© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
TERMODYNAMIKA PROCESOWA I
TECHNICZNA
Wykład VIII
Równania stanu typu van der Waalsa
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
2
Przypomnienie
Na poprzednim wykładzie omówiliśmy:
1. Równanie stanu gazu doskonałego.
2. Poprawione RSGD za pomocą współczynnika ściśliwości
z wykorzystaniem teorii stanów odpowiadających sobie.
3. Równania wirialne.
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
3
Wirialne równania stanu
1
2
3
4
5
6
1 2 3 4 5 6 7 8
p
r
T
r
Obszar zastosowań
obciętego
równania
wirialnego
?
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
4
Równania stanu typu
van der Waalsa
Przy modelowaniu obszaru oznaczonego znakiem zapytania, największy
sukces odniosły równania będące rozwinięciem idei zapoczątkowanej
przez holenderskiego fizykochemika van der Waalsa, który w roku 1878
w swojej pracy doktorskiej zaproponował pewne równanie uwzględniające
rzeczywiste własności cząsteczek gazu.
4. Równania stanu typu van der Waalsa.

© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
5
Równania stanu typu
van der Waalsa
Johannes Dideryk van der Waals 1837 – 1923
Laureat nagrody Nobla 1910
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
6
Równania stanu typu
van der Waalsa
Zarówno oryginalne równanie van der Waalsa jak i większość prostych
jego modyfikacji są równaniami algebraicznymi trzeciego stopnia względem
objętości molowej v i nazywane są one kubicznymi równaniami stanu
(cubic equations of state).
Niektóre jednak modyfikacje równania van der Waalsa są równaniami
algebraicznymi wyższych stopni, a do niektórych wprowadza się funkcje
niealgebraiczne (np. ekspotencjalne jak w przypadku równania BWR).
Główny sukces idei van der Waalsa polegał na tym, że równania tego typu
dobrze opisują nie tylko zależność między parametrami p – v – T w obszarze
gazu, ale przewidują istnienie punktu krytycznego oraz potrafią opisać
przemianę fazową para – ciecz !
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
7
Równania stanu typu
van der Waalsa
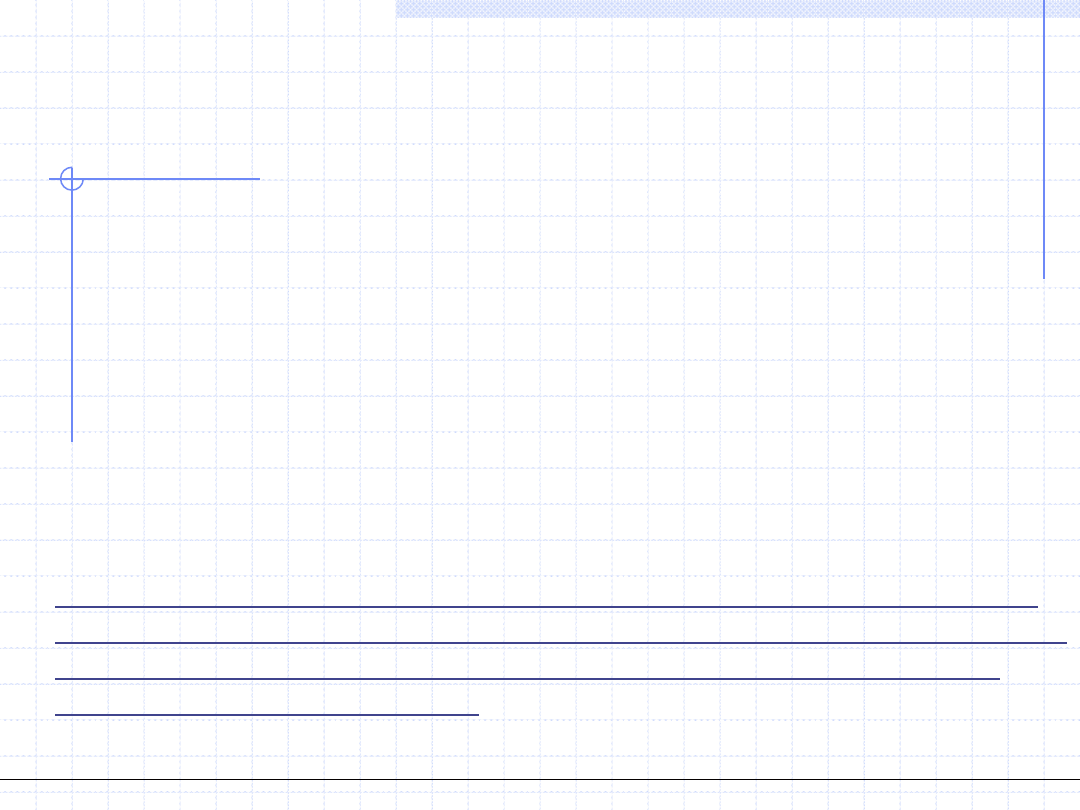
Równania stanu typu van der Waalsa mogą być formułowane i zapisywane
w różnych postaciach.
Najbardziej popularną postacią jest zależność ciśnienia od objętości molowej,
której zapis ma dwa człony: repulsywny (uwzględniający siły odpychania)
i atraktywny (uwzględniający siły przyciągania).
.
.
( , )
( , )
( , )
rep
att
p v T
p
v T
p
v T
Charakter poszczególnych sił określa znaki poszczególnych członów.
Człon repulsywny związany z odpychaniem jest dodatni (odpychanie się
cząsteczek zwiększa ciśnienie), natomiast człon atraktywny jest ujemny
(przyciąganie się cząsteczek zmniejsza ciśnienie).
Ściśle rzecz biorąc w członie repulsywnym zawarte są dwa efekty. Efekt
główny to ciśnienie ośrodka związane z termicznym chaotycznym
ruchem cząsteczek. Efekt dodatkowy natomiast uwzględnia odpychanie.
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
8
Równania stanu typu
van der Waalsa
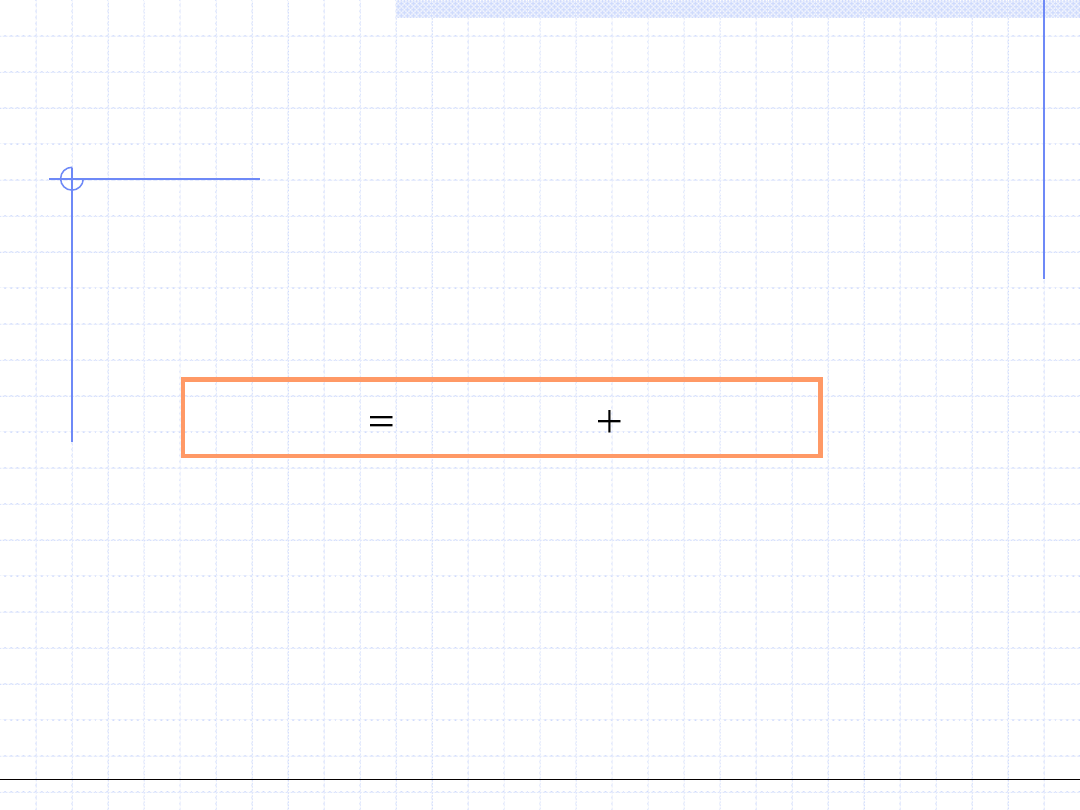
Fakt ten uwzględnia inna postać równań stanu typu van der Waalsa,
w której uzależniony jest współczynnik ściśliwości od temperatury i objętości
właściwej:
1
.
.
( , )
( , )
( , )
rep
att
z v T
z
v T
z
v T
gdzie:
.
( , )
rep
z
v T
poprawka uwzględniająca odpychanie się cząsteczek
.
( , )
att
z
v T
poprawka uwzględniająca przyciąganie się cząsteczek
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
9
Równanie van der Waalsa
4.1. Równanie van der Waalsa.
Równania kubiczne wywodzą się od bardzo słynnego równania zaproponowanego
w roku 1878 przez holenderskiego fizykochemika van der Waalsa. Równanie to jest
powszechnie znane jako równanie van der Waalsa (RS vdW). Równanie jest bardzo
proste a jednocześnie posiada ono dosyć mocne podstawy teoretyczne. Budowę tego
równania można uzasadnić wychodząc od równania opisującego gazy doskonałe,
które jak pamiętamy, spełniają dwa warunki:
-objętość własna cząsteczek jest równa 0,
-cząsteczki nie oddziaływują ze sobą.
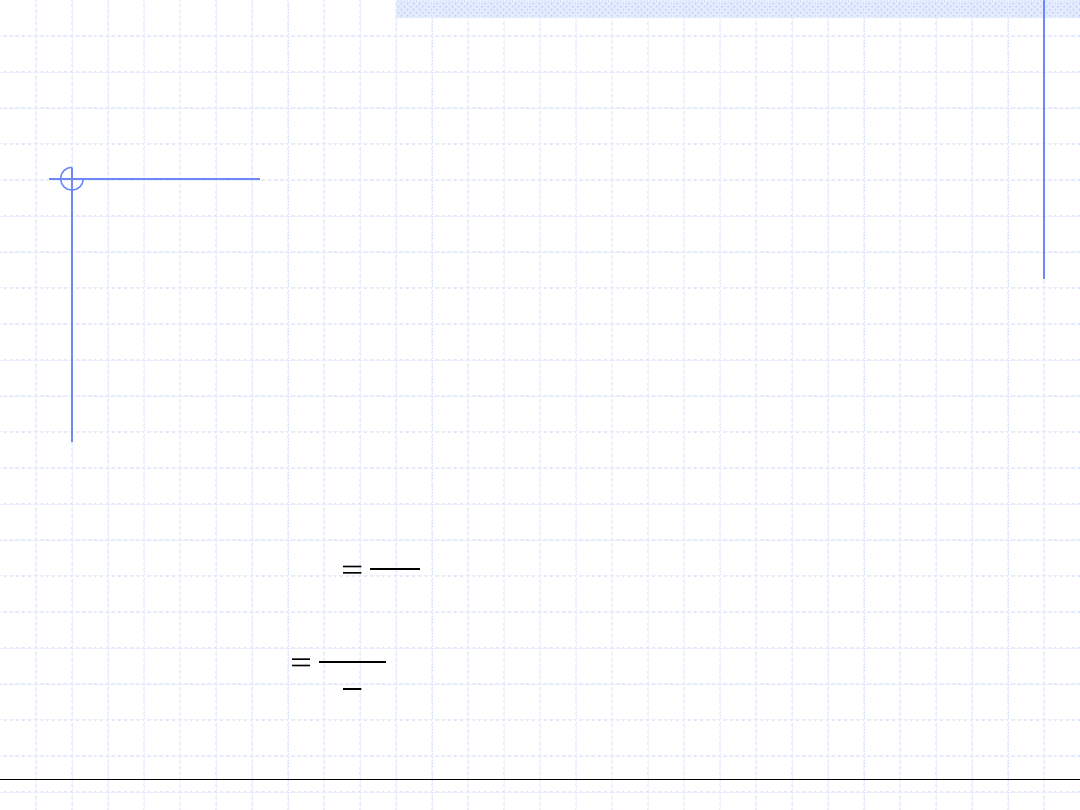
Gaz, którego cząsteczki spełniają
powyższe warunki będzie spełniał
również RSGD tzn.:
RT
pv
RT
p
v
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
10
Równanie van der Waalsa
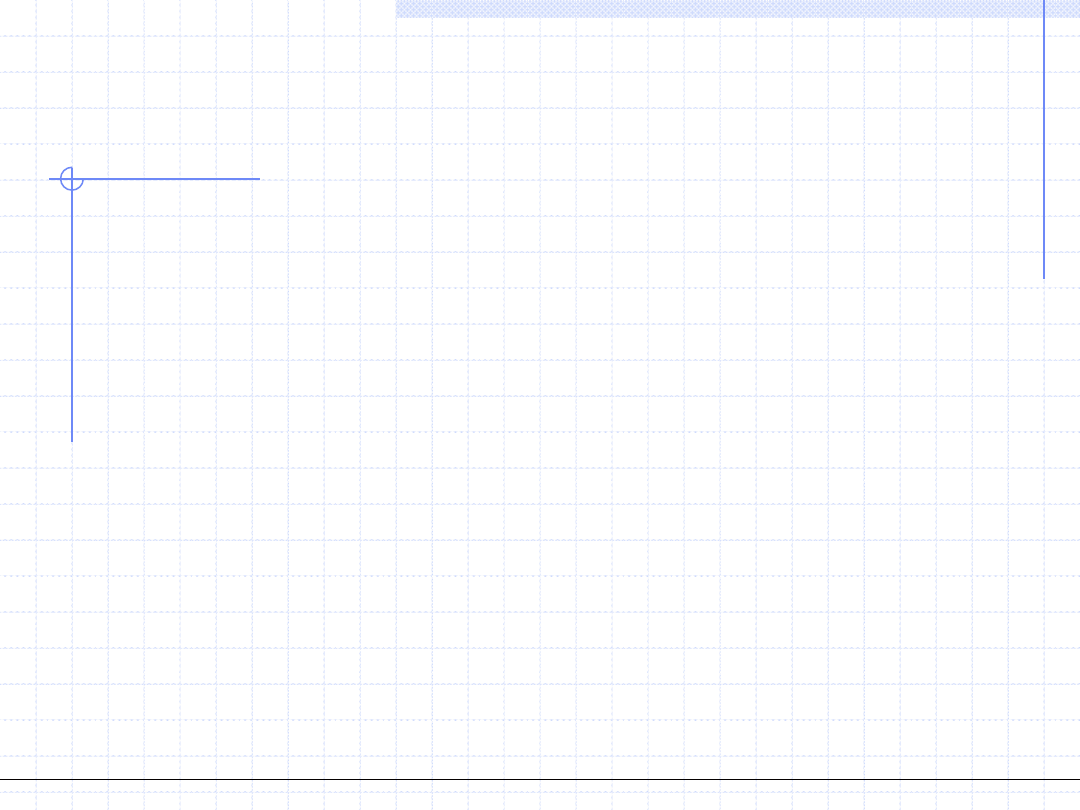
Cząsteczki substancji rzeczywistych oczywiście nie spełniają tych założeń.
Przyjęcie, że cząsteczki posiadają pewną własną objętość prowadzi do wniosku,
że objętość dostępna dla ruchu cząstek jest mniejsza o pewną wartość związaną
z ich własną objętością.
Zatem we wzorze opisującym ciśnienie należy zamiast v użyć (v-b) gdzie b jest
pewną stałą opisującą konieczne zmniejszenie objętości.
W rezultacie tej korekty otrzymamy:
GD
RS
v
RT
p
GD
RS
poprawione
b
v
RT
p
)
1
(
)
1
(
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
11
Równanie van der Waalsa
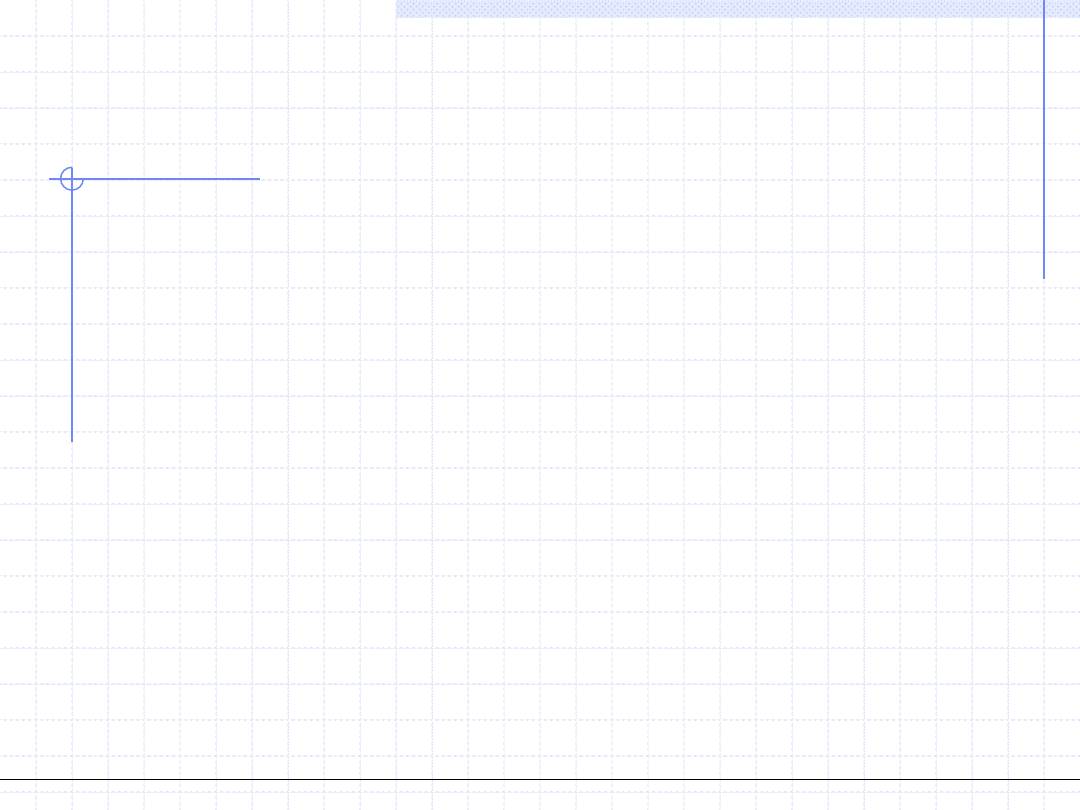
Drugim krokiem w konstrukcji równania vdW jest uwzględnienie faktu,
że rzeczywiste cząstki oddziaływują ze sobą a siły tego oddziaływania noszą
nazwę sił van der Waalsa.
W ogólnym przypadku oddziaływanie między cząsteczkami może mieć charakter
odpychający (repulsywny) wtedy gdy cząstki znajdują się bardzo blisko siebie
lub przyciągający (atraktywny) gdy cząstki są oddalone od siebie.
W oryginalnej teorii van der Waalsa uwzględniane są tylko oddziaływania
przyciągające (właśnie siły van der Waalsa). Skutkiem działania tych sił jest pewne
zmniejszenie ogólnego ciśnienia gazu.
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
12
Równanie van der Waalsa
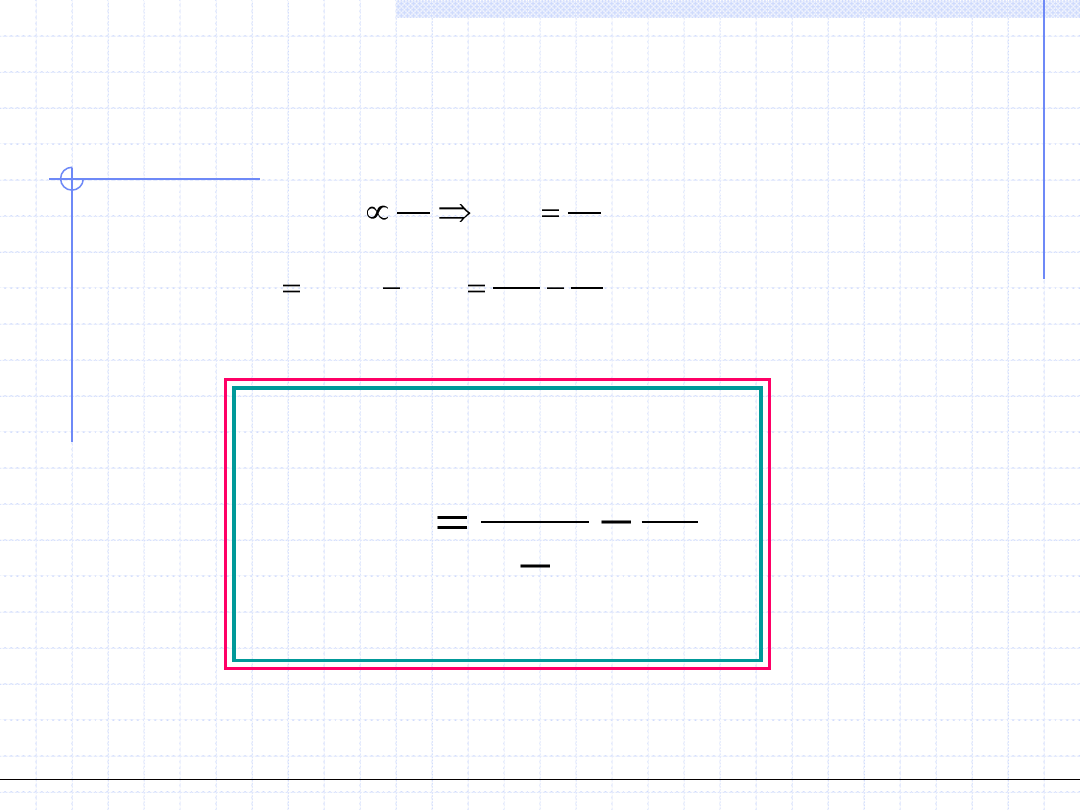
Zatem we wzorze opisującym ciśnienie ogólne należy wprowadzić pewną
poprawkę uwzględniającą to zmniejszenie:
zamiast p=p
(GD)
należy napisać p=p
(GD)
-p
(A)
.
Przy czym p
(A)
jest poprawką wynikającą z faktu działania sił przyciągających.
Uwzględniając fakt że siły van der Waalsa mają charakter kulombowski
(są pochodzenia elektromagnetycznego) można wywnioskować,
ze powinny być one odwrotnie proporcjonalne do kwadratu objętości.
Można zatem napisać:
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
13
Równanie van der Waalsa cd.
)
2
(
1
2
)
(
2
)
(
poprawka
v
a
p
v
F
A
A
Uwzględniając obydwie poprawki otrzymujemy równanie van der Waalsa:
2
)
,
(
v
a
b
v
RT
T
v
p
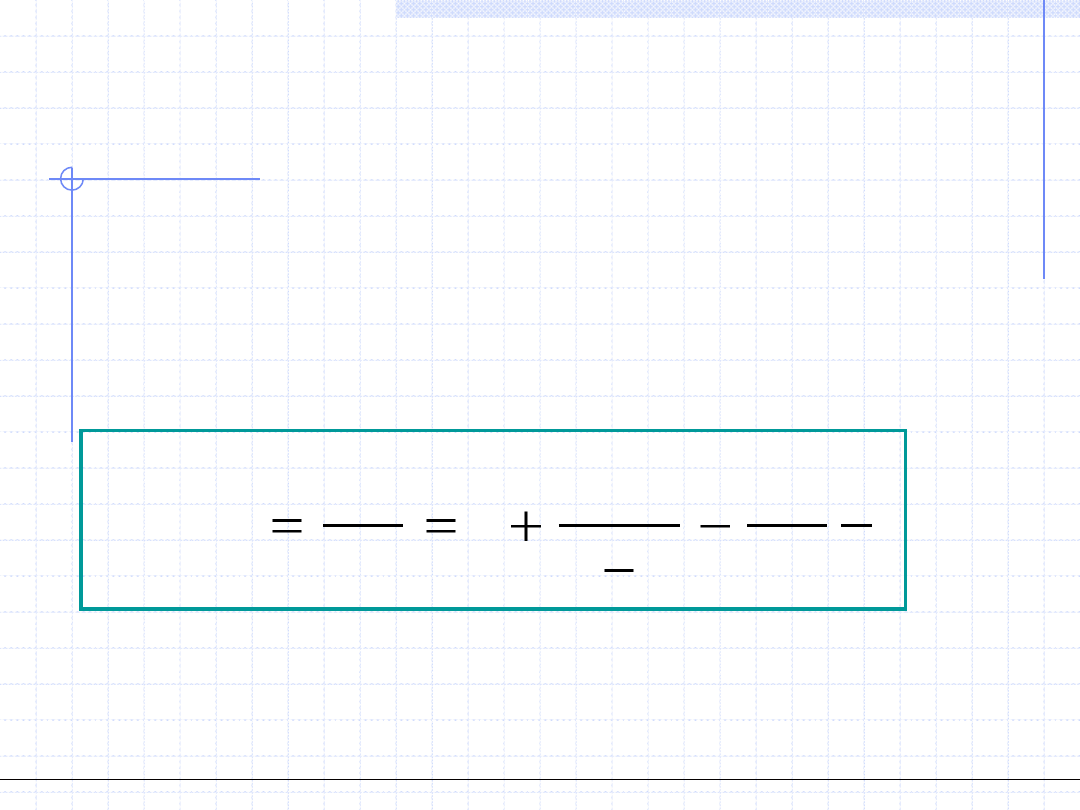
Wielkości oznaczone literami a i b są to tzw. stałe van der Waalsa. Zależą one
od substancji i w oryginalnym ujęciu powinny być wyznaczane w oparciu
o eksperymentalne dane dotyczące zależności p-v-T.
)
2
(
2
)
(
)
(
)
2
(
poprawką
z
RS
v
a
v
RT
p
p
p
A
GD
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
1
4
Równanie van der Waalsa cd.
Mnożąc obydwie strony równania przez v/(RT) możemy otrzymać
alternatywną postać równania van der Waalsa, w której mamy
opisany współczynnik ściśliwości jako funkcję objętości właściwej
i temperatury:
1
1
( , )
pv
b
a
z v T
RT
v
b
RT v
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
15
Równanie van der Waalsa cd.
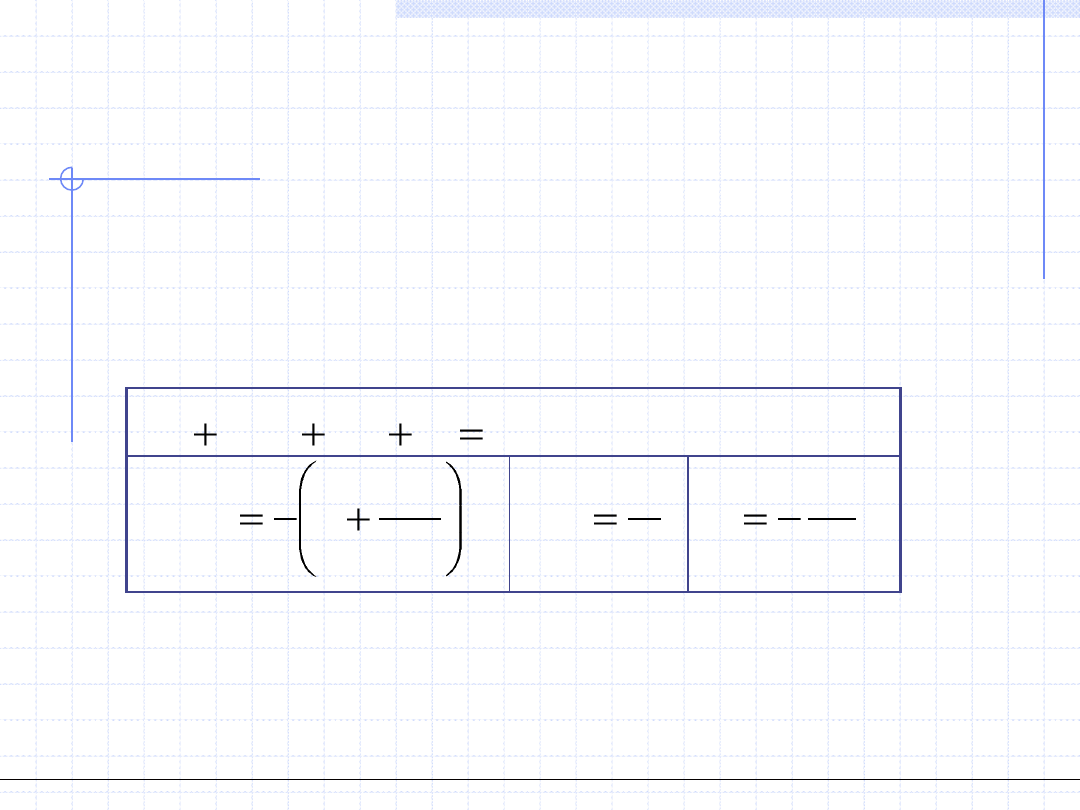
Równanie van der Waalsa można w prosty sposób przekształcić
do postaci wielomianu ze względu na objętość molową v:
p
ab
C
p
a
B
p
RT
b
A
gdzie
C
Bv
Av
v
0
2
3
Po ustaleniu temperatury i ciśnienia, w celu obliczenia objętości v powstaje
konieczność rozwiązania równania algebraicznego 3 – go stopnia.
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
16
Równanie van der Waalsa cd.
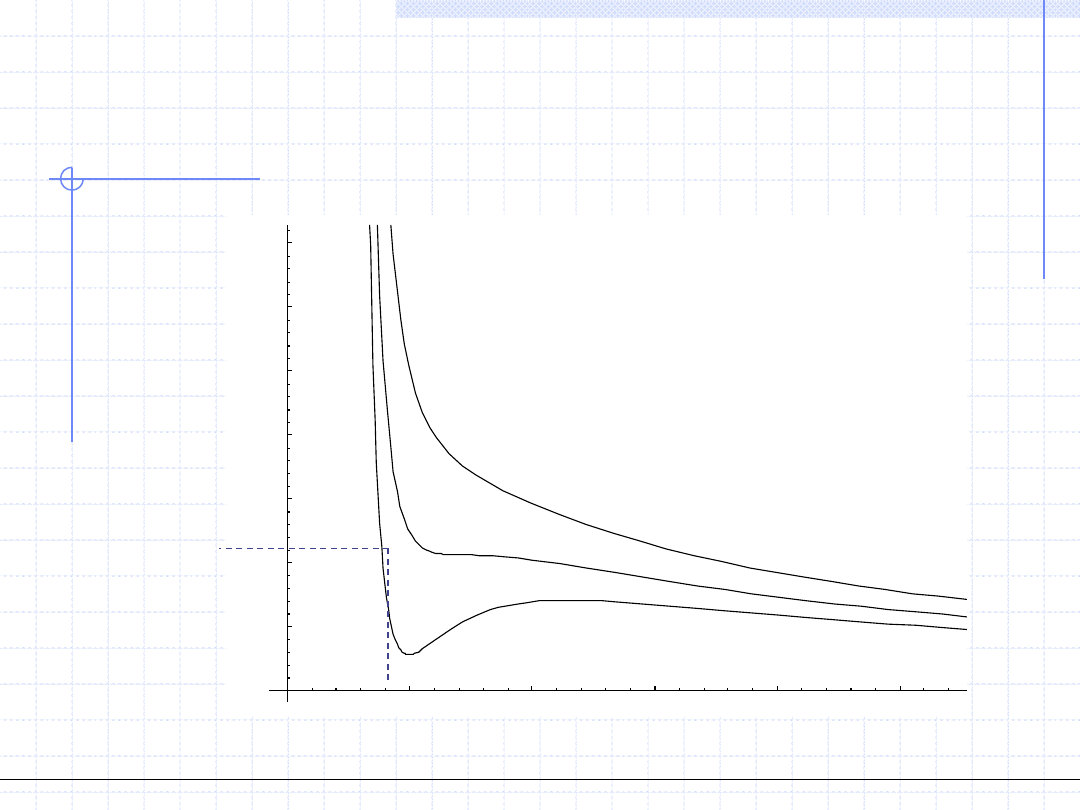
Na podstawie rozważań czysto matematycznych można stwierdzić, że
równanie to przy ustalonej temperaturze i ciśnieniu zawsze musi mieć
przynajmniej jeden pierwiastek rzeczywisty.
Okazuje się jednak że poniżej pewnej temperatury T
x
RS vdW
ma zawsze 3 pierwiastki rzeczywiste ! Fakt ten kłóci się z zachowaniem
substancji rzeczywistych. W określonych warunkach ciśnienia i temperatury
każda substancja posiada określoną gęstość a zatem również określoną
wartość v.
Dla temperatur większych od T
x
równanie zachowuje się
„porządnie” tzn. posiada jeden pierwiastek rzeczywisty. Typowe wykresy
izoterm w układzie p – v sporządzone na podstawie RS vdW (dla CO
2
)
mają postać:
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
17
Równanie van der Waalsa cd.
0.1
0.2
0.3
0.4
0.5
0.6
2.5
10
6
5
10
6
7.5
10
6
1
10
7
1.25
10
7
1.5
10
7
1.75
10
7
p [Pa]
v [m
3
/kmol]
T>T
x
T=T
x
T<T
x
CO
2
a=0.319113
.
10
6
[m
6
Pa/kmol
2
]
b=0.047595 [m
3
/kmol]
p=p
x
v
x
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
18
Równanie van der Waalsa cd.
Dla temperatury T=T
x
izoterma vdW zachowuje się bardzo podobnie
jak izoterma substancji rzeczywistej. W pewnym punkcie (p
x
,v
x
) posiada
ona punkt przegięcia identycznie jak izoterma rzeczywista. Nasuwa się
oczywista sugestia aby punkt ten (T
x
,p
x
,v
x
) zinterpretować jako punkt
krytyczny.
Przyjęcie, że jest to punkt krytyczny posiada doniosłe
konsekwencje. Pozwala ono na wyznaczenie stałych RS vdW
na podstawie doświadczalnych wartości parametrów krytycznych !
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
Równanie van der Waalsa cd.
Przyjęcie, że izoterma danego RS posiadająca punkt przegięcia jest izotermą
krytyczną oraz interpretacja tego punktu przegięcia jako punktu krytycznego
substancji będziemy nazywać uzgodnieniem równania stanu z punktem
krytycznym.
Na podstawie tego uzgodnienia można opracować procedurę, która pozwala
wyznaczać parametry równania stanu na podstawie wartości parametrów
krytycznych.
Poniżej przedstawię taką procedurę na przykładzie RS vdW.
Załóżmy że znamy wartości parametrów krytycznych T
kr
i p
kr
danej substancji.
Procedura uzgodnienia prowadzi do założenia T
x
=T
kr
, p
x
=p
kr
i v
x
=v
kr
.
Punkt przegięcia funkcji p(v,T) musi spełniać proste warunki różniczkowe:
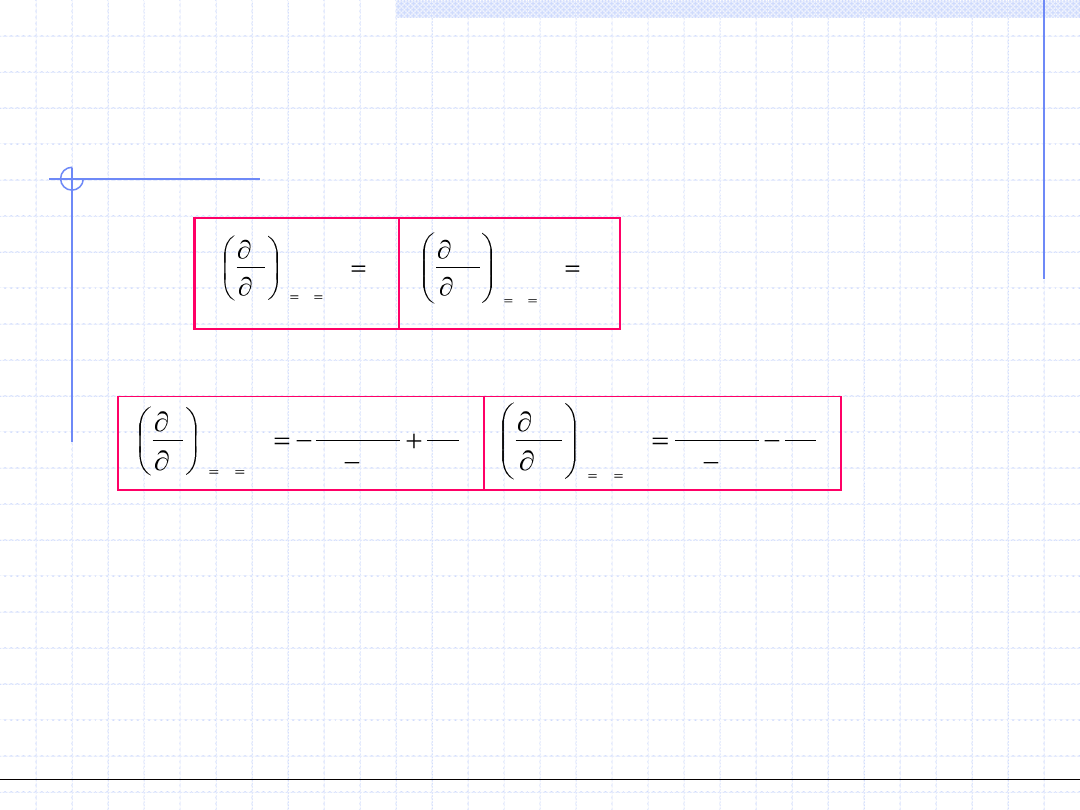
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
Równanie van der Waalsa cd.
0
0
2
2
kr
x
kr
x
T
T
T
T
T
T
v
p
v
p
Odpowiednie pochodne dla RSvdW będą miały postać:
4
3
2
2
3
2
6
)
(
2
2
)
(
v
a
b
v
RT
v
p
v
a
b
v
RT
v
p
kr
T
T
T
kr
T
T
T
kr
x
kr
x
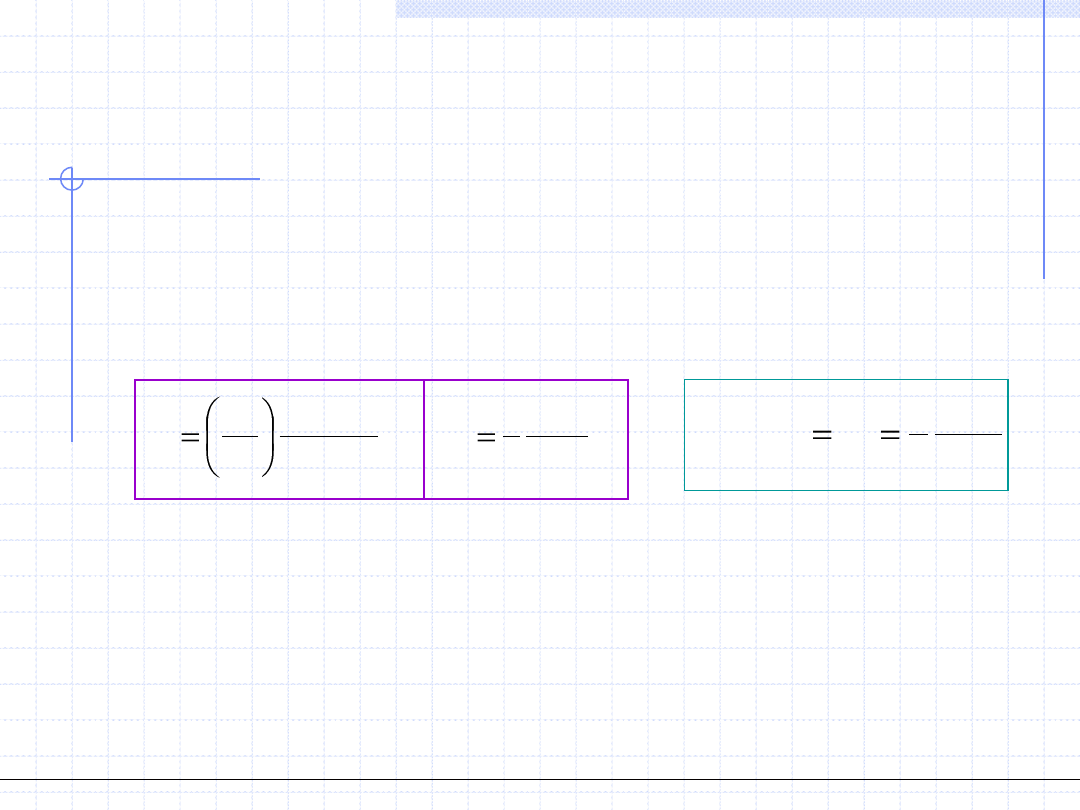
Przyjęcie warunków na punkt przegięcia daje dwa równania (1) i (2).
Trzecie równanie (3) otrzymujemy bezpośrednio z RS vdW.
W rezultacie otrzymujemy układ 3 równań:
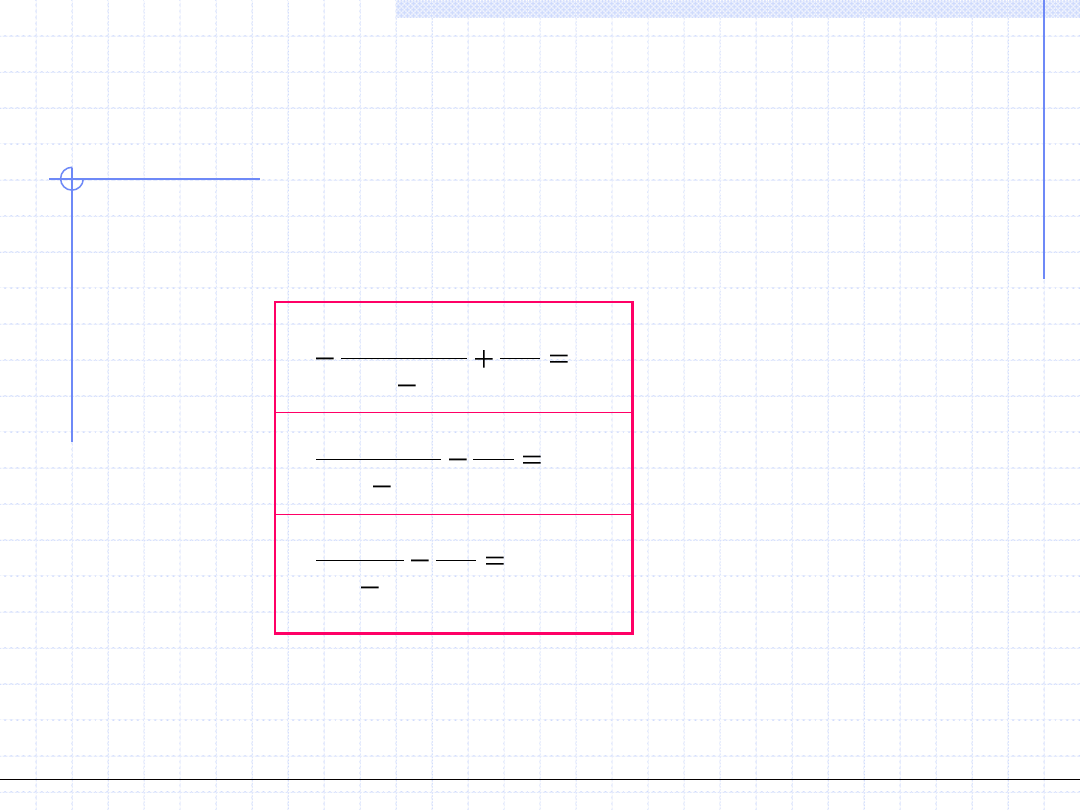
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
21
Równanie van der Waalsa cd.
kr
kr
kr
kr
kr
kr
kr
kr
kr
kr
p
v
a
b
v
RT
v
a
b
v
RT
v
a
b
v
RT
2
4
3
3
2
0
6
)
(
2
0
2
)
(
Równanie (1)
Równanie (2)
Równanie (3)
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
22
Równanie van der Waalsa cd.
kr
kr
kr
kr
p
RT
b
p
RT
a
8
1
)
(
64
27
2
Zakładając, że znamy T
kr
i p
kr
w powyższym układzie mamy 3 niewiadome:
a, b i v
kr
.
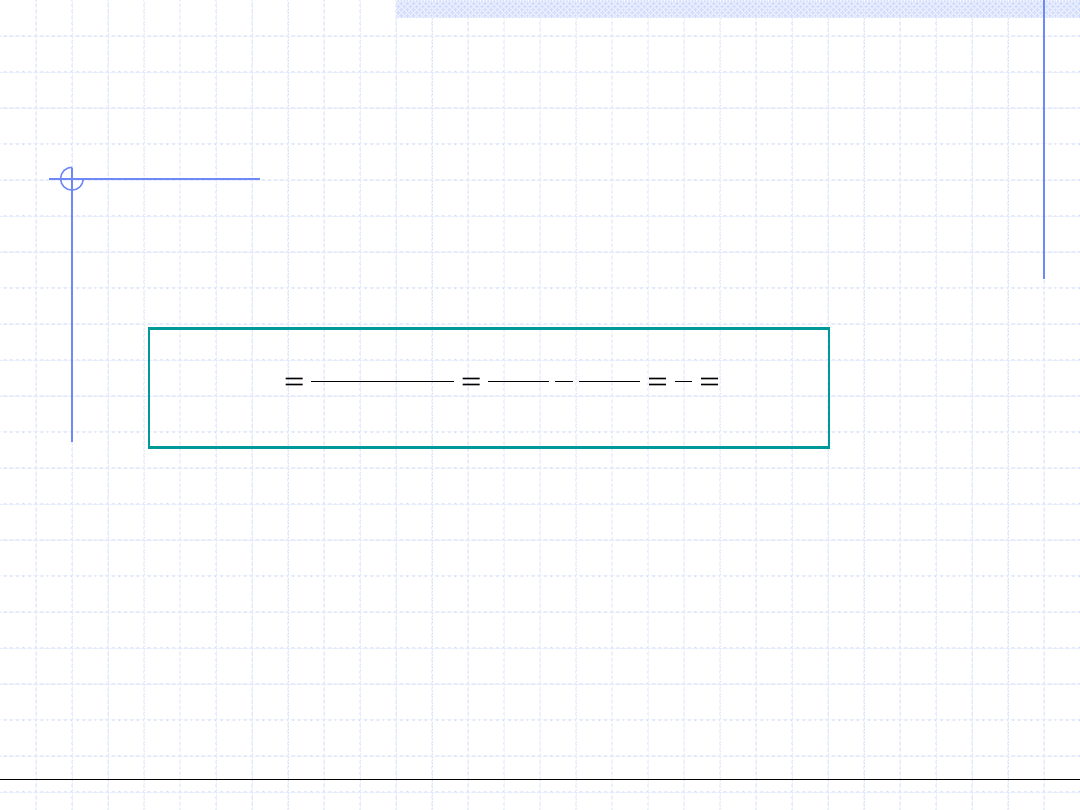
Rozwiązaniem układu są wzory określające stałe RS vdW oraz wzór określający v
kr
:
kr
kr
vdW
kr
p
RT
b
v
8
3
3
)
(
Powyższe uzgodnienie pozwala na opis rzeczywistej substancji przez
RS vdW przy zachowaniu wartości T
kr
i p
kr
. Należy zwrócić uwagę że
wartości stałych a i b otrzymane z powyższych wzorów będą na
ogół różne od stałych otrzymanych na podstawie danych p – v – T.
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
23
Równanie van der Waalsa cd.
Wartość v
kr
otrzymana z powyższego wzoru będzie na ogół różna od wartości
eksperymentalnej. W celu oszacowania tego odchylenia często korzysta się
z wartości z
kr
będącej cechą charakterystyczną danej substancji. Obliczmy tę
wartość dla RS vdW.
375
.
0
8
3
8
3
)
(
)
(
kr
kr
kr
kr
kr
vdW
kr
kr
vdW
kr
p
RT
RT
p
RT
v
p
z
Eksperymentalne wartości z
kr
dla substancji niepolarnych są zbliżone do 0.29,
natomiast dla substancji polarnych są jeszcze niższe. Widzimy więc, ze RS vdW daje
dosyć duże błędy przy opisie objętości krytycznych. Z tego też względu RS vdW przez
dłuższy czas było „w niełasce” tzn. nie było brane pod uwagę. Jednakże równanie
van der Waalsa posiada jeszcze inne zalety, które pozwoliły mu (po pewnych korektach)
osiągnąć ogromny sukces w drugiej połowie XX wieku. W celu rozpatrzenia zalet RS vdW
wróćmy do przebiegu izoterm generowanego przez to równanie dla CO
2
, ale przy
stałych a i b określonych przez parametry krytyczne.
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
24
Równanie van der Waalsa cd.
T=T
kr
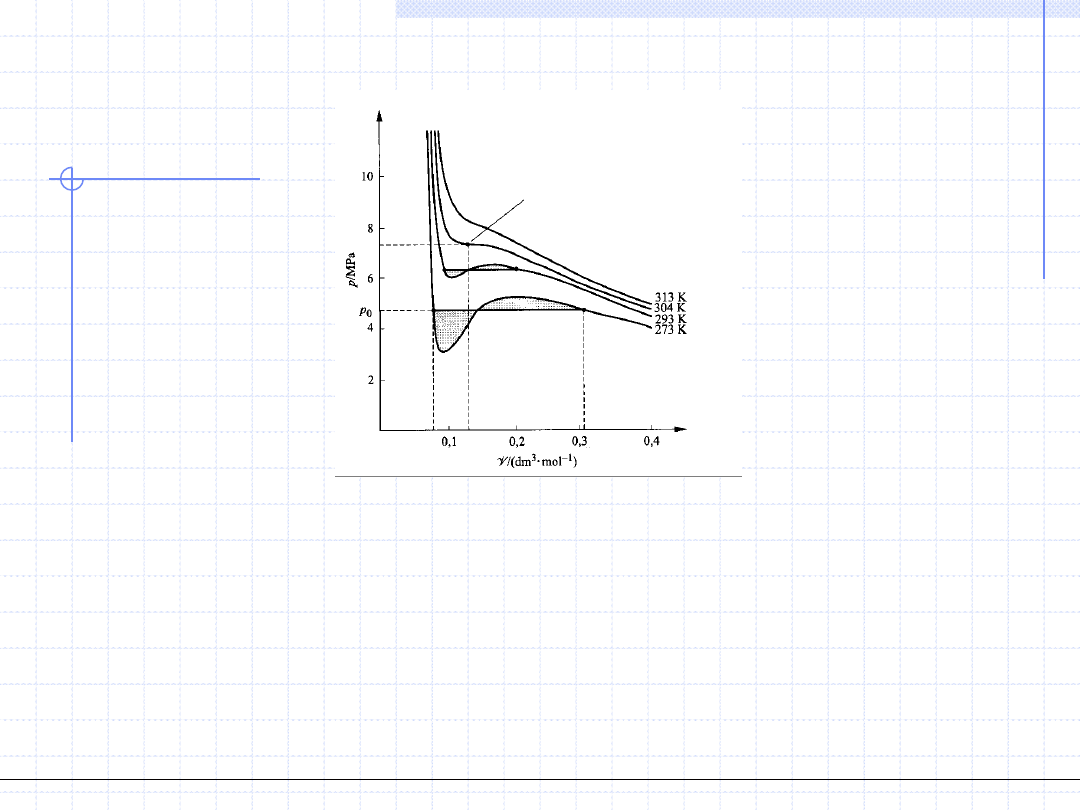
Przeanalizujmy przebieg izoterm dla temperatury niższej od krytycznej. Widzimy, ze
na wykresie występuje minimum i maksimum ciśnienia tzn. że istnieje zakres objętości,
w którym ciśnienie rośnie wraz z objętością. Oczywiście z punktu widzenia fizyki jest to
niemożliwe a więc przebieg izotermy w tych zakresach jest nierealny. Można jednak
założyć, że równanie w tych zakresach opisuje stany jednofazowe, cechujące się wyższą
entalpią swobodną niż stany z dwiema fazami – ciekłą i parową. Ponieważ wszystkie
układy termodynamiczne dążą do minimalizacji entalpii swobodnej substancja w takim
zakresie rozdzieli się na dwie fazy ciekłą i parową. Objętości molowe tych faz będą takie
aby spełniony był warunek równowagi układu dwufazowego tzn.:
Punkt krytyczny
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
25
Równanie van der Waalsa cd.
T=T
kr
"
'
0
g
g
dg
v’
v”
''
0
'
0
"
'
0
)
'
"
(
0
p
p
g
g
g
g
dg
vdp
vdp
dg
dT
0
)
(
)
'
"
(
)
(
|
)
(
"
'
0
"
'
"
'
"
0
'
0
v
v
v
v
p
p
dv
v
p
v
v
p
dv
v
p
vp
dp
p
v
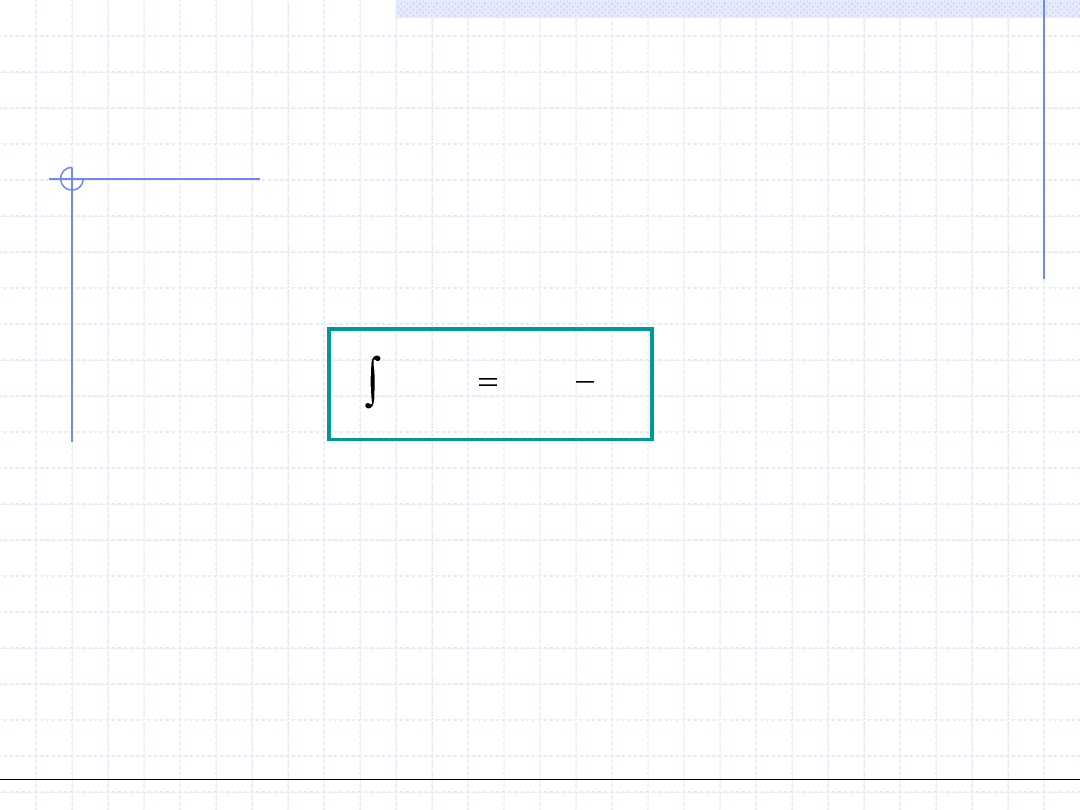
sdT
vdp
dg
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
26
Równanie van der Waalsa cd.
Ostatnia równość dostarcza warunek na to aby entalpie swobodne cieczy i pary
były sobie równe:
"
'
0
)
'
"
(
)
(
v
v
v
v
p
dv
v
p
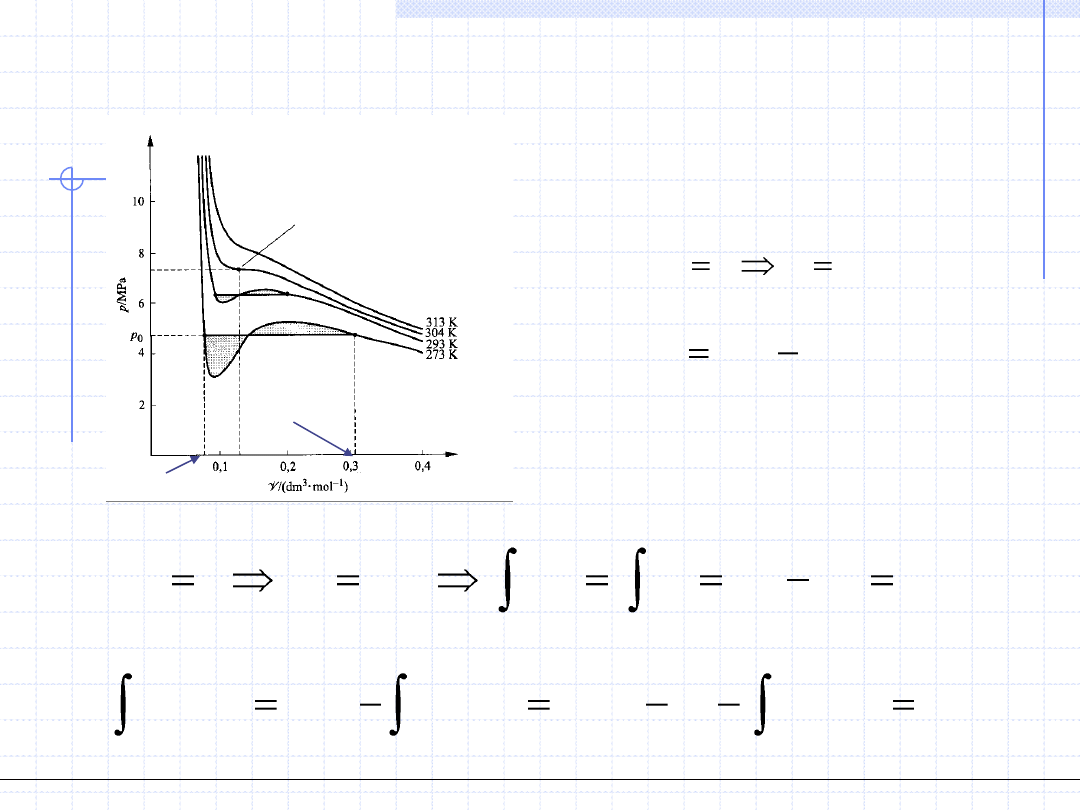
Powyższa zależność jest to analityczny zapis słynnej „reguły Maxwella”,
która mówi że przemiana fazowa ciecz – para zachodzi przy takim ciśnieniu p
0
dla którego pola zacienionych dwu figur na wykresie są sobie równe.

© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
27
Równanie van der Waalsa cd.
James Maxwell 1831 – 1879
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
Równanie van der Waalsa cd.
Uwzględnienie reguły Maxwella oraz przyjęcie, że skrajne pierwiastki równania p(v,T)=p
0
odpowiadają objętościom molowym cieczy i pary nasyconej, pozwala na opis przemiany
fazowej ciecz – para za pomocą równania van der Waalsa. Linia do punktu nasycenia cieczy
opisuje izotermę w zakresie cieczy.
Pomiędzy skrajnymi pierwiastkami (punktami nasycenia) równanie opisuje stan niestabilny,
który w rzeczywistości ulega samorzutnemu rozdziałowi na dwie fazy: parową i ciekłą.
Zamiast nierealnego przebiegu „sinusoidalnego” w rzeczywistości mamy linię prostą
odpowiadającą przemianie fazowej. Od punktu pary nasyconej (trzeci skrajny pierwiastek)
równanie z powrotem opisuje realny stan substancji a izoterma jest podobna do izotermy
gazu doskonałego.
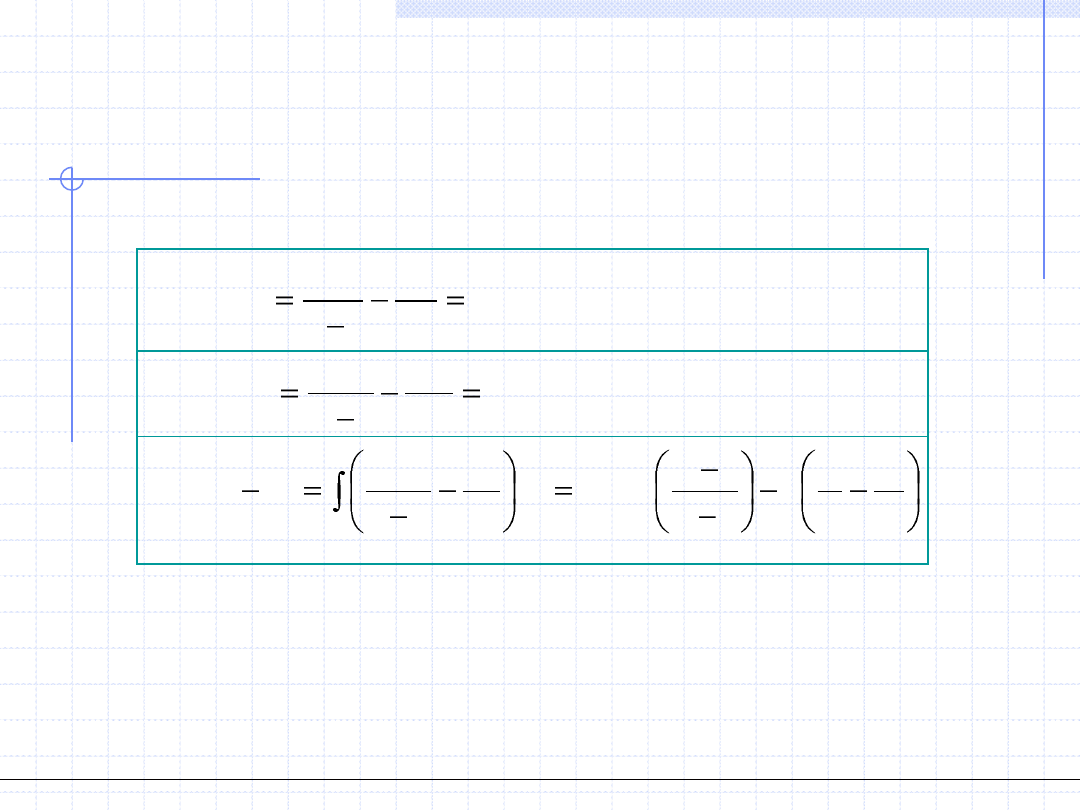
Reguła Maxwella łącznie z równaniem stanu pozwala na analityczne wyznaczenie ciśnienia
nasycenia oraz położenia obydwu punktów nasycenia dla danej temperatury
(niższej od krytycznej). Należy w tym celu rozwiązać układ 3 równań z trzema
niewiadomymi (p
0
,v’,v”):
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
29
Równanie van der Waalsa cd.
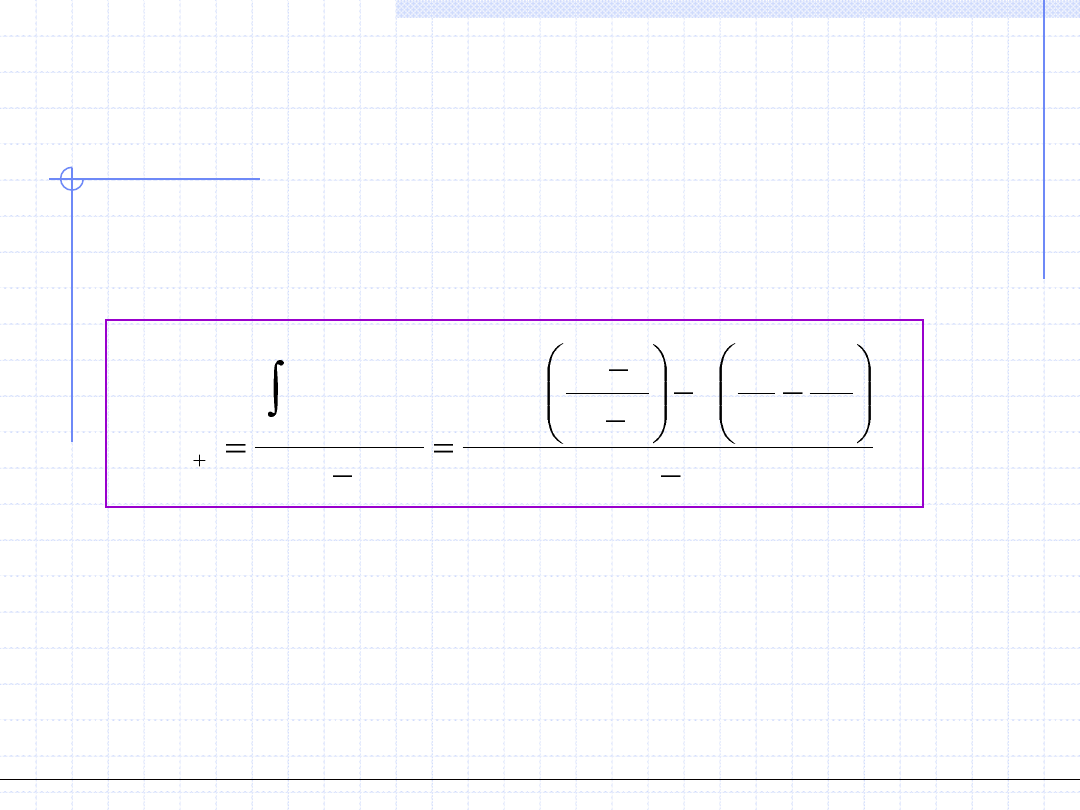
"
1
'
1
'
"
ln
)
'
"
(
"
"
)
,
"
(
'
'
)
,
'
(
"
'
2
0
0
2
0
2
v
v
a
b
v
b
v
RT
dv
v
a
b
v
RT
v
v
p
p
v
a
b
v
RT
T
v
p
p
v
a
b
v
RT
T
v
p
v
v
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
Należy zwrócić uwagę, że w przypadku równania van der Waalsa procedura powyższa
nie daje dokładnych wyników, szczególnie dla substancji polarnych. Natomiast idea
zastosowania reguły Maxwella oraz interpretacji pierwiastków równania jako punktów
nasycenia cieczy i pary zachowuje swoją ważność dla wszystkich równań typu van der Waalsa.
30
Równanie van der Waalsa cd.
Procedura rozwiązywania tego układu równań jest osobnym zagadnieniem numerycznym.
W szczególności można zastosować metodę kolejnych przybliżeń polegającą na:
-założeniu pewnej startowej wartości p
0
,
- rozwiązaniu równania p(v,T)=p
0
(znalezieniu trzech pierwiastków rzeczywistych v
1
,v
2
i v
3
),
- przyjęciu jako v’ pierwiastka najmniejszego a jako v” pierwiastka największego,
-zastosowaniu reguły Maxwella jako wzoru iteracyjnego w celu obliczenia kolejnej wartości p
0
:
i
i
i
i
i
i
i
i
v
v
i
v
v
v
v
a
b
v
b
v
RT
v
v
dv
T
v
p
p
i
i
'
"
"
1
'
1
'
"
ln
'
"
)
,
(
"
'
1
,
0
Procedurę iteracyjną przerywa się po osiągnięciu żądanej dokładności obliczeń. Pierwiastki
równania 3 – go stopnia można liczyć analitycznie lub numerycznie.
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
31
Inne kubiczne równania stanu wywodzące
się od równania van der Waalsa
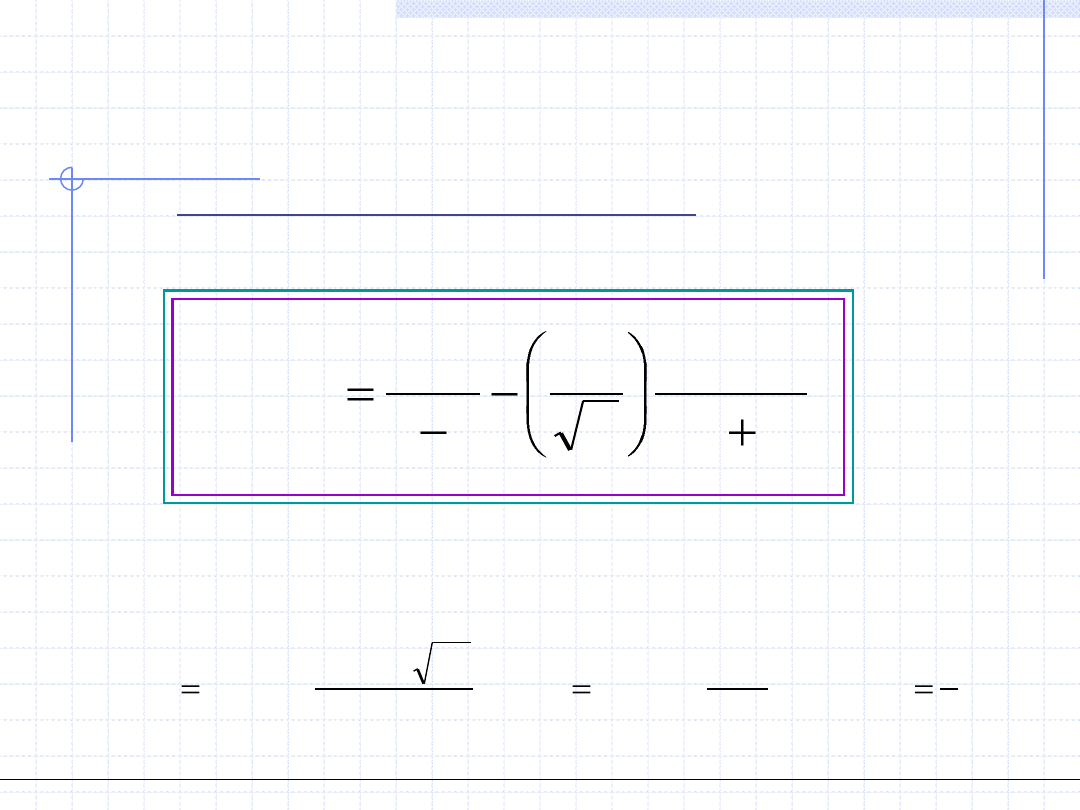
4.2. Równanie Redlicha – Kwonga (RS RK) 1949.
)
(
1
)
,
(
0
b
v
v
T
a
b
v
RT
T
v
p
3
1
)
(
08664
.
0
)
(
42748
.
0
2
0
RK
kr
kr
kr
kr
kr
kr
z
p
RT
b
p
T
RT
a
Uzgodnienie parametrów równania Redlicha – Kwonga z punktem krytycznym
prowadzi do wzorów:
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
32
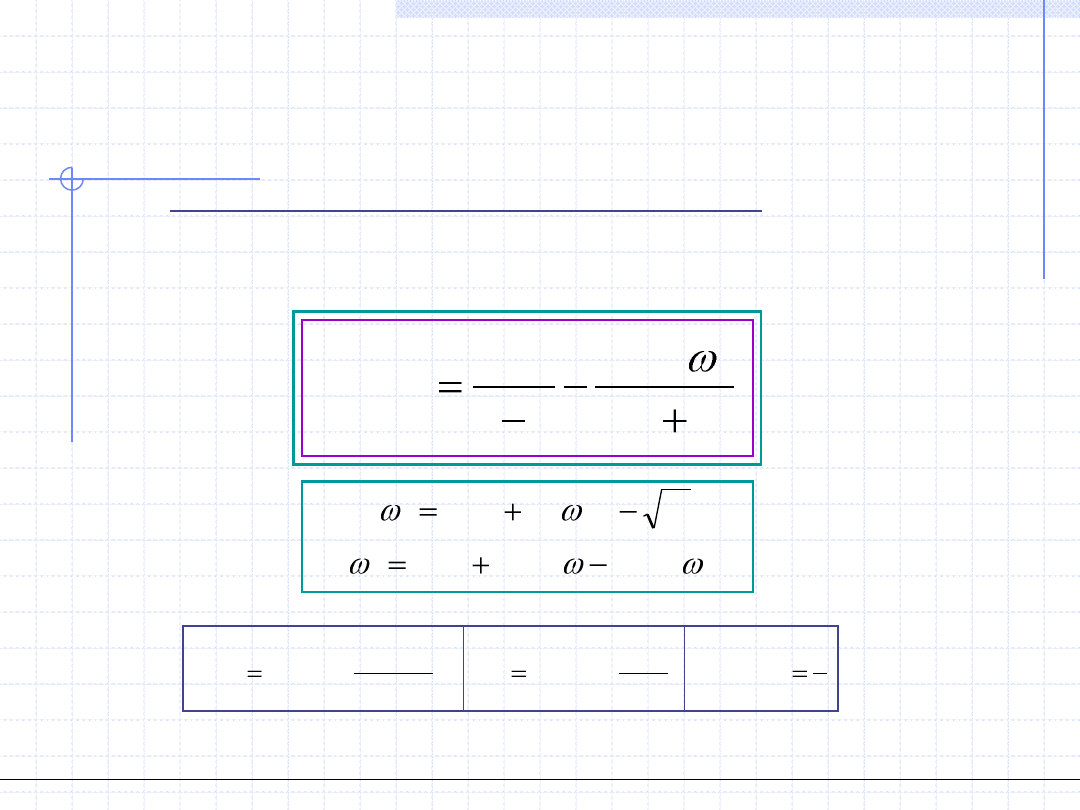
Równanie Soave – Redlicha - Kwonga
)
(
)
,
(
)
,
(
b
v
v
T
a
b
v
RT
T
v
p
r
2
2
176
.
0
574
.
1
48
.
0
)
(
)]
1
)(
(
1
[
)
,
(
m
T
m
a
T
a
r
kr
r
4.3. Równanie Soave - Redlicha – Kwonga (SRK) 1972.
gdzie
3
1
)
(
08664
.
0
)
(
42748
.
0
2
SRK
kr
kr
kr
kr
kr
kr
z
p
RT
b
p
RT
a
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
33
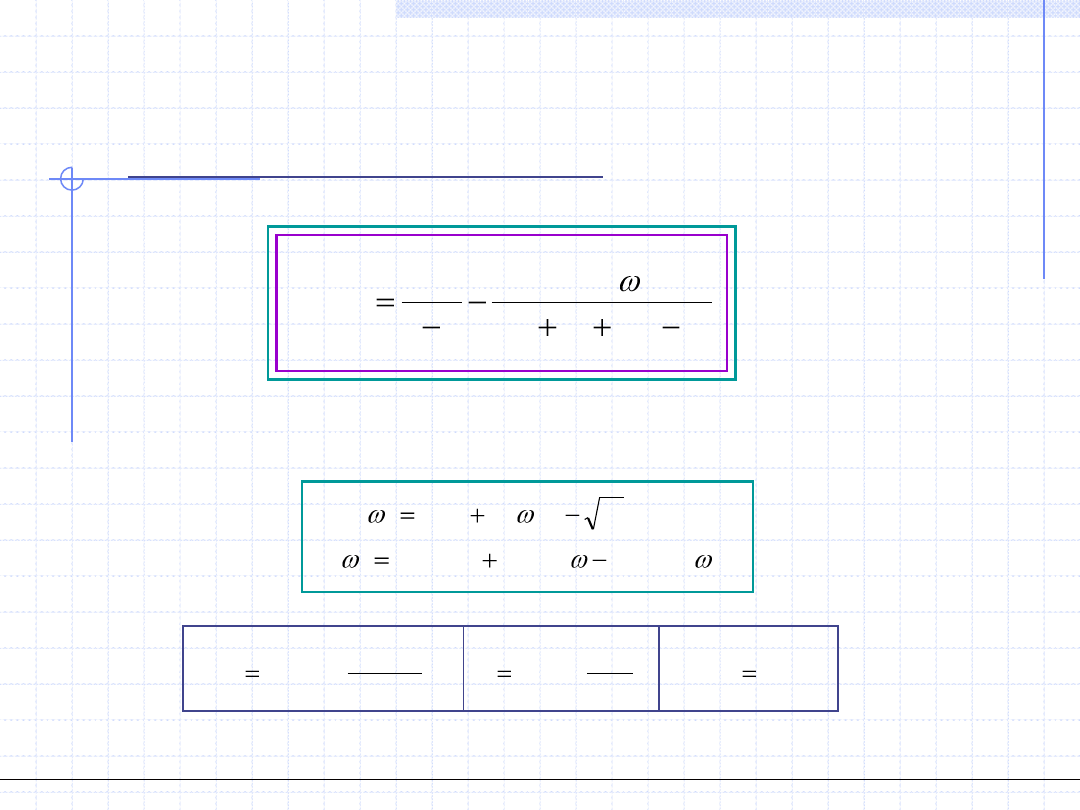
Równanie Penga - Robinsona
)
(
)
(
)
,
(
)
,
(
b
v
b
b
v
v
T
a
b
v
RT
T
v
p
r
2
2
26992
.
0
5422
.
1
37464
.
0
)
(
)]
1
)(
(
1
[
)
,
(
m
T
m
a
T
a
r
kr
r
4.4. Równanie Penga - Robinsona (PR) 1976.
gdzie
3074
.
0
)
(
0778
.
0
)
(
45724
.
0
2
PR
kr
kr
kr
kr
kr
kr
z
p
RT
b
p
RT
a
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
34
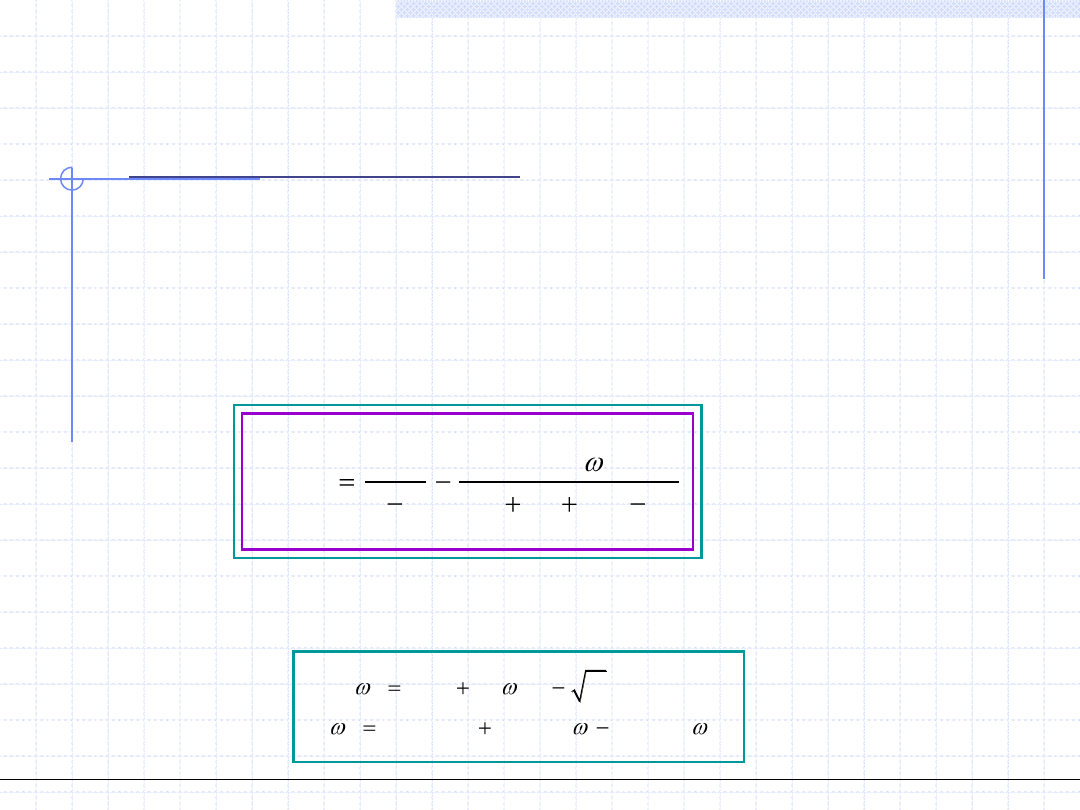
Równanie Patela - Teji
( , )
( , )
(
)
(
)
r
a T
RT
p v T
v
b
v v
b
c v
b
2
2
1
1
0 452413
1 30982
0 95937
( , )
[
( )(
)]
( )
.
.
.
r
kr
r
a T
a
m
T
m
4.5. Równanie Patela - Teji (PT) 1982.
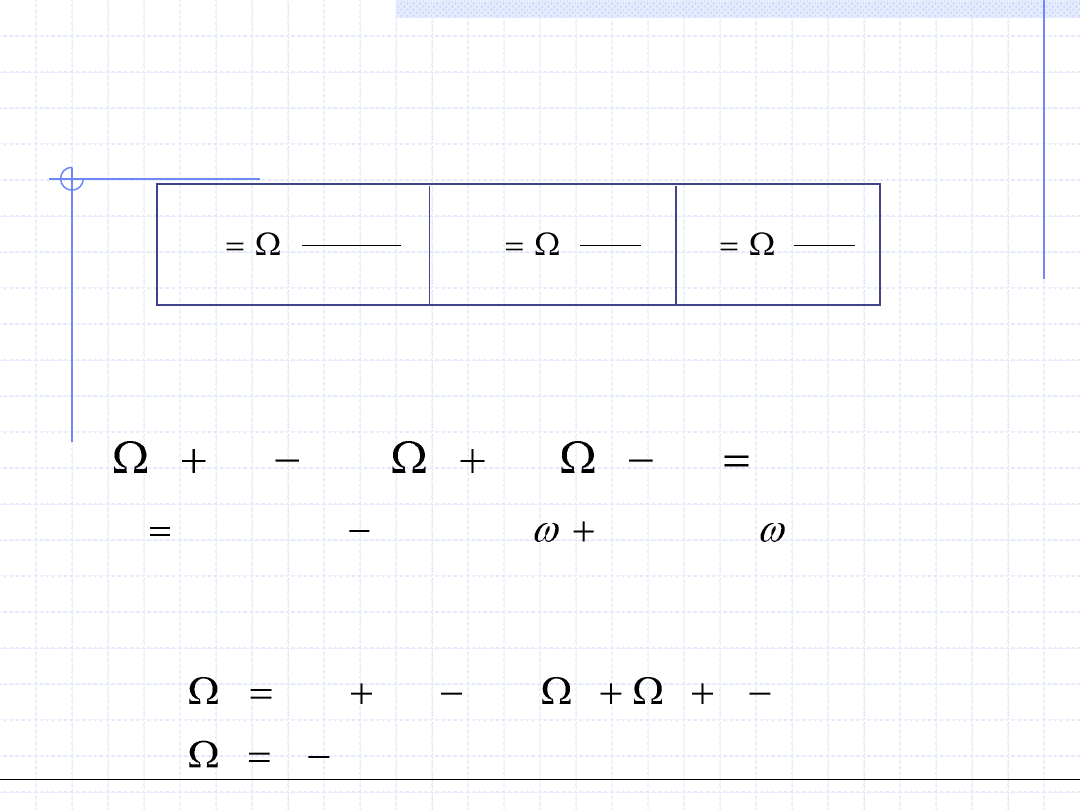
Zarówno oryginalne równanie van der Waalsa jak i jego modyfikacje Redlicha –
Kwonga, SRK i PR są równaniami 2 – parametrowymi. Parametry te można
wyznaczyć na podstawie eksperymentalnych wartości T
kr
, p
kr
i czynnika ω. Patel
i Teja zaproponowali korektę równania Penga – Robinsona poprzez
wprowadzenie trzeciego parametru „c”:
Parametr „a” zależy od temperatury i czynnika ω w sposób analogiczny jak
w równaniach SRK i PR:
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
35
Równanie Patela - Teji
2
(
)
kr
kr
kr
kr
a
b
c
kr
kr
kr
RT
RT
RT
a
b
c
p
p
p
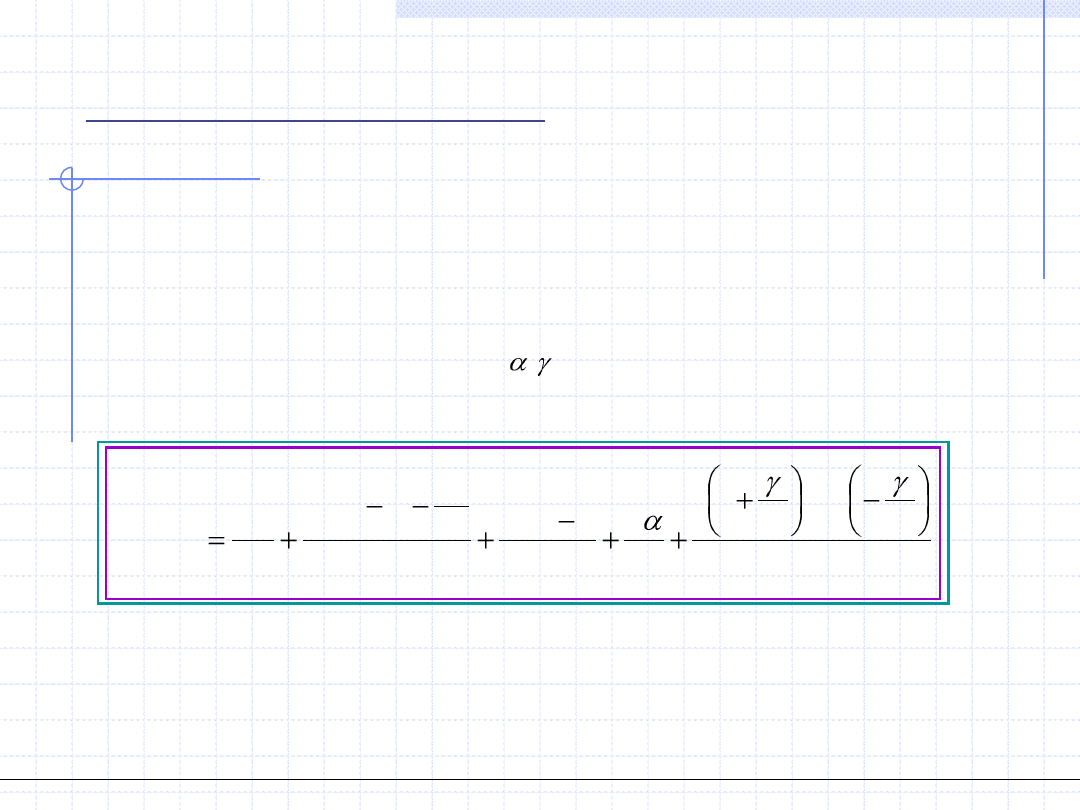
Uzgodnienie równania PT z punktem krytycznym prowadzi do wzorów
umożliwiających obliczenie parametrów a
kr
, b i c:
Bezwymiarowe parametry Ω
a
, Ω
b
i Ω
c
powinny być obliczane za pomocą
następującego algorytmu:
1. Wyznaczenie wartości Ω
b
jako najmniejszego dodatniego pierwiastka równania:
3
2
2
3
2
3
3
0
(
)
b
c
b
c
b
c
z
z
z
gdzie:
2
0 329032
0 076799
0 295937
.
.
.
c
z
Parametr z
c
ma wprawdzie interpretację jako współczynnik ściśliwości w punkcie
krytycznym, nie mniej jednak może on się różnić od eksperymentalnej wartości „z
kr
”.
2. Obliczenie wartości Ω
a
i Ω
c
na podstawie wzorów:
2
2
3
3 1
2
1
3
1
3
(
)
a
c
c
b
b
c
c
c
z
z
z
z
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
36
Niekubiczne równania stanu
2
3
2
2
6
3
2
2
exp
1
)
,
(
T
v
v
v
c
v
a
v
a
bRT
v
T
C
A
BRT
v
RT
T
v
p
,
,
,
,
,
,
,
c
b
a
C
B
A
5. Inne (niekubiczne) równania stanu.
Oprócz równań stanu 3 – go stopnia były i są stosowane równania wyższych
stopni a także równania zawierające różne nie algebraiczne funkcje. Przykładem
właśnie takiego nie algebraicznego równania jest opracowane w czasie II wojny
światowej równanie Benedicta – Webba – Rubina (BWR).
Punktem wyjścia do tego równania było równanie wirialne, zmodyfikowane za
pomocą funkcji ekspotencjalnej. Równanie BWR ma 8 stałych charakterystycznych
dla każdej substancji:
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
Niekubiczne równania stanu c.d.
W ostatnich latach pewną popularność uzyskują równania, w których zamiast
I członu van der Waalsa stosuje się pewne algebraiczne wyrażenie będące
aproksymacją (przybliżeniem) teoretycznych obliczeń wynikających z tzw.
modelu twardych kul (hard sphere model).
Przykładem takiego RS może być równanie Q5 opublikowane w roku 2008:
A. Kozioł,
Quintic equation of state for pure substances in sub- and supercritical
range
, Fluid Phase Equilib., 263 (2008) 18-25.
Pod względem matematycznym jest to równanie algebraiczne 5 – tego
stopnia zawierające 5 stałych: a(T), b, c, d i e.
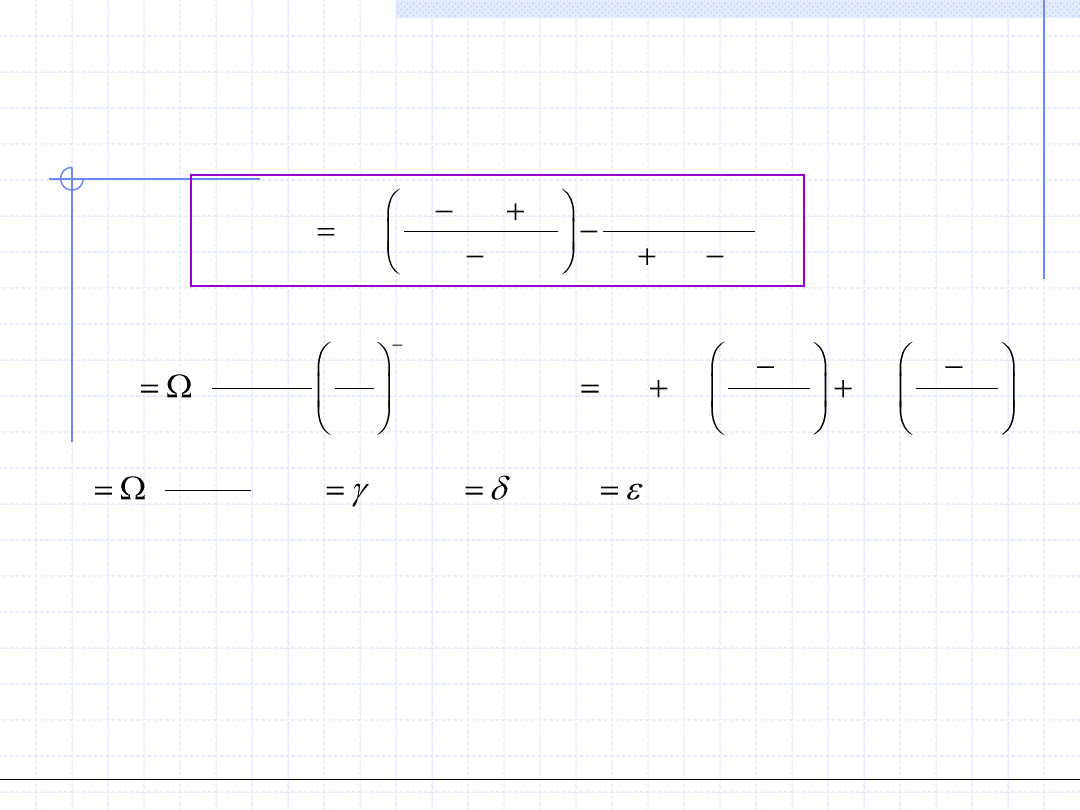
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
Niekubiczne równania stanu c.d.
2
2
3
2
(
)
( )
( , )
(
)
(
)
v d
e
a T
P v T
RT
v b
v
c v b
gdzie:
( )
3
2
0
0
0
1
2
(
)
( )
( )
(
)
m T
kr
a
kr
kr
kr
kr
kr
b
kr
RT
T
T
T
T
T
a T
m T
m
m
m
p
T
T
T
RT
b
c
b
d
b
e
b
p
Występujące w tym równaniu parametry Ω
a
, Ω
b
, m
0
,m
1
,m
2
,T
0
,γ, δ, ε dla 50 substancji
można znaleźć w oryginalnej publikacji.
Dla H
2
O parametry te wynoszą:
Ω
a
=0.629121, Ω
b
=0.0420773, m
0
=0.83638, m
1
=0.333144, m
2
=1.01617
T
0
=400 K, γ=15.1849, δ=1.4506, ε=1.13124
© Prof. Antoni Kozioł, Wydział Chemiczny Politechniki Wrocławskiej
Dziękuję bardzo Państwu za uwagę
Wyszukiwarka
Podobne podstrony:
Term proc i tech WYKLAD III
Term proc i tech W X
Term proc i tech W XII
Term proc i tech W XI
Term proc i tech W XIII
Elem. proc.tech, silniki spalinowe
Odlewnictwo Projekt, MBM, elnia, odbytki, oup, ppt, skrawanie karta, Choroszy, Proj.proc.tech, odlew
Narzędzia formierskie, MBM, elnia, odbytki, oup, ppt, skrawanie karta, Choroszy, Proj.proc.tech, odl
Spawalnictwo [1], MBM, uczelnia, VI semestr, odbytki, oup, ppt, skrawanie karta, Choroszy, Proj.proc
Proces technlogiczny, Proc. tech.wału.
Sp asp proc kom cz VIII 2010
zajęcia VIII
Instrumenty rynku kapitałowego VIII
wyklad3 tech bad
Prawo medyczne wykład VIII Obowiązek ratowania życia
Turystyka, wykład VIII, Agroturystyka
więcej podobnych podstron