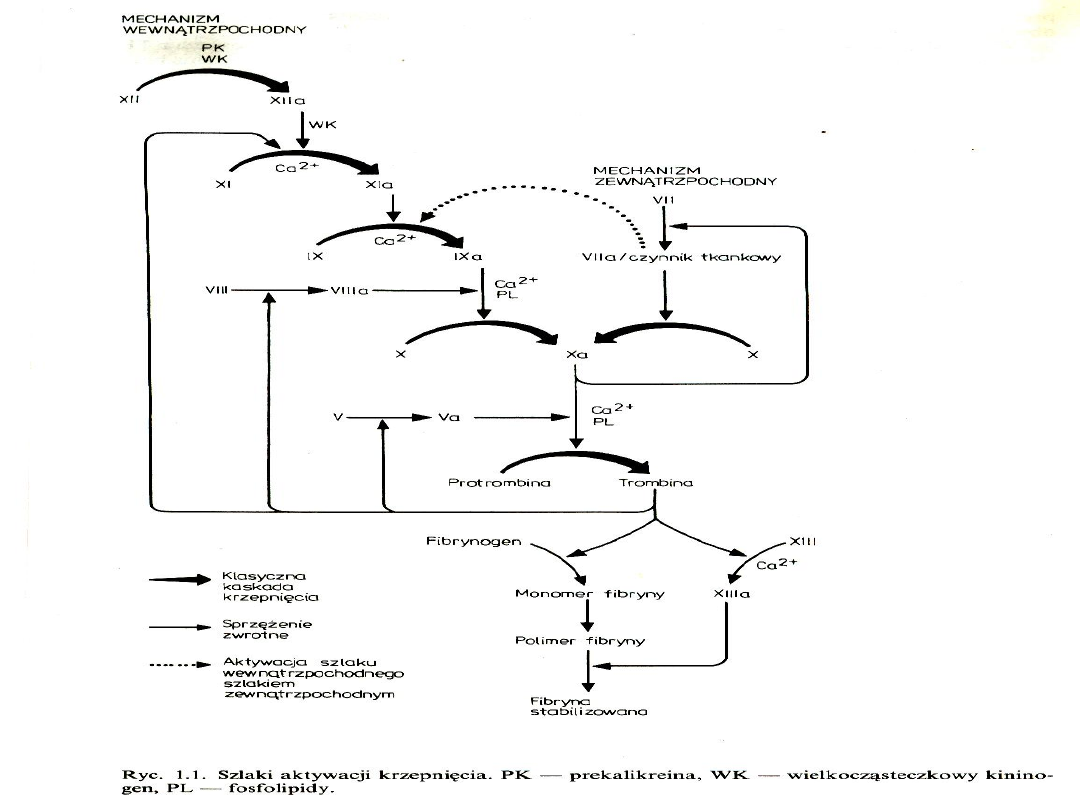
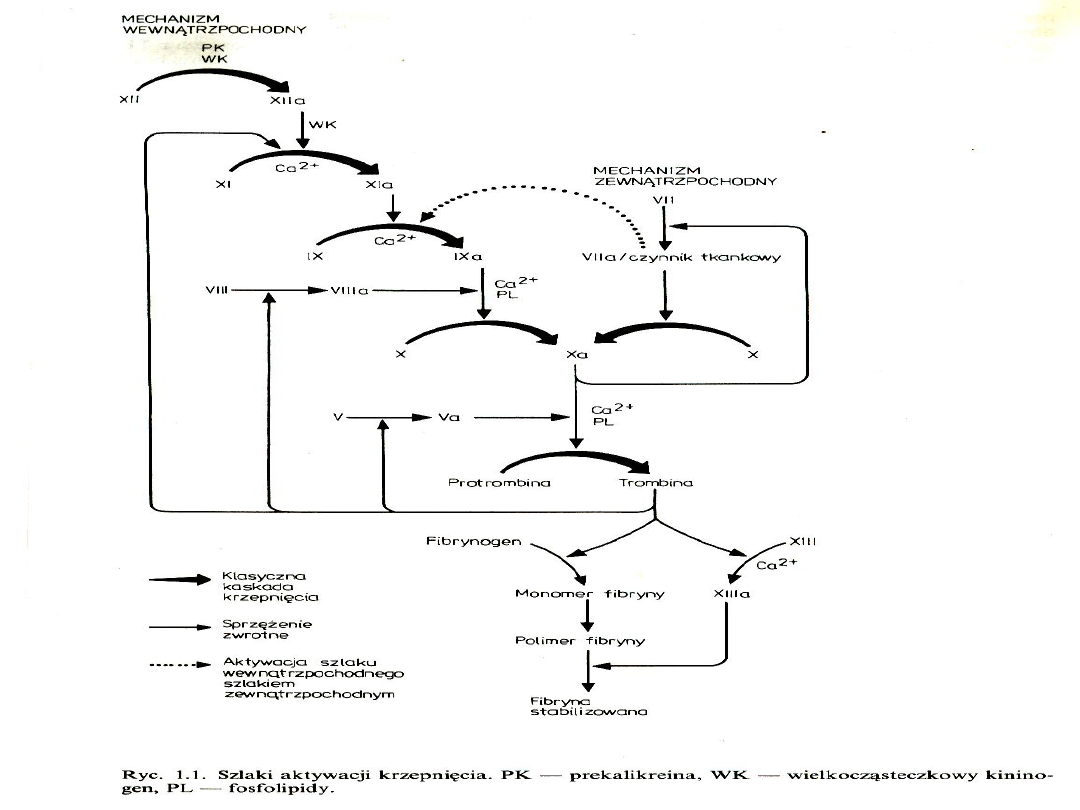
Składowe hemostazy
miejscowej
1.
Ściana naczynia
2.
Płytki krwi
3.
Osoczowy układ krzepnięcia

Hemostaza miejscowa
Hemostaza pierwotna (włośniczkowo-
płytkowa)
- skurcz naczynia
-adhezja i agregacja płytek krwiczop
płytkowy
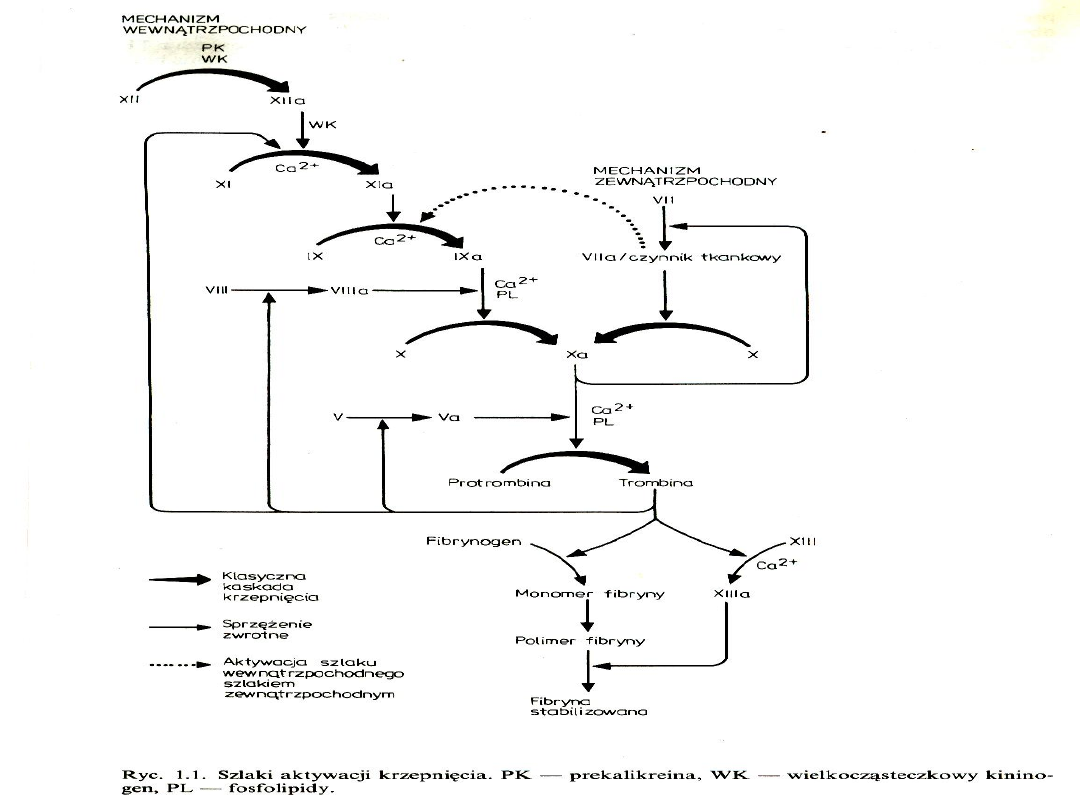
Hemostaza wtórna (osoczowa)
-aktywacja krzepnięcia z generacją
trombinypowstanie włóknika z fibrynogenu
czop hemostatyczny hamujący krwawienie

Podział skaz krwotocznych
Naczyniowe
Płytkowe
Osoczowe
Wrodzone:
Chorba Rendu-
Oslera
Małopłytkow
ości
Nadpłytkowoś
i
Wrodzone:
Hemofilie
Choroba von
Willebranda
Nabyte:
Choroba
Schönleina-
Henocha
Trombocytopa
tie
-wrodzone
-nabyte
Nabyte:
Niedobór witaminy
K
Choroby wątroby
Zespół DIC
Inhibitory
krzepnięcia

Najczęstsze wrodzone
skazy krwotoczne
Hemofilia A
1 : 16 000
Hemofilia B
1 : 110 000
Choroba von Willebranda 1%?

Hemofilia A
Brak, niedobór, lub nieprawidłowy
czynnik VIII
Gen dla czynnika VIII w dystalnej
części długiego ramienia
chromosomu X (Xq28)

Hemofilia B
Brak, niedobór, lub nieprawidłowy
czynnik IX
Gen dla czynnika IX w dystalnej
części długiego ramienia
chromosomu X
(Xq26-27)

Hemofilia A i B podlegają tym
samym prawom dziedziczenia i
mają taki sam obraz kliniczny.
Różnicowanie obu hemofilii jest
możliwe tylko za pomocą badań
laboratoryjnych

Dziedziczenie hemofilii
X
X XY
XX
X
Y
X
X XX
X
Y XY
X
X
X
X XY
XY

Historia hemofilii
II wiek – orzeczenie rabinackie zwalniające
chłopca z obrzezania jeśli jego dwóch braci
zmarło po tym zabiegu z powodu krwawienia
Albucasis (1013-1106) – lekarz arabski opisał
rodzinę w której mężczyźni umierali z powodu
krwawień nawet po małych urazach
Dr John Conrad Otto – pierwszy nowoczesny
opis hemofilii w 1803 r. Choroba dziedziczna,
dotycząca mężczyzn i objawiająca się
krwawieniami
Hemofilia – nazwa po raz pierwszy
wprowadzona przez Hopfa z Uniwersytetu w
Zurichu, w 1828 r.
1952 – odkrycie, że istnieją 2 różne formy
hemofilii (hemofilia A i B)

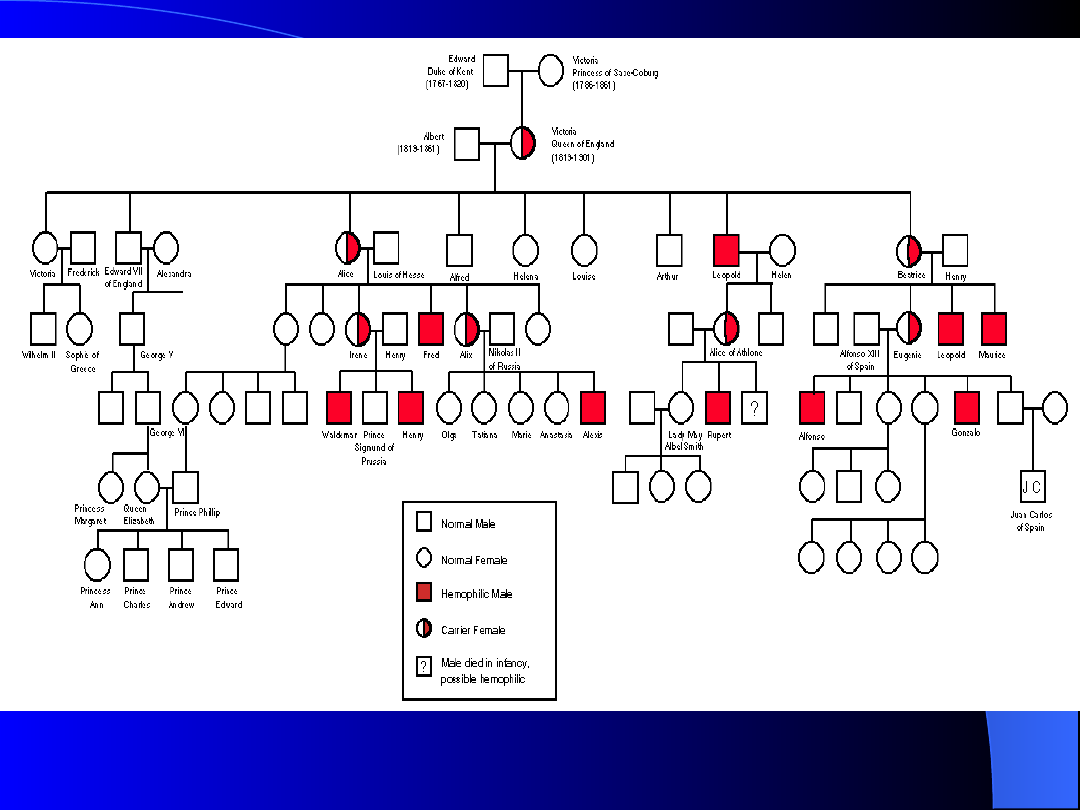
Hemofilia - choroba
królewska
Królowa Wiktoria – nosicielka

Postacie hemofilii
Ciężka - <1% cz.VIII lub
IX
Umiarkowana - 1 - 5%
Łagodna - 6 – 40%

Postać ciężka
50-60% chorych
Krwawienia samoistne

Objawy kliniczne ciężkiej postaci
hemofilii
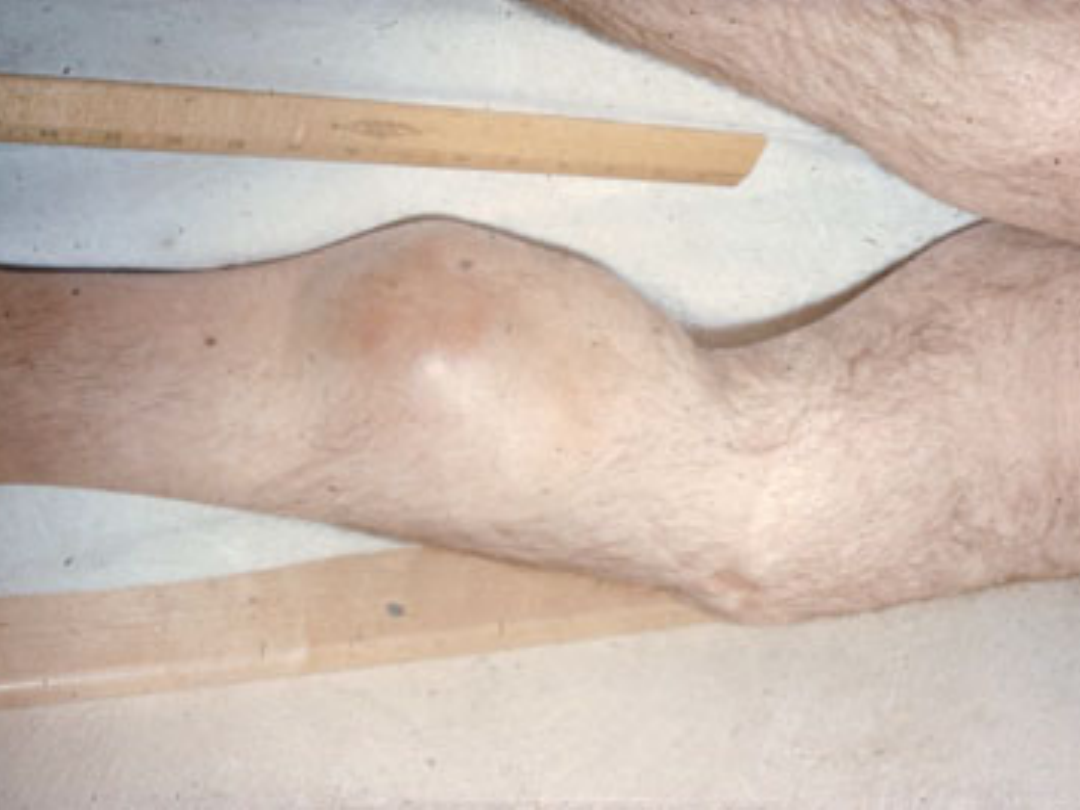
Krwawienia do stawów: 30-35/rok
staw kolanowy - 44%
staw łokciowy – 25%
staw skokowy – 15%
staw barkowy – 8%
staw biodrowy - 5%
inne stawy - 3%
Krwawienia do mięśni
Krwawienia do przestrzeni pozaotrzewnowej
Krwiomocz
Krwawienia z przewodu pokarmowego
Krwawienia śródczaszkowe

Hemofilia umiarkowana
Rzadko krwawienia samoistne
Krwawienia pourazowe równie
niebezpieczne jak w ciężkiej
hemofilii
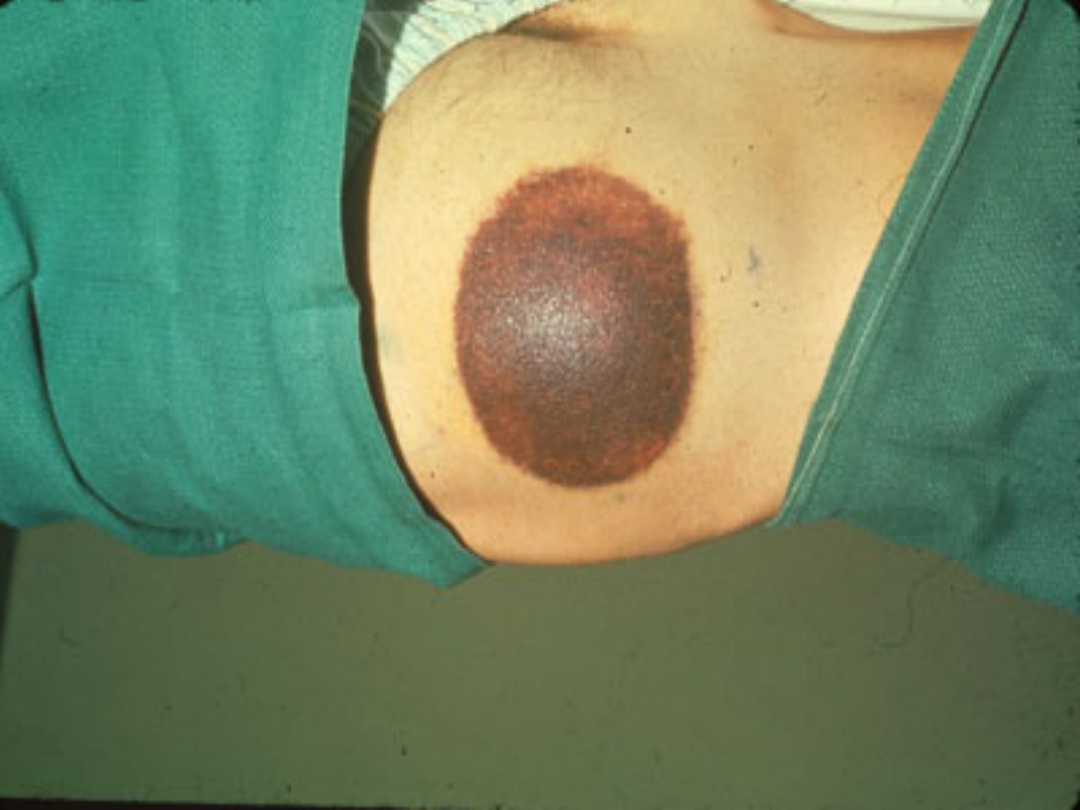

Hemofilia łagodna
Krwawienia samoistne nie
występują
Krwawienia pourazowe, mogą być
niebezpieczne

Rozpoznanie hemofilii
Wywiad rodzinny
Obraz kliniczny
APTT
N PT
cz.VIII – hemofilia A*
cz. IX – hemofilia B
*
należy wykluczyć chorobę von Willebranda

Opieka nad chorym na hemofilię
Profilaktyka
Leczenie krwawień
Leczenie powikłań

Historia leczenia hemofilii
A
Krew
1840 r. - Lane
Osocze 1923 r. - Feislly
Frakcja C-1 1950
Krioprecypitat, 1959 -1966 Pool,
Łopaciuk
Koncentraty cz.VIII z osocza
Koncentraty r-cz.VIII 1992 r.
Koncentraty r-cz.VIII trzeciej generacji
Terapia genowa

Preparaty cz. VIII
Krioprecypitat
Koncentraty cz.VIII z ludzkiego osocza
-średnio oczyszczone
-wysoko oczyszczone (monoklonalne)
Koncentraty r-cz.VIII
Koncentraty r-cz.VIII drugiej generacji
Koncentraty r-cz.VIII trzeciej generacji
(efektywność, bezpieczeństwo, stopień czystości, cena)

rVIII
Synteza rVIII – w komórkach jajnika
chomika chińskiego(CHO) lub w
komórkach nerki zarodka chomika
chińskiego (BHK)rVIII wydzielany z
komórek do podłoża hodowlanego
filtrowanie podłoża chromatografia
powinowactwa z użyciem
monoklonalnych mysich przeciwciał
swoiście wiążących cz.VIII usunięcie
przeciwciał i innych zbędnych białek za
pomocą technik chromatograficznych
oczyszczony rVIII+stabilizator
(albumina) liofilizacja preparat
handlowy

Preparaty rVIII
Recombinate (Baxter Biocsence) I gen.
ReFacto (Wyeth) III gen. (BDD
rVIII)
Kogenate FS (Bayer) III gen.
Advate rAHF PFM (Baxter Biocsence)
III gen.

Preparaty cz. IX
Świeżo mrożone osocze
Koncentraty zespołu protrombiny
Wysoko oczyszczone koncentraty
cz. IX
Koncentrat r-cz.IX (Benefix)

Dawkowanie koncentratów
czynników krzepnięcia
Zależy od okresu półtrwania
czynnika krzepniecia i stopnia
odzyskania in vivo
Cz.VIII 10-12 h 90-100%
1 j/kg - 2 j/dl
Cz.IX 16-18 h 50%
1 j/kg - 1 j/dl
Dawka koncentratów w
krwawieniach
Miejsce krwawienia
Optymal
na
aktywno
śc
czynnika
(j/dl)
Dawka
(j/kg/mc)
VIII
Dawka
j/kg/mc
IX
Częstośc
wstrzyknięć i
czas leczenia
24h dni
Do stawów
Do mięśni
Z przewodu
pokarmowego
Język/tylna ściana
gardła
Zaotrzewnowe
Śródczaszkowe
Krwiomocz
Małe krwawienia
30-50
30-50
30-50
40-60
30-50
60-80
30-50
20-30
15-25
15-25
15-25
20-30
15-25
30-40
15-25
10-15
30-50
30-50
30-50
40-60
30-50
60-80
30-50
20-30
1-2 1-2
1-2 1-2
1-2 2-3
1-2 3-4
1-2 3-4
1-3 7-10
1-2 1-2
1-2 1-2

Profilaktyka
pierwotna

Profilaktyka pierwotna
Cel:
-uchronienie chorych na ciężką
hemofilię przed kalectwem
powodowanym artropatią

Profilaktyka – hemofilia A
Wstrzyknięcia koncentratu
czynnika VIII w dawce 25-40
j.m./kg m.c. trzy razy w tygodniu,
począwszy od 1-2 r.ż.

Profilaktyka – hemofilia B
Wstrzyknięcia koncentratu
czynnika IX w dawce 25-40 j.m./kg
m.c. dwa razy w tygodniu,
począwszy od 1-2 r.ż.

Powikłania leczenia
Transmisja wirusów
Krążący antykoagulant

Krążący antykoagulant
Hemofilia A 20-25%
Hemofilia B 1-3%

Czynniki genetyczne
wpływające na pojawienie
się inhibitora
Związek z antygenami zgodności
tkankowej klasy II (DQA 1)
Defekty struktury genów cytokin
lub ich receptorów prowadzące do
zwiększonej odpowiedzi
immunologicznej

Charakterystyka
przeciwciał
IgG
Neutralizują aktywność VIII c
Miano inhibitora wyraża się w
jednostkach Bethesda
Low responders < 5 jB/ml
High responders > 5 jB/ml

Charakterystyka chorych:
low and high responders
5 jB/ml
A
B
C
D
E

Leczenie krwawień u chorych na
hemofilię A z krążącym
antykoagulantem
Duże dawki czynnika VIII
Aktywowane czynniki zespołu
protrombiny (FEIBA)
Rekombinowany czynnik VIIa
Plazmafereza
Zewnątrzustrojowa adsorpcja
przeciwciał na kolumnach sferozy-
białka A
Duże dawki czynnika VIII
40 j/kg + 20 j/kg
na każdą jB
inhibitora
bolus 5000 j,
500 – 1000 j/godz w
ciągłym wlewie
Aktywowane czynniki
zespołu protrombiny
FEIBA lub Autoplex
25 – 100 j/kg co 12 h
Rekombinowany czynnik
VII
90 g/kg co 3 godz
aż do poprawy

Chory na hemofilię A z krążącym
antykoagulantem
Czy aktualnie krwawi
tak nie
indukcja tolerancji
immunologicznej
HR LR
- aPCC - duże dawki
- r-VIIa cz.VIII
- plazmafereza+cz.VIII -PCC lub aPCC

FENOC STUDY
Blood 2007;109:546-551
Nie wykazano różnicy w
skuteczności leczenia krwawień do
stawów u chorych na hemofilię A
powikłaną inhibitorem pomiędzy
NovoSeven i FEIBA

Wywoływanie stanu tolerancji
immunologicznej
Przetaczając koncentraty czynnika VIII
codziennie lub co drugi dzień
obserwuje się u chorych powolne i
postępujące zmniejszenie miana
przeciwciał, a w końcu całkowite ich
zniknięcie z krążenia
Istotny jest czas, który upłynął od czasu
pojawienia się inhibitora do rozpoczęcia
leczenia, a także jego miano
Po osiągnięciu stanu TI należy wdrożyć
wtórną profilaktykę
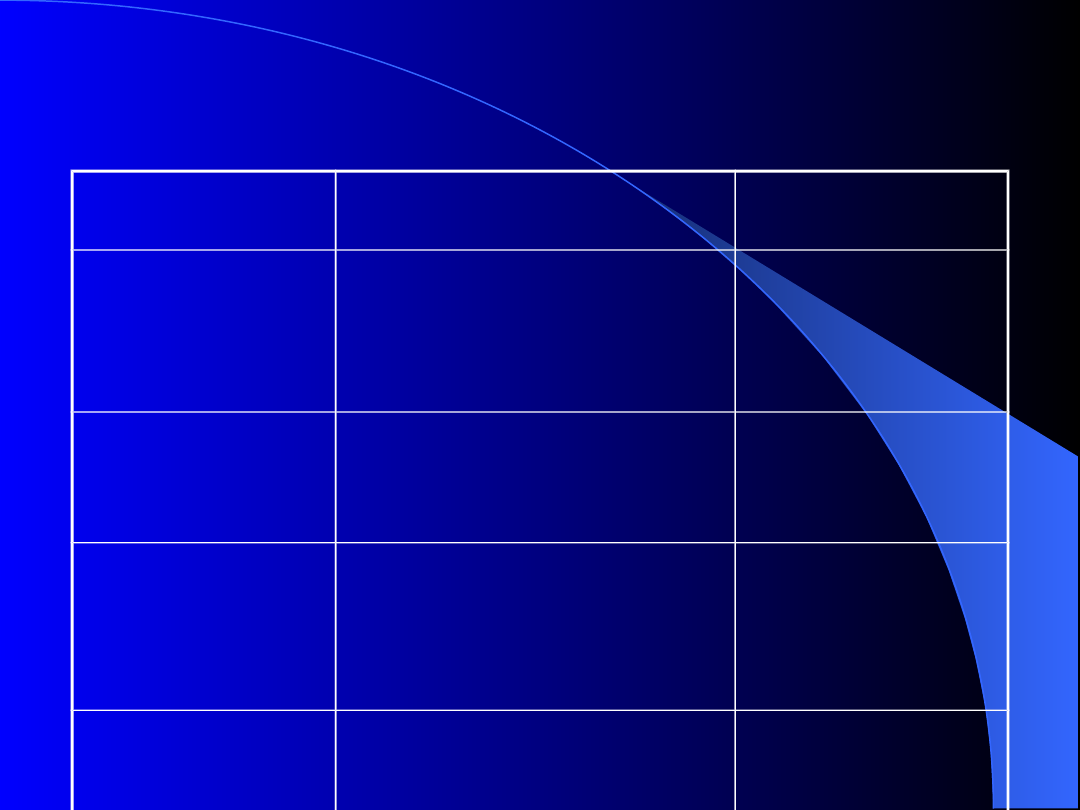
Metody wywoływania STI
Metoda (autorzy) Postępowanie
Średni czas
trwania
Duże dawki
cz.VIII
(Brackmann)
FVIII 200-300 j/kg
codziennie + FEIBA
1 – 3 lata
Małe lub średnie
dawki cz.VIII
FVIII 25 j/kg co drugi dzień
lub 50 j/kg codziennie
1 – 24 miesiące
Metoda Malmö
(Nilsson i wsp)
FVIII, cyklofosfamid, duże
dawki dożylnych IgG
(przed leczeniem
pozaustrojowa adsorpcja
przeciwciał)
1 miesiąc
Wieprzowy
czynnik VIII
20 – 60 j/kg co 2 dni lub na
żądanie
1 – 4 lata

Czynniki korzystnie wpływające na
osiągnięcie tolerancji
immunologicznej
Miano inhibitora przed
rozpoczęciem programu <10
j.B./ml
Miano inhibitora w pierwszej fazie
leczenia <500 j.B./ml
Historyczne miano <200 j.B./ml
Czas od wystąpienia inhibitora do
rozpoczęcia programu >2 lat

Inne czynniki mogące
wpływać na osiągnięcie
STI
Rodzaj koncentratu czynnika VIII
Dawka czynnika VIII
Współistniejące choroby
infekcyjne i procesy zapalne
Przerwy w programie >2 tyg.

Korzystne czynniki
prognostyczne
Uzyskanie STI u >80% chorych

Hemofilia B z inhibitorem
W obecności przeciwciał
przeciwko czynnikowi IX mogą
występować reakcje alergiczne po
wstrzyknięciu koncentratu cz.IX a
powikłaniem leczenia
substytucyjnego może być zespół
nerczycowy
NovoSeven w przypadku
krwawień

Choroba von Willebranda
(vWD)
Niedobór/defekt czynnika
von Willebranda
Dziedziczenie autosomalne
najczęściej dominujące

Czynnik von Willebranda
(vWf)
Glikoproteina występująca w
osoczu w postaci multimerów o
różnej masie cząsteczkowej i w
kompleksie z czynnikiem VIII
Gen kodujący vWf w chromosomie
12
Synteza – komórki śródbłonka i
megakariocyty

Rola vWf w hemostazie
Ułatwia adhezję płytek krwi do
warstwy podśródbłonkowej (wiąże
się z GP Ib-IX-V)
Tworzy kompleks z czynnikiem
VIII i ochrania go przed
degradacją proteolityczną

Klasyfikacja vWD
Typ 1 łagodny ilościowy niedobór vWf,
dziedziczenie autosomalne dominujące,
>70% objawowej vWD
Typ 2 jakościowy defekt vWf, podtypy
2A (10-15%), 2B (<5%), 2M i 2N (b.
rzadko), dziedziczenie autosomalne
dominujące, w typie 2N recesywne)
Typ 3 ciężki ilościowy niedobór vWf,
recesywny, 1-5 : 1000 000

vWD - objawy
Krwawienia z nosa
Skłonność do siniaczenia
Przedłużone i obfite krwawienia
miesiączkowe
Krwawienia do mięśni i stawów
(Typ 3)

Diagnostyka vWD
Testy przesiewowe
Przedłużony czas krwawienia
(może być prawidłowy)
Przedłużony APTT (może być
prawidłowy)
Przedłużony czas okluzji z
zastosowaniem analizatora PFA
100

Diagnostyka vWD
Rozpoznawanie typu
choroby
Antygen vWf (vWf:Ag)
Aktywność kofaktora rystocetyny
(R:Cof)
Aktywność prokoagulacyjna
czynnika VIII (czVIII:C)

Diagnostyka vWD
Rozpoznawanie podtypu
choroby
Aglutynacja płytek pod wpływem
rystocetyny (RIPA)
Analiza multimerów vWf
(elektroforeza w żelu agarozy)
Wiązanie czynnika VIII do vWf
Zawartość vWf w płytkach krwi
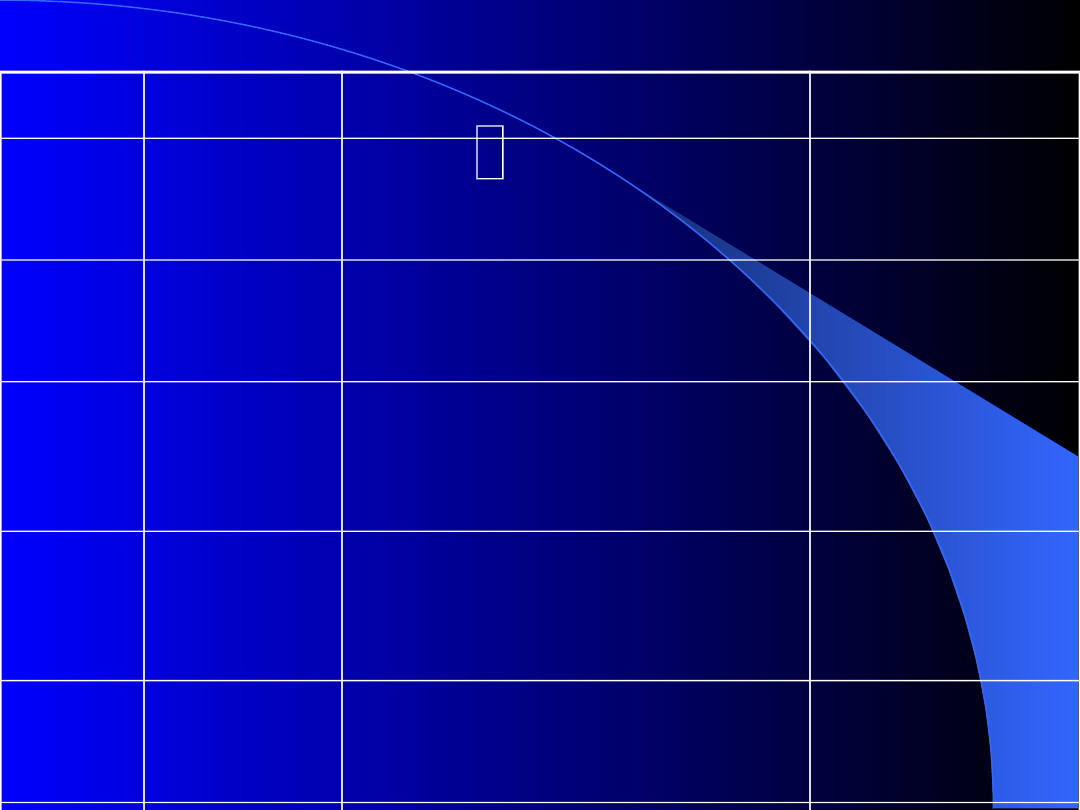
Klasyfikacja choroby von Willebranda
Typ
(podtyp)
Skaza
krwotoczna
Charakterystyka laboratoryjna
Podłoże
molekularne
Typ1
Łagodna lub
umiarkowana
vWF:Ag, R:Cof, VIII:C
proporcjonalnie zmniejszone
(<50%), prawidłowy rozkład
multimerów
Niewielki ilościowy
niedobór vWF
Typ 2A
Łagodna lub
umiarkowana
vWF:Ag, R:Cof, VIII:C zmniejszone
w różnym stopniu (R:Cof/vWF:Ag
<0.3), brak dużych i pośrednich
multimerów
Defekt uwalniania
vWF z komórki lub
nadmierna
proteoliza w osoczu
Typ 2B
Łagodna lub
umiarkowana
vWF:Ag, R:Cof, VIII:C zmniejszone
w różnym stopniu (R:Cof/vWF:Ag
ok. 0.5), brak dużych multimerów,
zwiększenie RIPA, łagodna
małopłytkowość
Zwiększone
powinowactwo vWF
do GP Ib płytek
Typ 2M
Łagodna lub
nasilona
vWF:Ag i VIII:C zmniejszone w
różnym stopniu, zmniejszenie R:Cof
większe niż antygenu pomimo
obecności dużych i pośrednich
multimerów
Zmniejszone
powinowactwo vWF
do GP Ib płytek
Typ 2N
Łagodna lub
nasilona
Zmniejszenie VIII:C większe niż
vWF:Ag, prawidłowy rozkład
multimerów
Brak lub
zmniejszenie
zdolności vWF do
wiązania czVIII
Typ 3
Ciężki
przebieg
kliniczny
vWF:Ag, R:Cof, VIII:C znacznie
zmniejszone (<5%), brak lub
śladowa ilość wszystkich
multimerów
Znaczny ilościowy
niedobór vWF
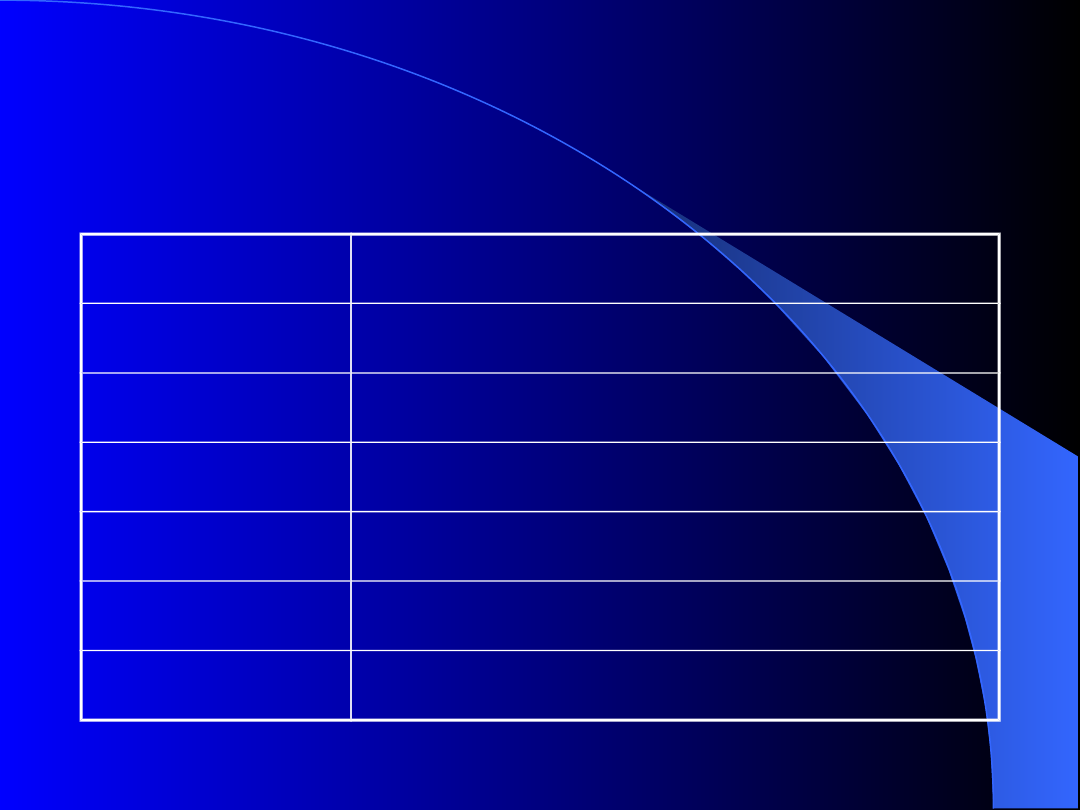
Leki stosowane w różnych
typach vWD
Typ/podtyp Lek z wyboru
1
Dezmopresyna
2A
Czynnik VIII-vWf
2B
Czynnik VIII-vWf
2M
Czynnik VIII-vWf
2N
Dezmopresyna
3
Czynnik VIII-vWf

Wrodzone defekty
fibrynogenu
Afibrynogenemia
Hypofibrynogenemia
Dysfibrynogenemia

Afibrynogenemia - objawy
Krwawienia z przewodu
pokarmowego
Krwawienia z dróg rodnych
Krwawienia śródczaszkowe
Krwawienia dostawowe (20%)
Powikłania zakrzepowo-zatorowe o
chorych leczonych koncentratami
fibrynogenu

Afibrynogenemia –
badania laboratoryjne
Przedłużenie – APTT, PT, TT
Brak fibrynogenu w osoczu i
płytkach krwi
Łagodna małopłytkowość?
Przedłużony czas krwawienia
(30%)

Afibrynogenemia -
leczenie
Leczenie substytucyjne –
krioprecypitat, koncentrat
fibrynogenu (ostre krwawienia,
zabieg operacyjny, profilaktyka w
ciąży)

Dysfibrynogenemia
Defekt dotyczy konwersji
fibrynogenu w fibrynę a
najczęściej polimeryzacji
monomerów fibryny
Dziedziczenie – autosomalne
dominujące

Dysfibrynogenemia -
objawy
Krwawienia (40%) – łagodna skaza
krwotoczna, łatwe siniaczenie,
krwawienia do tkanek miękkich,
obfite krwawienia miesiączkowe
Zakrzepy (30%) – oporność na
działanie fibrynolityczne
Poronienia, upośledzone gojenie
ran
Defekty bezobjawowe (30%)

Niedobór czynnika VII
Dziedziczenie – autosomalne,
recesywne
Objawy – krwawienia z nosa, krwiaki
podskórne, krwawienia z przewodu
pokarmowego, moczowo-płciowego i do
stawów
Rozpoznanie – PT , aktywność/stężenie
czVII (wykluczenie nabytych
niedoborów – doustne antykoagulanty,
choroby wątroby, niedobór wit.K)
Leczenie – FFP, koncentraty cz.VII

Niedobór czynnika X
Dziedziczenie – autosomalne,
recesywne
Objawy jak w niedoborze cz.VII
Rozpoznanie – przedłużenie APTT
i PT, zmniejszenie stężenia cz.X
Leczenie – FFP, koncentrat
czynników zespołu protrombiny

Niedobór czynnika XI
Dziedziczenie autosomalne
recesywne
Objawy – rzadko krwawienia
samoistne, opóźnione krwawienia
pourazowe
Rozpoznanie – APTT , stężenie
czXI 5-15j/dl
Leczenie - FFP

Niedobór czynnika XIII
Częstość – 1 : 1000 000
Dziedziczenie – autosomalne,
recesywne
Objawy – krwawienia do tkanek
miękkich, do stawów, pseudotorbiele,
opóźnione krwawienia pourazowe i po
zabiegach, upośledzone gojenie ran
Rozpoznanie – test rozpuszczalności
skrzepu w 5M roztworze mocznika,
stężenie antygenu lub aktywność
cz.XIII (<1%)
Leczenie – FFP, krioprecypitat,
koncentrat cz.XIII (T1/2 = 9-19 dni)

Niedobory czynników XII,
PK, HMWK
Tzw. anomalie
Defekty bezobjawowe
Kierowani do hematologa po
wykryciu izolowanego
przedłużenia APTT

Podział skaz krwotocznych
Naczyniowe
Płytkowe
Osoczowe
Wrodzone:
Chorba Rendu-
Oslera
Małopłytkow
ości
Nadpłytkowoś
i
Wrodzone:
Hemofilie
Choroba von
Willebranda
Nabyte:
Choroba
Schönleina-
Henocha
Trombocytopa
tie
-wrodzone
-nabyte
Nabyte:
Niedobór witaminy
K
Choroby wątroby
Zespół DIC
Inhibitory
krzepnięcia

Witamina K
Witamina K
1
– dostarczana z
pokarmem roślinnym
Witamina K
2
– wytwarzana przez
bakterie flory jelitowej

Wchłanianie witaminy K
Witamina K jest związkiem
rozpuszczalnym w tłuszczach i
wchłania się z przewodu
pokarmowego w obecności
kwasów żółciowych

Działanie witaminy K na
układ krzepnięcia
Konieczna do potranslacyjnej
modyfikacji czynników zespołu
protrombiny
(czynniki: II, VII, IX i X)
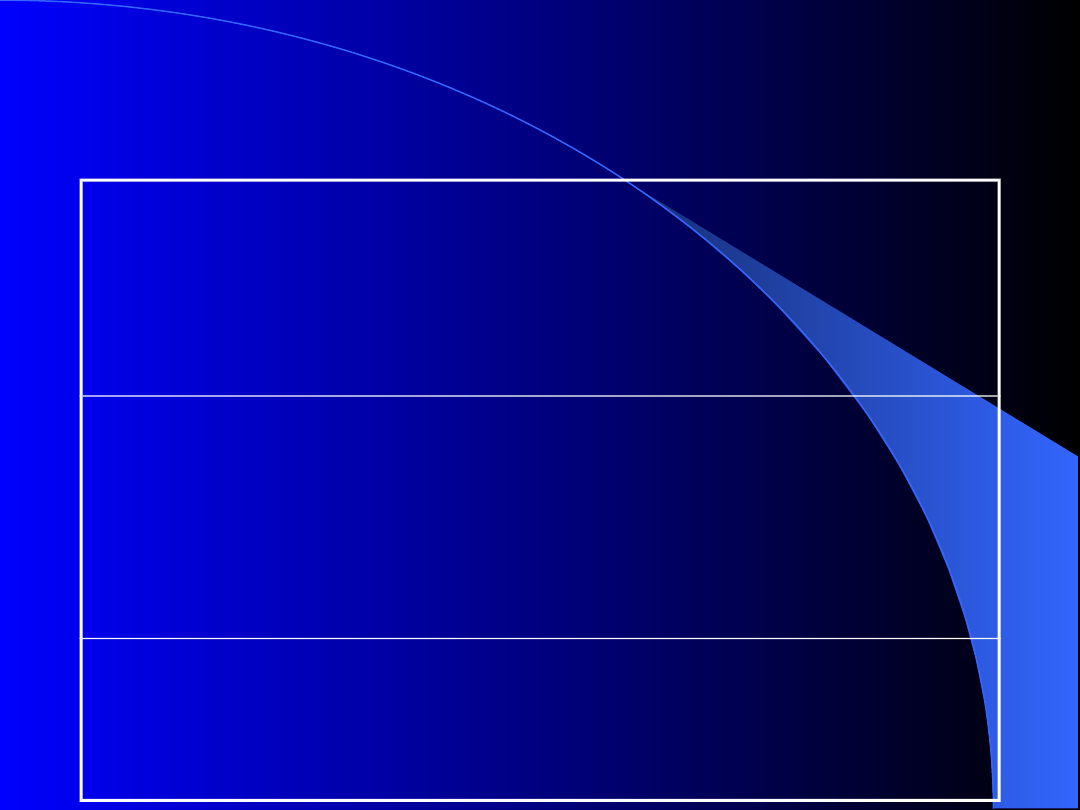
Przyczyny niedoboru witaminy K
Upośledzone wytwarzanie witaminy K
brak flory bakteryjnej jelit
wyjałowienie jelit przez stosowanie
antybiotyków
Upośledzone wchłanianie witaminy K
zahamowanie wydzielania żółci do światła
jelita
zespoły upośledzonego wchłaniania
wpływ leków (kolestyramina)
Upośledzone wykorzystanie witaminy K
antagonistyczny wpływ doustnych
antykoagulantów

Kliniczne objawy
niedoboru witaminy K
Podskórne wylewy krwi
Krwawienia z nosa i dziąseł
Krwiomocz
Krwawienie z przewodu
pokarmowego

Rozpoznanie niedoboru
witaminy K
Przedłużenie PT (INR )
APTT N/
Zmniejszona aktywność
czynników: II, VII, IX, X przy
prawidłowej aktywności cz. V

Zaburzenia hemostazy w
chorobach wątroby
cz. II, V, VII, IX, X
N/ Fibrynogen
AT, alfa2-antyplazmina
alfa2-makroglobulina
/N liczba płytek

DIC- uwagi ogólne
Mechanizm patofizjologiczny zaburzeń
krzepnięcia krwi występujący w różnych
stanach chorobowych
Charakteryzuje się systemową aktywacją
krzepnięcia, zużyciem czynników
hemostatycznych a także uszkodzeniem
tkanek i narządów
Śmiertelność ~ 60%, w ciężkich
postaciach > 80 %
11532 publikacje w PubMed (1951-2007)

• Defibrination (Soulier et al.,1952)
• Intravascular defibrination (Schneider,
1952)
• Disseminated intravascular
coagulation (Hardaway and
McKay, 1959)
• Consumption coagulopathy (Lasch et
al.,1961)
• Intravascular coagulation with
fibrinolysis (C. Owen et al., 1969)
Ann Surg. 1959 April; 149(4): 462–470.
Disseminated Intravascular Coagulation A Cause
of Shock
Robert M. Hardaway III and Donald G. McKay

Rozsiane krzepnięcie
śródnaczyniowe (DIC)
charakteryzuje się patologiczną,
uogólnioną aktywacją krzepnięcia
prowadzącą do odkładania fibryny
w łożysku naczyniowym z
wytworzeniem mnogich
zakrzepów w mikrokrążeniu i –
rzadziej – w obrębie większych
naczyń krwionośnych. Proces ten
prowadzi do niedokrwiennego
uszkodzenia tkanek i narządów
oraz do skazy krwotocznej.

Przyczyny krwawień w DIC
Zużycie czynników krzepnięcia (I,
V, XIII) i płytek krwi
Degradacja czynników krzepnięcia
przez plazminę (I, V, VIII, IX, XI)
Zaburzenia czynności płytek (FDP)
Obecność we krwi
antykoagulantów (FDP)

Ostry vs przewlekły DIC
ekspozycja czynnika wywołującego masywna i
w krótkim okresie czasu (np.poważny uraz,
posocznica) - pokonanie mechanizmów
regulujących, zużycie czynników krzepnięcia,
krwawienia i/lub powikłania zakrzepowe –
ostry/zdekompensowany/jawny DIC
ekspozycja czynnika wywołującego niewielka i
w dłuższym okresie (nowotwory, naczyniaki) –
brak lub niewielkie objawy kliniczne (zazwyczaj
zakrzepica), obecność cech laboratoryjnych –
przewlekły/zkompensowany DIC

Epidemiologia
1: 1000 przyjęć do szpitala
wieloprofilowego
Najczęściej w przebiegu ? infekcji,
urazów, powikłań położniczych, chorób
nowotworowych, ukąszeń węży

Choroby i stany kliniczne
związane z DIC
Zakażenia: bakteryjne, grzybicze, wirusowe,
pierwotniakowe
Urazy: wielonarządowe, zabiegi operacyjne,
oparzenia
Choroby nowotworowe: guzy lite,
mielo/limfoproliferacje
Powikłania ciąży i porodu: zator płynem
owodniowym, przedwczesne odklejenie łożyska,
ciąża obumarła
Ostra niewydolność wątroby
Anomalie naczyniowe: zespół Kasabach-Merritt,
duże tętniaki
Reakcje toksyczne lub immunologiczne: reakcje
potransfuzyjne, przełomy hemolityczne, ukąszenia
węży, odrzucenie przeszczepu
Patogeneza DIC w
posocznicy
Mechanizm
Patofizjologia
Zwiększona generacja
trombiny
Stłumienie układu
inhibitorów
Zahamowanie fibrynolizy
Aktywacja reakcji
zapalnej
Aktywacja
zewnątrzpochodnego szlaku
krzepnięcia (TF/VII)
Zwiększone zużycie,
zmniejszona synteza w
wątrobie, degradacja przez
elastazę, zmniejszenie
ekspresji trombomoduliny
przez cytokiny prozapalne
Zwiększone stężenie PAI-1
Poprzez aktywowane czynniki
krzepnięcia i zahamowanie
układu białka C
Sepsa cytokiny
Aktywacja krzepnięcia
TROMBINA
tworzenie złogów osłabienie fibrynolizy zużycie czynników
aktywacja
fibryny
poprzez PAI-1, TAFI hemostatycznych receptorów
komórkowych
Zużycie inhibitorów
Zużycie czynników
krzepnięcia krzepnięcia i płytek Zmiana kształtu
komórek
tworzenie zakrzepów
naczyniowych
generacja FDP – ów
Przeciek
naczyniowy
uszkodzenie narządów
skaza krwotoczna
Obrzęk

Mechanizmy odpowiedzialne za
podtrzymywanie generacji
trombiny
Aktywacja wewnątrzpochodnej drogi krzepnięcia
Zmniejszona aktywność inhibitorów krzepnięcia
stężenie białka C, S i antytrombiny
ekspresji receptora białka C i trombomoduliny na
sródbłonku
degradacja poprzez elastazę
Zwiększona dostępność fosfolipidów
externalizacja błony komórkowej podczas aktywacji
i/lub apoptozy
powstanie mikrocząsteczek z płytek, monocytów,
komórek śródbłonka
stężenia lipoprotein (VLDL)
oczyszczanie przez układ siateczkowo-śródbonkowy
Obecność krążacego czynnika tkankowego na
monocytach i mikrocząsteczkach

Obraz kliniczny DIC
Krwawienia
Zakrzepy
!!!
+ Objawy mikroangiopatycznej niedokrwistości
hemolitycznej

Obraz kliniczny DIC - cd
Krwawienia
Wybroczyny, sińce
Pęcherze krwotoczne
Krwawienie z ran
pourazowych/pooperac
yjnych
Sączenie z miejsc
wkłucia do żył i tętnic
Krwiaki podskórne
Krwawienia z dziąseł,
nosa
Krwawienia z przewodu
pokarmowego
Krwiomocz
Zmiany
zakrzepowe i
uszkodzenie
narządów
Skóra
Płuca
Nerki
Wątroba
Nadnercza
Serce
OUN
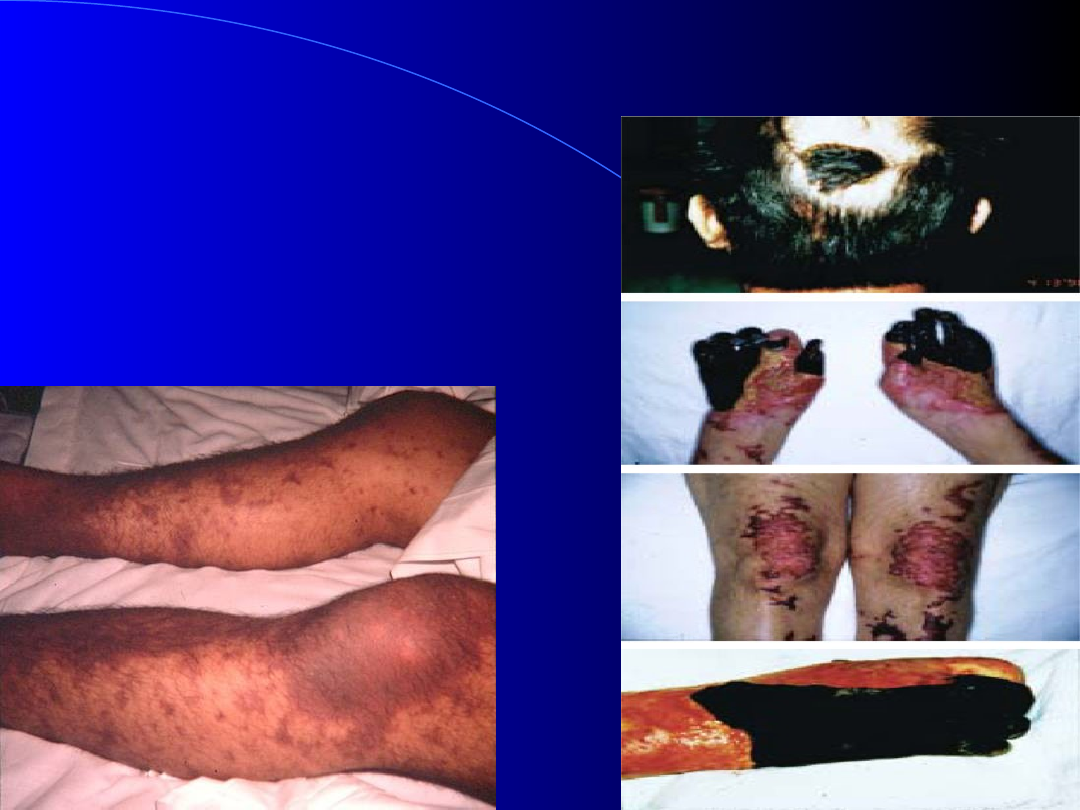
Plamica
piorunująca
Plamica w przebiegu
zespołu
Waterhouse-
Friderichsena
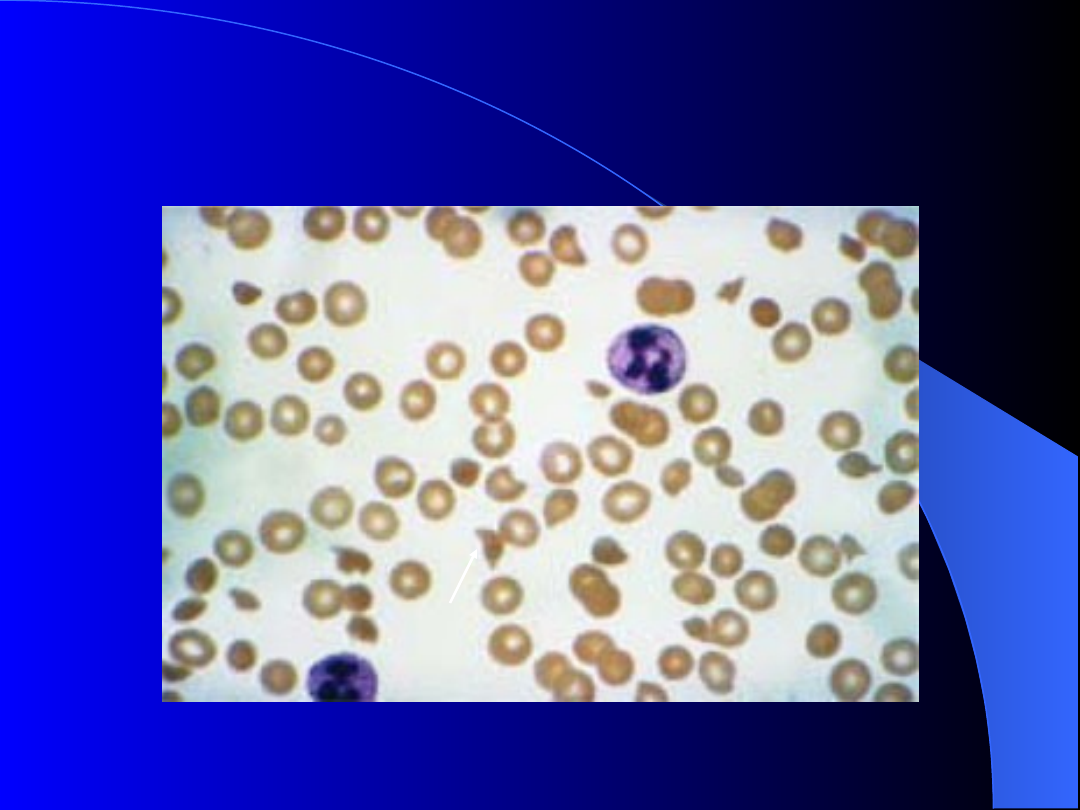
Schizocyty w rozmazie krwi
obwodowej
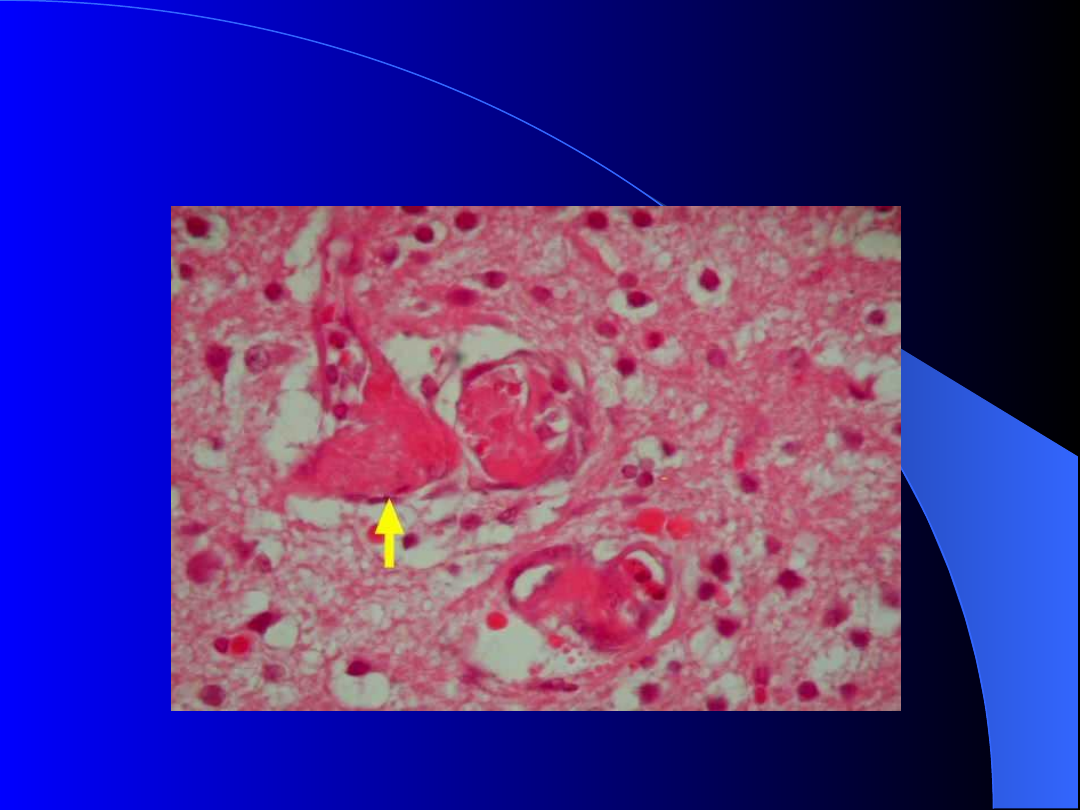
Złogi fibryny w naczyniach mózgowych
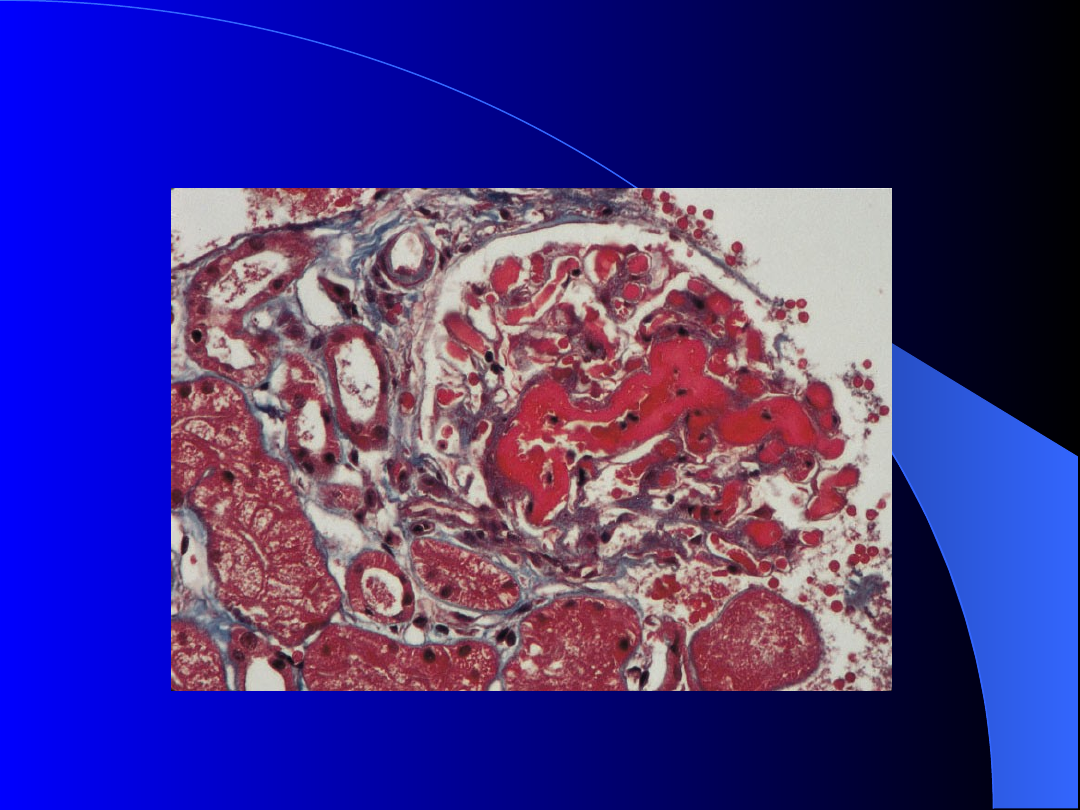
Złogi fibryny w naczyniach nerkowych

Diagnostyka DIC
Nie ma zespołu DIC bez wywołującej go
przyczyny
Nie ma specyficznego testu
laboratoryjnego dla rozpoznania DIC
Diagnostyka laboratoryjna DIC opiera
się na jednoczesnej interpretacji kilku
testów krzepnięcia
Zespół DIC rozwija się i przebiega
dynamicznie dlatego badania
laboratoryjne muszą być odpowiednio
często powtarzane

Nieprawidłowości w badaniach
laboratoryjnych w przebiegu
ostrego DIC
Badania podstawowe hemostazy(testy
globalne)
Czasy krzepnięcia (APTT, PT, TT)
przedłużone
Stężenie fibrynogenu
Liczba płytek krwi
Inne badania
Stężenie produktów degradacji fibryny
– FDP
Aktywność AT
Stężenie innych czynników krzepnięcia
(II, V, XIII)
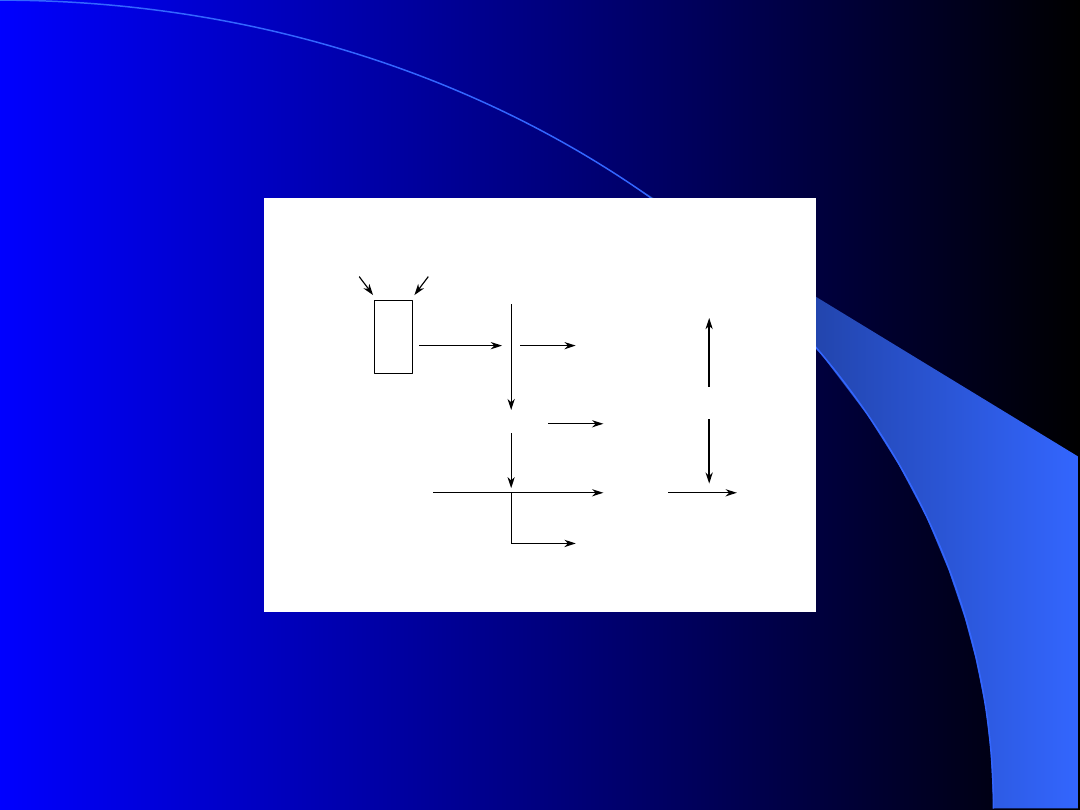
VIIa-TF
IXa
Xa
Va
PL
Ca
2+
Protrombinaza
Fibrynogen
Protrombina
Trombina
F1+2
TAT
FPA
Fibryna
PAP
Plazmina
DD
Generacja markerów aktywacji
krzepnięcia i fibrynolizy

Wyniki specyficznych markerów
krzepnięcia i fibrynolizy w DIC
Fibrynopeptyd A
Fragment protrombiny 1+2
Kompleks trombina-antytrombina
Kompleks plazmina-antyplazmina
D-dimery

Kryteria rozpoznania DIC
wg ISTH
5 etapowy algorytm rozpoznania
DIC

I.
ocena ryzyka
wystąpienia DIC
– czy występuje choroba
odpowiedzialna za rozwój DIC?

II.
Wykonanie badań
diagnostycznych
liczba płytek krwi
czas protrombinowy
stężenie fibrynogenu
stężenie markerów fibryny

III.
Ocena wyników badań
hemostazy
Liczba płytek
>100 G/l = 0, <100 = 1, <50 = 2
Markery fibryny
norma=0, umiarkowany wzrost=2, duży
wzrost=3
Przedłużenie czasu protrombinowego
<3 s = 0, >3 s i <6 s = 1, >6 s = 2
Stężenie fibrynogenu
>1.0g/l = 0, <1.0g/l = 1

IV. Obliczenie wskaźnika

V. Rozpoznanie
5 : jawny zespół DIC
Czułość i swoistość >90%

Różnicowanie
Choroby wątroby
Liczba płytek , Fibrynogen ,
FDP , PT, APTT ,
D-dimery N, cz.VIII N,
Pierwotna fibrynogenoliza
Fibrynogen , FDP , APTT, PT TT
, cz.V iVIII
Liczba płytek N, D-dimery N

Leczenie DIC
Leczenie choroby podstawowej – usunięcie
przyczyny wywołującej DIC
Przetoczenia preparatów krwiopochodnych –
zabezpieczenie chorego przed groźnym dla
życia krwawieniem, przywrócenie
czynnościowo sprawnego składu krwi
Leki hamujące krzepnięcie krwi –
zahamowanie patologicznej aktywacji
krzepnięcia
Naturalne inhibitory krzepnięcia –
przywrócenie prawidłowego działania
układów inhibitorowych
Inne leki
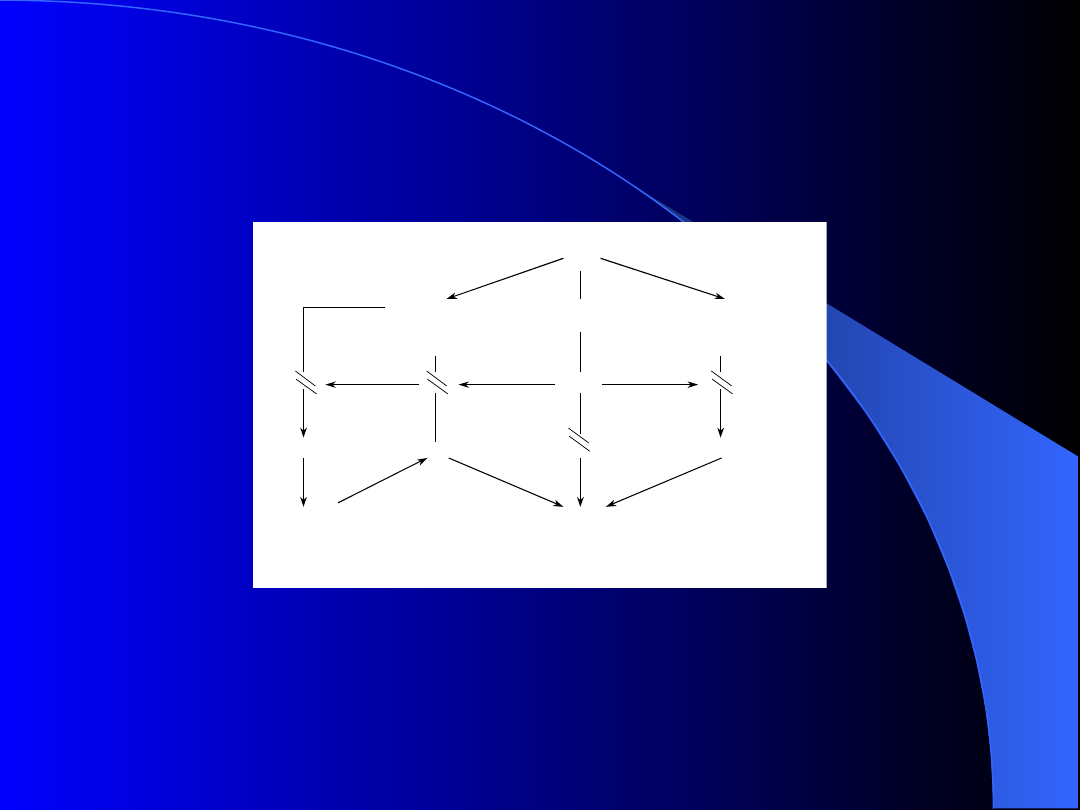
OBP
PROKOAGULANTY
-
TF
-
CP
-
IL
-
1,TNF, VPF
ANTYKOAGULANTY
-
anneksyna
VIII
AKTYWNOŚĆ PROTEOLITYCZNA
-
u
-
PA, t
-
PA,
anneksyna
II
-
inhibitory PAI
-
1, PAI
-
2
-
elastaza
,
katepsyna
G
NADKRZEPLIWOŚĆ
ZAKRZEPICA
i/lub
DIC
ATRA
KRWAWIENIE
i/lub
ZAKRZEPICA
FIBRYNOLIZA
Mechanizmy działania ATRA w
modulowaniu koagulopatii w OBP

Leczenie uzupełniające
KKP (1-2j/10kg)– liczba płytek <20
000/l lub <50 000 przy skazie
krwotocznej
FFP (15-20 ml/kg co 12-24h) –
stężenie fibrynogenu <1g/l i/lub
przedłużenie czasów krzepnięcia
+ krwawienia
Krioprecypitat 1 j/10 kg co 24 h
Koncentrat fibrynogenu (2-3 g)
KKCz – przy znacznej utracie krwi

Leczenie opornych
krwawień w przebiegu DIC
rVIIa (NovoSeven)
Leki antyfibrynolityczne – kwas
tranexamowy, EACA – tylko w
przypadku krwawień związanych z
nadmierną fibrynolizą (OBP, rak
prostaty)

rVIIa w DIC
Franchini M i wsp. Potential role
of recombinant activated factor
VII for the treatment of severe
bleeding associated with
disseminated intravascular
coagulation: a systematic
review. Blood Coagulation and
Fibrinolysis 2007; 18: 589-593.

rVIIa w DIC
Przyczyna DIC i
krwawienia
Liczba
choryc
h
Dawka
wstępna
(g/kg)
Liczba
dawek
*Odpowied
ź
Powikłania
położnicze
Nowotwory złośliwe
Urazy
Sepsa
Uszkodzenie wątroby
Gorączka Dengua
OZT
OBP
32
18
7
3
3
7
7
1
60 – 170
90
120
90 - 120
40 – 180
100
18.5 -
120
90
1 – 19
3 - 10
1 - 3
1 - 3
16.5-33
g/kg/h
1 – bd
1
2
32/32
15/18
7/7**
3/3
3/3
4/7
6/7
1/1
*
Nie stwierdzono żadnych objawów niepożądanych a w szczególności powikłań
zakrzepowo-zatorowych
** 3 zgony z przyczyn innych niż krwawienie lub zakrzepica

rVIIa w leczeniu DIC
Wnioski
Krwawienia w przebiegu DIC
oporne na terapię substytucyjną
(FFP, krioprecypitat, KF, kkp,
kkcz) i inne metody leczenia mogą
być wskazaniem do zastosowania
rVIIa

rVIIa w DIC
Pytania i problemy do
rozwiązania
Konieczne są badania kliniczne z
randomizacją w celu:
potwierdzenia skuteczności rVIIa
w opornych krwawieniach w
przebiegu DIC
określenia bezpieczeństwa
stosowania leku
ustalenia optymalnego
dawkowania

Leki hamujące krzepnięcie
krwi
Heparyna standardowa/heparyny
drobnocząsteczkowe
Rekombinowana hirudyna

Heparyna w leczeniu DIC
5 –10 j/kg/h
WSKAZANIA
Plamica
piorunująca
Nowotwory
złośliwe
Obumarły płód
PRZECIWWSKAZ
ANIA
Piorunująca
niewydolność
wątroby
Krwawienie do
OUN
Czynne krwawienie
Ciężka
małopłytkowość

Koncentraty inhibitorów
krzepnięcia
Antytrombina
-działanie antytrombinowe
-działanie przeciwzapalne - CRP,
IL-6
Rekombinowane aktywowane
białko C (rAPC)
Rekombinowana Trombomodulina
Rekombinowany TFPI

Antytrombina
Badanie KyberSept (n=2314) Wysokie
dawki AT (30 000 jm/4d) w leczeniu
chorych z ciężką sepsą. Badanie
randomizowane kontrolowane placebo.
Wyniki: Brak wpływu na śmiertelność w
28, 56 i 90 dniu
Znamiennie mniejsza śmiertelność w 90
dniu w grupie chorych otrzymujących
AT bez heparyny w stosunku do
chorych leczonych AT i heparyną
(44.9% vs 52.5%)
(Warren B i wsp. JAMA 2001, 286,1869)

Antytrombina w DIC
Badanie KyberSept
Analiza 563 chorych, którzy
spełniali kryteria DIC wg ISTH a
nie otrzymywali heparyny. Grupa
AT (286) vs placebo (277)
Wyniki: znamienne zmniejszenie
śmiertelności już w 28 dniu w
grupie leczonej AT (redukcja o
14.6%, p = 0.02)
(Kienast J i wsp. J Thromb Haemost 2006, 4,
90)

APC w leczeniu sepsy
Badanie PROWESS i ENHANCE
Drotrecogin alfa (Xigris; Eli Lilly
and Company) 24g/kg/h przez 96
h
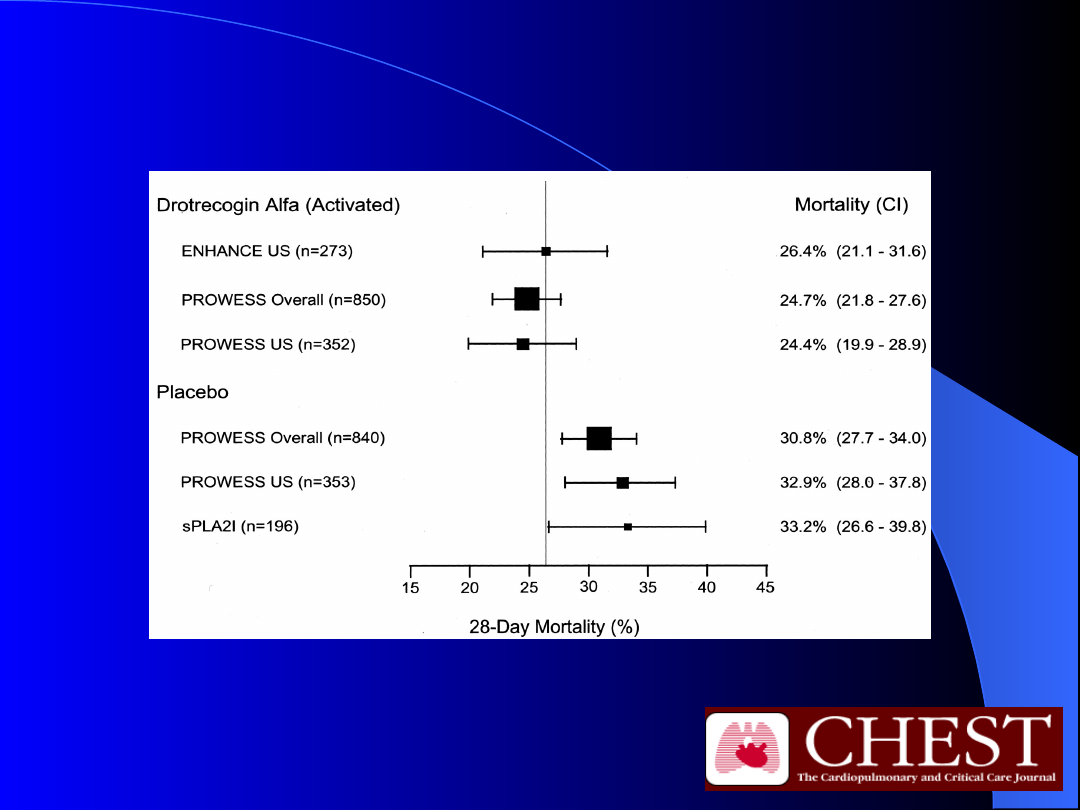
Bernard, G. R. et al. Chest 2004;125:2206-2216
The 28-day mortality rates in trials of severe sepsis

APC w leczeniu chorych z ciężką
sepsą i małym ryzykiem zgonu
(APACHE II<25 lub uszkodzenie
1 narządu) n=2613
Drotrecogin alfa 24g/kg/h przez
96 h vs placebo
Brak różnic w śmiertelności
(18.5% vs17%)
Częściej poważne krwawienia w
grupie APC (3.9 vs 2.2%)
N Engl J Med. 2005, 353, 1332

Rekombinowana
Trombomodulina w
leczeniu DIC
Badanie III fazy,randomizowane z
podwójnie ślepą próbą na 234 chorych
z DIC w przebiegu nowotworowych
chorób krwi i ciężkich infekcji
Heparyna 8j/kg/h vs ART -123
0.06mg/kg przez 6 dni
Wyniki: znamiennie częstsze ustąpienie
DIC w grupie z ART-123
(66.1%vs49.9%)
(
Saito H i wsp. J Thomb Haemost 2007,5,31)

rTFPI w DIC
Zmniejszenie śmiertelności w
przebiegu sepsy na modelu
zwierzęcym
Trend w kierunku zmniejszenia
śmiertelności wraz z poprawą
czynności narządów – badania II
fazy u chorych z ciężką sepsą
Brak wpływu na przeżycie chorych
z ciężką sepsą – badania III fazy

Inne potencjalne metody
leczenia DIC
Przeciwciała przeciwko selektynie
rIL-10
Przeciwciała monoklonalne anty-TNF
Inhibitor MAPK (kinaza białkowa
aktywowana mitogenem p38)
Przeciwciała anty-CD14
rNAPc2
Przeciwciała anty-TF/cz.VIIa

Patologiczne inhibitory
krzepnięcia
Autoprzeciwciała przeciwko
czynnikowi VIII
- bez uchwytnej przyczyny
- u kobiet po porodzie
- w przebiegu chorób
autoimmunologicznych
- w chorobach nowotworowych

Diagnostyka nabytych
inhibitorów czVIII
Przedłużone APTT
Brak korekcji po dodaniu
prawidłowego osocza
Zmniejszenie aktywności czVIII

Leczenie
rVIIa lub FEIBA – krwawienie
Prednizon + cyklofosfamid
Rituximab
Document Outline
- Slide 1
- Skaza krwotoczna Definicja
- Składowe hemostazy miejscowej
- Hemostaza miejscowa
- Slide 5
- Slide 6
- Podział skaz krwotocznych
- Najczęstsze wrodzone skazy krwotoczne
- Hemofilia A
- Hemofilia B
- Slide 11
- Dziedziczenie hemofilii
- Historia hemofilii
- Hemofilia - choroba królewska
- Slide 15
- Postacie hemofilii
- Postać ciężka
- Objawy kliniczne ciężkiej postaci hemofilii
- Slide 19
- Hemofilia umiarkowana
- Slide 21
- Hemofilia łagodna
- Rozpoznanie hemofilii
- Opieka nad chorym na hemofilię
- Historia leczenia hemofilii A
- Preparaty cz. VIII
- rVIII
- Preparaty rVIII
- Preparaty cz. IX
- Dawkowanie koncentratów czynników krzepnięcia
- Dawka koncentratów w krwawieniach
- Profilaktyka pierwotna
- Profilaktyka pierwotna
- Profilaktyka – hemofilia A
- Profilaktyka – hemofilia B
- Powikłania leczenia
- Krążący antykoagulant
- Czynniki genetyczne wpływające na pojawienie się inhibitora
- Charakterystyka przeciwciał
- Charakterystyka chorych: low and high responders
- Slide 41
- Slide 42
- Slide 43
- Chory na hemofilię A z krążącym antykoagulantem
- FENOC STUDY Blood 2007;109:546-551
- Wywoływanie stanu tolerancji immunologicznej
- Metody wywoływania STI
- Slide 48
- Inne czynniki mogące wpływać na osiągnięcie STI
- Korzystne czynniki prognostyczne
- Hemofilia B z inhibitorem
- Choroba von Willebranda (vWD)
- Czynnik von Willebranda (vWf)
- Rola vWf w hemostazie
- Klasyfikacja vWD
- vWD - objawy
- Diagnostyka vWD
- Diagnostyka vWD
- Diagnostyka vWD
- Klasyfikacja choroby von Willebranda
- Leki stosowane w różnych typach vWD
- Wrodzone defekty fibrynogenu
- Afibrynogenemia - objawy
- Afibrynogenemia – badania laboratoryjne
- Afibrynogenemia - leczenie
- Dysfibrynogenemia
- Dysfibrynogenemia - objawy
- Niedobór czynnika VII
- Niedobór czynnika X
- Niedobór czynnika XI
- Niedobór czynnika XIII
- Niedobory czynników XII, PK, HMWK
- Podział skaz krwotocznych
- Witamina K
- Wchłanianie witaminy K
- Działanie witaminy K na układ krzepnięcia
- Przyczyny niedoboru witaminy K
- Kliniczne objawy niedoboru witaminy K
- Rozpoznanie niedoboru witaminy K
- Zaburzenia hemostazy w chorobach wątroby
- DIC- uwagi ogólne
- Slide 82
- Slide 83
- Rozsiane krzepnięcie śródnaczyniowe (DIC)
- Przyczyny krwawień w DIC
- Ostry vs przewlekły DIC
- Epidemiologia
- Choroby i stany kliniczne związane z DIC
- Patogeneza DIC w posocznicy
- Slide 90
- Slide 91
- Mechanizmy odpowiedzialne za podtrzymywanie generacji trombiny
- Slide 93
- Obraz kliniczny DIC - cd
- Slide 95
- Slide 96
- Slide 97
- Slide 98
- Diagnostyka DIC
- Slide 100
- Slide 101
- Wyniki specyficznych markerów krzepnięcia i fibrynolizy w DIC
- Slide 103
- I. ocena ryzyka wystąpienia DIC
- II. Wykonanie badań diagnostycznych
- III. Ocena wyników badań hemostazy
- IV. Obliczenie wskaźnika
- V. Rozpoznanie
- Różnicowanie
- Leczenie DIC
- Slide 111
- Leczenie uzupełniające
- Leczenie opornych krwawień w przebiegu DIC
- rVIIa w DIC
- rVIIa w DIC
- rVIIa w leczeniu DIC Wnioski
- rVIIa w DIC Pytania i problemy do rozwiązania
- Leki hamujące krzepnięcie krwi
- Heparyna w leczeniu DIC 5 –10 j/kg/h
- Koncentraty inhibitorów krzepnięcia
- Antytrombina
- Antytrombina w DIC
- APC w leczeniu sepsy
- Slide 124
- Slide 125
- Rekombinowana Trombomodulina w leczeniu DIC
- rTFPI w DIC
- Inne potencjalne metody leczenia DIC
- Patologiczne inhibitory krzepnięcia
- Diagnostyka nabytych inhibitorów czVIII
- Leczenie
Wyszukiwarka
Podobne podstrony:
Skazy krwotoczne osoczowe
Definicja i podzia skazy krwotocznej
skazy krwotoczne
Skazy krwotoczne 9
Skazy krwotoczne
3.Skazy krwotoczne, Farmacja, Farmakologia(1), Hemostaza, Układ krwionośny
Skazy krwotoczne, Pediatria
Pediatria - SKAZY KRWOTOCZNE - wikad I, - PIERWSZA POMOC - ZDROWIE, - Ratownictwo Medyczne, Semestr
Hematologia, Skazy krwotoczne
Krzepnięcie krwi skazy krwotoczne
Skazy krwotoczne i zasady transfuzji krwi
Skazy krwotoczne
skazy krwotoczne 7
Pediatria skazy krwotoczne cz II
interna skazy krwotoczne
więcej podobnych podstron