
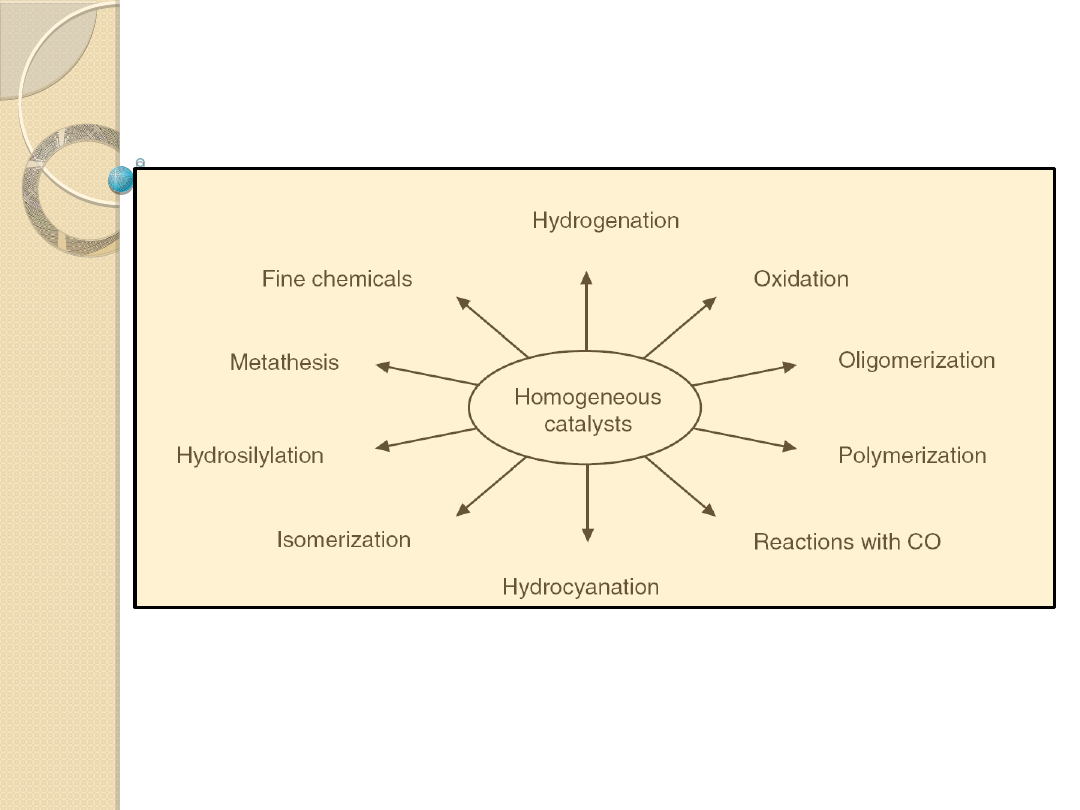
HOMOGENOUS CATALYSIS
Kataliza
homogenic
zna
Kwasowo
-
zasadowa
Enzymatyc
zna
Kompleks
ami
metali
Nukleofil
owa
ElektrofIlo
wa
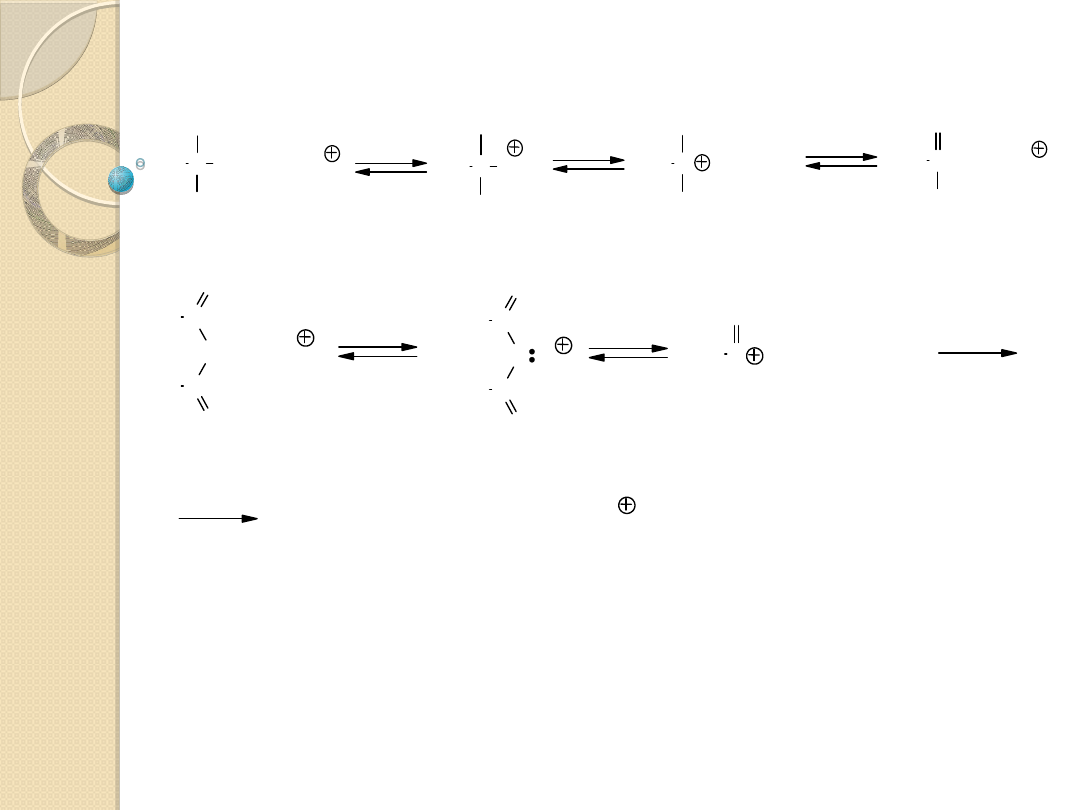
KATALIZA NUKLEOFILOWA
Przykłady katalizatorów:
jony halogenków I
-
, Br
-
, F
-
,
jony alkoholanowe RO
-
, fenolanowe Ar
-
,
wodorowęglanowe HCO
3
-
,
aminy R
3
N, R
2
NH, RNH
2
, C
5
H
5
N i imidazol, a także
niektóre inne jony NO
2
-
, CN
-
, ClO
-
, HOO
-
.
Katalizowane reakcje:
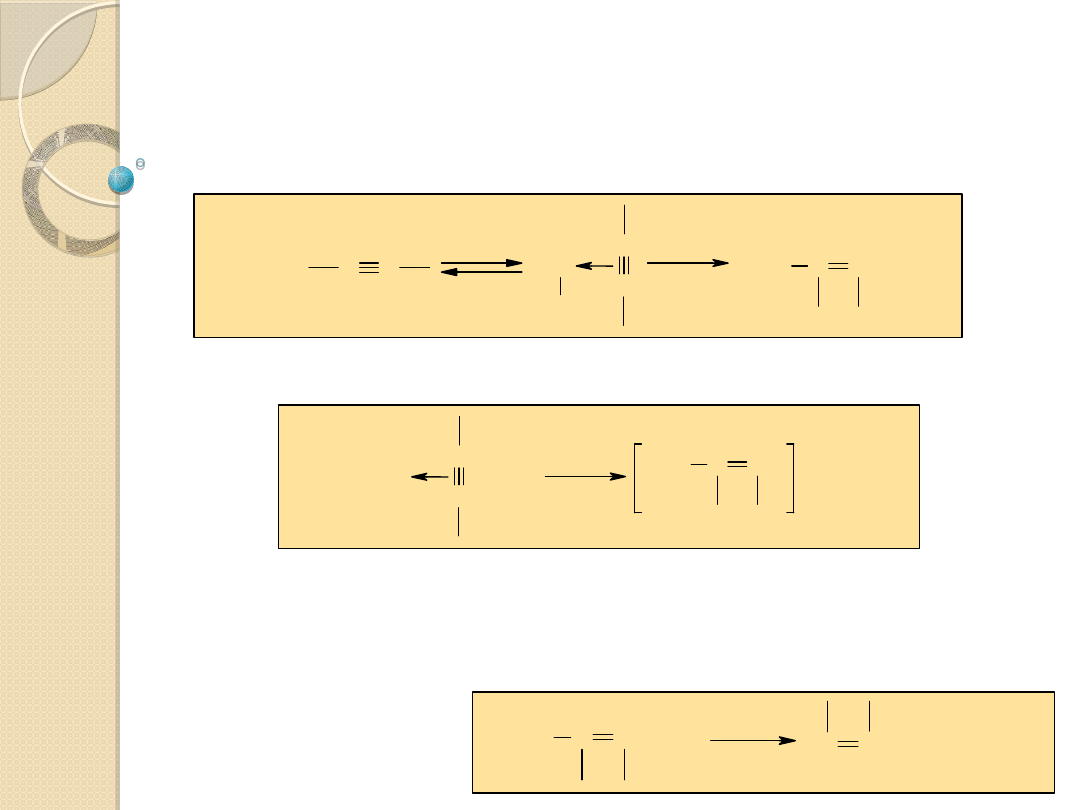
reakcje przyłączenia
reakcje podstawienia
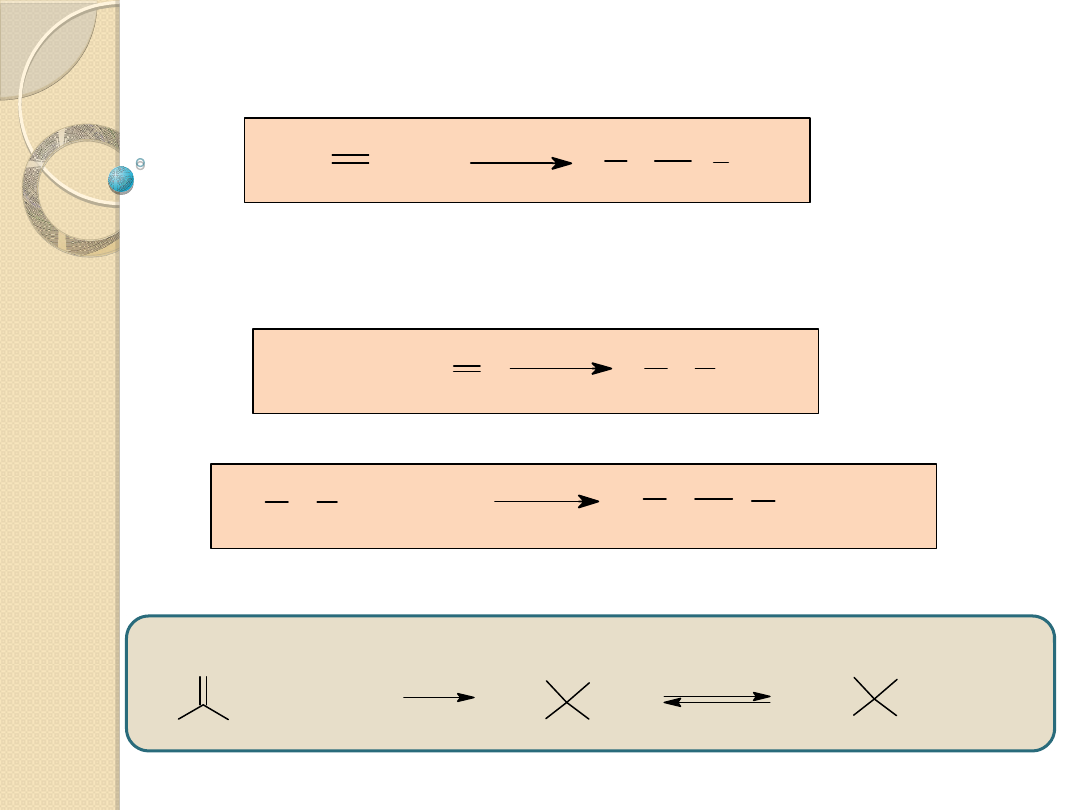
X Z + YH
Y X ZH
Y
-
+X
Y X
Z
Z
-
Y X Z
-
+ YH
Y X Z
+ Y
-
H
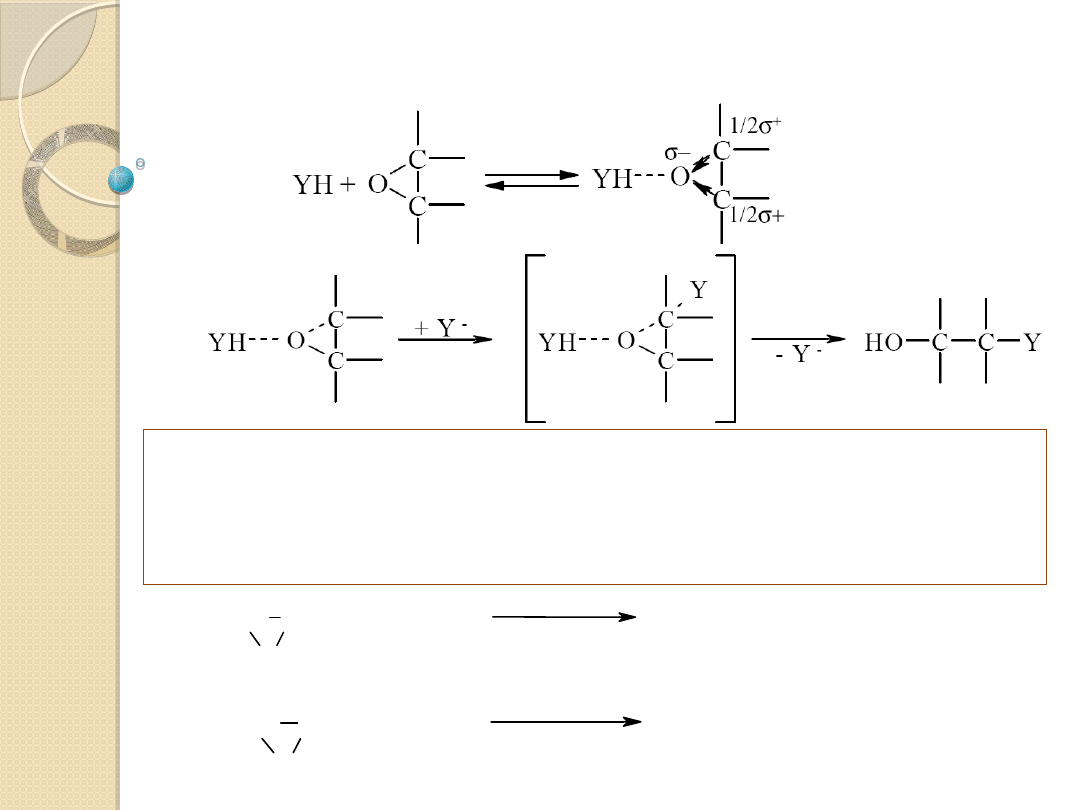
Reakcje przyłączenia nukleofilowego do
wiązania podwójnych
Regeneracja katalizatora w reakcji z YH:
Katalizator: jon Y
-
- sprzężona zasada substratu;
wykazuje znacznie silniejsze właściwości nukleofilowe,
niż YH i łatwo przyłącza się do wiązania podwójnego
Przykłady:
C
H
3
CH
3
O
+ CN
-
C
H
3
CN
O
C
H
3
_
+ HCN
C
H
3
CN
OH
C
H
3
+ CN
-
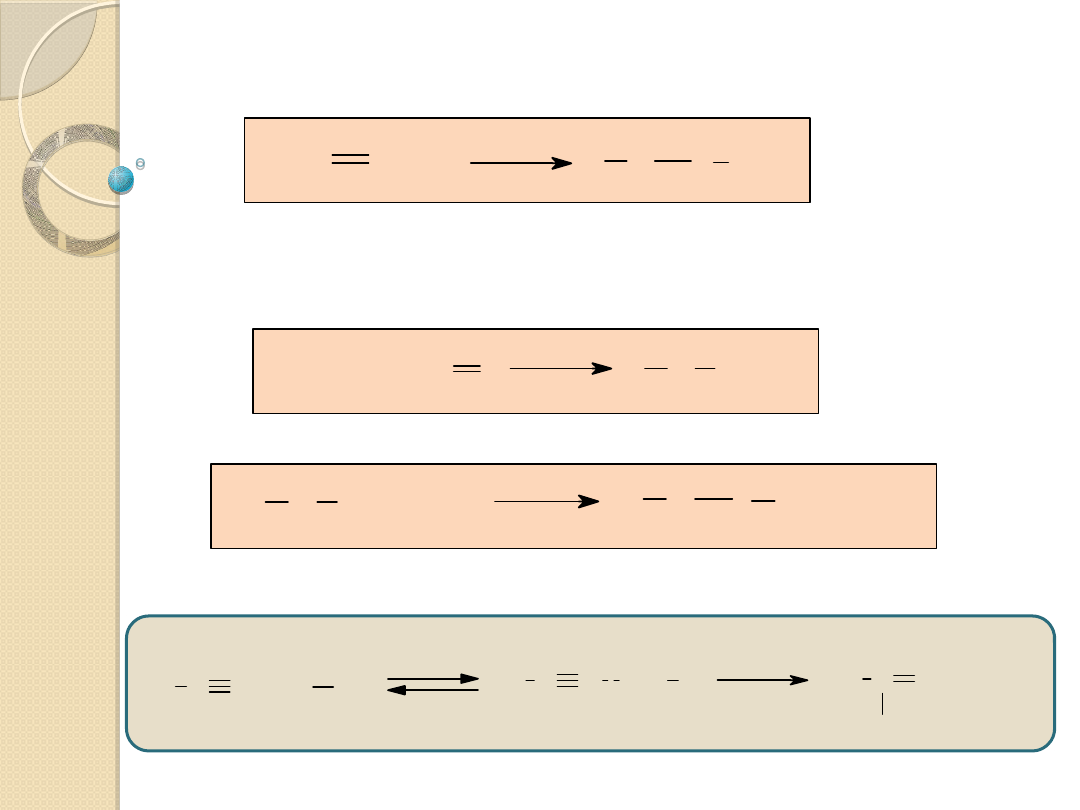
X Z + YH
Y X ZH
Y
-
+X
Y X
Z
Z
-
Y X Z
-
+ YH
Y X Z
+ Y
-
H
Reakcje przyłączenia nukleofilowego do
wiązania podwójnych
Regeneracja katalizatora w reakcji z YH:
Katalizator: jon Y
-
- sprzężona zasada substratu;
wykazuje znacznie silniejsze właściwości nukleofilowe,
niż YH i łatwo przyłącza się do wiązania podwójnego
Przykłady:
R' C N R OH
+
R' C N HO R
+ RO
-
R' C
OR
NH+ RO
-
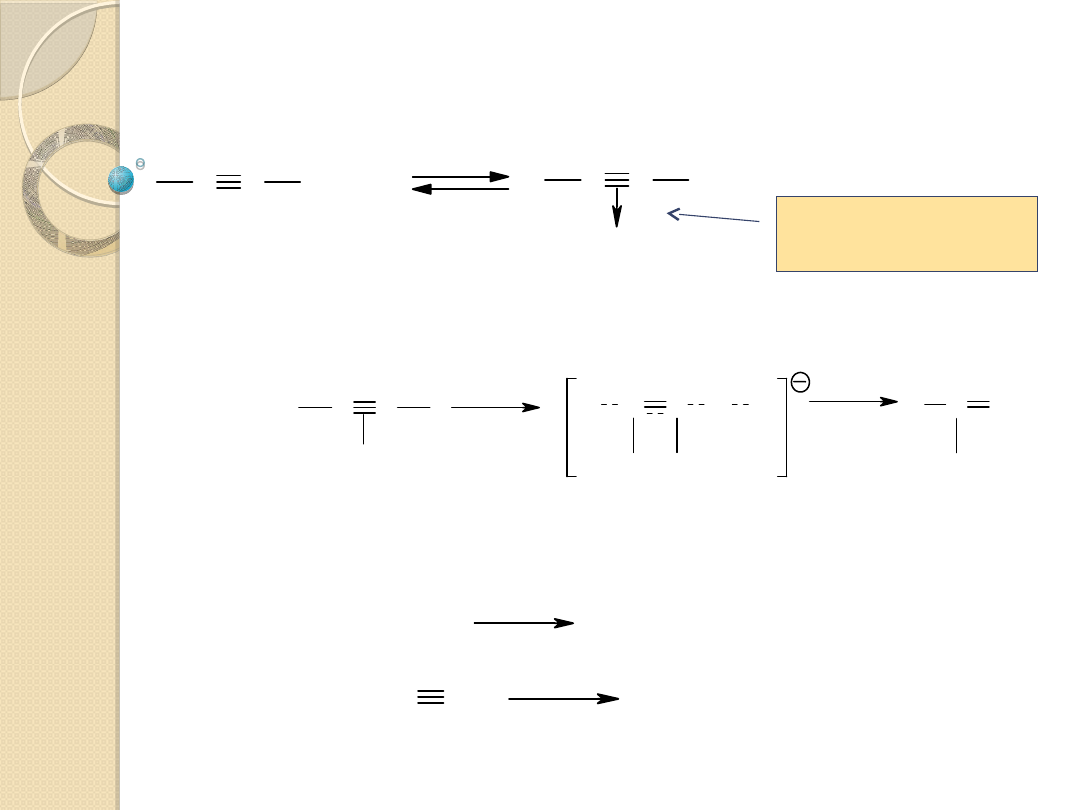
C C
C C
+ YH
YH
C C
Y C C H Y
+ Y
-
YH
- Y
-
Y C CH
Reakcje przyłączenia YH do olefin z podstawnikami
elektroakceptorowymi przy wiązaniu podwójnym (CH
2
=CHZ, gdzie
Z - COOR, NO
2
, CF
3
, COR, F lub CN) i pochodnych acetylenu.
Przyłączenie nukleofilowe katalizatora Y
-
z jednoczesną jego
regeneracją z cząsteczki YH:
Aktywacja wskutek
tworzenia
p-kompleksu
Pary reagent (YH) - katalizator (Y
-
): ROH-RO
-
, H
2
O-HO
-
, H
2
S-HS
-
,
ArOH-ArO
-
itp.
H
2
O + CH
2
=CHCN HOCH
2
CH
2
CN
OH
-
C
H
CH
CH
3
OH +
CH
3
OCH=CH
2
CH
3
O
-
Reakcje przyłączenia YH do związków heterocyklicznych z
małymi pierścieniami
Kataliza sprzężonymi zasadami jest najbardziej efektywna
dla reakcji przyłączania alkoholi, kwasów karboksylowych,
fenoli, merkaptanów, siarkowodoru i cyjanowodoru do
oksiranów.
CH
2
CH
2
O
+ CH
3
COOH CH
3
COOCH
2
CH
2
OH
CH
3
COO
-
CH
2
CH
2
O
+ C
6
H
5
OH C
6
H
5
-O-CH
2
CH
2
OH
C
6
H
5
O
-
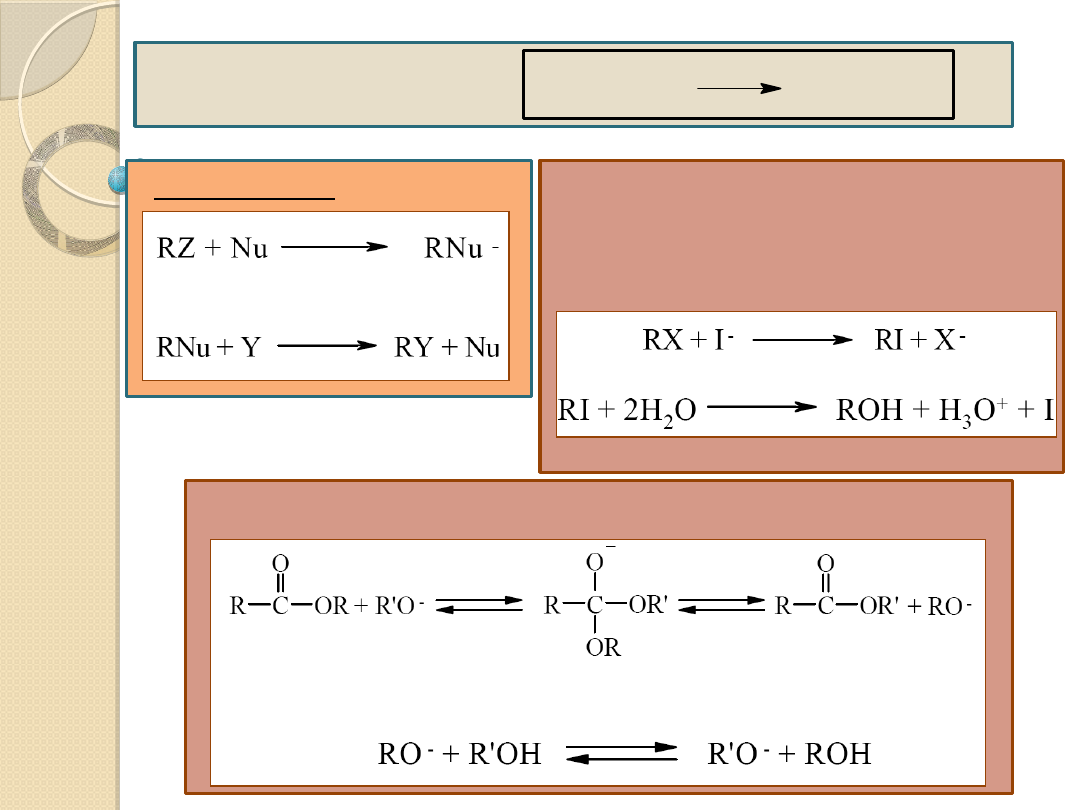
Reakcja
podstawienia:
RZ + Y RY + Z
Mechanizm:
Reakcje hydrolizy chlorków lub
bromków alkilowych katalizowane
anionem jodkowym
Reakcje transestryfikacji
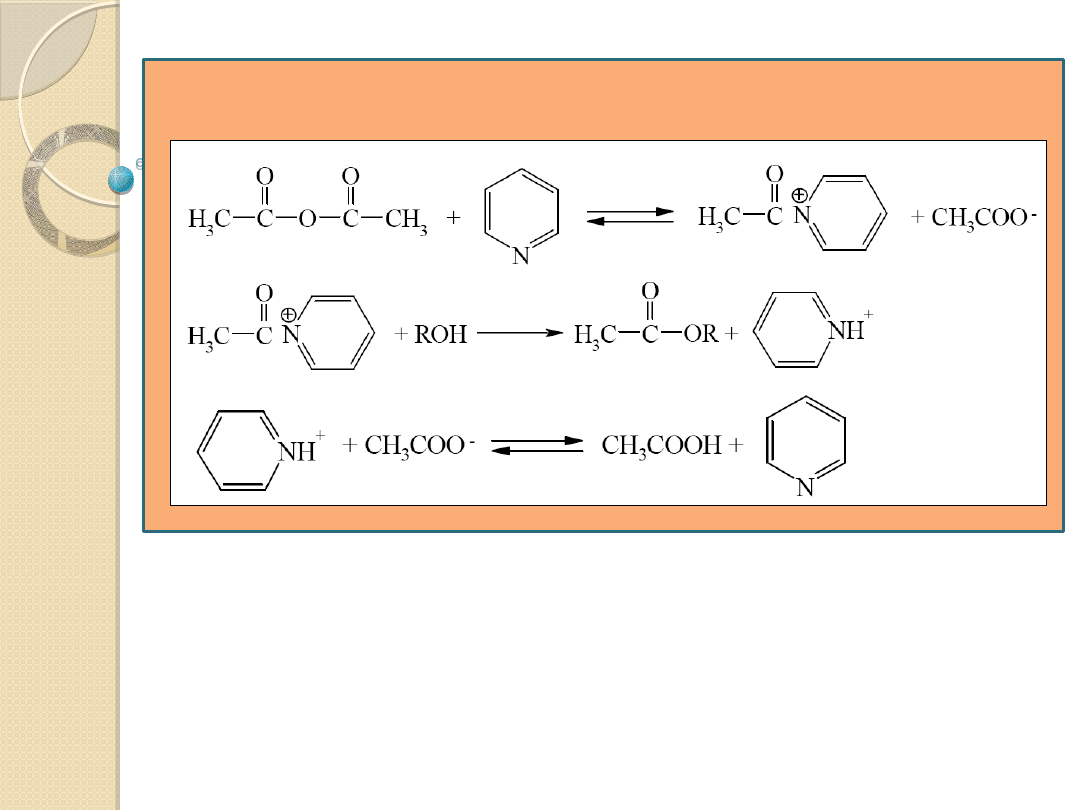
Reakcje acylowania
H
P
k
k
O
H
Nu
)
log(
2
O
H
Nu
k
k
2
)
/
log(
2
O
H
Nu
R
R
P
)
/
log(
2
O
H
B
Nu
B
K
K
H
]
[
]
][
[
Nu
HO
NuH
K
Nu
B
74
,
15
log
2
O
H
B
K
Ilościowy związek nukleofilowości z zasadowością i
polaryzowalnością nukleofila:
gdzie:
- nukleofilowość nikleofila względem wody - równa
stosunkowi stałych szybkości;
- parametr charakteryzujący względną
polaryzowalność nukleofila względem
wody (R - refrakcja);
-
parametr
charakteryzujący
zasadowość nukleofila względem
wody,
, - parametry charakteryzujące czułość danej reakcji w
stosunku do zmiany polaryzowalności (P) i zasadowości
(H) nukleofila.
(równanie Edwards'a)
Kiedy P<< H to czynnikiem określającym nukleofilowość
jest zasadowość i równanie Edwards'a przyjmuje postać
równania Bröndsteda:
)
log(
)
log(
2
2
O
H
B
Nu
B
O
H
Nu
K
K
H
k
k
Oryginalne równanie Bröndsteda
)
(
Nu
B
Nu
K
G
k
lub
G
K
k
Nu
B
Nu
log
log
log
O
H
B
O
H
k
k
G
2
2
log
log
log

Aktywność nukleofila jako katalizatora, a wpływ
rozpuszczalnika
W większości przypadków nukleofil występujący w roli
katalizatora stanowi parę jonową M
+
Y
-
.
W rozpuszczalnikach protonowych o porównywalnej
polarności (woda, etanol, metanol) aktywość nukleofilową
wolnego Y
-
obniża się o 2-5 rzędy wskutek specyficznej
solwatacji z utworzeniem wiązań wodorowych [Y
-...
(HOR)
n
]. Przy tym silniej solwatowane są jony o
mniejszym rozmiarze i z dużym ładunkiem, co może
doprowadzić odwrócenia rzędu nukleofilowości anionów w
rozpuszczalnikach protonowych.
Maksymalna szybkość reakcji osiągana jest w polarnych
aprotonowych rozpuszczalnikach (np. DMF, DMSO,
acetonitryl, tetrametylosulfon), w których sól dysocjuje,
a swobodny anion Y
-
praktycznie nie oddziaływuje z
rozpuszczalnikiem.
M
+
Y
-
M
+
+ Y
+
W rozpuszczalnikach o umiarkowanej polarności
(15<<40, wyższe alkohole, aceton) równowaga
dysocjacji przesunięta jest w kierunku pary jonowej:
Asocjacja anionów Y
-
z kationem zmniejsza nukleofilową
aktywność anionu podobnie jak specyficzna solwatacja,
wskutek wiązań wodorowych.
Wzrost
rozmiaru
kationu
zawsze
sprzyja
wzrostowi
nukleofilowości anionu.
W celu zwiększenia aktywności oraz zapewnienia
rozpuszczalności w rozpuszczalnikach
organicznych (szczególnie niepolarnych)
katalizatory nukleofilowe stosuje się w postaci soli
z objętościowymi kationami (np. (C
2
H
5
)
3
C
6
H
5
CH
2
N
+
,
(n-C
4
H
9
)
3
P
+
) lub dodaje specjalne ligandy zdolne do
wiązania kationu nieorganicznego, w szczególności
cykliczne etery poliglikoli, tzw. etery koronowe,
odpowiednie do rozmiarów kationów:
O
O
O
O
Mg
Np. dodatek dicykloheksano-18-krown-6-
dobenzenu umożliwia rozpuszczenie w nim
równomolowych ilości KMnO
4
lub KCl, które
nie rozpuszczają się w czystym benzenie.
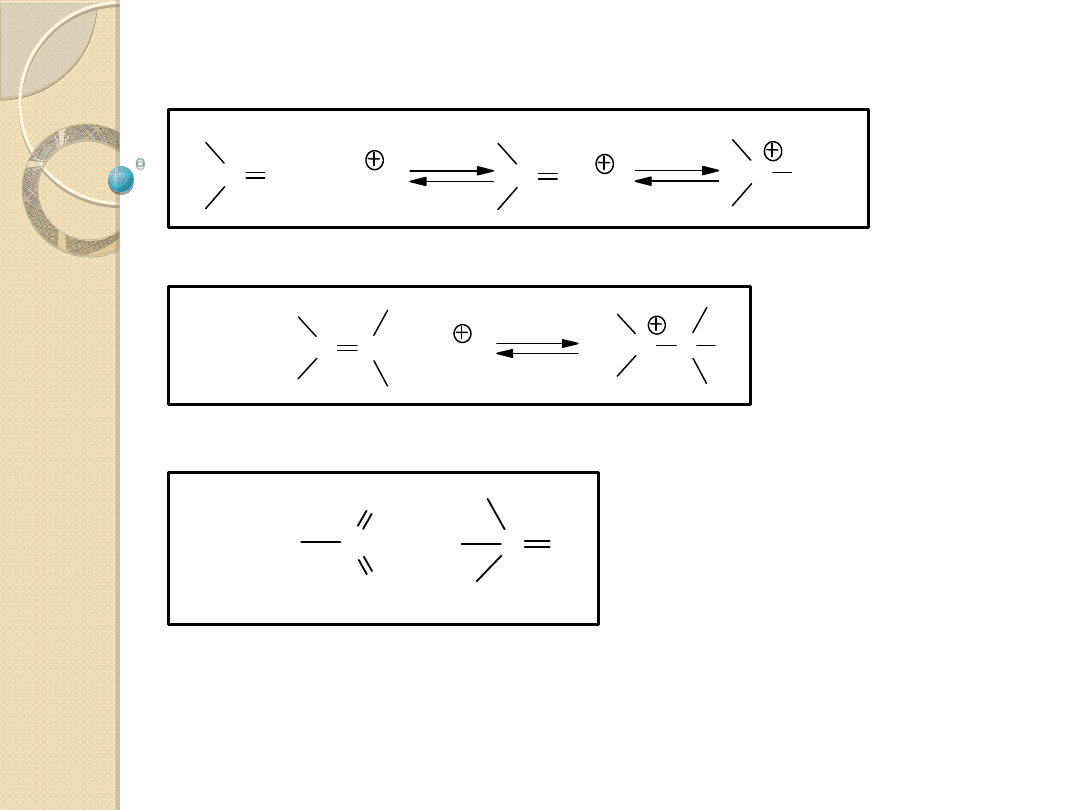
KATALIZA KWASOWA
Najbardziej rozpowszechnione katalizatory kwaśne :
H
2
SO
4
, HCl, H
3
PO
4
, ArSO
3
H, HCOOH
Przykłady procesów katalizowanych kwasami protonowymi:
hydratacja,
eteryfikacja,
estryfikacja,
alkilowanie,
kondensacja związków karbonylowych.
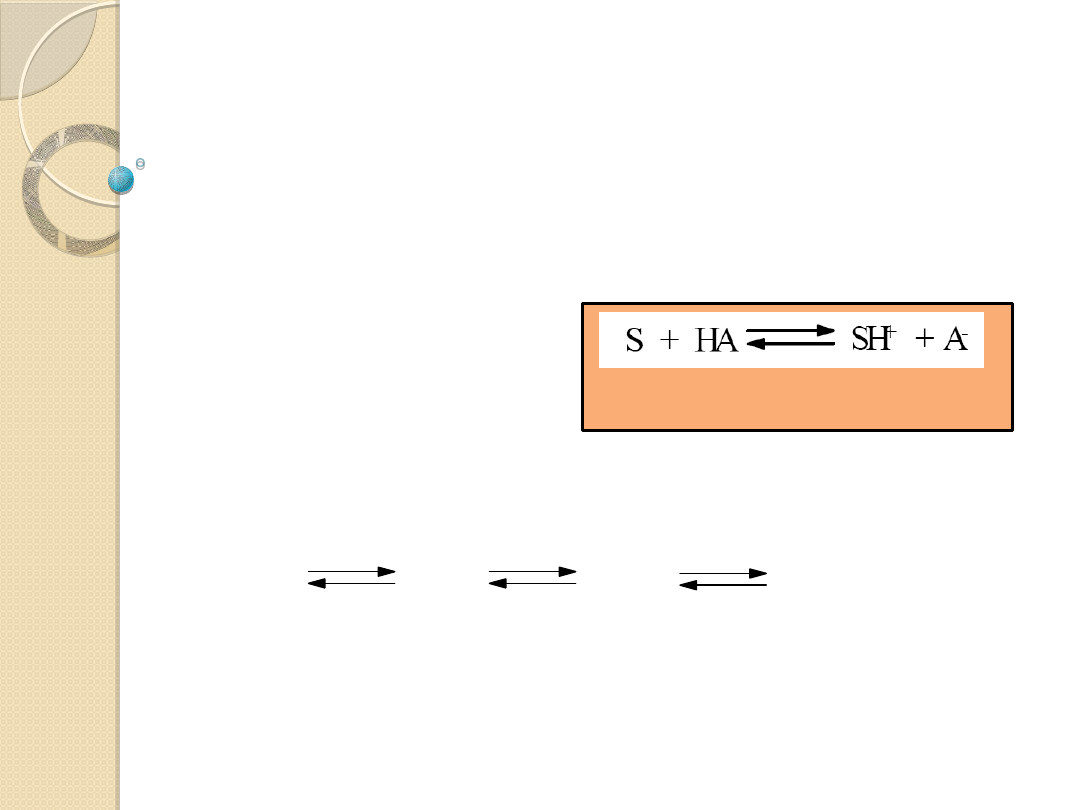
Równowaga kwasowo-zasadowa (protonolityczna):
AH + B
A
-
+ HB
AH
...
B
A
:
...
HB
+
-
+
Substancje organiczne zdolne do przyłączenia
protonów:
związki tlenu, azotu (ROH, RCOOH, R
2
CO,
R
3
N),
substancje z nienasyconymi -elektronami
(C=C, CC) lub związki aromatyczne.
np. H
3
O
+
,
2
ROH
.
RX + H
+
RXH
R
+
+ HX
+
szybko
wolno
R
+
+ :YH
RY + H
+
szybko
C C
H
+
+ B
C C
+ BH
+
+
CH
3
CH
2
CH
2
+
CH
3
CHCH
3
Mechanizm A-1
lub
Uboczna izomeryzacja karbokationu w bardziej stabilny:
CH
3
C OH
CH
3
CH
3
+ H
CH
3
C OH
2
CH
3
CH
3
CH
3
C
CH
3
CH
3
+ H
2
O
CH
2
C
CH
3
CH
3
+ H
3
O
CH
3
C
O
O
C
O
CH
3
+ H
CH
3
C
O
O
C
O
CH
3
H
CH
3
C
O
+ CH
3
COOH
+ROH
CH
3
COOH + CH
3
COOR + H
Przykłady reakcji przebiegających zgodnie z mechanizmem A-1:
+ H
C X
C XH
C XH
+ H
C C
C C H
S
O
O
,
P O
Mechanizm A-2
a także do wiązań podwójnych (C=C)
-N=O ,
lub wiązań
Dalsze przekształcenie
protonowanej substancji jest
zawsze dwucząsteczkowe.
CH
3
CH
2
O CH
2
CH
3
CH
3
CH
2
OH
2
CH
3
CH
2
O CH
2
CH
3
H
+ H
3
O
+ H
2
O
CH
3
CH
2
OH +
CHCH
3
CH
2
+ H
+ H
2
O
CH
CH
3
CH
3
CH
CH
3
CH
3
OH
2
CH
CH
3
CH
3
OH
-H
+
Przykłady:
CH
CH
3
CH
3
-H
+
+
H
C
3
H
7
+
CH
CH
3
CH
3
CH
CH
3
CH
3
+ CH
2
=CH-CH
3
(CH
3
)
2
CHCH
2
CHCH
3
itd.
+ H
3
O
+
O
C
R
OR
OH
C
R
OR + H
2
O
OH
C
R
OR
OH
2
+ H
2
O
OH
C
R
OR
OH
+ H
3
O
OH
C
R
OR
OH
H
- H
2
O
OH
C
R
OH
-ROH
+ H
2
O
RCOOH + H
3
O
C X + HA
C X
...
HA
C X + HA
C X
...
HA
O
C
CH
3
OH
2
C
H
3
C
OH
O
O
O
C CH
3
H
+ CH
3
COOH
CH
3
COOCH
3
+ CH
3
COOH + H
2
O
Dla słabych reagentów zasadowych, słabych kwasów
katalizatorów i niepolarnych rozpuszczalników:
Przykład:
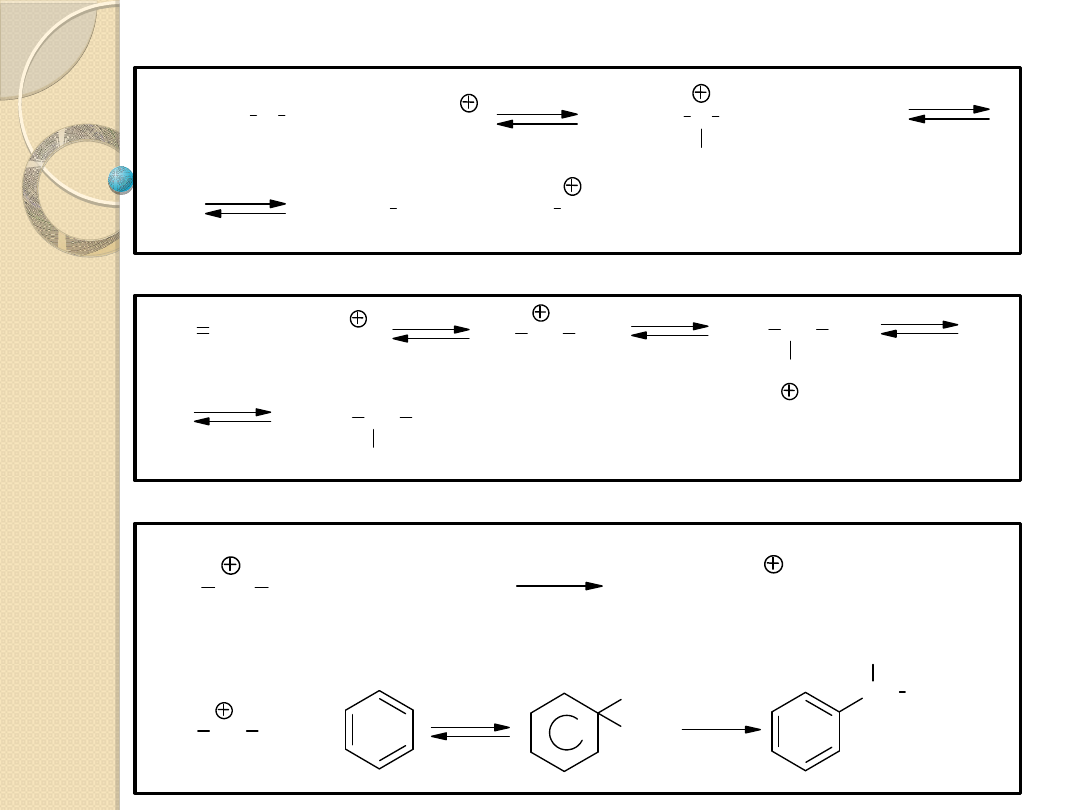
KATALIZA ELEKTROFILOWA
Cl
2
+ FeCl
3
Cl
Cl:FeCl
3
C X
C X
C
-
+ E
E
XE
X X
X X
-
X
+ E
E
XE
Katalizatory - kwasy Lewisa:
sole niektórych metali (ZnCl
2
, AlCl
3
, FeCl
3
, TiCl
4
,
SnCl
4
, BF
3
),
jony metali (Li
+
, Ag
+
, Hg
2+
),
inne cząsteczki obojętne i jony, jak SO
3
, P
2
O
5
,
R
+
, NO
2
+ itd.
E = AlCl
3
, TiCl
4
, SnCl
4
, FeCl
3
, Ag
+
, Mg
2+
, Hg
2+
, Th
4+
i in.
-
C C
Cl C C
Cl C C Cl
Cl
2
+ FeCl
3
Cl
Cl:FeCl
3
+ FeCl
4
+ FeCl
3
-
H
C
2
H
5
+ AlCl
4
C
2
H
5
Cl + AlCl
3
CH
3
CH
2
Cl:AlCl
3
C
6
H
6
+
C
6
H
5
C
2
H
5
+ AlCl
3
+ HCl
HF + SbF
5
HSbF
6
BF
3
+ HF
HBF
4
-
RCH
2
CH
3
+ HF
.
TaF
5
RCHCH
3
+ H
2
+ TaF
6
Kompleksy niektórych kwasów Lewisa z protonowymi kwasami
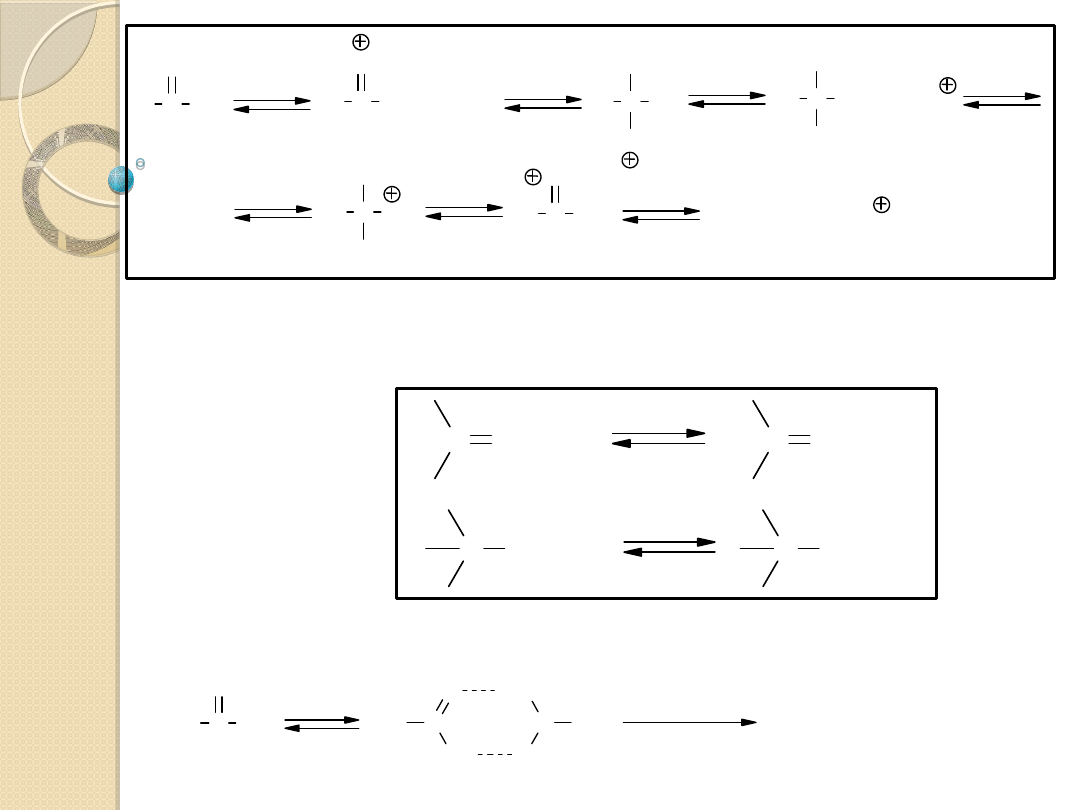
KATALIZA ZASADOWA
-
-
SH + B
S + BH
-
-
AH + B
A + BH
-
AH + B
AH
...
B
Katalizator:
aniony (HO
-
, RO
-
, NH
2
-
, i inne),
cząsteczki obojętne (NH
3
, (CH
3
)
3
N, C
6
H
5
N
itp.)
gdzie: SH=H
2
O, ROH, RCOOH, itp.
-
O
C
XCH
2
OH
+ B
- BH
+
O
C
XCH
2
O
- CO
2
XCH
2
-
+ BH
+
XCH
3
+ B
-
-
-
CH
3
CHO + HO
CH
2
CHO + H
2
O
-
-
CH
2
CHO + CH
3
CHO
O
CH
C
H
3
CH
2
CHO
-
O
H
CH
C
H
3
CH
2
CHO + OH
Przykłady:
B + R
2
NH
B
...
HNR
2
RCOX
C
X
O
R
N
R
R
... ...H...B
-
RCONR
2
+ BH + X

TEORIA TWARDYCH I MIĘKKICH KWASÓW I ZASAD
PEARSONA
(HSAB - HARD AND SOFT ACIDS AND BASES)
Twarde kwasy są to kwasy Lewisa, których elektrony
walencyjne są słabo polaryzowalne, a więc trudne do
usunięcia. Są nimi kationy o małym promieniu
jonowym i dużym ładunku, a także cząsteczki, których
podstawniki indukują na atomie centralnym wysoki
ładunek dodatni (H
+
, Li
+
, Na
+
, K
+
, Mg
2+
, Cu
2+
, Fe
3+
,
Th
4+
, Ti
4+
,
BF
3
, AlCl
3
, AlR
3
).
Miękkie kwasy są to akceptory o małym ładunku
dodatnim, dużym promieniu jonowym i łatwo
wzbudzanych zewnętrznych elektronach. Są nimi
łatwo polaryzowalne kationy metali (Cu
+
, Hg
2+
, Pd
2+
,
Pt
2+
, Tl
+
, Cd
2+
) i substancje organiczne zdolne do
przyjmowania pary elektronowej na zdelokalizowany
orbital p, np. tetracyjanoetylen.

Twarde zasady są to związki stanowiące donory
elektronów
o
niskiej
polaryzowalności,
wysokiej
elektroujemności i słabej zdolności do utlenienia. Do nich
należą przede wszystkim zasady posiadające wolną parę
elektronową na atomach O, N, F (F
-
, HO
-
, C
2
H
5
O
-
, NO
3
-
,
PO
4
3-
, NH
2
-
, NH
3
, R
3
N).
Miękkie zasady są to donory elektronów wykazujące
dużą
polaryzowalność,
niską
elektroujemność
i
podatność na utlenienie. Do tego typu zasad należą
aniony (HS
-
, I
-
, Br
-
, CH
3
COS
-
, R
-
, S
2
O
3
2-
, SCN
-
), obojętne
cząsteczki (R
2
S, H
2
S, PR
3
, P(OR)
3
CO) i cząsteczki, w
których ze swobodnym orbitalem kwasu oddziaływają
elektrony p (olefiny, związki aromatyczne itp.).
Szereg względnej miękkości zasad B:
I
-
>Br
-
>Cl
-
>S
2-
>RS
-
>CN
-
>H
2
O>C
5
H
5
N>AcO
-
>F
-
>NH
3
>HO
-
Szereg względnej miękkości kwasów M
+
:
Hg
2+
>Ag
+
>CH
3
Hg
+
>Cd
2+
>Zn
2+
>Ni
2+
>Fe
2+
>Cu
2+
>Cr
3+
>Z
r
4+
>H
+
Specyficzna kataliza kwasowo-zasadowa - taki
typ katalizy, przy którym szybkość reakcji zależy tylko
od wartości pH (H) środowiska lub stężenia jonów H
3
O
+
lub OH
-
i nie zależy od stężeń innych kwasów lub
zasad obecnych w mieszaninie reakcyjnej.
Ogólna kataliza kwasowo-zasadowa - taki typ
katalizy, przy którym szybkość reakcji zależy i od pH
środowiska i od stężeń i struktury kwasów (lub zasad),
znajdujących się w roztworze buforowym.
k
G K
ef
A
A
k
G K
ef
B
B
Ilościowy związek między stałą szybkości reakcji
katalitycznej, a stałą kwasowości kwasu lub zasadowości
zasady (równanie Brönsteda):
lub
KOMPLEKSY METALI PRZEJŚCIOWE
W KATALIZIE REAKCJI ORGANICZNYCH
Metale przejściowe posiadają 9 orbitali bliskich co do
energii: trzy wolne np oraz jeden ns i pięć (n-1)d
częściowo zapełnione. Z nimi wiąże się właściwości
katalityczne kompleksów.
Najbardziej
rozpowszechnione
liczby
koordynacyjne kompleksów - katalizatorów to 4,
5 i 6.
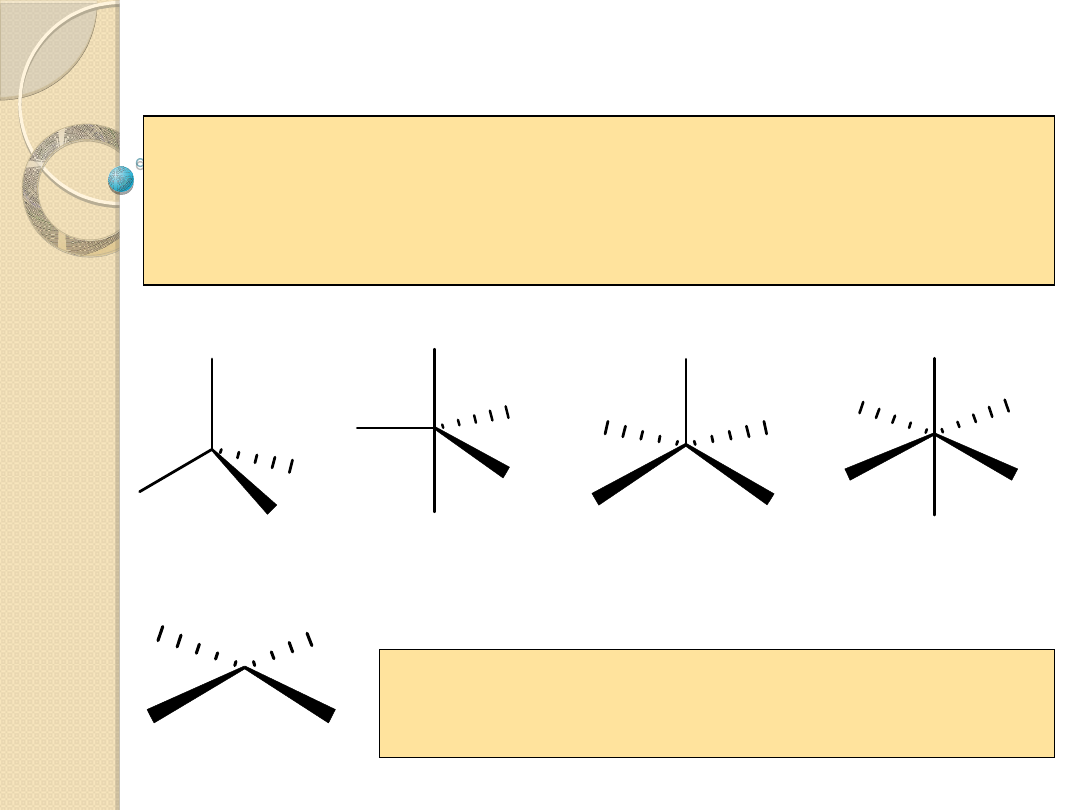
Sposoby przestrzennego rozmieszczenia ligandów w
kompleksach
piramida
tetragonalna
kwadrat
oktaedr
bibiramida
trygonalna
tetraedr

Konfiguracja kompleksów metali przejściowych
Podział ligandów według typu wiązania z
metalem:
ligandy s-donorowe
ligandy p-donorowe.
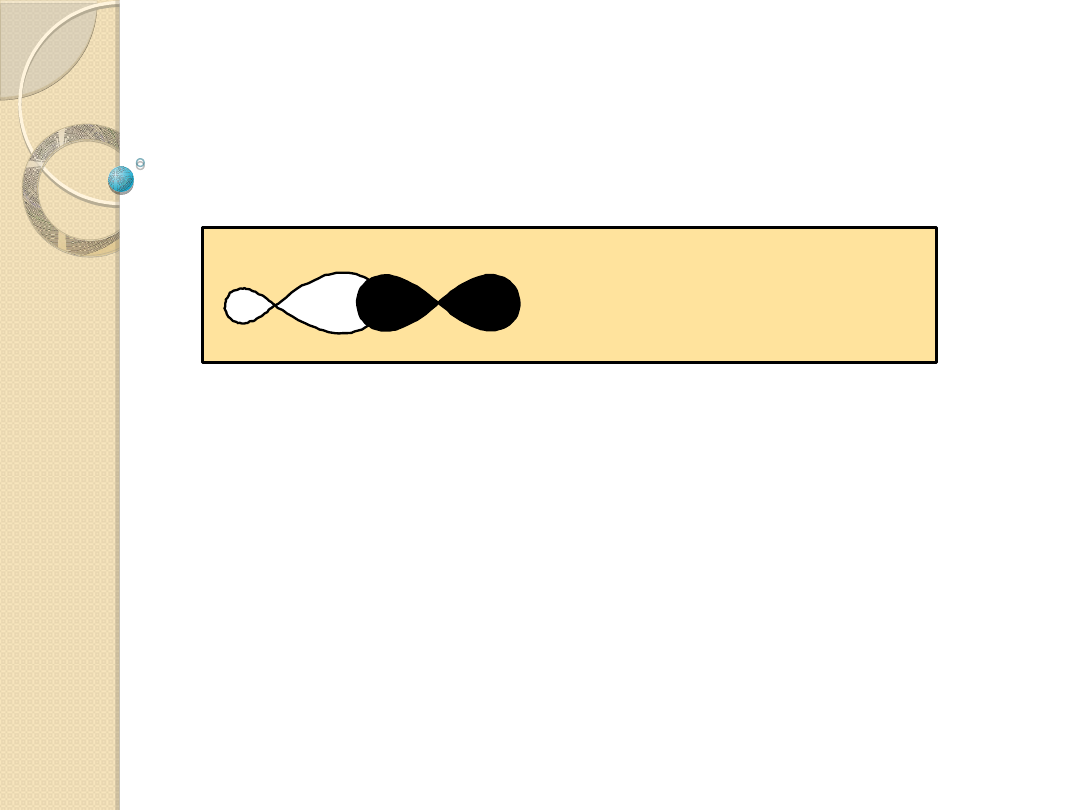
Ligandy s-donorowe:
ligandy s-donorowe posiadające tylko jedną wolną parę
elektronową i nie mające dostępnych wolnych orbitali
(H:
-
, H
3
N:, H
3
C:
-
, R
3
C:
-
, itp.); tworzą jedyne wiązanie s,
zapełniając jeden z wymienionych w tabeli orbitali
zhybrydyzowanych metalu.
+ +
-
-
.
.
M
N
wiązanie metalu M
z
ligandem
s-
donorowym
HO , Cl
:
..
.. -
..
..
: :-
F , Br
:
..
.. -
..
..
: :-
, :
+ +
-
-
.
M
Cl
-
.
wiązanie
p
x
+
+
-
-
.
M
Cl
.
wiązanie
p
y
d
xy
+
-
+
+
-
-
.
wiązanie
+
-
+
-
-
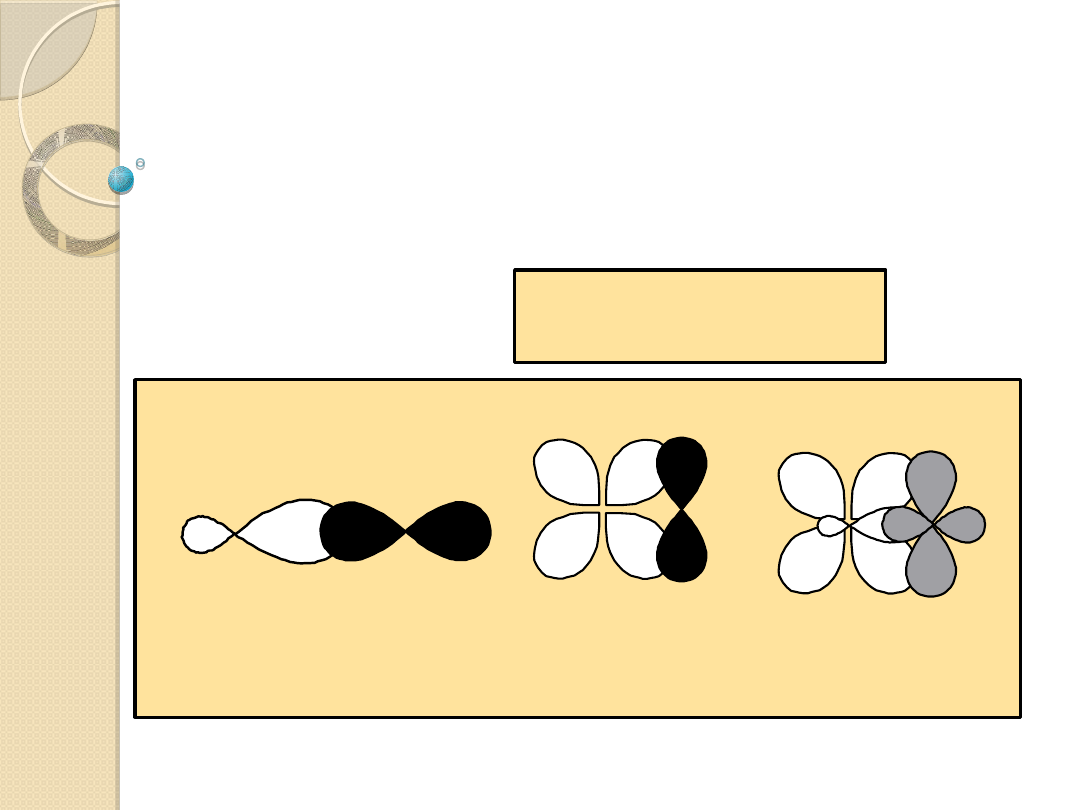
ligandy s-donorowe posiadające kilka wolnych par
elektronowych, uczestniczących w tworzeniu wiązań z
metalem . Ligandy tego typu oprócz wiązań s tworzą
wiązanie p. Wiązanie s z metalem tworzy jeden z
zapełnionych orbitali ligandu p
x
. Druga wolna para
elektronów liganda, prostopadła do osi metal - ligand
(p
y
), tworzy wiązanie p niemające symetrii obrotowej.
Takie ligandy s nazywa się p - donorowymi.
Wiązania metalu M z ligandem s-donorowym /p-
donorowym.
C O
+
-
.
M
wiązanie
C O
-
+
+
-
-
M
.
wiązanie
d
xy
+
-
+
orbital *
C O
+
+
-
-
.
wiązanie CO
+
-
+
-
-
+
M
+
ligandy s-donorowe posiadające obok wolnej pary
elektronowej energetycznie dostępny wolny orbital
(:CO, :NO, :PR
3
, Br:
-
, I:
-
, R
2
S:
-
, R
2
C:, itp.). Ligandy tego
typu tworzą dodatkowo wiązanie p poprzez
oddziaływanie wolnych par elektronowych metalu z
pustymi orbitalami p akceptorowych ligandów. Może
to być wolny orbital np w karbenie R
2
C: lub nitrenie
R
2
N: albo antywiążący orbital p* w CO i NO.
Wiązania metalu M z s-donorowym/p-akceptorowym
ligandem (CO).

-
+
-
.
M
wiązanie
C
C
+
+
+
-
-
.
+
-
+
M
wiązanie
C
C
-
-
d
xy
orbital
*
+
+
C
C
-
-
+
+
-
-
.
wiązanie
-
+
-
+
Ligandy p-donorowe:
substancje z węglowodorowymi z wiązaniami
podwójnymi i potrójnymi,
pochodne związków aromatycznych,
dieny.
Wiązania M z ligandem p-donorowym
(C=C).
Niektóre ligandy mogą występować w roli ligandów s i p-
donorowych (CO, N
2
).
Reguła
Tolmana
(16
i
18
elektronów
walencyjnych)
Diamagnetyczne kompleksy metali przejściowych istnieją w
kinetycznie lub spektroskopowo określonych ilościach tylko
w tym przypadku, jeśli liczba elektronów walencyjnych w
strefie koordynacyjnej metalu jest równa 16 lub 18.
Wszystkie przekształcenia kompleksu metalu w trakcie
reakcji przebiega przez tworzenie związków przejściowych,
posiadających tylko 16 i 18 elektronów walencyjnych metalu,
a liczba elektronów walencyjnych w każdym etapie zmienia
się o 0, +2 lub –2.
Numer
grupy
Stary/nowy
IV
A/
4
VA/5
VIA/6 VIIA/7
VIII
Ib/1
1
3d
Ti
V
Cr
Mn
Fe
Co
Ni
Cu
4d
Zr Nb
Mo
Tc
Ru
Rh
Pd
Ag
5d
Hf Ta
W
Re
Os
Ir
Pt
Au
Stopień
utlenienia
0
I
II
III
IV
4
3
2
1
0
5
4
3
2
1
6
5
4
3
2
7
6
5
4
3
8
7
6
5
4
9
8
7
6
5
10
9
8
7
6
-
10
9
8
7

18 – elektronowy kompleks (8
elektronów Fe
0
(d
8
) + 10 (2*5)
16 – elektronowy kompleks
pentakarbonylżelazo(0) Fe
0
(d
8
) + 8 (2*4)
DYSOCJACJA
Koordynacyjnie nienasycone 16 – elektronowe kompleksy mogą
tworzyć się w wyniku termicznego lub fotochemicznego
odszczepienia jednego z ligandów:
Fe
0
(CO)
5
Fe
0
(CO)
4
+ CO
1 6 0
o
C
HCo(CO)
4
HCo(CO)
3
+ C
O
PODSTAWIENIE
podstawienie cząsteczki rozpuszczalnika S substratem Y:
np. w kwadratowych d
8
-kompleksach Rh
I
Cl(CO)[P(C
5
H
5
)
3
]
2
,
Pt
II
HCl[P(CH
3
)
3
]
2
wolne miejsce koordynacyjne jest zwykle zajęte
przez słabo związaną cząstkę rozpuszczalnika.
S
M
L
L
L
L
Y
M
L
L
L
L
+ Y
+ S
podstawienie ligandu w nasyconym 18 – elektronowym
kompleksie
L
n
MX +Y L
n
MY + X
Mechanizmy podstawienia
L
n
MX X + L
n
M L
n
MY
wolno
+ Y, szybko
r k
[L MX]
n
dysocjacyjny (D) (S
N
1)
Szybkość reakcji:
X
Y
L
n
MX + Y
L
n
M
L
n
MY + X
wolno
szybko
r k
[L MX][Y]
n
asocjacyjny (A) (S
N
2, przyłączenia-eliminacji)
Mechanizmy
dysocjacyjny
i
asocjacyjny
są
przypadkami
skrajnymi.
Najczęściej
reakcja
zachodzi według mechanizmu pośredniego.
Szybkość reakcji:
X
Y
L
n
M
X
Y
L
n
M
asocjacyjno-dysocjacyjny - następuje
jednoczesne rozrywanie i powstawanie wiązań
wymienno-asocjacyjny I
a
- wymiana między
wiązaniami X i Y następuje w wyniku utworzenia
wiązania z grupą wchodzącą
wymienno-dysocjacyjny I
d
- wymiana między
wiązaniami X i Y następuje w wyniku rozerwania
wiązania z grupą wychodzącą

L
n
MX + S
wolno
L
n
MS + Y
L
n
MY + S
szybko
L
n
MS + Y
r k
k
1
2
[L MX]
[L MX][Y
n
n
Czynniki komplikujące obraz podstawienia:
Podstawienie z udziałem rozpuszczalnika niekiedy konkuruje z
bezpośrednim podstawieniem typu S
N
2. Wówczas równanie kinetyczne
zawiera dwie składowe:
działanie rozpuszczalnika

Powstawanie kompleksów zewnętrznosferowych (z
ang. outer-sphere complexes)
[L
5
MX]
n+
+ Y
m-
{[L
5
MX]Y}
n-m
W wypadku tworzenia kompleksów zewnątrzsferowych
równanie kinetyczne jest drugiego rzędu, nie zależnie, czy
mechanizm etapu określającego szybkość obejmuje
aktywację asocjacyjną czy dysocjacyjną
Zachodzi, gdy reagujący kompleks i ligand są jonami, w
których w wyniku przyciągania elektrostatycznego
następuje stabilizacja wchodzącego ligandu na obrzeżu
sfery koordynacyjnej kompleksu [L
5
MX]
n+
.
W przypadku reakcji, w których nie uczestniczą jony
wchodząca grupa Y może zostać związana na obrzeżu
kompleksu metalu, np. za pomocą wiązania wodorowego.
szybko
[Co(NH
3
)
5
Cl]
2
+
+ OH
-
[Co(NH
3
)
4
(NH
2
)Cl]
+
+ H
2
O
wolno
[Co(NH
3
)
4
(NH
2
)Cl]
+
[Co(NH
3
)
5
Y]
2+
+ Cl
-
+ Y
-
potem H
+
powstawanie sprzężonej
zasady.
Szereg ligandów według zdolności ułatwienia podstawienia w
pozycji trans:
H
2
O, HO
-
, NH
3
< Cl
-
<I
-
< C
6
H
5
-
< CH
3
-
< H
-
, PR
3
< C
2
H
4
, CO, CN
-
Przykład:
Pt
X
P(C
2
H
5
)
3
Cl
P
(C
2
H
5
)
3
Pt
X
P(C
2
H
5
)
3
NC
5
H
5
P
(C
2
H
5
)
3
+ C
5
H
5
N
+ Cl
-
+
(II)
(II)
dla X=H
-
szybkość podstawienia jest 10 000 razy większa, niż dla
kompleksu w którym X=Cl
-
.
Wpływ struktura ligandu L na szybkość
podstawienia
elektronowy
sferyczny.
Bardziej objętościowe ligandy zwykle sprzyjają
dysocjacji kompleksu.
Efekty elektronowe najsilniej przejawiają się u ligandów
położonych w pozycji trans względem ligandu podstawowego
(tak zwany
efekt trans
; spowodowany udziałem jednego
orbitalu metalu w tworzeniu wiązania z dwoma ligandami
znajdującymi się w położeniu trans względem siebie.
Zachodzi pomiędzy dwoma kompleksami lub między
kompleksem metalu i cząsteczką organiczną.
C
6
H
5
CH
3
+ Co
III
(OAc)L
5
[C
6
H
5
CH
3
]
+
+ Co
II
(OAc)L
5
-
.
C
6
H
5
CH
2
+ HOAc + Co
II
L
5
.
Reakcje przeniesienia elektronu przebiegają w niektórych
utleniająco-redukujących katalitycznych procesach i są
zazwyczaj etapem regeneracji katalizatora.
PRZENIESIENIE ELEKTRONU
W reakcjach między dwoma kompleksami elektron
przekazywany jest z jednego na drugi kompleks i stopień
utlenienia metalu zmienia się w obu kompleksach. W drugim
przypadku zmienia się stopień utlenienia metalu, a cząsteczka
organiczna ulega przemianie chemicznej.
Fe
II
(CN)
6
4-
+ Ir
IV
Cl
6
2-
Fe
III
(CN)
6
3-
+ Ir
III
Cl
6
3-
e
-
Cr
II
(H
2
O)
6
2+
+ ClCo
III
(NH
3
)
5
2+
[(H
2
O)
6
Cr Cl Co
III
(NH
3
)
5
]
4+
Cr
III
(H
2
O)Cl
2
+ + Co
II
(NH
3
)
5
(H
2
O)
2+
mechanizm zewnętrznosferowy - obydwa kompleksy
zachowują swoją otoczkę koordynacyjną, a elektron
przekazywany jest przez obie sfery koordynacyjne
mechanizm wewnętrznosferowy - dwa kompleksy tworzą
produkt pośredni w obrębie wewnętrznej sfery koordynacyjnej,
z jednym wspólnym ligandem, pełniącym rolę “przewodnika”
elektronów

Dysocjacyjna koordynacja cząsteczek X-Y
Typy przyłączania:
utleniające,
homolityczne,
heterolityczne.
Zachodzi z rozerwaniem wiązania s
Przyłączenie utleniające
L
m
M
(n+2)+
(X)(Y)
L
m
M
n+
+ X-Y
W niektórych przypadkach reakcja bywa odwracalna.
Odwrotny proces nazywa się redukcyjną lub redukującą
eliminacją.
Polega na przekazaniu dwóch elektronów metalu cząsteczce X-
Y, w rezultacie czego zachodzi rozerwanie wiązania i
utworzenie dwóch ligandów (X:
-
i Y:
-
), które zajmują wolne
miejsca koordynacyjne w nowym kompleksie.
Utleniające przyłączenie możliwe jest w przypadku istnienia
wyższego stopnia utlenienia metalu (n+2), przy obecności w
metalu pary niewiążących elektronów i dwóch wolnych miejsc
koordynacyjnych dla wchodzących ligandów X i Y.
Rh
I
CO
CO
I
Rh
I
CO
CO
I
CH
3
I
+ CH
3
I
(I)
(III)
Ir
Cl
P(C
6
H
5
)
3
CO
P
(C
6
H
5
)
3
Ir
Cl
P(C
6
H
5
)
3
H
P
(C
6
H
5
)
3
CO
H
+ H
2
(I)
(III)
Przyłączenie utleniające jest najbardziej typowe dla
koordynacyjnie nienasyconych 16-elektronowych
kwadratowych d
8
-kompleksów, np.
Reakcje utleniającego przyłączenia są znane również
dla d
6
- i d
10
-kompleksów, np.:
Pd
0
Pd
II
(d
10
d
8
)
Ru
II
Ru
IV
(d
6
d
4
)
Związki ulęgające reakcjom przyłączenia utleniającego:
halogenki alkilowe,
halogenki, halogenowodory,
chlorki kwasowe,
bezwodniki kwasów karboksylowych (z udziałem wiązania
C-O),
związki aromatyczne (z udziałem wiązań C-H, C-X i in.),
cząsteczki zawierające wiązania wielokrotne C=O, CO,
O=O i in.
Ir
Cl
P(C
6
H
5
)
3
CO
P
(C
6
H
5
)
3
Ir
Cl
P(C
6
H
5
)
3
O
P
(C
6
H
5
)
3
CO
O
+ O
2
(I)
(III)
Reakcji przyłączenia utleniającego ulegają także koordynacyjnie
nasycone 18‑elektronowe kompleksy. W tym przypadku przyłączenie
X i Y poprzedza odszczepienie jednego z ligandów (utleniające
podstawienie)
C
6
H
5
Pt
II
L
3
Br + L
Pt
0
L
4
+ C
6
H
5
Br
Mechanizmy
X
Y
L
m
M
n+
+ X-Y
L
m
M
L
m
M
(n+2)+
XY
synchroniczne przyłączenie utleniające (charakterystyczny
dla reagentów niepolarnych (H
2
) i niektórych polarnych w
niepolarnym środowisku)
ze wstępnym hetero- lub homolitycznym rozpadem wiązania
w cząsteczce X‑H (charakterystyczny m.in. dla substancji
kwasowych):
o ze wstępną jonizacją kwasu w polarnym
rozpuszczalniku
HX H
+
(solw.)
+ X:
-
(solw.)
L
m
M
(n+2)+
HX
-
L
m
M:
n+
+ H
+
(solw.)
L
m
M:H
(n+2)+
+ X:
-
C X
L
m
M:
n+
+ R
3
CX
L
m
M:
(n+2)+
CR
3
+ X:
-
L
m
M
(n+2)+
(CR
3
)(X)
L
m
M:
n+
wolno
szybko
o w wyniku nukleofilowego ataku metalu (jak w reakcjach S
N
2)
L
m
M
(n+1)+
R
L
m
M
(n+2)+
(X)(R) + R
.
L
m
M
n+
+ R
.
M
L
L
L
L
R
X
mechanizm rodnikowy (dla niektórych metali istniejących na
pośrednich stopniach utlenienia; przyspieszane przez inicjatory
reakcji rodnikowych)
Dla wszystkich etapowych mechanizmów w odróżnieniu od
synchronicznego charakterystyczne jest tworzenie produktów
przyłączenia trans.
M
(n+1)+
XL
m
+ M
( n+1)+
YL
m
2M
n+
L
m
+ X-Y
Co
2
0
(CO)
8
+ H
2
2Co
I
H(CO)
4
Co
2
II
(CN)
10
6-
2Co
III
H(CN)
5
2Co
II
(CN)
5
3-
Homolityczne przyłączenie
Np.
Wiąże się ze wzrostem stopnia utlenienia metalu o
jeden i zachodzi przy oddziaływaniu cząstki X-Y z
dwoma atomami metalu
M
n+
L
m-1
X + Y
+
+ L
-
M
n+
L
m
+ X-Y
Rh
III
Cl
6
3+
+ H
2
Rh
III
Cl
5
H
3+
+ H
+
+ Cl
-
Heterolityczne przyłączenie
Reakcja ta przyspieszana jest przez zasady, wiążące tworzący
się HCl (RNH
2
, HO
-
i inne).
Heterogeniczne przyłączenie jako suma
utleniającego przyłączania i redukującej eliminacji
Pt
II
Cl
2
[P(C
2
H
5
)
3
]
2
+ H
2
Pt
IV
Cl
2
H
2
[P(C
2
H
5
)
3
]
2
Pt
II
ClH[P(C
2
H
5
)
3
]
2
+ HCl
Przebiega bez zmiany stopnia utlenienia i liczby
koordynacyjnej i stanowi podstawienie ligandów poprzedzone
heterogenicznym rozpadem wiązania w X-Y.
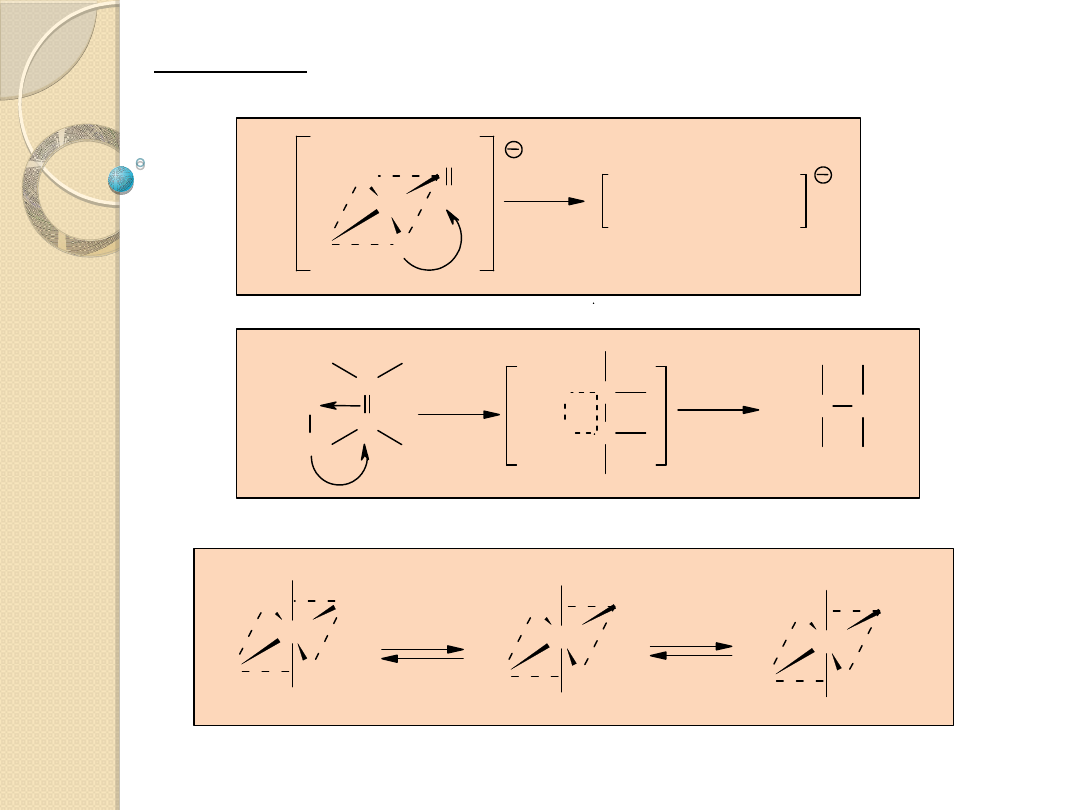
Migracja atomów lub grup z metalu do ligandu
(insercja, wstawienie)
M
Y
X
L
m
L
m
-M-X-Y
Etap charakterystyczny dla większości reakcji katalizowanych
kompleksami metali przebiegających z udziałem związków
nienasyconych i CO.
Tworzenie wiązania chemicznego między ligandami X i Y
znajdującymi się w sferze koordynacyjnej metalu.
Wewnątrzcząsteczkowy atak nukleofilowy ligandu Y na
aktywowaną w wyniku koordynacji cząsteczkę X, w roli której
występuje zwykle związek nienasycony lub CO.
CH
2
CH
2
Pd
Cl
OH
Cl
Cl
2
PdCH
2
-CH
2
OH
Ti
R
C
C
Ti
R C
C
=
/
C C
L
m
L
m
L
m
Ti
R
Mn
OC
CO
CH
3
OC
CO
CO
Mn
OC
CO
OC
CO
COCH
3
Mn
OC
CO
OC
CO
COCH
3
14
CO
+
14
CO
(I)
(I)
(I)
Przykłady:
(CO)
5
W C
O
CH
3
Li
LiCH
3
+ W(CO)
6
O
CH
2
NH
2
COCH
3
N
H
2
CH
2
COOH
L
2
Cu
II
+ H
2
O + 2L
L
4
Cu
II
+
+ CH
3
OH
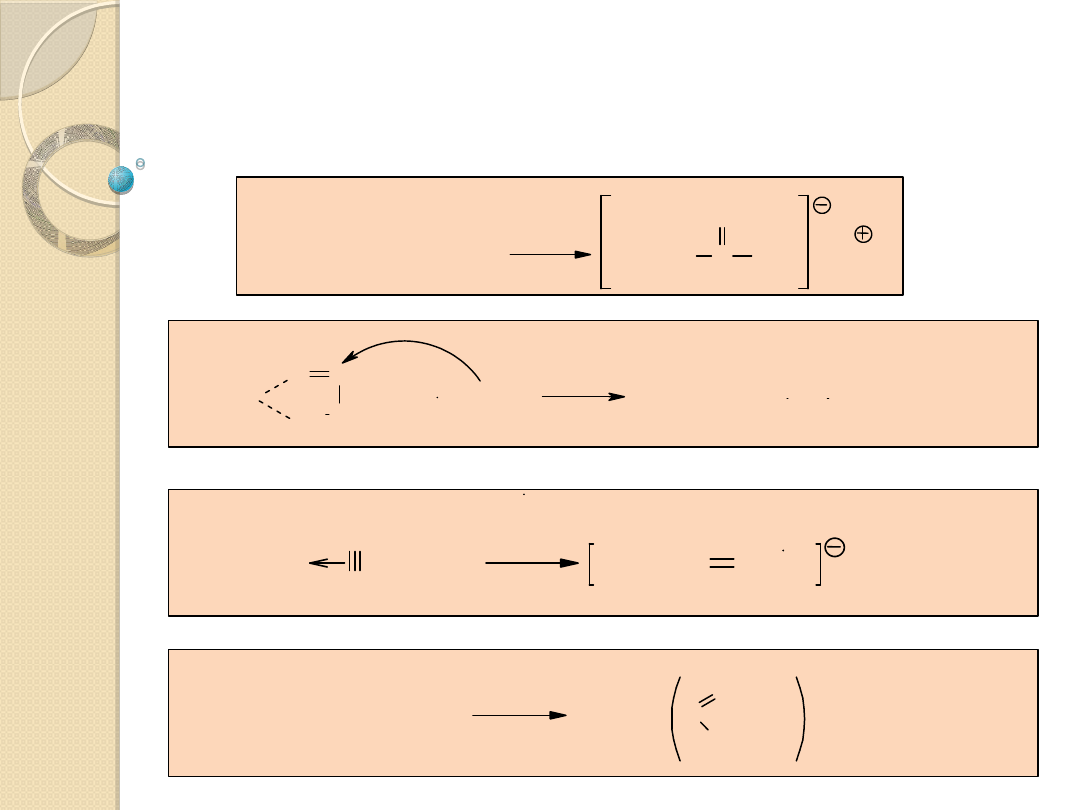
Zewnętrzny atak ligandów
C
O
COCH
3
Ir
I
(CO)
3
[P(C
6
H
5
)
3
]
2
+
Ir
I
(CO)
2
[Pt(C
6
H
5
)
3
]
2
+ CH
3
O
-
CH
CH
CH
2
CHOH
L
n
Cu
I
+ H
2
O
L
n
Cu
I
+ H
+
(solw.)
atak nukleofilowy
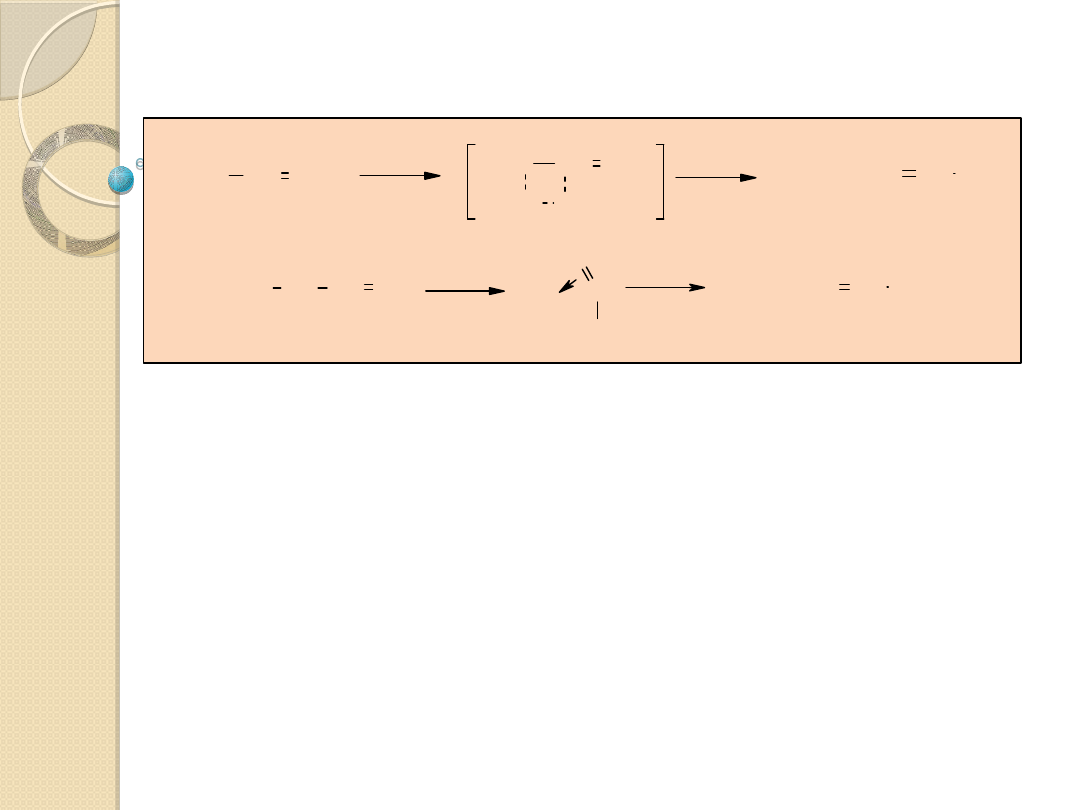
CH
2
CH CH
2
CH
CH
3
CH
2
CH
2
CH CH
3
L
n
Mo
L
n
M
L
n
Mo
+
+
+ H
+
CH CHCl
Cu
Cl H
CH CHCl
CH
2
CH CH
3
L
n
Cu
II
L
n
L
n
Mo
+
+
Atak elektrofilowy
CH
2
CH
2
R
Ti
(IV)
H CHR
CH
2
Ti
H
CH
2
CH
2
L
n
Ti
IV
L
n
L
n
TiH + CH
2
=CHR
L
n
W
(IV)
C CH
3
H
H
Cl
3
CL
3
W C CH
3
H
H
a - i b - Eliminacja
M
X
Y
L
m
M
(n-2)+
+ X-Y
L
m
n+
Rh
Cl
P(C
6
H
5
)
3
C
(C
6
H
5
)
3
P
H
S
C
Rh
Cl
P(C
6
H
5
)
3
S
(C
6
H
5
)
3
P
H C C
(II)
(I)
+
Redukująca eliminacja
Przykłady:
Rh
(III)
H
H
Rh
(III)
H
H
Rh
(I)
L
3
Cl
L
2
Cl
L
3
Cl
+ H
2
- L
Rh
H
H
Rh
H
H
C
H
2
CH
2
Rh
H
C
2
H
5
L
2
Cl
L
2
Cl
L
2
Cl
C
2
H
6
+ Rh
I
L
2
Cl
Rh
I
L
2
Cl + H
2
H
2
Rh
III
L
2
Cl
+ C
2
H
4
Homogeniczne odwodornienie
np., uwodornienie katalizowane kompleksem Wilkinsona
(kwadratowy d
8
-kompleks rodu Rh
I
[P(C
6
H
5
)
3
]
3
Cl).
MECHANIZMY REAKCJI KATALIZOWANYCH
KOMPLEKSAMI METALI
RCH
2
CH CHR'
M
H
CH
CHR'
CH
2
R
M
H CHR'
CH CH
2
R
M CH CH
2
R
CH
2
R'
H
M C
H
CHR
CH
2
R'
M
H
CHR
C
H
CH
2
R'
RCH CH CH
2
R'
MH +
MH +
C
H
CHR'
CH
2
R
M
M
H
CHR
CH
CHR'
CHR
C
H
CH
2
R'
RCH CH CH
2
R'
n+
(n+2)+
M
n+
M
n+
+
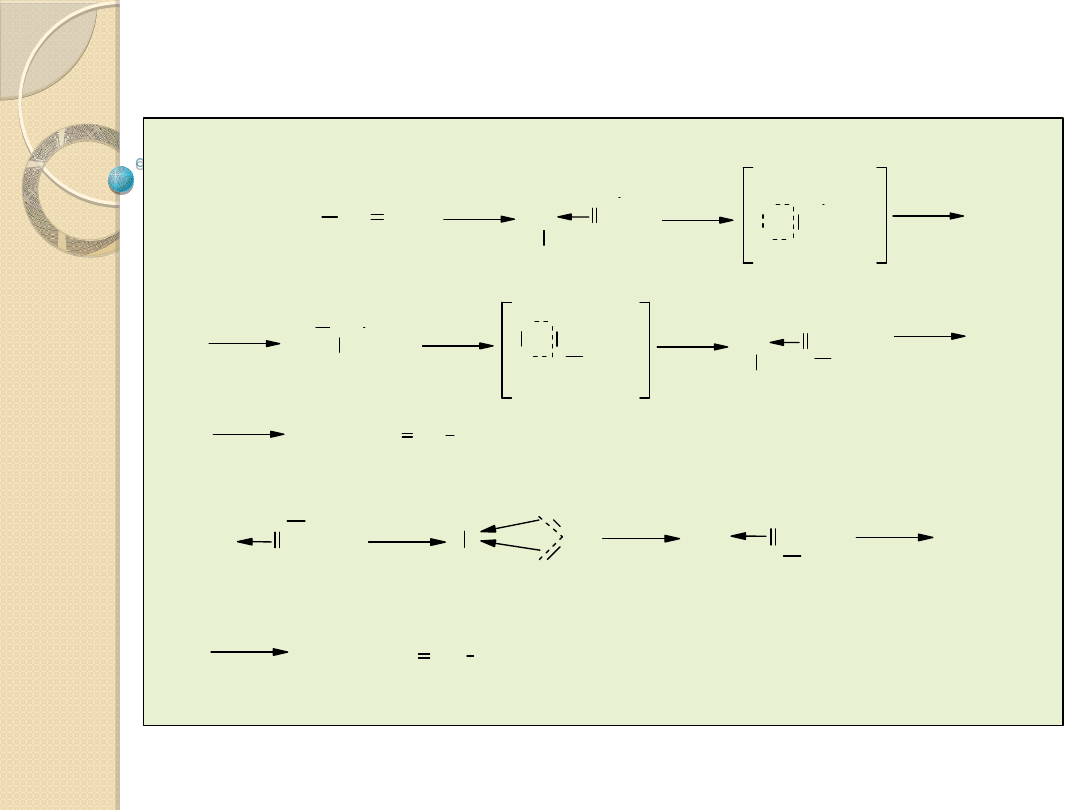
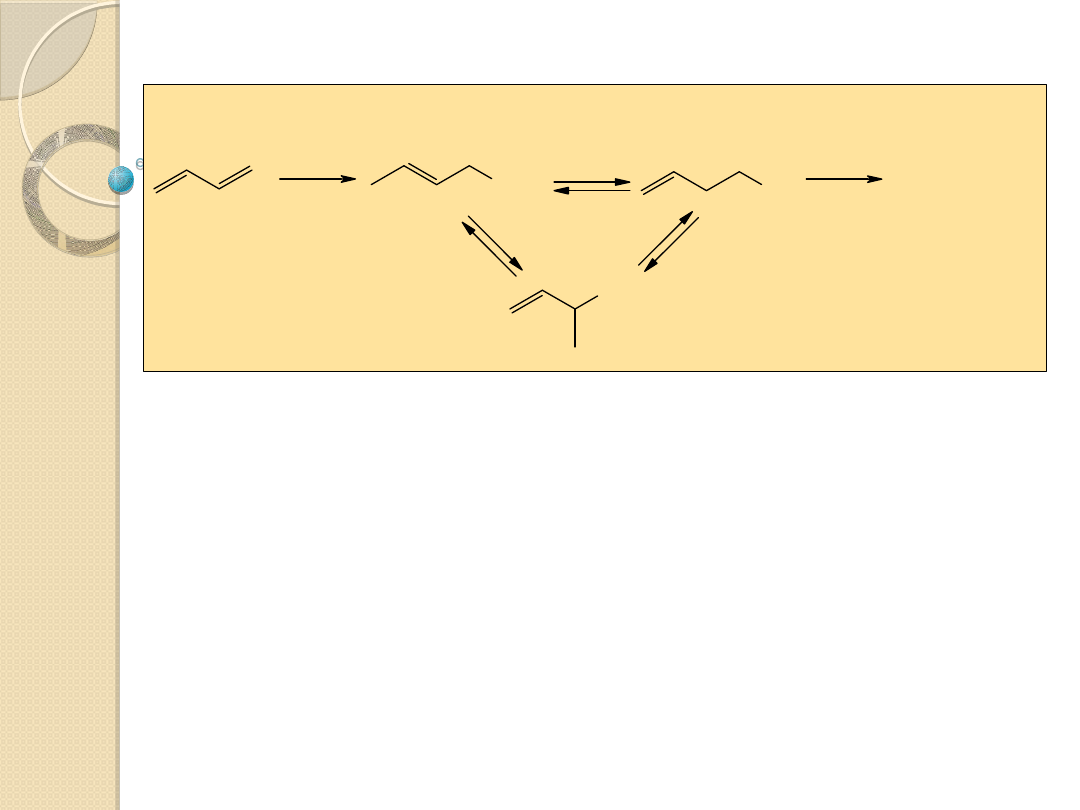
Izomeryzacja olefin
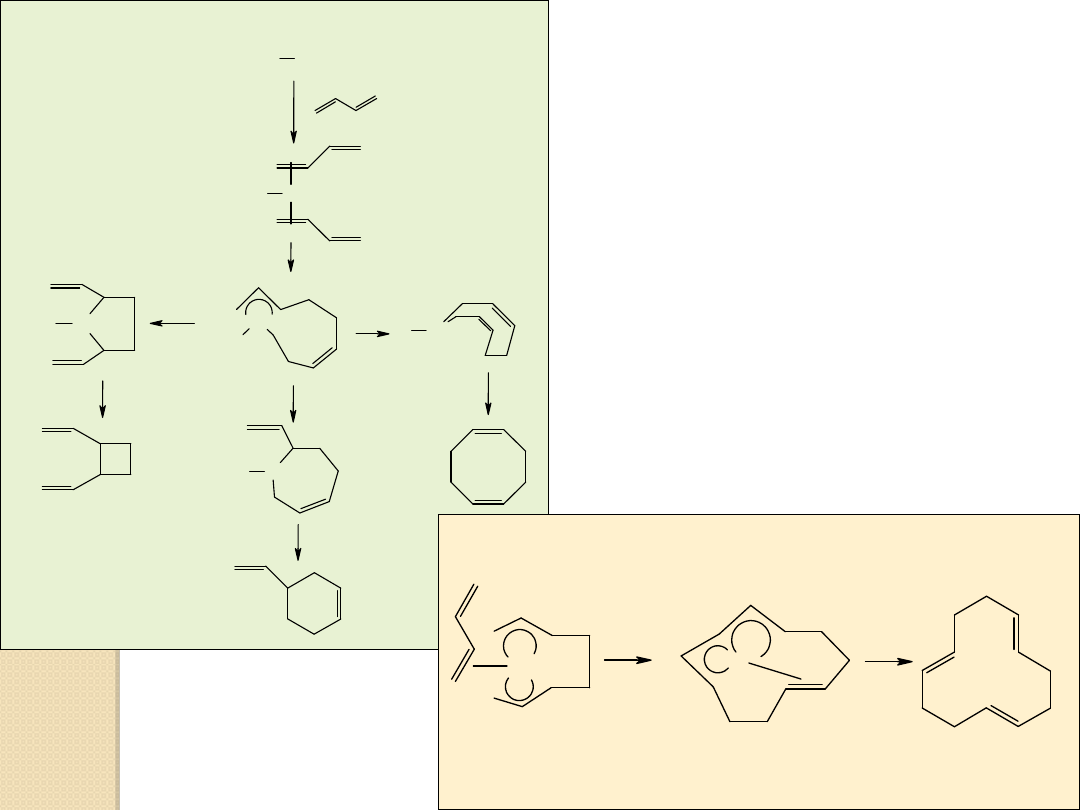
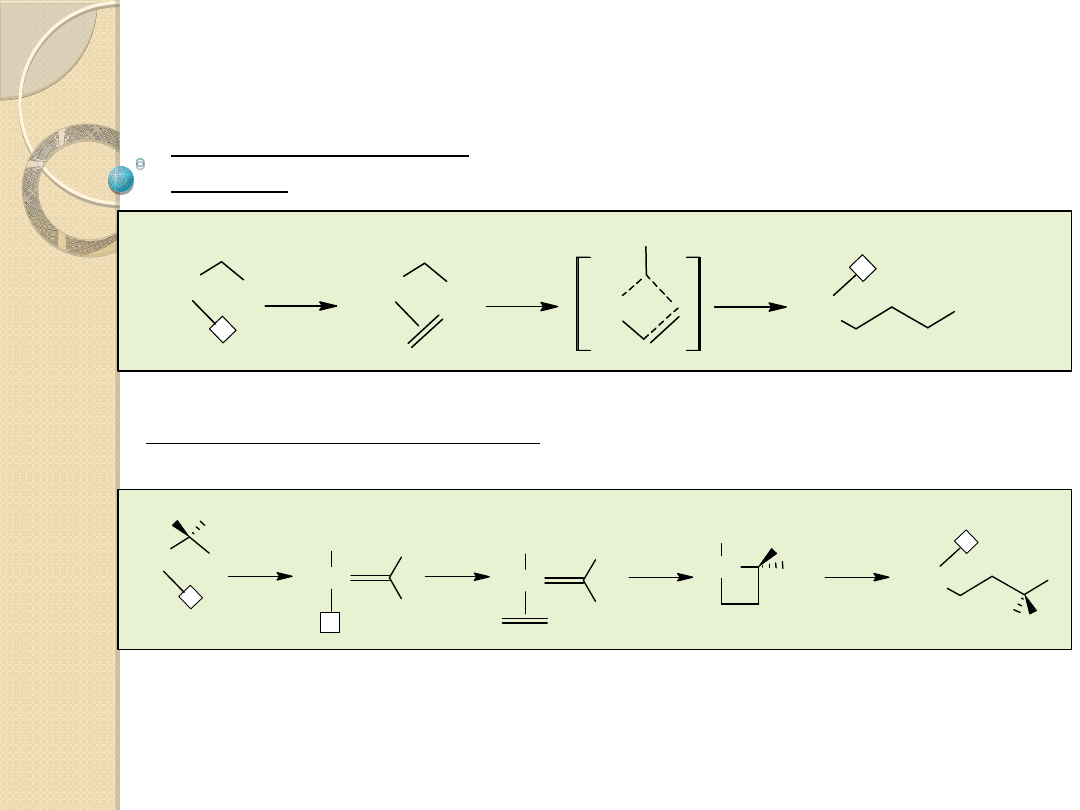
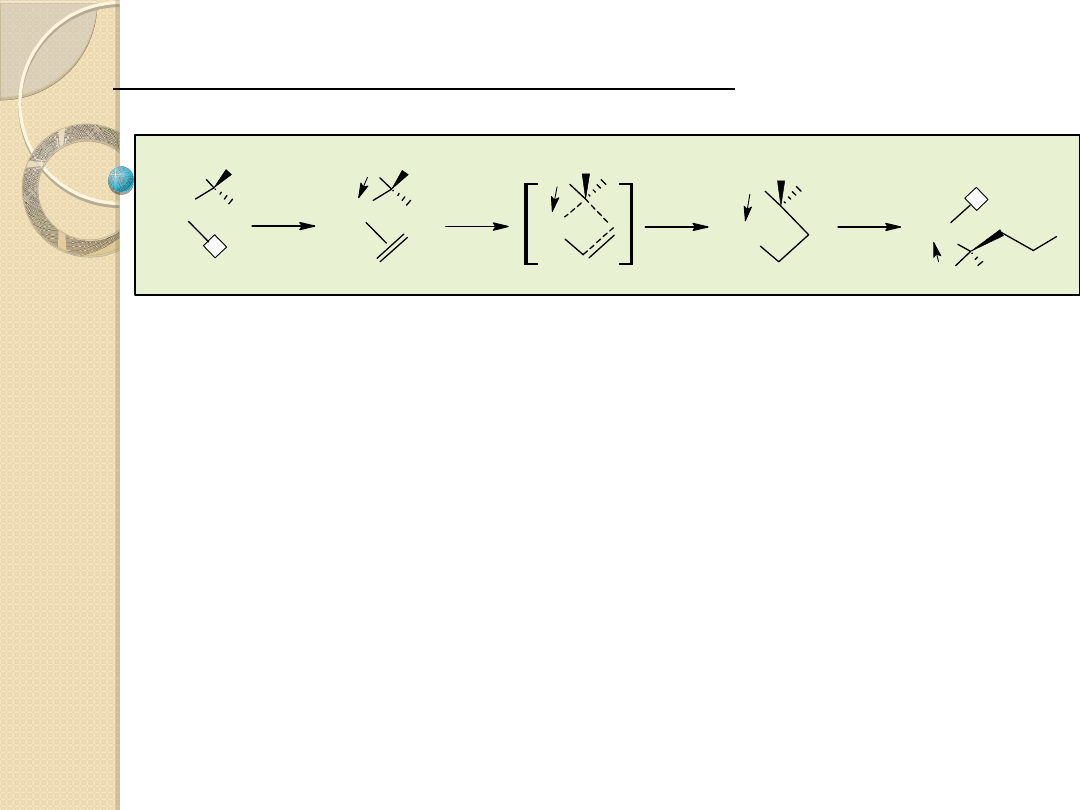
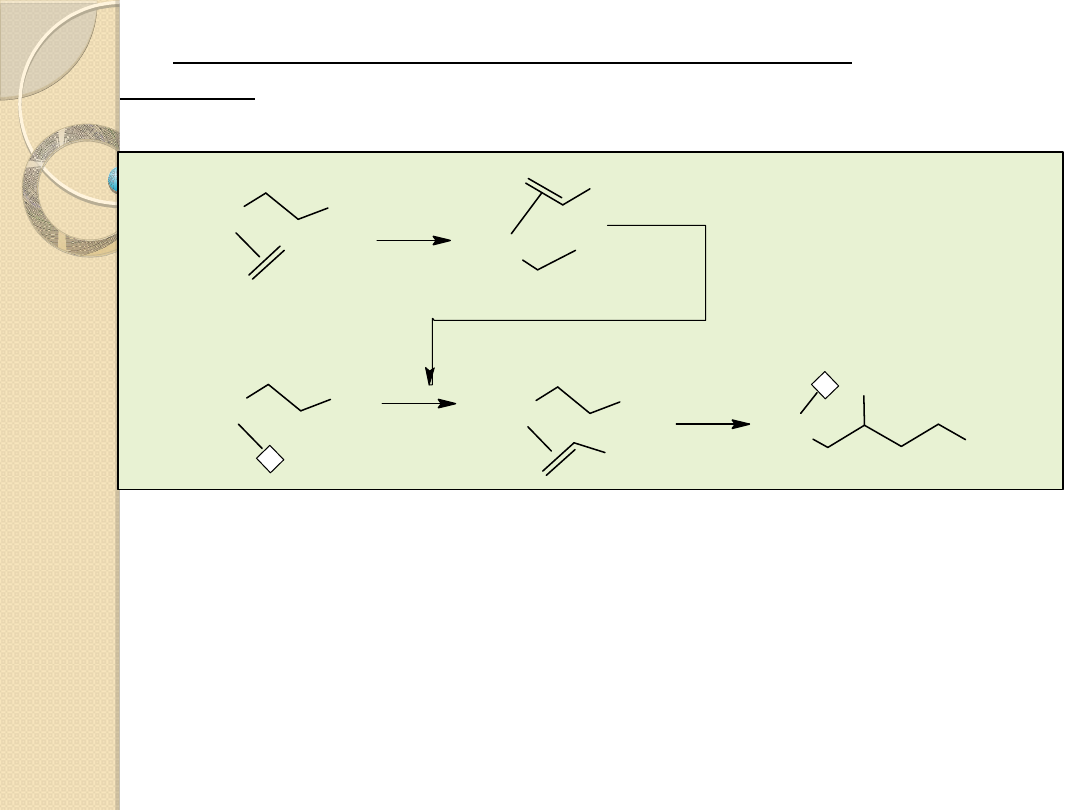
Dimeryzacja i trimeryzacja butadienu
L Ni
(0)
L Ni
Ni
L
Ni
L
Ni
L
Ni
L
2
- LNi
- LNi
- LNi
L=R
3
P lub (RO)
3
P
Cyklotrimeryzacja
butadienu w obecności
acetyloacetonianu niklu (II)
zredukowanego
Al(C
2
H
5
)
2
(OC
2
H
5
)
Ni
Ni
-Ni
ttt
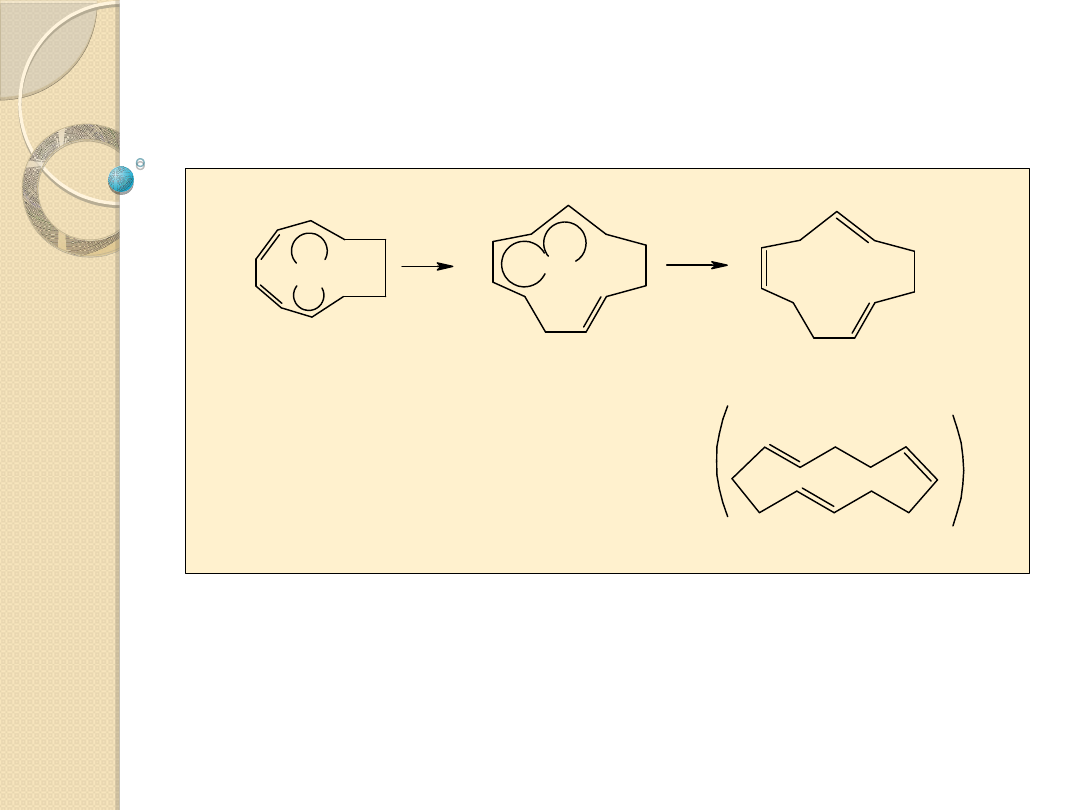
Cyklotrimeryzacja
butadienu
w
obecności
katalizatora otrzymanego przez zmieszenie TiCl
4
z nadmiarem Al
2
Cl
3
Et
3
w benzenie
Ti
Ti
-Ti
ctt
inaczej

H Al
H
CH
2
CH
2
Al
n
H(CH
2
CH)
n-1
CH=CH
2
H(CH
2
CH
2
)
n
OH
- AlH
[O]
H
2
O
Oligomeryzacja etylenu
w obecności: organicznych związków glinu,
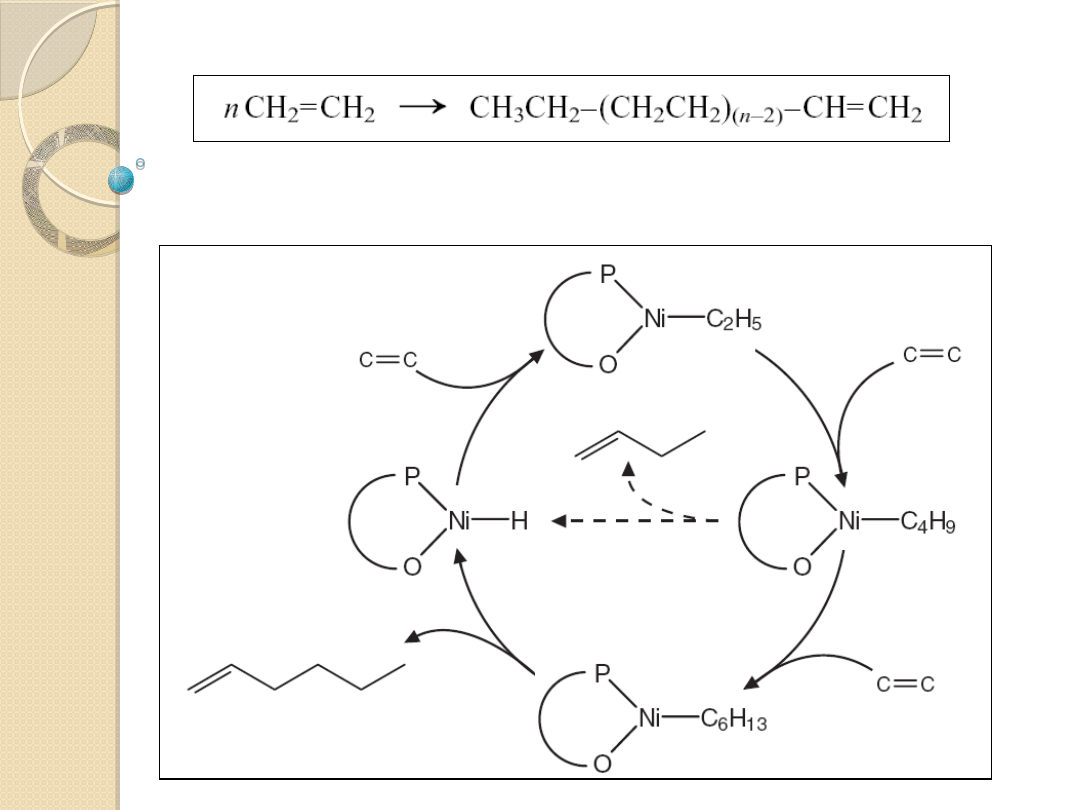
Oligomeryzacja etylenu (Process SHOP – Shel higher olefin
process)
Katalizatory niklowe z ligandami fosfinowymi, np. Ph
2
PCH
2
COOK
Produkty: mieszanina olefin C4–10, C12–18, and C20+
[M]
P
[M]
P
[M]
P
[M]
P
[M]
P
H
H
[M]
P
H H
[M]
P
H
H
[M]
P
H
H
[M]
H
H
P
Oligomeryzacja i polimeryzacja olefin
Mechanizm Green-Rooney
Mechanizm Cossée-
Arlman
[M]
P
H H
[M]
P
H
H
[M]
P
H
H
[M]
H
P
H
[M]
H
P
H
Modyfikowany mechanizm Green-Rooney
P = łańcuch polimerowy; - wolne centrum kwasowe
Typowe zakończenie łańcucha i przeniesienie
łańcucha
[M]
P
[M]
P
P'
[M]
P
[M]
P'
[M]
P'
P
1
2
P, P’= łańcuch polimerowy; - wolne centrum kwasowe
C
H
2
C
H
2
Ti
Cl
Cl
C
2
H
5
L
L
Al
C
2
H
5
Cl
Ti
Cl
R
Cl
L
L
Al
R
R
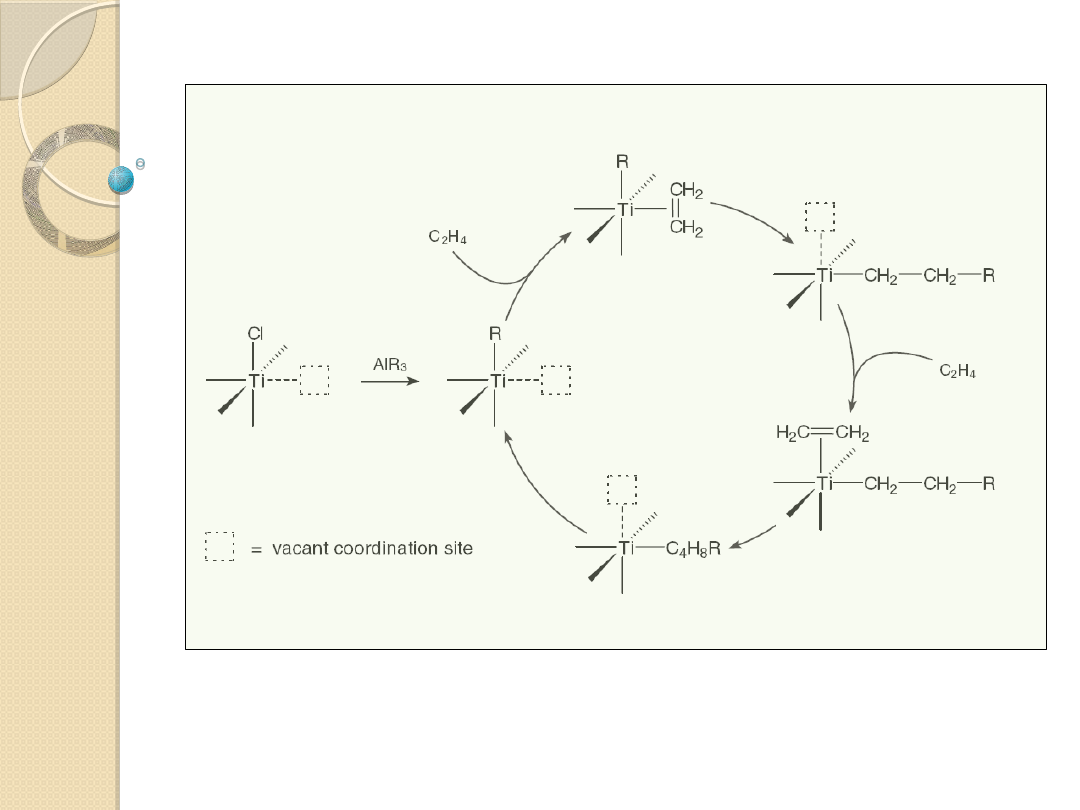
Katalizatory Ziglera-Natty (odkryte przez Ziglera w
1953 r.)
Związki kompleksowe metali przejściowych oraz związki
metaloorganiczne pierwiastków grup głównych (zwykle 1,2 i
13): MX
n
+ M’R
m
X
r
’ , gdzie M - metal przejściowy grup 4‑10, X -
halogen, acetyloaceton, RO, NCS i itp.; R - alkil, aryl, itp.; X’ -
halogen, OR.
Przykłady katalitycznie aktywnych kompleksów utworzonych z
LTiCl
2
i C
2
H
5
AlCl
2
:
Nagroda Nobla z Chemii – 1963 r.
„Za odkrycia na polu chemii i
technologii polimerów
(katalizatory polimeryzacji)”
Karl Ziegler
(1898-
1973)
Giulio Natta
(1903-
1979)
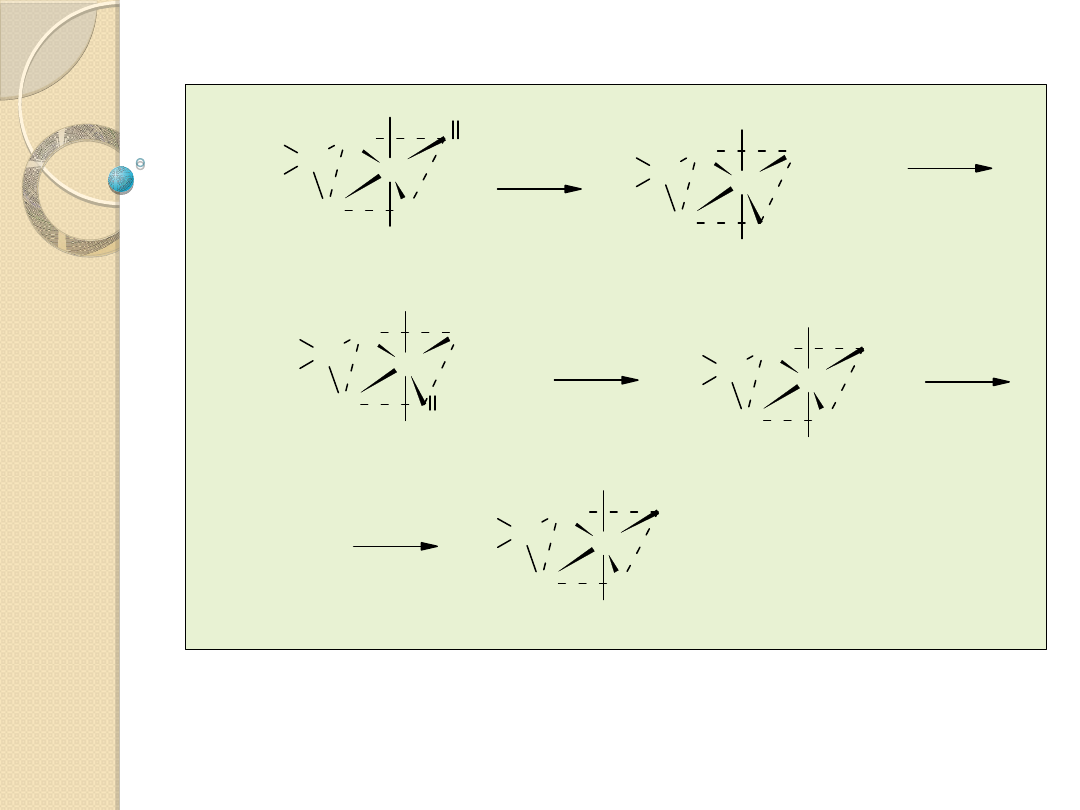
CH
2
CH
2
CH
2
CH
2
Ti
Cl
CH
2
CH
2
R
Cl
L
L
Al
R
R
Ti
Cl
R
Cl
L
L
Al
R
R
Ti
Cl
CH
2
CH
2
R
Cl
L
L
Al
R
R
Ti
Cl
(CH
2
CH
2
)
2
R
Cl
L
L
Al
R
R
Ti
Cl
(CH
2
CH
2
) R
Cl
L
L
Al
R
R
. . .
+ C
2
H
4
+ C
2
H
4
n
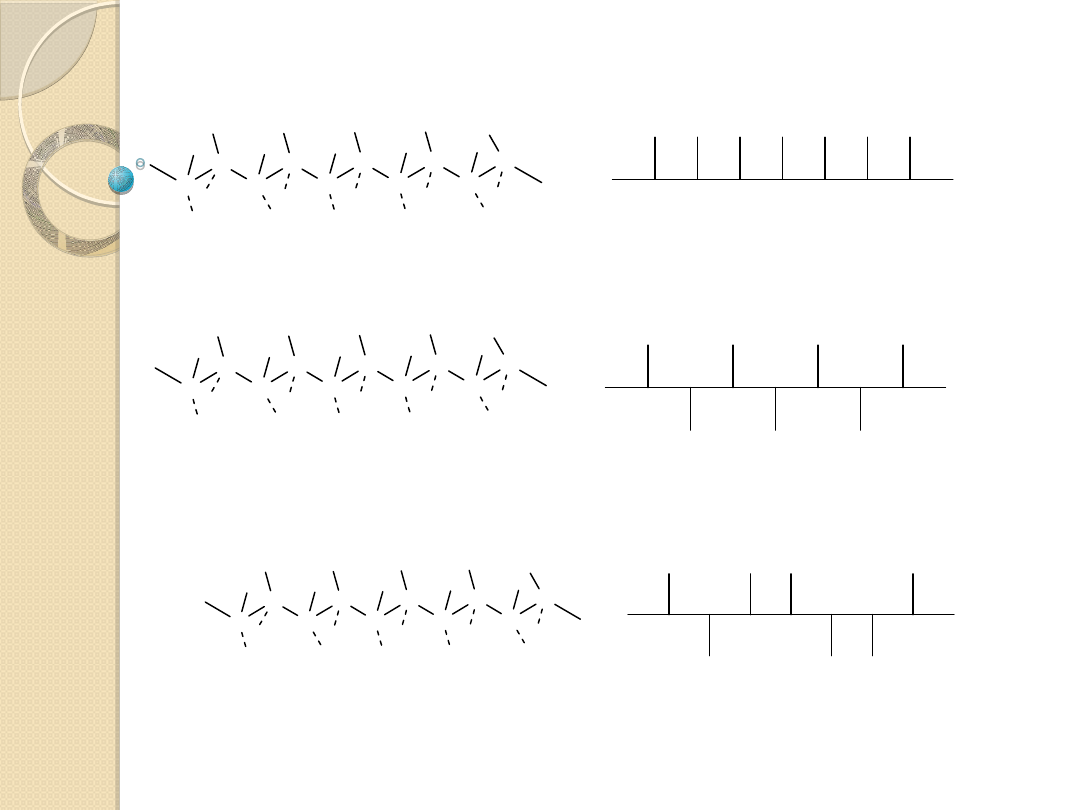
C
C
C
C
C
C
C
C
C
C
R
H
R
H
R
H
R
H
R
H
H
H
H
H
H
H
H
H
H
H
izotaktyczny
C
C
C
C
C
C
C
C
C
C
R
H
H
H
R
H
H
H
R
H
H
H
R
H
H
H
R
H
H
H
syndiotaktyczny
Możliwe produkty polimeryzacji:
•
C
C
C
C
C
C
C
C
C
C
R
H
R
H
H
H
H
H
R
H
H
H
H
H
R
H
R
H
H
H
ataktyczny
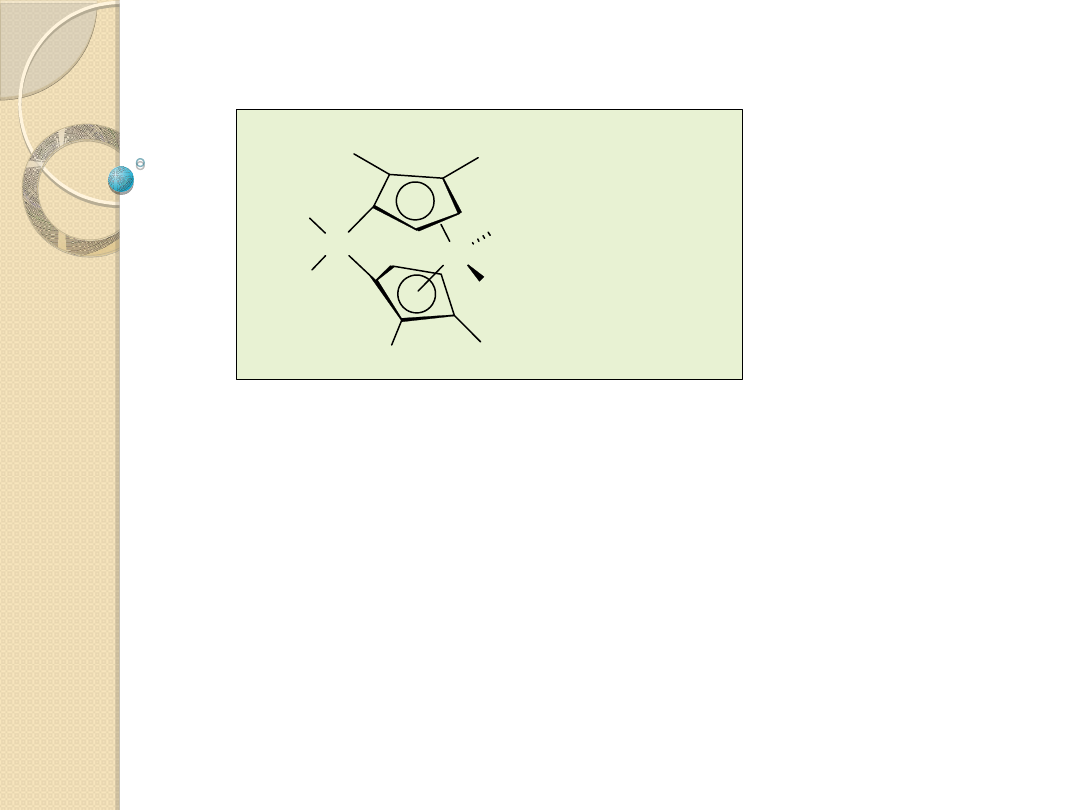
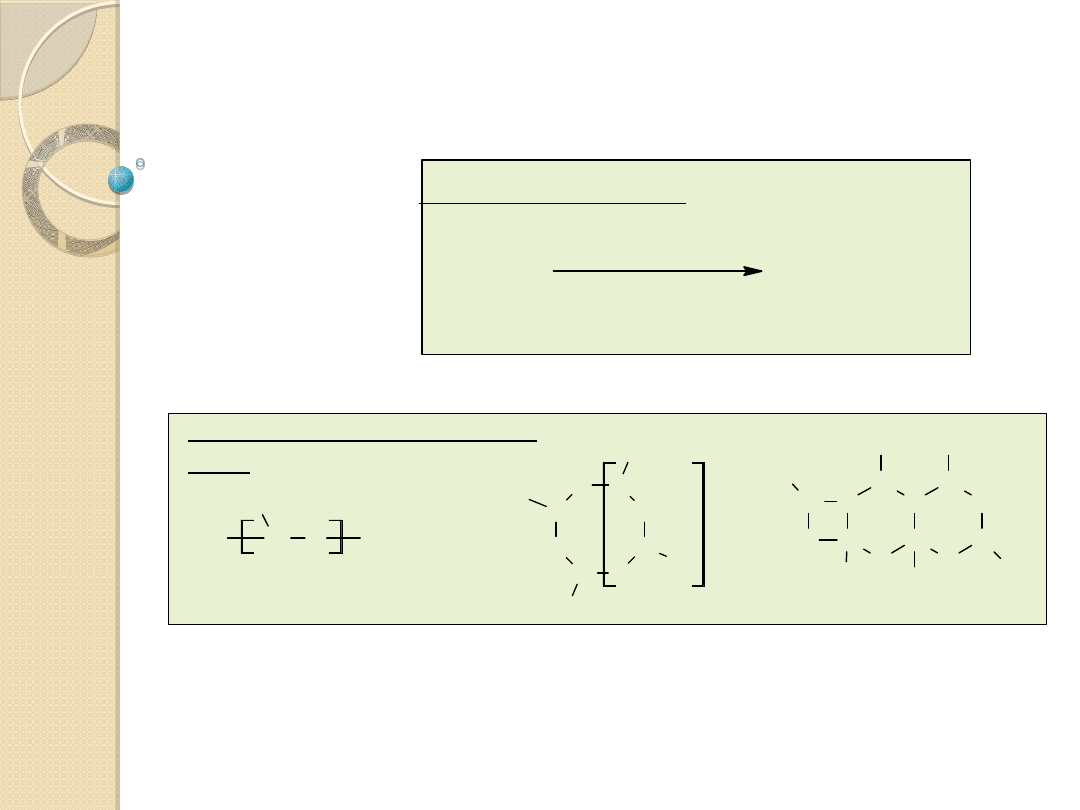
Katalizatory metalocenowe
A
R
R
R
R
R
R
M
X
X
M=Ti, Zr, Hf
X - skoordynowane do atomu M to atomy chlorowca (na ogół Cl)
lub grupy alkilowe.
A - atomy węgla lub krzemu.
R, R’ - grupy alkilowe lub inne grupy węglowodorowe
(CH
3
)
3
Al
[-Al-(CH
3
)
3
-O-]
n
n~5-20
MAO
hydroliza
Me
Al
Me
O
AlMe
2
n
n
Al
O
O
Al
Al
O
O
Al
Me
Me
Me
Me
O
Al
Al
O
O
Al
O
Al
Al
O
Al
O
Me
Me
Me
Me
Me
Me
Metyloaluminoksan (MAO)
Prawdopodobne struktury
MAO
( najważniejszy aktywator katalizatorów
metalocenowych)
Otrzymywanie MAO
Mt
P
H
H
H CH
3
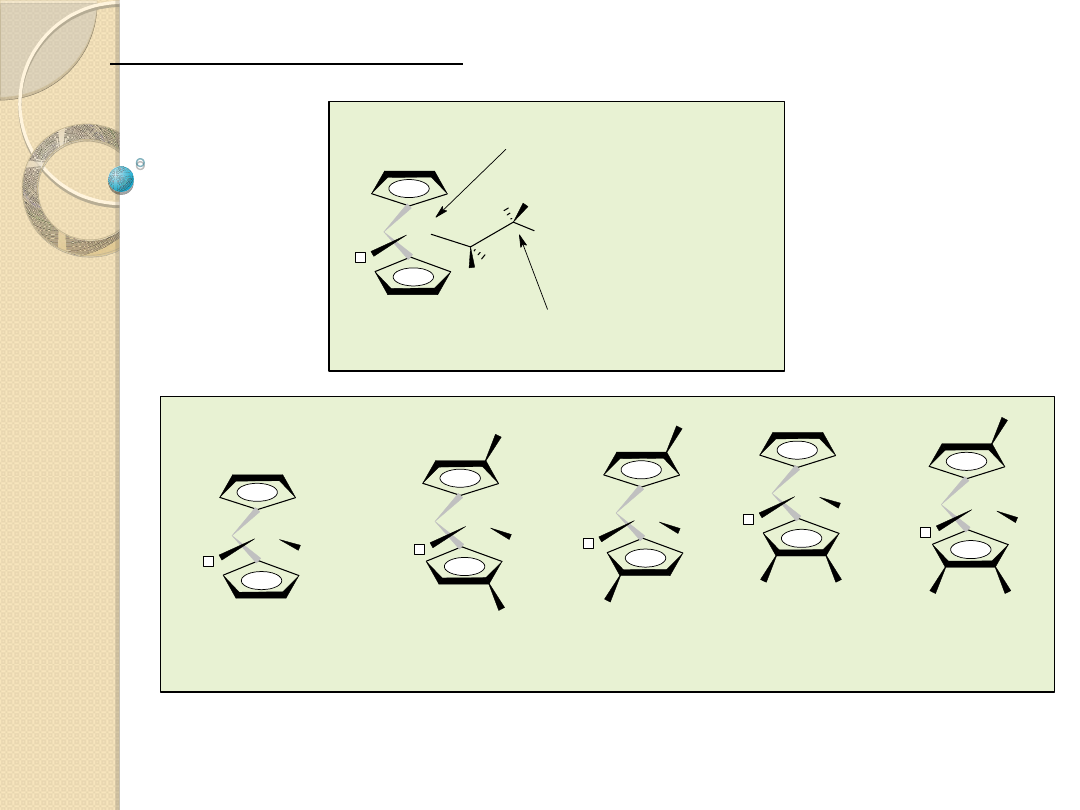
centrum kontroli enancjometrycznej
(enantiomorphic-site-control)
kontrola "ko�ca �
a�cucha"
(chain-end control)
Mt
P
Mt
P
Mt
P
Mt
P
Mt
P
Reguły symetrii Ewen’a
C
2v
, achiralny
ataktyczny PP
C
s
, achiralny
ataktyczny PP
C
2
, chiralny
izotaktyczny PP
C
s
, prochiralny
syndiotaktyczny PP
C
1
, chiralny
hemiizotaktyczny PP
CH
3
Zr
R
w�
�
czenie propylenu
w�
�
czenie etylenu
CH
3
Zr
R
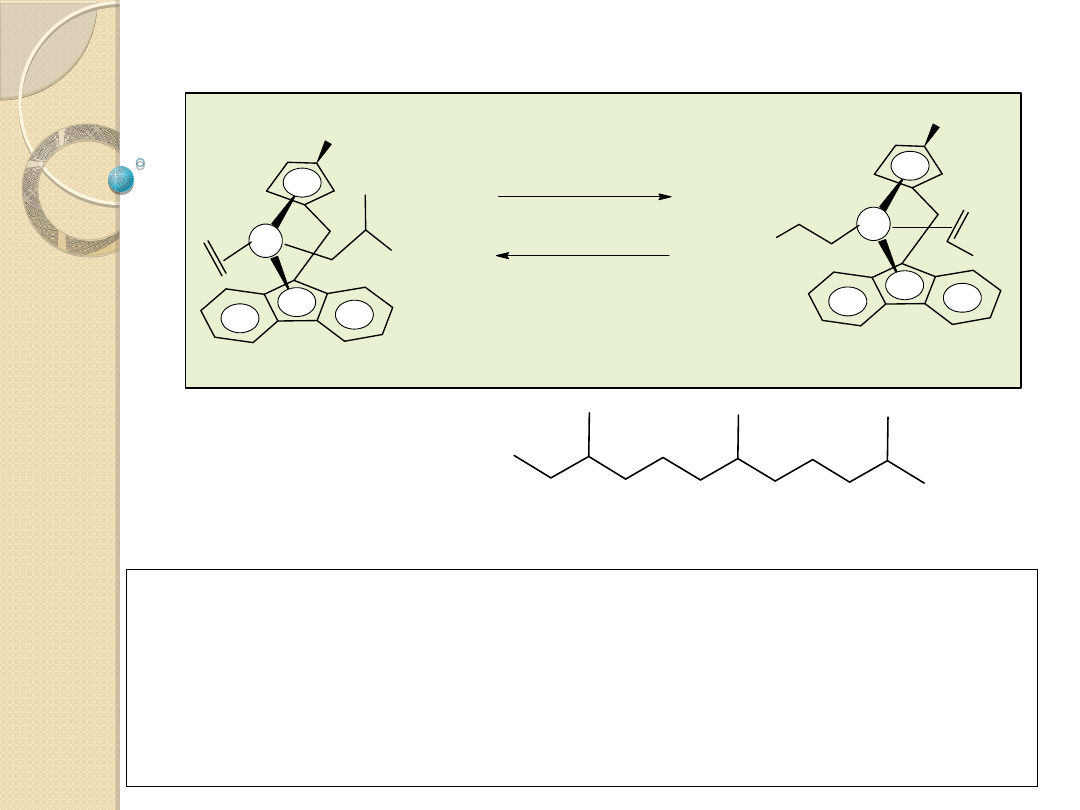
Kopolimeryzacja etylenu i propylenu
R – powstający polimer
poli(etylen-alt-propylen)
Inne katalizatory oligomeryzacji alkenów i dienów oraz
kooligomeryzacji alkenów i kooligomeryzacji alkenów z dienami:
związki niektórych metali szlachetnych, np. PdCl
2
,
[PdCl
2
(PhCN)
2
], RhCl
3
.
3H
2
O, RuCl
3
, IrCl
3
.
4H
2
O.
kompleksy metali grup 8-10 (np. Ni, Pd)
N
N
R
R
R
R
R
R
N
N
R
R
N
N
O
O
R R
R
R
R
R
R
R
N
N
N
N
R R
N
N
R
R
Ligandy dla metali przejściowych grup
8-10
N-N ligandy
O
P
O
N
R
O
N
O
O
N
R
R
N
N
MeO
OMe
Ligandy N-O
P
N
R
N
P
R
R
R
R
R
R
R
R
ligandy N-X
R
R
R
R
R
R R
R
R
R
R
R R
R
R
R
+
+
1
2
3
4
5
6
7
8
2
3
4
5
6
7
8
1
CH
3
CH W
(VI)
Cl
5
H
CH
3
CH W
(VI)
Cl
4
WCl
6
+ C
2
H
5
AlCl
2
C
5
H
5
W
VI
Cl
5
+ AlCl
3
C
2
H
5
W
VI
Cl
5
-HCl
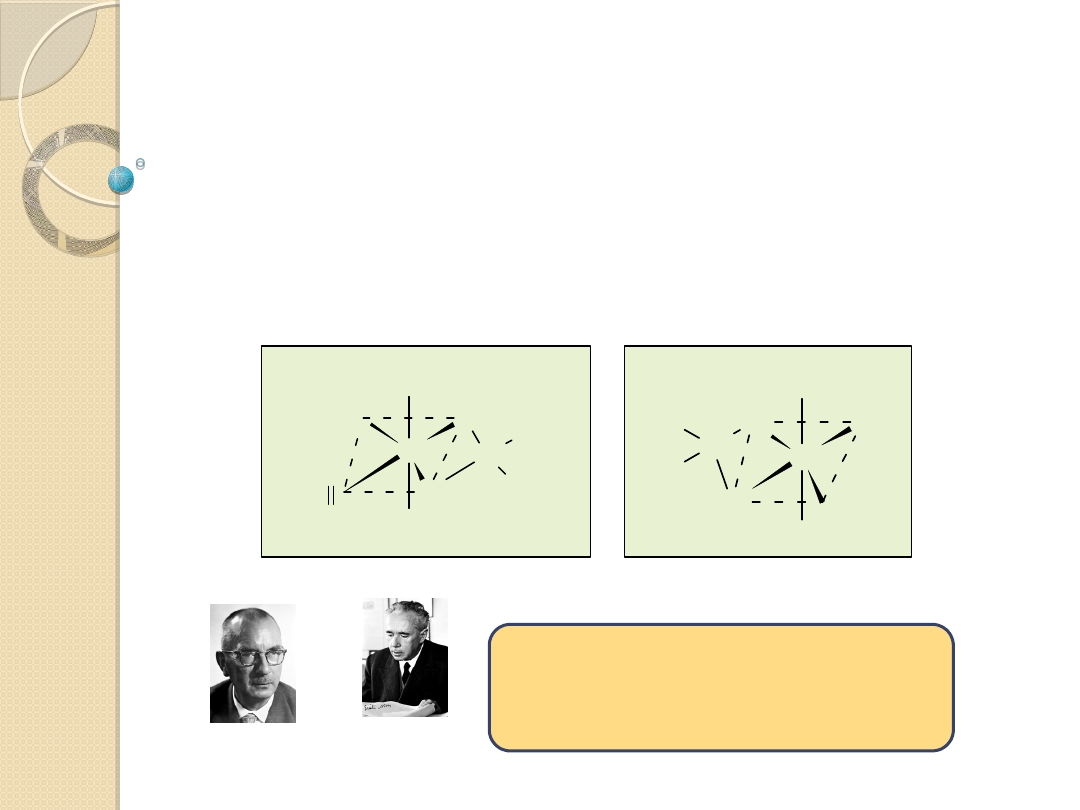
Metateza olefin (dysproporcjonowanie,
dysmutacja)
.
Nagroda Nobla
z chemii - 2005
„za rozwój
metod
metatezy”
Yves Chauvin
Richard R. Schrock
Robert H. Grubbs
Cechy reakcji metatezy to:
termoneutralność,
niska energia aktywacji rzędu 30 kJ/mol (dla
aktywnych katalizatorów),
duża szybkość reakcji,
w przybliżeniu pierwszy rząd reakcji zarówno
względem kompleksu jak i alkenu.
C
M
R
R
C
C
R
R
R
R
C
M C
C
R
R R
R
R
R
=
/
C C
R
R R
R
M C R
R
M C
R
R
C C
R
R R
R
C
M
R
R
C
C
R
R
R
R
+
1
2
3
4
5
6
1
2
3
4
5
6
+
1
2 3
4
5
6
1
1
2
2
3
3
4
4
5
5
6
6
PdCl
2
+ CH
2
=CH
2
+ H
2
O CH
3
CHO + Pd
o
+ 2HCl
CH
2
=CH
2
+ CH
3
COOH + 1/2O
2
CH
3
COOCH=CH
2
+ H
2
O
CH
2
=CH
2
+ 2CH
3
COOH + 1/2O
2
CH
3
COOCH
2
CH
2
OOCCH
3
+ H
2
O
Utlenianie
CH
2
=CH-CH
3
+ CH
3
COOH + 1/2O
2
CH
2
=CH-CH
2
OOCCH
3
+ H
2
O
+ 2 CH
3
COOH + O
2
CH
3
COOCH
2
-CH=CH-CH
2
OOCCH
3
CH
3
C CH
3
O
CH
3
-CH=CH
2
+ 1/2O
2
+ 0,5 O
2
CH
3
CHO
CH
2
CH
2
PdCl
2
PdCl
4
2-
+ C
2
H
4
PdCl
3
(C
2
H
4
)
-
+ Cl
-
PdCl
3
(C
2
H
4
)
-
+ H
2
O PdCl
2
(H
2
O)(C
2
H
4
) + Cl
-
PdCl
2
(H
2
O)(C
2
H
4
) + H
2
O PdCl
2
(OH
-
)(C
2
H
4
) + H
3
O
+
CH
2
CH
2
Pd
Cl
Cl
Cl
Pd
Cl
Cl
CH
2
CH
2
OH
Cl
+ H
2
O
(II)
(II)
H
2
O Pd CH
2
Cl
Cl
CH
O
H
H
CH
3
CHO + PdCl
-
+ HCl + H
2
O
Mechanizm utleniania etylenu
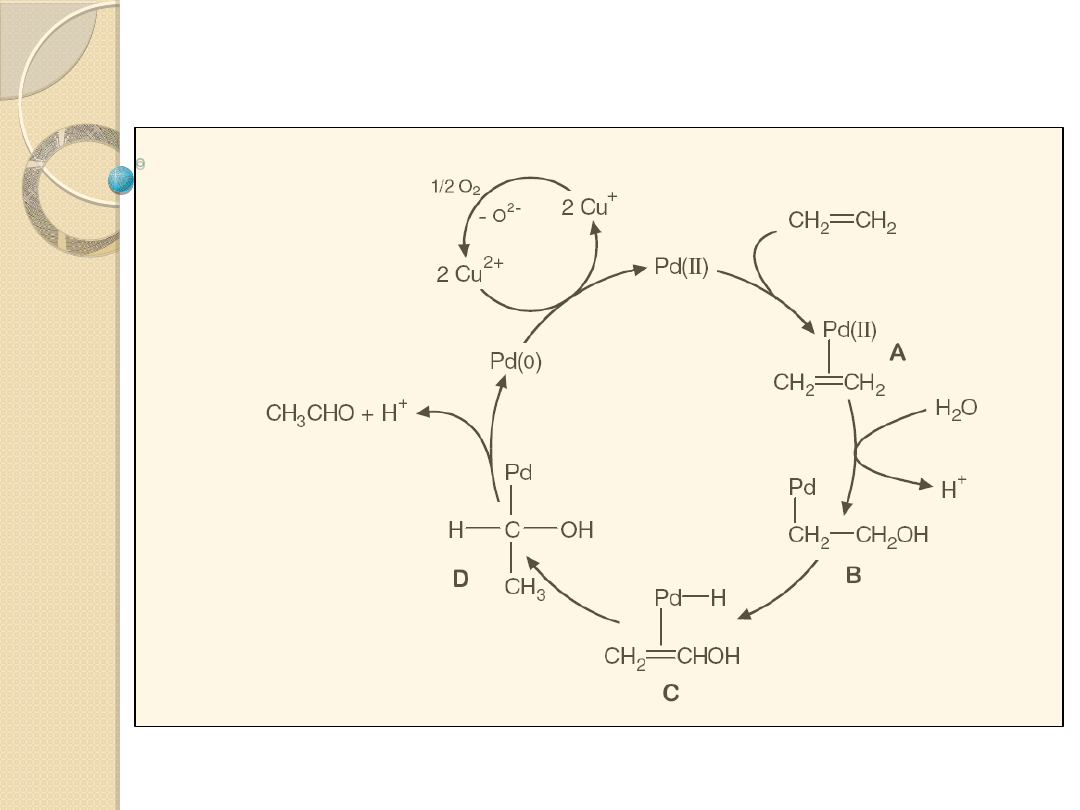
Mechanizm utleniania etylenu
C C
C
C
O
+ ROOH
+ ROH
Mo O
+
OH
R
O
O
+
C
C
Mo O
+
R
O
O
H
O
C
C
Mo
OH
OR
O
C
C
O
+
C
C
O
+
ROH
Mo
OH
O
+
VI
.
Utlenienie propylenu wodoronadtlenkiem tert-butylu lub
wodoronadtlenkiem etylobenzenu w obecności naftenianów i
żywiczanów molibdenu
CH-CH
3
CH
2
O
CH
3
-CH=CH
2
+ ROOH
+ ROH
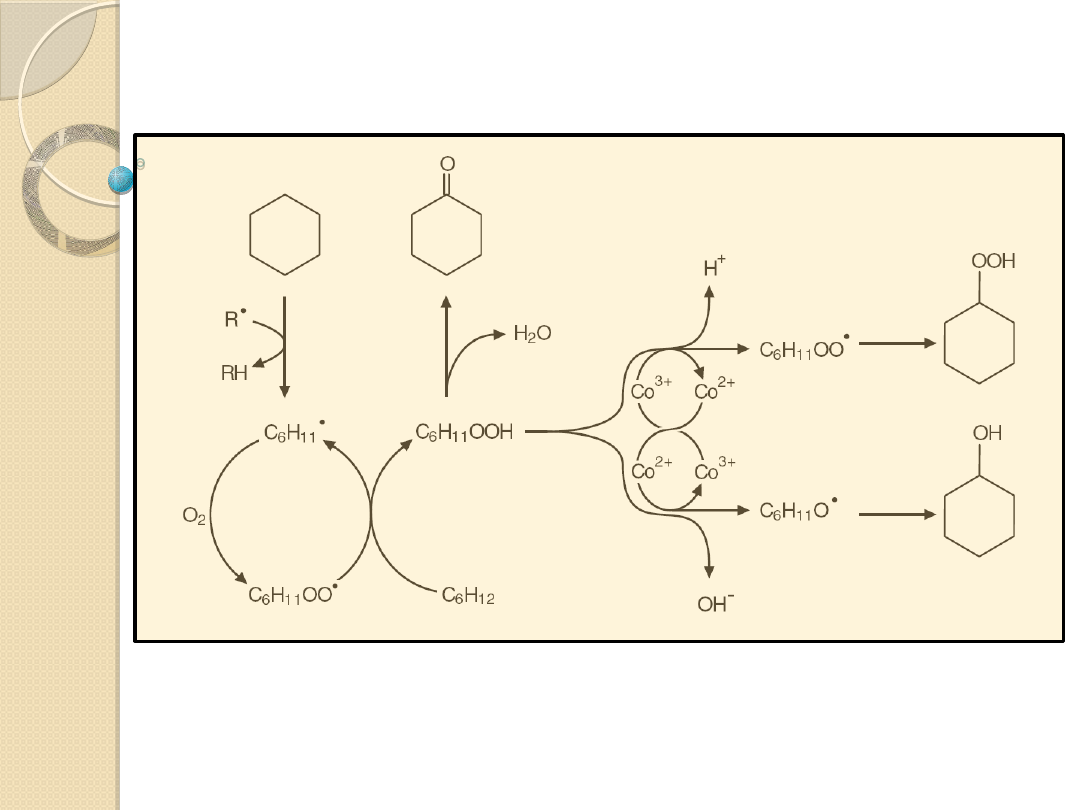
Utlenienie cykloheksanu
C C
C
H
CX
+ HX
X=OH, OR, OCOR, NR
2
, Cl, Br, F, CN, SiR
3
C C
C
H
CX
+ HX
Przyłączenie HX do alkinów i
olefin
Reakcję katalizują kompleksy Hg(II), Cu(I), Zn(II), Cd(II), Rh(I),
Pd(II).
Reakcję przyspieszają kwasy protonowe i aprotonowe.
Niektóre związki zawierające słabo kwaśny wodór (np. HCN)
lub atom wodoru typu wodorkowego (HSiR
3
) przyłączają się
do olefin tylko w obecności kompleksów metali.
C C
M
X
C
C
M C CX
L
n
MX +
L
n
L
n
C
C
M C CX
L
n
M
L
n
+ HX
-
+ H
+
M C CX
C
H
CX
L
n
+ H
+
+ L
n
M
+
Przyłączenie HX do acetylenu i jego pochodnych
lub zewnętrzny atak nukleofilowy.
Tworzenie produktu reakcji i regeneracja katalizatora
zachodzi w wyniku protonolizy metaloorganicznego
produktu przejściowego (zewnętrzny atak elektrofilowy)
Obejmuje tworzenie -kompleksów, po którym następuje
insercja w wiązanie M-X
Katalizatory reakcji hydrocyjanowania (addycja HCN do
C=C lub CC):
karbonylki żelaza, kobaltu i niklu (zwłaszcza w
obecności fosfin), [Ni{R
2
P(CH
2
)
4
PR
2
}
2
],
[M{P(OR)
3
}
4
] (M=Ni,Pd),
[Ni(CN)
4
]
4-
,
CuCl itp.
W wypadku nieaktywnych alkenów m.in., [Co
2
(CO)
8
],
Co
2
(CO)
6
(PPH
3
)
2
], [Co
2
(CO)
6
{P(OPh)
3
}
2
],
Mo(CO)
3
{P(OPh)
3
}
3
]+TiCl
3
, [W(CO)
3
{P(OPh)
3
}
3
]+BPh
3
.
M
H
CN
L
n
L
n
M + HCN
L
n
M + HCN
L
n
MNCH
[HM(CN)L
n
+ CH
2
=CHR
HM(CN)L
n
RCH
2
CH
2
H(CN)L
n
CH
2
=CHR
ML
n
+ RCH
2
CH
2
CN
Hydrocyjanowanie butadienu
CN
CN
CN
NC(CH
2
)
4
CN
+ HCN
+ HCN
Reakcja ta katalizowana homogenicznymi kompleksami
Ni(0) z triarylofosfatem Ni[P(OAr)
3
]
4

Czynniki hydrosililujące:
wodoroalkilosilany SiR
3
H
SiHX
3
SiHR
n
X
3-n
(X=halogen, OR)
Hydrosililowanie, tj. addycja Si-H do C=C, C=O, jako efektywna
droga otrzymywania alkilosilanów, monomerów do tworzyw
krzemoorganicznych.
Hydrosililowanie,
Reagenty sililowane:
alkeny,
alkiny
ketony.
Katalizatory hydrosililowania:
wodorkowe kompleksy metali przejściowych, np. RhH(CO)
(PPh3)3],
kompleksy z których mogą powstawać wodorki, tj. karbonylki,
hydrokarbonylki, kompleksy fosfinowe itd., np. [Ni(CO)
4
],
[Fe(CO)
5
], [Fe
2
(CO)
9
], [Co
2
(CO)
8
], i [IrCl(CO)(PPh
3
)
2
związki koordynacyjne nie zawierające grup karbonylowych,
przede wszystkim kompleksy halogenkowe i fosfinowe, np.
H2PtCl6
, IrCl
3
, H
2
IrCl
6
, RhCl
3
, RuCl
3
, [RuX
2
(PR
3
)
3
], [PtX
2
(PR
3
)
2
],
RhX(PR
3
)
3
], kompleksy alkenowe tych pierwiastków
SiHR
3
+ R'CH=CH
2
Si(CH
2
CH
2
R')R
3
SiHR
3
+ R'
2
CO R'
2
CHOSiR
3
CoH(CO)
4
CoH(CO)
3
R'Co(CO)
3
R
3
SiH
-SiR
3
R'
R'CoH(CO)
3
(SiR
3
)
-CO
alken
Mechanizm hydrosilinowania
Przykłady hydrosilinowania
RCH CH
2
RCH CH
3
CHO
+ CO +H
2
RCH
2
CH
2
CHO
C
H
CH + CO + H
2
O CH
2
=CHCOOH
CH
2
CH
2
+ CO + H
2
O CH
3
CH
2
COOH
CH
3
OH + CO CH
3
COOH
Reakcje tlenku węgla
karboksylowanie acetylenu i olefin,
karbonylowanie alkoholi.
hydroformylowanie olefin
Aktywność
katalityczna
w
reakcji
hydroformylowania:
Rh>>Co>Ir~Ru>Os>Pt>Pd>Fe>Ni
Kompleks Wilkinsona - [RhHCO(PPh
3
)
3
] – bardzo aktywny katalizator
formylowania olefin.
Hydroformylowanie propylenu w obecności katalizatora
rodowego (L=trifenylofosfina)
L
2
Rh
CO
CO
C
3
H
7
L
2
Rh
C
3
H
7
CO
CO
L
2
Rh
H
CO
CH
2
CH
2
L
2
Rh
H
H
CO
COC
3
H
7
CO
L
2
Rh
C
O
C
3
H
7
L
HRh(CO)
2
L
C
3
H
7
CHO
CH
2
=CHCH
3
H
2
HRh(CO)L
3
Mechanizm hydroformylowania w obecności
karbonylku kobaltu
HCo(CO)
4
HCo(CO)
3
+ C
2
H
4
HCo(CO)
3
(C
2
H
4
)
C
2
H
5
Co(CO)
3
+ CO
C
2
H
5
Co(CO)
4
C
2
H
5
COCo(CO)
3
+ HCo(CO)
4
C
2
H
5
CHO +Co
2
(CO)
7
Co
2
(CO)
7
Co
2
(CO)
8
+ H
2
2HCo(CO)
4
- CO
+ CO
Hydrokarboksylowanie alkoholi
CH
3
OH + CO + H
2
CH
3
CH
2
OH
Najbardziej aktywnymi katalizatorami w tej reakcji są
związki rutenu i układy rutowo-kobaltowe.
C
H
CH + CO + ROH CH
2
=CHCOOR
CH
2
CH
2
+ CO + EtCOOH (EtCO)
2
C
H
CH + CO + RSH CH
2
=CHCOSR
CH
2
CH
2
+ CO + H
2
O CH
3
CH
2
COOH
C
H
CH + CO + HNR
2
CH
2
=CHCOONR
2
Reakcje karboksylowania alkinów i alkenów
Katalizatorami hydrokarboksylowania są karbonylki
kobaltu, niklu, żelaza, rutenu, rodu oraz kompleksy
palladu.
Hydrokarboksylowanie alkoholi
CH
3
OH + CO + H
2
CH
3
CH
2
OH
Najbardziej aktywnymi katalizatorami w tej reakcji są
związki rutenu i układy rutenowo-kobaltowe.
Mechanizm syntezy kwasu propionowego lub jego estru
C
2
H
4
(CO)
3
Co
H
CH
2
CH
2
(CO)
3
Co C
O
C
2
H
5
C
2
H
5
C OR
O
HCo(CO)
3
(CO)
3
CoC
2
H
5
(CO)
4
CoC
2
H
5
CO
ROH
HCo(CO)
4
Co
2
(CO)
8
H
2
CO
(R=H lub alkin):
Karbonylowanie metanolu
[Rh
I
(CO)
2
I
2
]
-
CH
3
I
[CH
3
Rh
III
(CO)
2
I
3
]
-
CO
C I
O
C
H
3
C Rh(CO)
2
I
3
O
C
H
3
-
C Rh(CO)I
3
O
C
H
3
-
HI
CH
3
OH
CH
3
COOH
H
2
O
CH
3
I
CH
3
COI
CO
HI + CH
3
OH CH
3
I + H
2
O
CH
3
COI + H
2
O CH
3
COOH + HI
MeOH + CO MeCOOH
Karbonylowanie metanolu
[Rh
I
(CO)
2
I
2
]
-
CH
3
I
[CH
3
Rh
III
(CO)
2
I
3
]
-
CO
C I
O
C
H
3
C Rh(CO)
2
I
3
O
C
H
3
-
C Rh(CO)I
3
O
C
H
3
-
HI
CH
3
OH
CH
3
COOH
H
2
O
CH
3
I
CH
3
COI
CO
HI + CH
3
OH CH
3
I + H
2
O
CH
3
COI + H
2
O CH
3
COOH + HI
MeOH + CO MeCOOH
Heterogeniczne rozszczepienie najczęściej zachodzi pod
wpływem kompleksów Ru(II), Ru(III), Rh(III) itd., np.
Homolitycznie rozszczepiają wodór kompleksy kwadratowe d
8
-
elektronowe Rh(I), Ir(I) oraz [Co(CN)
3
]
3-
, [CoH(N
2
)(PPh
3
)
3
]
3
2M(I) + H
2
M(III)
H
H
2M(II) + H
2
2M(III)H
MA
2
+ H
2
MH + H
+
+ A
-
Redukcja związków organicznych . Uwodornienie
alkenów i alkinów
Sposoby aktywowania cząsteczki wodoru
[RhCl(PPh
3
)
3
] + H
2
RhCl(H)
2
(PPh
3
)
3
[IrCl(CO)(PPh
3
)
2
] + H
2
[IrCl(H)
2
(CO)(PPh
3
)
2
]
2[Co(CN)
5
]3-
+ H
2
2[Co(CN)
5
H]
3-
[RhCl
6
]
3-
+ H
2
[RhCl
5
H]
3-
+ H
+
+ Cl
-
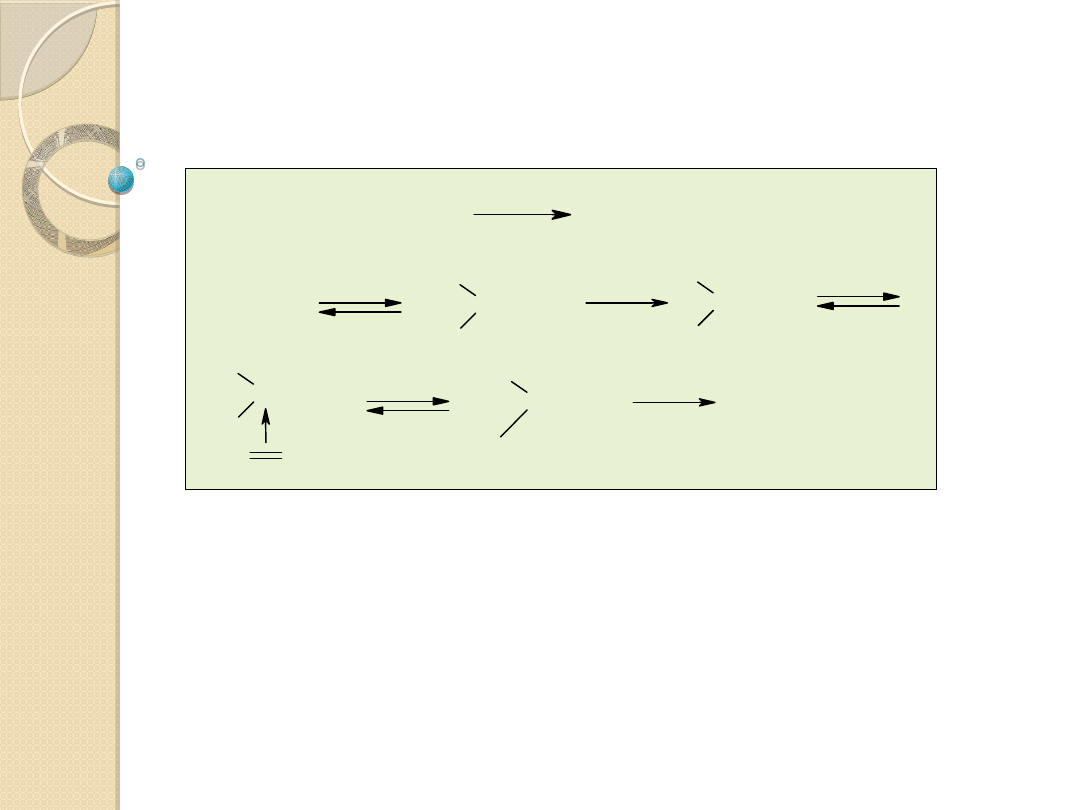
Rh L
3
Cl
H
Rh L
3
Cl
H
H
Rh L
2
Cl
H
H
Rh L
2
Cl
H
C
H
2
CH
2
H
Rh L
2
Cl
C
2
H
5
CH
3
CH
3
+ Rh L
2
Cl
H
2
-L
+C
2
H
4
I
III
III
I
Rh L
2
Cl + H
2
H
2
Rh L
2
Cl
I
III
Mechanizm uwodornienia olefin z udziałem kompleksu
Wilkinsona (Rh
I
(PPh
3
)
3
Cl)
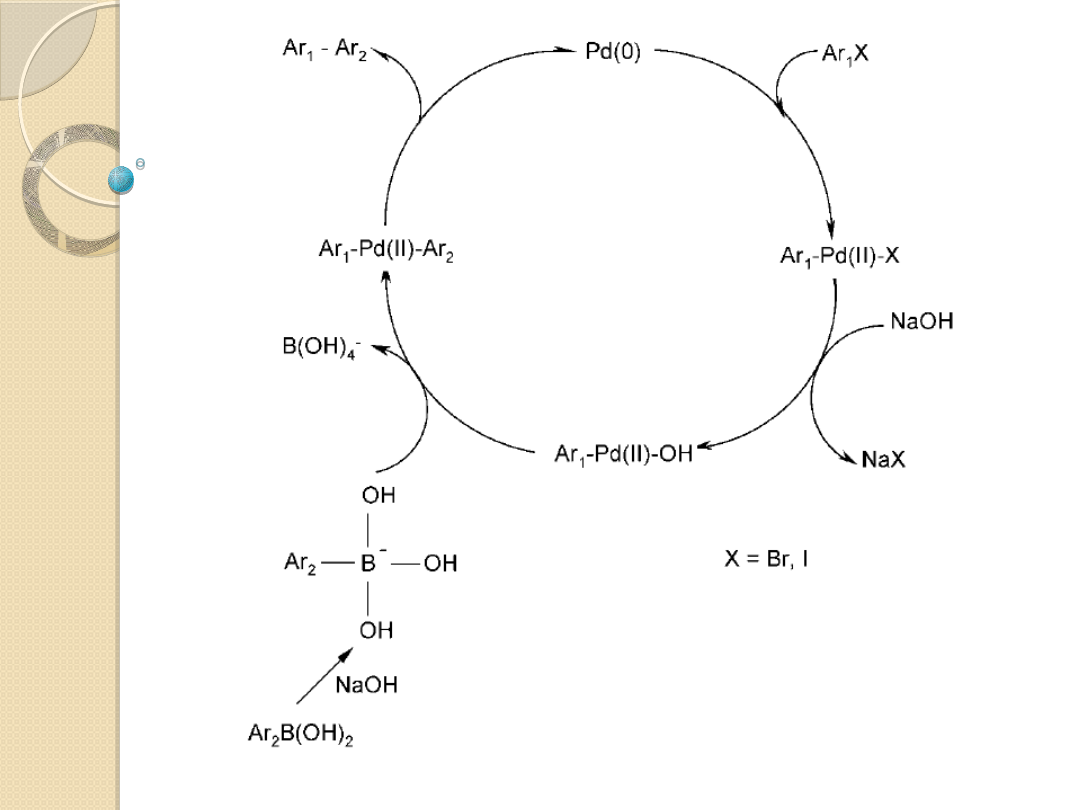
Reakcja Suzuki
Suzuki coupling
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
Wyszukiwarka
Podobne podstrony:
KATALIZA HOMOGENICZNA - REFERAT, KATALIZATORY- to substancje, które modyfikują kinetykę reakcji chem
kataliza wstep 2009
Zdefiniuj i porównaj katalizę homogeniczną i heterogeniczną
Adsorpcja Grzybek zaliczenie 2009 BoocaTM, sem 1, Kataliza (magdapliki), EGZ
kataliza heterogeniczna cz 3 2009
katalizatory heterogeniczne cz 1 2009
Wykład 6 2009 Użytkowanie obiektu
Przygotowanie PRODUKCJI 2009 w1
Wielkanoc 2009
przepisy zeglarz 2009
Kształtowanie świadomości fonologicznej prezentacja 2009
zapotrzebowanie ustroju na skladniki odzywcze 12 01 2009 kurs dla pielegniarek (2)
perswazja wykład11 2009 Propaganda
Wzorniki cz 3 typy serii 2008 2009
2009 2010 Autorytet
Cw 1 Zdrowie i choroba 2009
download Prawo PrawoAW Prawo A W sem I rok akadem 2008 2009 Prezentacja prawo europejskie, A W ppt
więcej podobnych podstron