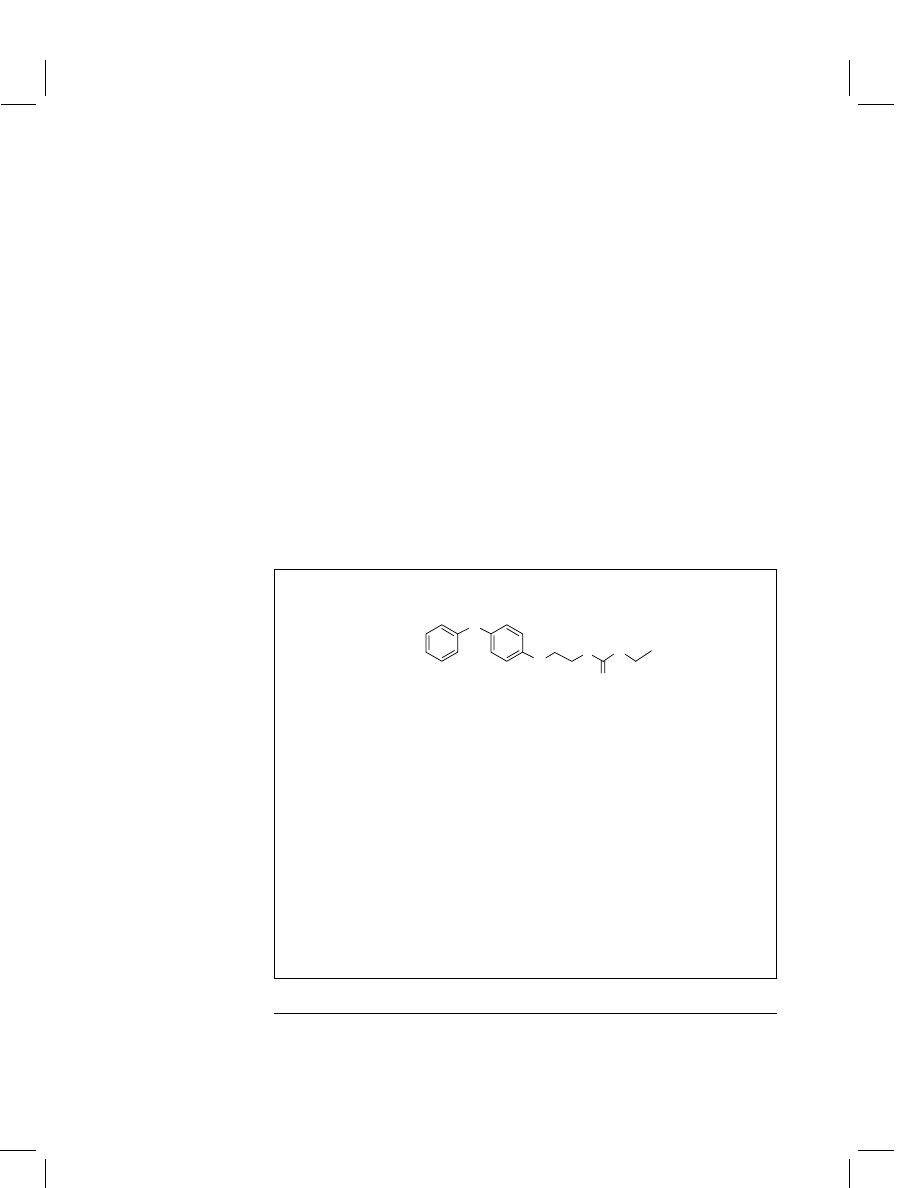
Fenoxycarb
Materials to be
analyzed
Air, water, soil, and plant (pasture grass hay, forage, cu-
curbits, citrus, pome fruit, tree nuts, fruiting vegetables,
and cotton) and animal materials (tissues, milk, blood,
and eggs)
Instrumentation
Liquid
chromatography/ultraviolet
detection,
three-
column switching liquid chromatography with fluo-
rescence detection, two-column switching liquid chro-
matography/ultraviolet detection, gas chromatography/
thermionic specific detection, gas chromatography/mass
spectrometry, and liquid chromatography/atmospheric
pressure ionization/mass spectrometry.
1
Introduction
Chemical name
(IUPAC )
Carbamic acid, [2-(4-phenoxyphenoxy)ethyl]-, ethyl
ester
Structural formula
O
O
O
O
H
N
Empirical formula
C
17
H
19
NO
4
Molar mass
301.34
Melting point
53.6
◦
C
Vapor pressure
6.5
× 10
−9
mbar at 25
◦
C
Solubility (25
◦
C)
Soluble in water: 5.66 mg L
−1
Readily soluble in organic solvents: ethanol 51, acetone
77, toluene 63, n-octanol 13, n-hexane 0.53 g per 100 mL
Other properties
Colorless to white solidified melt, no dissociation con-
stant in an accessible pH range, octanol/water partition
coefficient (log K
ow
) 4.07 at 25
◦
C.
Use pattern
An insect growth regulator, used to control early instar
larvae of Homoptera, Lepidoptera, and Coleoptera in
citrus, cotton, and vines and fruiting vegetables
Regulatory position
The residue of concern is for the parent, fenoxycarb,
only
Handbook of Residue Analytical Methods for Agrochemicals.
C
2003 John Wiley & Sons Ltd.

Fenoxycarb
1295
2
Outline of methods
1
Air was sampled for a specific rate and time and the analyte collected on XAD-2
resin. The analyte was eluted from the resin using methanol followed by concen-
tration of the eluate and analysis using gas chromatography/nitrogen–phosphorus
detection (GC/NPD). Water was extracted by passing the sample through XAD-2
resin followed by elution of the analyte using ethyl acetate. Further purification of the
extract was obtained using Florisil chromatography and final analysis was obtained
using liquid chromatography/ultraviolet detection (LC/UV). Pond water was parti-
tioned into hexane followed by evaporation of the solvent and analysis using LC/UV.
Soil was extracted with acetone, acetonitrile was added, and the mixture partitioned
with hexane. After discarding the hexane, the acetone–acetonitrile was adjusted to
basic pH and re-partitioned with hexane. The hexane fraction was reduced and sub-
jected to analysis using gas chromatography (GC)/thermionic specific detection. Plant
and animal samples were extracted with acetone, filtered, and partitioned with hex-
ane. After discarding the hexane, the acetone–acetonitrile fraction was adjusted to
basic pH and re-partitioned with hexane followed by further purification using silica
and Florisil SPE cartridges. Final analysis was accomplished using gas chromatog-
raphy/mass spectrometry (GC/MS). Certain citrus samples were blended with C
8
or
C
18
SPE packing material and the mixture was loaded into a glass column. The an-
alyte was eluted using dichloromethane–acetonitrile for final analysis using liquid
chromatography/atmospheric pressure ionization/mass spectrometry (LC/API/MS).
3
Apparatus
3.1
Air
Air sampler, Alpha-1 (Messgerate-Werk Lauda, RMT 20, Germany)
Circulation cooler
Mini-Buck calibrator, Model M-5 (A.P. Buck, Inc.)
OSHA versatile sampler (OVS) sorbent tubes (SKC, Inc., Cat. No. 226-30-16)
Teflon vacuum pump (Analytichem International, N726.3FT.18)
Ultrasonic bath (Branson, Model 2200)
3.2
Water
Carbon filter tube (Fisher 08-261B)
Chromatography columns: 100
× 15-mm i.d. with Teflon stopcock
Glass-fiber filter (Whatman GF/D, 11-cm)
Rotary evaporator (B¨uchii) with water-bath temperature 50
◦
C
3.3
Soil
Buchner funnels, 7-cm
Evaporation flasks, 250- and 500-mL

1296
Individual compounds
Glass-microfiber filters, 7-mm (Whatman GF/D)
Reciprocating shaker
Separatory funnels, 125- and 250-mL
Side-arm flask, 500-mL
Wide-mouthed bottles with Teflon-lined lids, 250-mL
3.4
Pasture grass hay, forage, cucurbits, citrus, pome fruit, tree nuts,
fruiting vegetables, and cotton substrates
Bottles, 8-oz
Round-bottom flasks, 50-, 100-, 250- and 1000-mL
Side-arm flasks, 500-mL
Florisil, 1000-g/6-cm
3
(J.T. Baker, 7213-07)
Homogenizer, Polytron (or equivalent)
PrepSep Florisil (Fisher Scientific, Cat. No. P476)
PrepSep Silica (Fisher Scientific, Cat. No. P478)
Silica, 100-g/6-cm
3
(J.T. Baker, 7086-07)
Sep-Pak, C
18
(Waters, 43365)
3.5
Animal tissues, milk, blood, and eggs
Acrodisc filter, liquid chromatography (LC), poly(vinylidene fluoride) (PVDF),
0.2-µm, 13-mm
Bottles, 32-oz, wide-mouthed
Side-arm flasks, 500-mL
Flat-bottom boiling flasks, 250-mL
Glass-wool (Fisher, 11-390)
Silica gel solid-phase-extraction (SPE) cartridge, 1-g/6-cm
3
(J.T. Baker, 7086-07)
Other items as in Soil and Plant material lists
4
Reagents
Acetone, glass distilled
Acetonitrile, LC grade
Ammonium hydroxide, ACS reagent grade
Anhydrous sodium sulfate, ACS reagent grade
Dichloromethane, HPLC grade
Ethyl acetate, glass distilled
Florisil (Fluka), 60–100-mesh
Hexane, glass distilled and residue analysis grade
Methanol, glass distilled and analytical grade
Phosphoric acid, ACS grade
Potassium dihydrogenphosphate, analytical grade
Potassium phosphate, monobasic, ACS grade

Fenoxycarb
1297
Sea sand, purified with acid and calcined
Sodium carbonate, ACS grade
Sodium sulfate, ACS grade
Toluene, high-purity
Water, glass distilled and LC grade
XAD-2 resin (0.15–0.20-mm particle size), research grade
5
Sample preparation
5.1
Air
Air was sampled by passing air at 0.5 L min
−1
for 4 h (via vacuum) through an OVS
tube containing a glass-fiber filter (to trap aerosols and particulates) and XAD-2 resin
(to trap vapors). A second XAD-2 section in the sampling tube provided a means of
checking for overloading of the first XAD-2 section. After sampling, the glass-fiber
filter and first section of XAD-2 resin were transferred to a 10-mL round-bottom
flask. The second portion of XAD-2 resin was transferred to a second 10-mL
round-bottom flask. A volume of 5 mL of methanol was added to each flask followed
by ultrasonication for 5 min. The solids were allowed to settle and the methanol was
transferred by pipet to respective 25-mL round-bottom flasks. The extraction process
using another 5-mL portion of methanol was repeated for each sample. The pooled
methanol fractions for each sample were evaporated to dryness via rotary evaporation
and the residues were reconstituted in 12 mL of hexane for analysis by GC/NPD.
5.2
Water
Water was extracted for fenoxycarb by passing 1 kg of water sample through a glass-
microfiber filter into a 1-L dropping funnel. A chromatography column containing 5 g
of XAD resin supported by 5 g of sea sand was successively conditioned with 80 mL
of methanol and then 80 mL of acetone. Drying was accomplished by passing dry
nitrogen through the column. The filtered water sample was passed through the XAD
resin at a rate of 10 mL min
−1
. After passage of the water sample through the resin,
the remaining water was forced out of the column using dry nitrogen. The analyte was
eluted from the column using 100 mL of ethyl acetate at 1 mL min
−1
and collected
in a 250-mL flask. The eluate was reduced to dryness using a rotary evaporator
and reconstituted in 2 mL of ethyl acetate. Another column was prepared by adding
10 mL of hexane–ethyl acetate (23 : 2, v/v) in a chromatography column plugged with
glass-wool. Next, 10 g of Florisil in a solvent slurry mixture were slowly added to the
column by gently tapping the sides and the solvent level was allowed to drain to the
top of the Florisil. A 2-mL fraction from the previous column step was quantitatively
transferred to the Florisil column and the column was washed with 100 mL of ethyl
acetate. The analyte was eluted using 100 mL of hexane–ethyl acetate (17 : 3, v/v)
and collected in a 250-mL flask. This fraction was reduced to dryness and the residue
was reconstituted using 0.5 mL of mobile phase for analysis using LC/UV.
Pond water was analyzed for fenoxycarb by partitioning 1 L of filtered (Whatman
No. 2 filter) water sample with 75 mL of hexane. The partitioning step was repeated

1298
Individual compounds
twice and the pooled hexane fraction was dried through a bed of anhydrous sodium
sulfate. The dried hexane was reduced to about 1 mL using rotary evaporation and
quantitatively transferred to a concentration tube. This fraction was again reduced to
about 1 mL and the sides of the concentration tube were rinsed with 2 mL of methanol.
This fraction was then reduced to dryness and reconstituted in an appropriate volume
of mobile phase for analysis using three-column switching LC/UV.
5.3
Soil
Soil was extracted for fenoxycarb by placing 20 g of sample in a 250-mL extraction jar
with a Teflon-lined lid containing 20 mL of 1% phosphoric acid. The jar was allowed
to stand for 20 min before adding 200 mL of acetone followed by mechanical shaking
for 30 min. The extract was filtered through glass-fiber filters into a 500-mL side-arm
filtering flask using 2
× 15 mL of acetone to rinse the extraction jar. This fraction
was quantitatively transferred to a 500-mL evaporation flask and reduced in volume
to 20–25 mL at a water-bath temperature of
<35
◦
C to remove all traces of acetone.
Acetonitrile (30 mL) was added to the flask and swirled to mix before transfer to a
250-mL separatory funnel. A volume of 2
× 10 mL of acetonitrile–water (3 : 2, v/v)
was used to rinse the evaporation flask. The acetonitrile–water mixture was partitioned
with 50 mL of hexane (1 min). The hexane phase was separated from the aqueous
phase and partitioned twice (for 1 min each time) with 10 mL of acetonitrile–water
(3 : 2, v/v). The two acetonitrile–water solvent extracts were returned to the original
acetonitrile–water extract and the hexane was discarded. The acetonitrile–water frac-
tion was reduced to about 40 mL via rotary evaporation at a water-bath temperature
of
<35
◦
C to remove the acetonitrile. To the remaining aqueous fraction was added
2 mL of concentrated ammonia solution and 20 mL of deionized water followed by
transfer to a 125-mL separatory funnel. A volume of 50 mL of hexane was added to
the evaporation flask for rinsing purposes before transfer to the separatory funnel. Af-
ter phase separation, the hexane phase was dried through a bed of anhydrous sodium
sulfate and collected in a clean 250-mL evaporation flask. The aqueous fraction was
partitioned twice more, each time with 50 mL of hexane. The pooled and dried hexane
fraction was reduced to dryness using rotary evaporation prior to reconstitution in an
appropriate volume of hexane for GC analysis.
5.4
Plant material
5.4.1
Pasture grass hay, forage, and cucurbits
Fenoxycarb was extracted from pasture grass hay, forage, and cucurbits (cucum-
bers, squash, and cantalope) by weighing a 25-g representative sample into a 16-oz
wide-mouthed jar followed by the addition of 20 mL of 1% phosphoric acid. After
waiting for 20 min, 200 mL of acetone were added. After waiting for 1 min, the sample
was homogenized using a Polytron at a rate of 17 000–20 000 rpm for 2 min. The sam-
ple was then filtered using a 500-mL side-arm flask equipped with a Buchner funnel
containing a glass-microfiber filter. The sample bottle was rinsed with 50 mL of ace-
tone and the solvent was filtered. The extract was transferred to a 1-L round-bottom

Fenoxycarb
1299
flask and the acetone volume was reduced to about 18 mL using rotary evaporation
at a water-bath temperature of
<35
◦
C. The volume of the remaining aqueous frac-
tion was measured and enough acetonitrile was added to obtain an acetonitrile : water
ratio of 3 : 2 (v/v). This fraction was transferred to a 250-mL separatory funnel using
2
× 10 mL of acetonitrile–water (3 : 2, v/v) solvent mixture for rinsing purposes. The
1-L round-bottom flask was rinsed with 50 mL of hexane and the rinsate was added
to the separatory funnel, which was then shaken for 1 min. The hexane fraction was
removed and back-partitioned with 2
× 10 mL of acetonitrile–water (3 : 2, v/v) and
these two portions were returned to the original aqueous acetonitrile fraction. The
hexane layer was discarded. Acetonitrile was removed via rotary evaporation until
the first drops of water were observed in the condenser (the aqueous volume should be
similar to that previously, about 18 mL). Concentrated ammonia solution (2 mL) and
water (20 mL) were added and the mixture was transferred to a 125-mL separatory
funnel. The flask was rinsed with 50 mL of hexane and the rinsings were also added to
the separatory funnel. After shaking for 1 min, the hexane phase was removed and
transferred to a 250-mL round-bottom flask. The aqueous portion was partitioned
twice more with 50 mL of hexane. The aqueous phase was discarded and the pooled
hexane fraction was reduced to dryness using rotary evaporation. This fraction was
reconstituted in 2 mL of hexane and analyzed by LV/UV for crop samples. Pasture
grass and forage samples were subjected to further purification by preconditioning a
silica gel SPE cartridge with 2–3 mL of hexane. The sample was loaded on to the SPE
cartridge and the 250-mL round-bottom flask was rinsed with 2
× 2-mL portions of
hexane, the rinsings also being added to the cartridge. The column was first eluted with
7 mL of dichloromethane–hexane–tetrahydrofuran (49 : 50 : 1, v/v/v) solvent mixture,
which was discarded after passage through the cartridge. The analyte was eluted using
an appropriate volume of the same elution solvent (typically 15–25 mL) and collected
in a 50-mL round-bottom flask. Note that the fenoxycarb elution volume on each lot
of silica gel SPE cartridges was profiled due to lot-to-lot variability. The eluate was
reduced to dryness followed by reconstitution in 2 mL of hexane. A 5-mm deep layer
of sodium sulfate was added to the top of a Florisil PrepSep cartridge and was then
preconditioned using 2–3 mL of hexane–ethyl acetate (23 : 2, v/v). The 2-mL hexane
fraction was loaded on to the Florisil PrepSep cartridge using 2
× 2-mL portions of
hexane for rinsing purposes. The analyte was eluted with 50 mL of hexane–ethyl
acetate (23 : 2, v/v) and collected in a 100-mL round-bottom flask. This fraction was
reduced to dryness and reconstituted in an appropriate volume of mobile phase for
LC analysis or in an appropriate volume of acetonitrile for GC/MS analysis.
5.4.2
Citrus, pome fruit, tree nut, fruiting vegetables, and cotton substrates
A 10-g representative sample (5-g sample for citrus oil or cotton substrates) was
extracted by adding 150 mL of acetonitrile–water (4 : 1, v/v) to the sample in an 8-oz
bottle and homogenized with a Polytron at high speed for 2 min. The extract was
filtered through a Whatman No. 1 filter-paper into a 500-mL side-arm flask. The
extraction bottle was rinsed with 50 mL of acetonitrile–water (4 : 1, v/v) for citrus
and cottonseed oil (for molasses use 10 mL of water followed by 40 mL of acetoni-
trile for rinsing). The extract was transferred to a 500-mL separatory funnel and
partitioned twice, each time with 50 mL of hexane for 1 min. The hexane fractions

1300
Individual compounds
were discarded. The extract was transferred to a 500-mL boiling flask and the acetoni-
trile was removed using rotary evaporation at a water-bath temperature of 35–40
◦
C
(removal of all the acetonitrile is critical). For tree nut samples only, a Waters C
18
Sep-Pak cartridge was conditioned with 10 mL of acetonitrile–water (3 : 2, v/v) and
10 mL of water at a flow rate of 1–3 drops per second (avoiding column dryness).
The sample was loaded followed by washing with 25 mL of acetonitrile–water (1 : 4,
v/v) and then 10 mL of acetonitrile–water (3 : 2, v/v). The analyte was eluted using
40 mL of acetonitrile–water (3 : 2, v/v). For all other samples (excluding tree nuts),
the remaining aqueous portion after removing the acetonitrile was transferred to a
250-mL separatory funnel to which 50 mL of 0.5% sodium carbonate and 25 mL of
water saturated with sodium chloride were added. For tree nuts, the eluate from the
Waters Sep-Pak cleanup step was transferred to a 250-mL separatory funnel, and
30 mL of 0.5% sodium carbonate and 10 mL of water saturated with sodium chloride
were added. All the samples were partitioned twice, each time with 50 mL of hexane.
The pooled hexane fraction was dried through anhydrous sodium sulfate. The re-
maining aqueous portion was discarded. For tree nuts only, the dried hexane fraction
was reduced to dryness and reconstituted in 2 mL of hexane for Florisil purification.
The dried hexane fraction for all other samples was reduced to 5–15 mL using rotary
evaporation for silica gel purification. A silica gel SPE cartridge was conditioned
with 6 mL of ethyl acetate–hexane (1 : 4, v/v) and 6 mL of hexane at a flow rate of 1–
2 drops per second. The hexane fraction was loaded on the column followed by ad-
dition of 6 mL of ethyl acetate–hexane (1 : 4, v/v) for washing purposes. The analyte
was eluted using 12 mL of ethyl acetate–hexane (1 : 4, v/v) and collected in a 50-mL
concentration tube. This fraction was reduced to dryness and reconstituted in 2 mL of
hexane for Florisil cleanup, except for cucurbit and fruiting vegetable samples, which
were reconstituted in acetonitrile–0.05 M potassium dihydrogenphosphate (1 : 1, v/v)
for LC analysis. All other samples (including tree nuts) were further purified by con-
ditioning a Florisil SPE cartridge with 3–5 mL of ethyl acetate–hexane (2 : 23, v/v) at
a rate of 1–2 drops per second (avoiding column dryness). The hexane fraction was
loaded on to the column and the concentration tube was rinsed in 2
× 2 mL of hexane,
the rinsings being added to the column. The analyte was eluted with 50 mL of ethyl
acetate–hexane (2 : 23, v/v) and collected in a 125-mL boiling flask. This fraction
was reduced to dryness and reconstituted in 1 mL of acetonitrile–0.05 M potassium
dihydrogenphosphate (1 : 1, v/v) for LC analysis.
5.4.3
Oranges, onions, grapes, and tomatoes
A 0.5-g portion of sample was weighed into a mortar and gently blended with 0.5 g
of silica-based sorbent containing C
8
or C
18
functional groups (45–55-µm particle
diameter range) to obtain a homogeneous mixture. The mixture was introduced into
a 100
× 9-mm i.d. glass column. A 10-mL volume of dichloromethane–acetonitrile
(3 : 2, v/v) was added to the column and allowed to elute dropwise under slight
vacuum into a 15-mL conical-shaped graduated cylinder. This fraction was reduced
to 0.5 mL using a gentle stream of nitrogen prior to analysis using LC/MS [with
atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI)].
This method was applicable to the analysis of 13 carbamate residues, including
fenoxycarb.
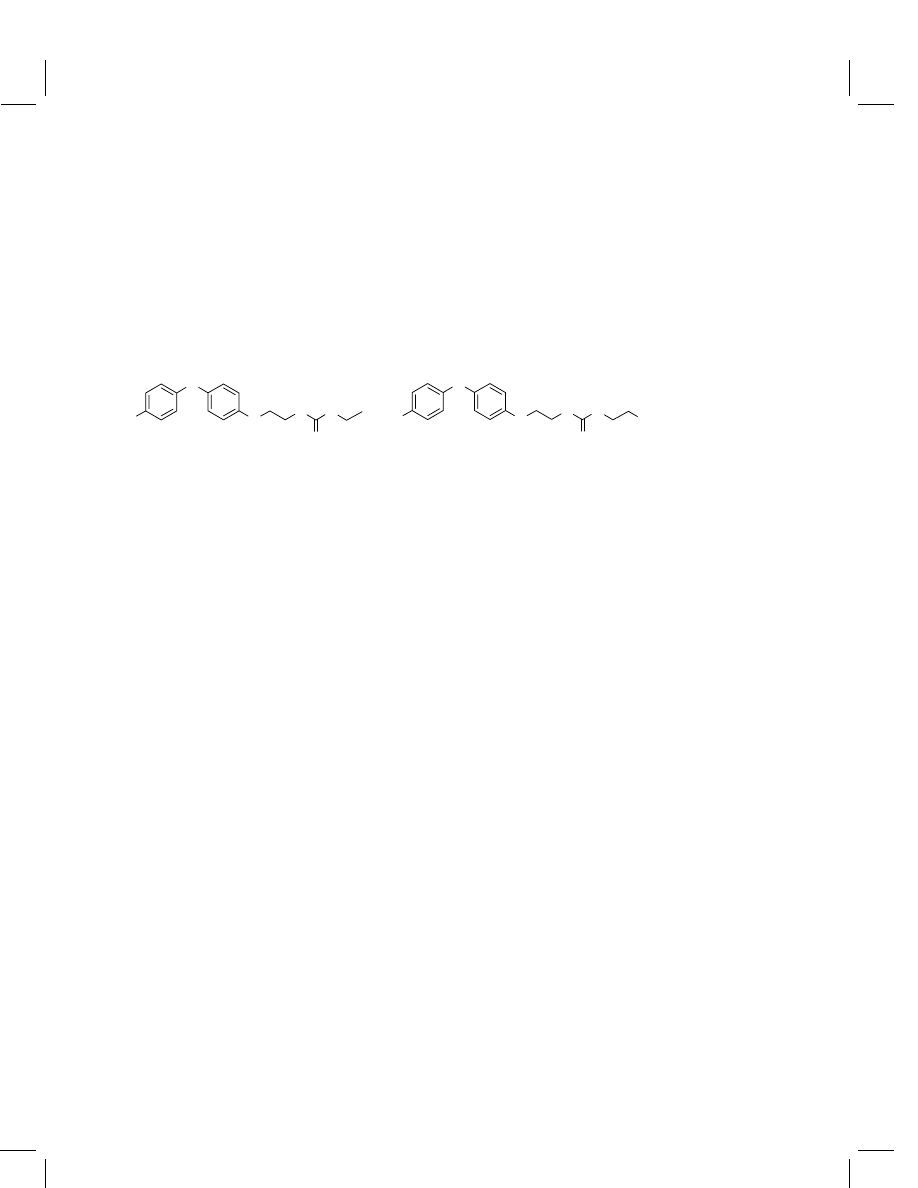
Fenoxycarb
1301
5.5
Animal material
5.5.1
Meat, milk, blood, and eggs
In addition to the parent fenoxycarb, residue methods for the two major
metabolic products Ro-16-8797
{CGA-294850, ethyl N-2-[4-(4-hydroxyphenoxy)
phenoxyethyl]carbamate, MW
= 317.3} and Ro-17-3192 {CGA-294851, (2-
hydroxyethyl)-N -2-[4-(4-hydroxyphenoxy)phenoxyethyl]carbamate, MW
= 333.3}
in animal by-products were also developed.
O
O
N
H
O
O
HO
Ro-16-8797
O
O
N
H
O
O
HO
OH
Ro-17-3192
Fat, muscle, and blood samples (or liver, kidney, well-mixed milk, and egg ho-
mogenate) were extracted for fenoxycarb and its hydroxylated metabolites Ro-16-
8797 and Ro-17-3192 by homogenizing 10 g of tissue or whole blood in 200 mL of
acetonitrile [or 200 mL of water–acetonitrile (1 : 4, v/v)] for 2 min using a Polytron
at high speed. The extract was then filtered through Whatman No. 1 filter-paper into
a 500-mL side-arm flask. A volume of 50 mL of acetonitrile [or 50 mL of water–
acetonitrile (1 : 4, v/v)] was used to rinse the extraction bottle and filter pad. This
fraction was transferred to a 500-mL separatory funnel and partitioned twice using
50 mL of hexane each time. The hexane was discarded. For fat, muscle, and blood, the
acetonitrile was removed using rotary evaporation at a water-bath temperature of 35–
40
◦
C. For liver, kidney, milk, and eggs, 25 mL of water saturated with sodium chloride
and 50 mL of toluene were added to the remaining portion of water–acetonitrile (1 : 4,
v/v). The mixture was shaken for 1 min. The aqueous phase was discarded and the
toluene phase was transferred to a 500-mL flat-bottom flask. This fraction was re-
duced to dryness. For all samples, 25 mL of 0.5% sodium carbonate were added and
this fraction was swirled and sonicated to ensure complete dissolution and mixing
prior to transfer to a 125-mL separatory funnel. A second 25-mL portion of 0.5%
sodium carbonate was used for rinsing purposes. A volume of 2
× 25 mL of ethyl
acetate–hexane (1 : 1, v/v) was used for rinsing purposes and also as partitioning sol-
vent by shaking the separatory funnel for 1 min. After phase separation, the organic
solvent was dried through a bed of prerinsed [using 25 mL of ethyl acetate–hexane
(1 : 1, v/v)] sodium sulfate and collected in a 250-mL flat-bottom boiling flask. The
aqueous portion was partitioned a second time using 50 mL of ethyl acetate–hexane
(1 : 1, v/v) and dried through the same bed of sodium sulfate. The pooled and dried
ethyl acetate–hexane fraction was reduced just to dryness using rotary evaporation
and a water-bath temperature of 35–40
◦
C. A silica gel SPE cartridge was conditioned
using 15 mL of ethyl acetate and 10 mL of dichloromethane (avoiding column drying)
at a rate of 1–2 drops per second via gravity (a vacuum system was not used). To the
residue contained in the flat-bottom flask were added 5 mL of dichloromethane, which
was shaken vigorously and sonicated to ensure complete dissolution and mixing. This
fraction was loaded on to the preconditioned silica column. A second 5-mL portion
of dichloromethane was added to rinse the flask and also added to the silica gel SPE

1302
Individual compounds
cartridge. First, 10 mL of hexane and then 6 mL of ethyl acetate–hexane (1 : 4, v/v)
were added to the flask, followed by transfer to the silica column. These wash sol-
vents were discarded. Fenoxycarb and Ro-16-8797 were eluted with 8 mL of ethyl
acetate–hexane (1 : 1, v/v) and collected in a 50-mL concentration tube. Ethyl acetate
(15 mL) was then added to elute Ro-17-3192 and collected in a separate 50-mL con-
centration tube. The two fractions were reduced to dryness separately and each was
reconstituted in acetonitrile–0.02 M potassium dihydrogenphosphate (1 : 1, v/v) for
analysis using column switching LC.
6
Instrumentation
The following instrumental conditions have been shown to be suitable for the analysis
of fenoxycarb. Other operating parameters may be employed provided that fenoxycarb
is separated from sample interferences and the response is linear over the range of
interest.
Operating conditions for air
Gas chromatograph
Hewlett-Packard 5890A Series II with HP-7673A
autosampler and nitrogen–phosphorus detector.
Column
HP-17 fused silica, 10 m
× 0.53-mm i.d., 2.0-µm film
thickness
Temperatures
Injector 250
◦
C, detector 300
◦
C, oven 245
◦
C
Gas flow rates
Carrier gas, He, 11 mL min
−1
; make-up gas, N
2
, 22 mL
min
−1
; H
2
, 2.7 mL min
−1
; air, 108 mL min
−1
Volume injected
5 µL
Retention time for
3.4 min
fenoxycarb
Operating conditions for water
High-performance
Kontron 640
liquid chromatograph
Kontron Uvikon 735 LC/UV detector
Column
125
× 4.6-mm i.d., SS, Nucleosil C
18
, 5-µm particle
size
Mobile phase
Isocratic, acetonitrile–0.05 M potassium dihydrogen-
phosphate (1 : 1, v/v, pH 4.5)
Detector wavelength
228 nm
Flow rate
1 mL min
−1
Retention time for
10 min
fenoxycarb
Three-column switching
High-performance
Perkin-Elmer Series 410
liquid chromatograph
(pump 1)
Injector
Perkin-Elmer ISS-100 autosampler
Pumps 2 and 3
Waters M6000A
Switching valves
Valco Instruments, Model EL6W six-port with elec-
tronic actuator
Detector
Perkin-Elmer LS 40 fluorescence detector

Fenoxycarb
1303
Column 1
Phase-Sep S5 C1, 10 cm
× 4.6-mm i.d., 5-µm particle
size
Column 2
Phase-Sep S3 ODS-2, 10 cm
× 4.6-mm i.d., 3-µm par-
ticle size
Column 3
Hamilton PRP-1, 15 cm
× 4.1-mm i.d., 10-µm particle
size
Mobile phase 1
Acetonitrile–0.05 M phosphate buffer (2 : 3, v/v) at
1 mL min
−1
Mobile phase 2
Acetonitrile–0.05 M phosphate buffer (1 : 1, v/v) at
1 mL min
−1
Mobile phase 3
Acetonitrile–0.05 M phosphate buffer (13 : 7, v/v) at
1 mL min
−1
Injection volume
200 µL
Excitation/emission
230
/300 nm
wavelengths
Retention time for
fenoxycarb
Column 1, 8.9 min; column 2, 17.3 min; column 3,
23.6 min
Operating conditions for soil
Gas chromatograph
Varian 3400 with thermionic specific detector
Column
Restek Rtx-5, 30-m
× 0.53-mm i.d., 1.5-µm film thick-
ness
Temperatures
Injector: initial 50
◦
C (held 0 min), increased at
200
◦
C min
−1
to 250
◦
C (held 17.5 min)
Detector: 300
◦
C
Oven: initial 50
◦
C, increased at 25
◦
C min
−1
to 250
◦
C
Gas flow rates
Air, 180 mL min
−1
; He, 30 mL min
−1
; H
2
, 4.5 mL
min
−1
Volume injected
4–5 µL
Retention time for
1.25 min
fenoxycarb
Operating parameters for pasture grass hay, forage, cucumbers, squash, and
cantalope
High-performance
liquid chromatograph
Perkin-Elmer Model ISS-200 automatic HPLC sam-
pler
Kratos Spectroflow 400 LC pumps
ABI Model 783 variable-wavelength ultraviolet (UV)
detector
Valco Instruments, six-port switching valve
Column 1
Spherisorb C1, 100
× 4.6-mm i.d., 5-µm particle size
(Fisher, Cat. No. 05-692-547)
Column 2
Spherisorb ODS2, 150
× 4.6-mm i.d., 3-µm particle
size (Fisher, Cat. No. 05-692-536)
Mobile phase 1
Acetonitrile–0.05 M potassium dihydrogenphosphate
(2 : 3, v/v), 1 mL min
−1
Mobile phase 2
Acetonitrile–0.05 M potassium dihydrogenphosphate
(2 : 3, v/v), 1 mL min
−1

1304
Individual compounds
Detector wavelength
228 nm
Injection volume
40 µL
Retention time for
Column 1, 7 min; column 2, 12 min
fenoxycarb
Gas chromatograph/
mass spectrometer
Hewlett-Packard 6890 Series gas chromatograph with
Model 5973 mass-selective detector
Column
DB-1701, 30 m
× 0.25-mm i.d., 0.15-mm film thick-
ness (J&W Scientific, Cat. No. 1220731)
Temperatures
Injector : 200
◦
C
Detector : 280
◦
C
Oven : initial 50
◦
C (held 1 min), ramp A 50
◦
C min
−1
to 250
◦
C (held 0 min), ramp B 10
◦
C min
−1
to 300
◦
C
(held 5 min)
Gas flows rates
Pressure 10 psi, EPP mode, column (He) 1.2 mL min
−1
;
purge 60 mL min
−1
, purge time 0.6 min
Volume injected
2 µL
Retention time for
1.25 min
fenoxycarb
Selected ion monitoring
Target ion m
/z 116, qualifier ions m/z 186 and 301
Citrus, pome fruit, tree nuts, fruiting vegetables, and cotton substrates
High-performance
liquid chromatograph
Two Waters Model 501 pumps
Perkin-Elmer Model ISS-100 automatic sampler
Kratos ABI Spectroflow Model 783 ultraviolet/visible
(UV/VIS) detector
Column 1
Phase Separation Spherisorb C1, 100
× 4.6-mm i.d.,
S5
Column 2
Phase Separation Spherisorb ODS2, 150
× 4.6-mm
i.d., S3
Mobile phase 1
Acetonitrile–0.05 M potassium dihydrogenphosphate
(2 : 3, v/v)
Mobile phase 2
Acetonitrile–0.05 M potassium dihydrogenphosphate
(1 : 1, v/v)
For tree nuts: acetonitrile–methanol–0.05 M potassium
dihydrogenphosphate (8 : 5 : 7, v/v/v)
Detector wavelength
225 nm
Flow rate
1 mL min
−1
Injection volume
50 µL
Retention time of
fenoxycarb
About 13 min for tree nut substrates and 20 min for all
other substrates
Operating parameters for oranges, onions, grapes, and tomatoes
High-performance
liquid chromatograph
Hewlett-Packard 1100 Series LC-MSD equipped with
an atmospheric pressure ionization (API) source (APcI
or ESI)
Column
Spherisorb C
8
, SS, 150
× 4.6-mm i.d., 3-µm particle
size (and a LiChrosorb RP-8 guard column)

Fenoxycarb
1305
Mobile phase (APCI)
Methano–water (1 : 1, v/v), isocratic for 5 min, linear
to 60% methanol for 5 min, held for 5 min, then to 90%
methanol in 5 min, held for 7 min, 1 mL min
−1
(other
conditions may be more appropriate if analyzing solely
for fenoxycarb)
Mobile phase (ESI)
Methanol–water, (1 : 1, v/v), isocratic for 15 min, to
70% methanol in 5 min, held for 5 min, then to 90%
methanol in 5 min, held for 5 min
APcI (
+ mode)
302 [M
+ H]
+
, 230 [M
+ H − (CH
3
)
2
NCO]
+
Vaporizer temperature, 325
◦
C; nebulizer gas, N
2
, at
4.1 bar; drying gas, N
2
, at 4 L min
−1
and temperature
350
◦
C; capillary voltage, 4000 V; corona current, 4 µA
ESI (
+ mode)
302 [M
+ H]
+
Gas temperature, 350
◦
C at 13 L min
−1
; nebulizer gas
pressure, 30 psi; capillary voltage, 4000 V
Injection volume
5 µL
Retention time for
26–30 min
fenoxycarb
Operating parameters for meat, milk, blood, and eggs
High-performance
liquid chromatograph
Two Waters Model 501 pumps
Perkin-Elmer Model ISS-100 automatic sampler
Kratos ABI Spectroflow Model 783 UV/VIS detector,
VICI EQ60 LC switching valve
Column 1
Supelcosil LC-CN, 33
× 4.6-mm i.d., 5-µm particle
size
Column 2
Supelcosil LC-1, 250
×4.6-mm i.d., 5-µm particle size
Mobile phase 1
0.02 M potassium dihydrogenphosphate (adjusted to
pH 3 with phosphorous acid)–methanol (7 : 3, v/v)
Mobile phase 2
0.02 M potassium dihydrogenphosphate (adjusted to
pH 3 with phosphorous acid)–methanol–acetonitrile
(12 : 5 : 3, v/v/v)
Detector wavelength
235 nm
Flow rate
1.5 mL min
−1
Injection volume
50 µL
Retention time ranges
Fenoxycarb: 14–27 min
Ro16-8797: 10–16 min
Ro-17-3192: 5.6–8 min
7
Evaluation
7.1
Method
Quantification was performed in all cases using the external calibration method.
A series of standards were injected and the responses plotted against their known
concentrations. Peak responses in samples were compared with the calibration plots

1306
Individual compounds
to obtain the amount found (nanograms). A fresh calibration plot was generated with
each analytical set of samples.
7.2
Recoveries, limit of detection (LOD) and limit
of quantitation (LOQ)
The lower practical level of quantitation for fenoxycarb in air is 10 µg m
−3
using the
described sampling rates and times. The average recovery obtained from fortifying
and extracting the OVS tubes was 94%.
The recoveries of fenoxycarb (CGA-114597) from water ranged from 71 to 90%
at fortification levels from 0.05 to 1 µg L
−1
. The LOQ was 0.05 µg L
−1
.
The average recovery obtained for fenoxycarb when the analysis of pond water
was performed using three-column switching LC/fluorescence detection was 100%
at fortification levels ranging from 0.001 to 10 µg L
−1
. The LOQ and LOD were
0.001 µg L
−1
and 0.4 ng injected, respectively.
The average recoveries for fenoxycarb in soil were 89, 105, and 104% for soil
collected in California, Washington, and Georgia, respectively, at fortification levels
ranging from 0.01 mg kg
−1
to 1.0 mg kg
−1
. The LOQ and LOD were 0.01 mg kg
−1
and 0.2 ng injected, respectively.
The average recovery obtained for fenoxycarb in pasture grass at the method LOQ
was 82%. At all fortification levels, the average recovery for pasture grass was 81%.
For cucurbits (cucumbers, squash, cantalope) the average recovery at the method
LOQ were 102% and for all fortification levels is 99%. The LOQ and LOD were
0.01 mg kg
−1
and 2 ng injected, respectively, using LC. The average recovery for
the analysis of pasture grass forage and pasture grass hay using GC/MS were 97%
and 85%, respectively. The recovery data obtained using LC/UV and GC/MS were
comparable. However, confirmatory evidence was obtained using GC/MS.
The average recoveries and standard deviations for the many citrus, pome fruit,
tree nut, fruiting vegetables, and cotton substrate sample types were acceptable when
fortified at concentration levels ranging from 0.01 to 4 mg kg
−1
. The LOQ of the
method was 0.01 mg kg
−1
, except for citrus oil (0.02 mg kg
−1
), and the LOD was
1.25 ng injected.
The fenoxycarb recoveries for orange, onion, grape, and tomato samples ranged
from 63 to 70%. The LOQ and LOD were 0.01 mg kg
−1
and 0.005 mg kg
−1
, respec-
tively, when using liquid chromatography/electrospray ionization mass spectrometry
(LC/ESI/MS).
Average recoveries for fenoxycarb, Ro-16-8797, and Ro-17-3192 for all animal
sample substrates ranged from 80% (beef kidney) to 111% (goat kidney), 76% (goat
milk) to 93% (beef omental fat), and 56% (dairy milk) to 76% (beef perirenal fat),
respectively. The LOQ and LOD were 0.01 µg g
−1
and 2.5 ng injected, respectively.
7.3
Calculation of residues
Water
µ
g kg
−1
= AC100/B DR

Fenoxycarb
1307
where A
= ng found (from the calibration plot), B = injection volume, C = final
fraction volume, D
= weight of sample extracted, and R = percentage recovery (ex-
pressed as a decimal).
Pond water
ppb
= ng found/mL injected
where ng found is taken from the calibration plot,
g injected
= gV
i
/V
f
where g
= weight of sample (1.0 mL = 1.0 g), V
i
= volume of sample injected into
the LC system, and V
f
= final fraction volume.
Soil
ppm (µg g
−1
)
= (µg g
−1
equivalents from calibration plot) (V
f
/W
s
) (dilution
factor)
where V
f
= final fraction volume (mL) and W
s
= sample weight (g).
Plant and animal sample substrates
mg injected
= GV
a
V
i
/[(V
e
+ W(M/100)]V
f
where G
= mg sample extracted, V
a
= aliquot volume, V
e
= extraction volume,
V
i
= injection volume, M = sample moisture (%), and W = sample weight (g).
ppm
= ng found from calibration plot/mg sample injected
Reference
1. M. Fernandez, Y. Pico, and J. Manes, J. Chromatogr. A, 871, 43 (2000).
Robert A. Yokley
Syngenta Crop Protection, Inc., Greensboro, NC, USA
Document Outline
- Front Matter
- Table of Contents
- Volume I
- Volume II
- Recent Advances in Analytical Technology, Immunoassay and Other Nonchromatographic Methods
- Best Practices in the Generation and Analyses of Residues in Environmental Samples
- Compound Class
- Individual Compounds
- Azoxystrobin
- Famoxadone
- Fluthiacet-Methyl
- Flutolanil
- Hymexazol
- Imibenconazole
- Mepanipyrim
- Mepronil
- Tebuconazole
- Acetamiprid
- Alanycarb
- Azinphos-Methyl
- Benfuracarb
- Buprofezin
- Cyfluthrin
- Fenothiocarb
- Fenoxycarb
- Fenpyroximate
- Hexythiazox
- Imidacloprid
- Isoxathion
- Milbemectin
- Pyrimidifen
- Pyriproxyfen
- Index
Wyszukiwarka
Podobne podstrony:
91942 08w
91942 abb
91942 04m
91942 05d
91942 03d
91942 01e
91942 08j
91942 05b
91942 08p
91942 04b
91942 08u
91942 06c
91942 04i
91942 01c
91942 03c
91942 01a
91942 08x
91942 03f
91942 toc
więcej podobnych podstron