



SLL/CLL (chłoniak z małych limfocytów/przewlekła białaczka
limfatyczna)
Nowotwór zbudowany z monomorficznych małych limfocytów B (z
domieszką prolimfocytów i paraimmunoblastów).
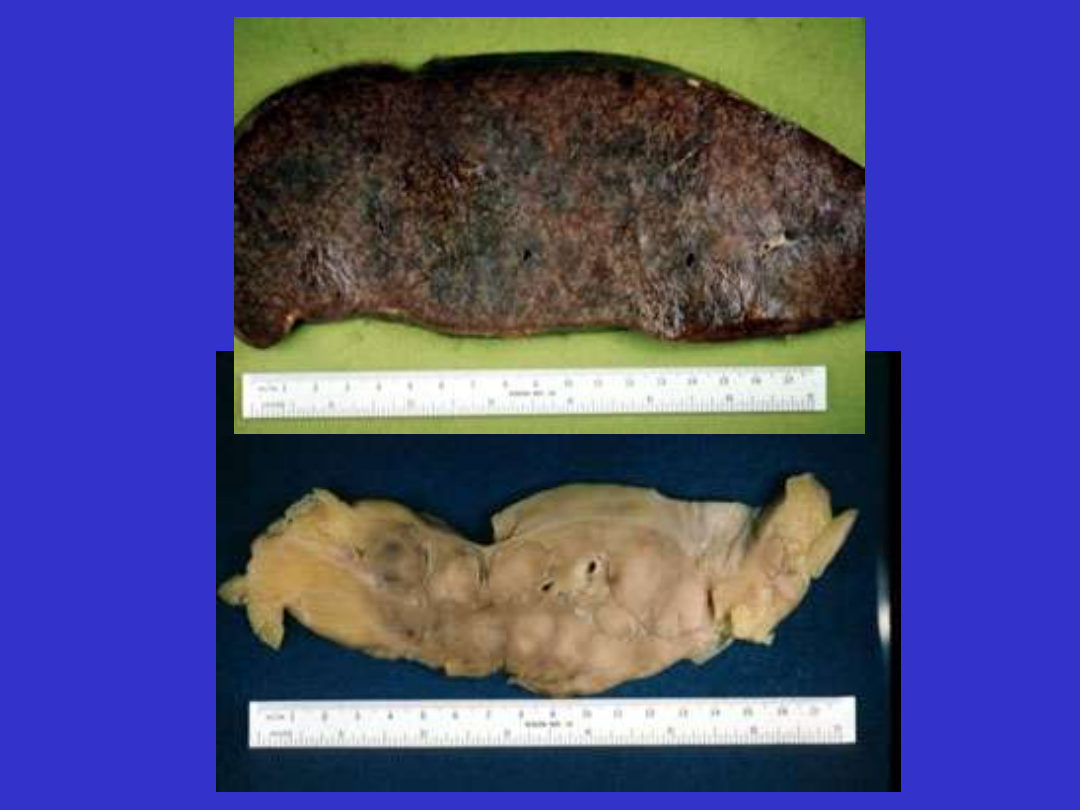
CLL – zajęcie szpiku, krwi obwodowej, węzłów, często wątroby,
śledziony (90% przewlekłych białaczek limfatycznych, ok. 6,7%
chłoniaków).
SLL – forma bezbiałaczkowa.
Występowanie powyżej 50 r.ż. (średnio ok. 65 r.ż.), M/K = 2/1;
Klinicznie:
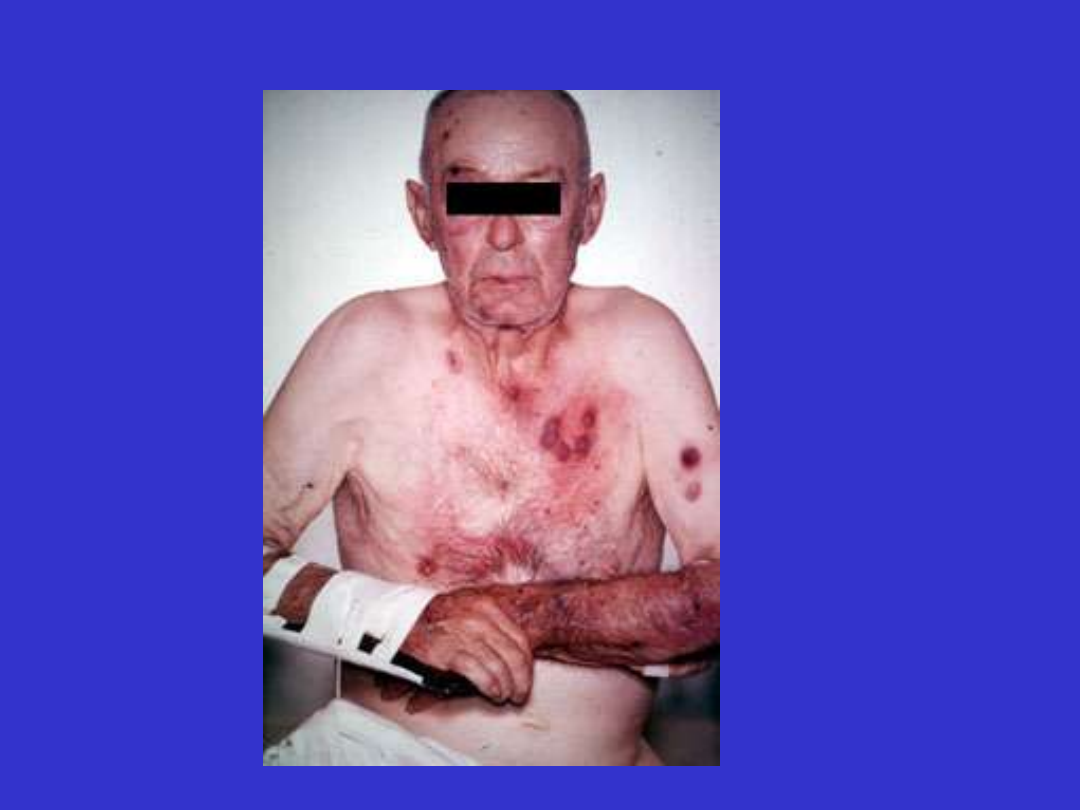
Zwykle brak charakterystycznych objawów klinicznych, osłabienie,
łatwe męczenie, anemia hemolityczna autoimmunologiczna,
infekcje, splenomegalia, hepatomegalia, limfadenopatia, nacieki
pozalimfatyczne (skóra, sutek, narząd wzroku).

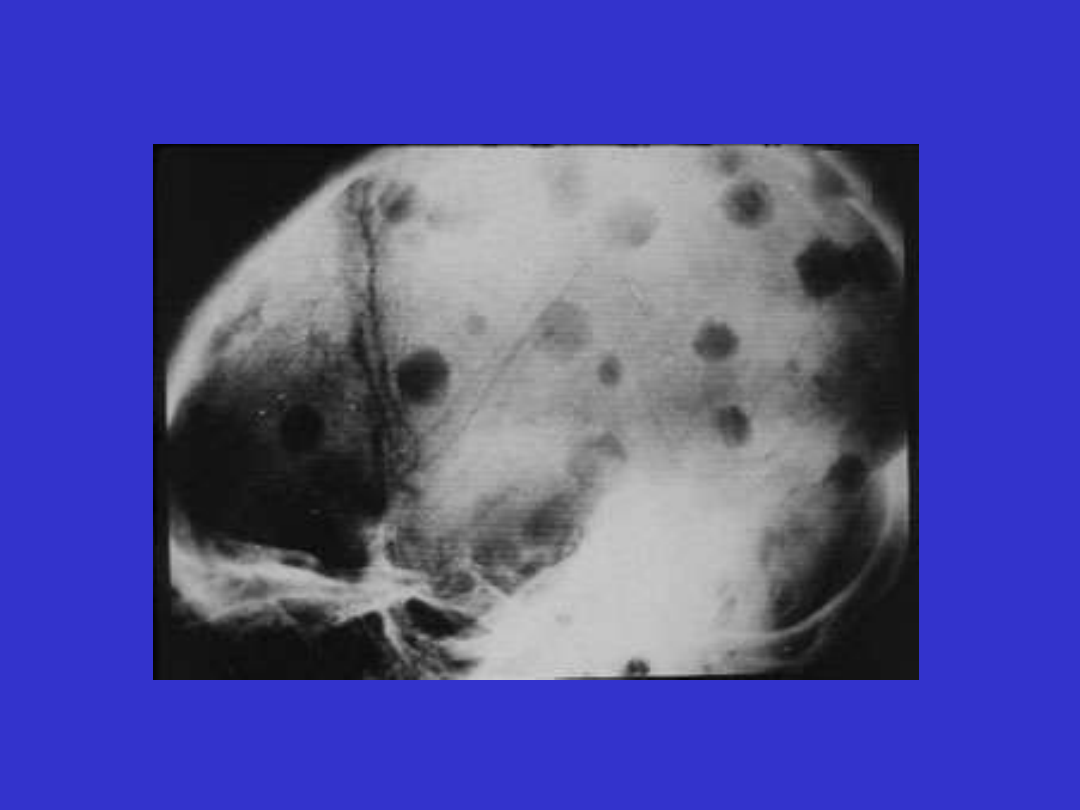
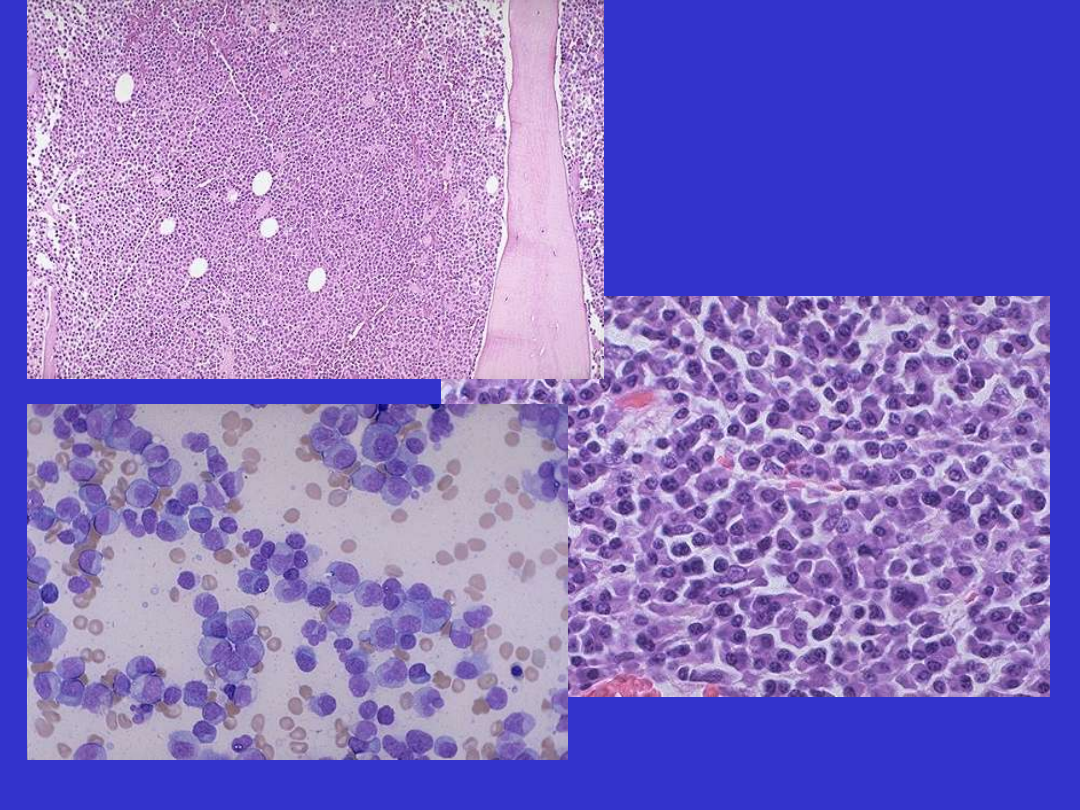
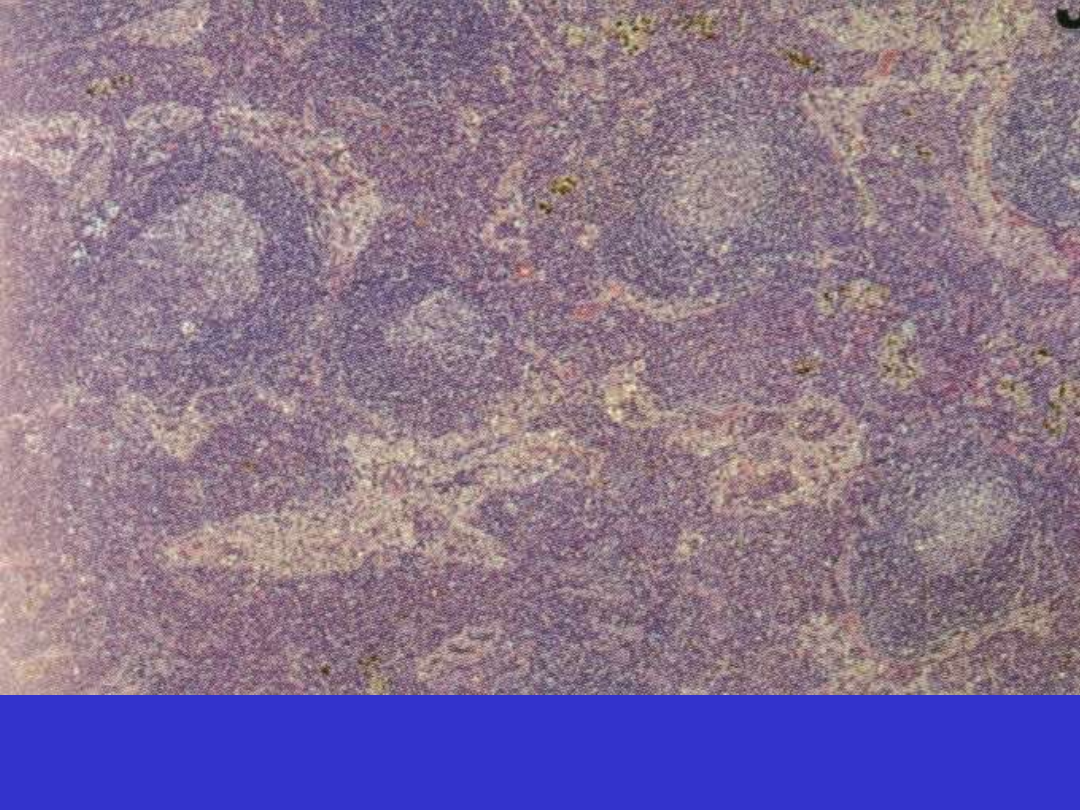
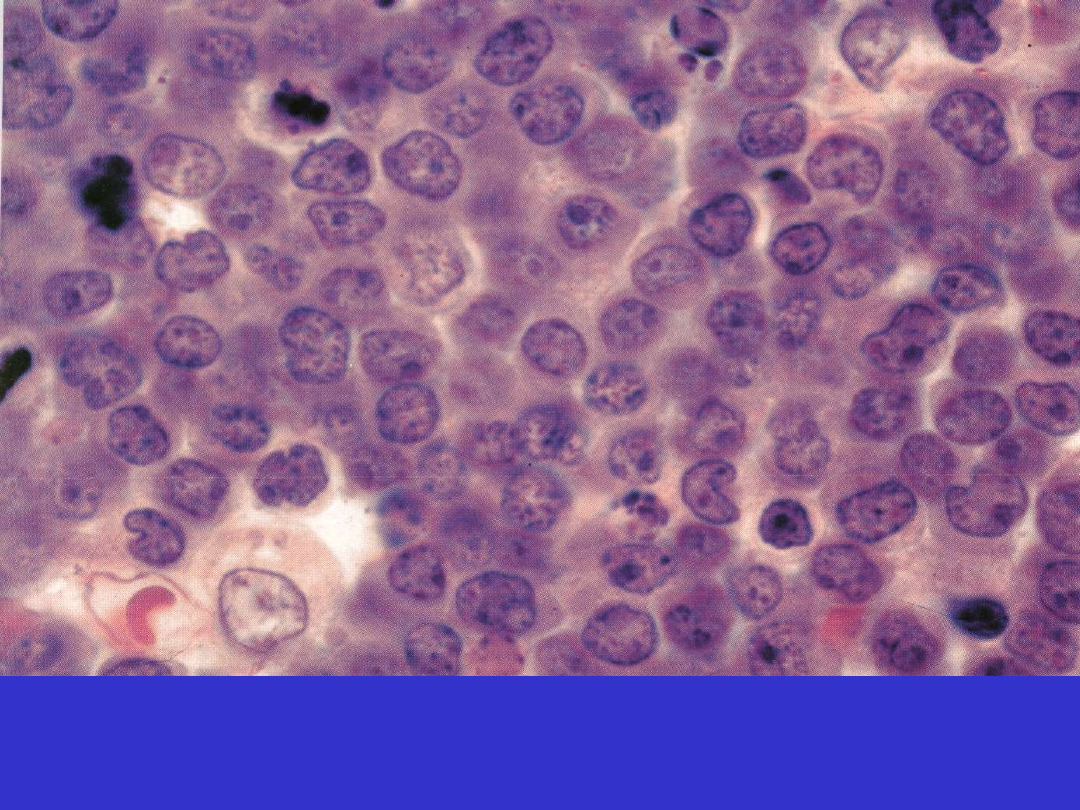
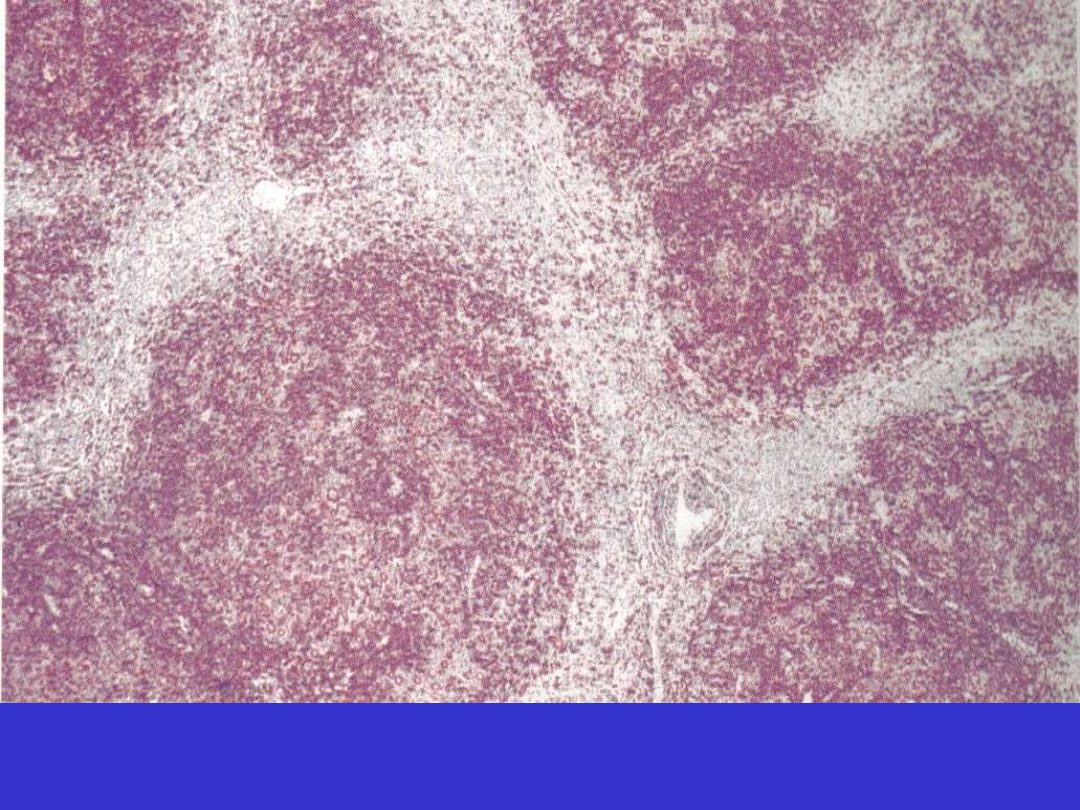
SLL/CLL
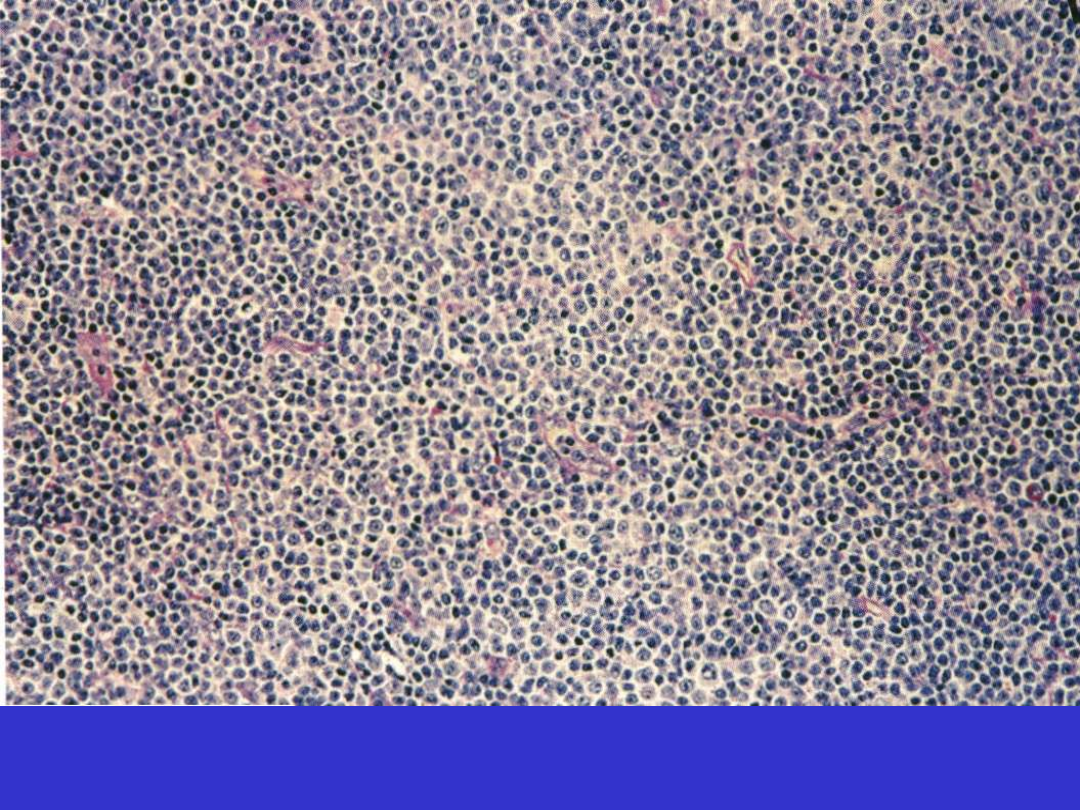
SLL/CLL; Centrum proliferacyjne
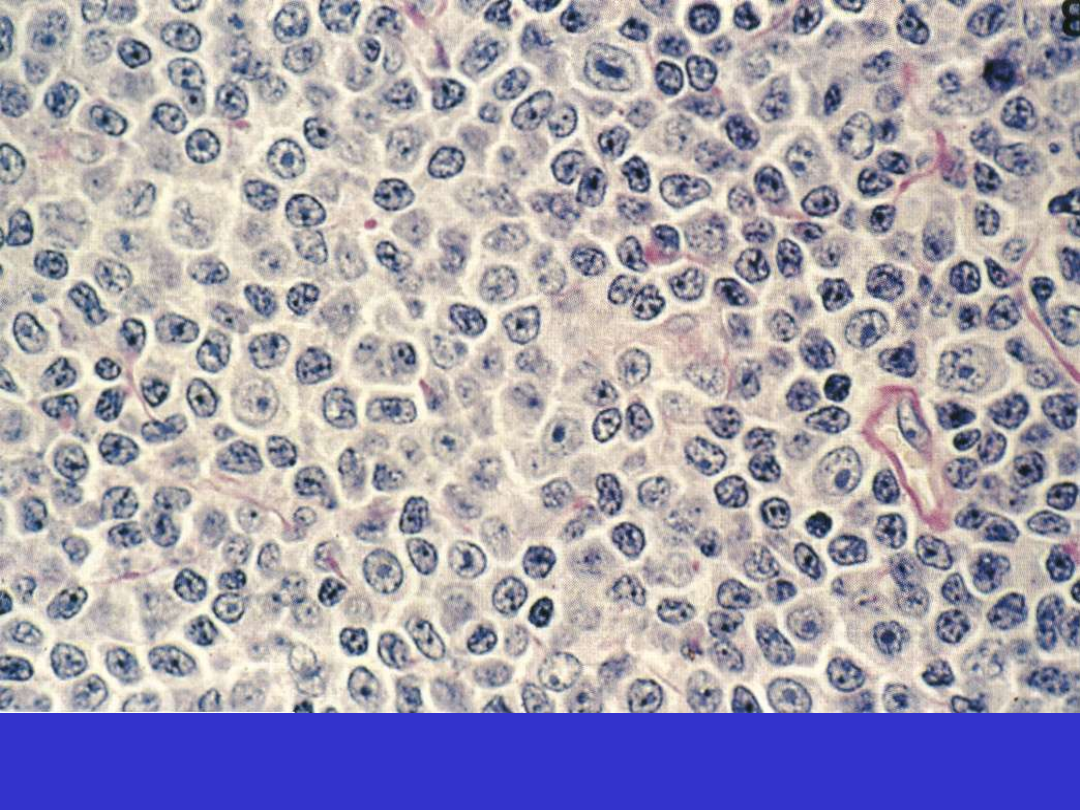
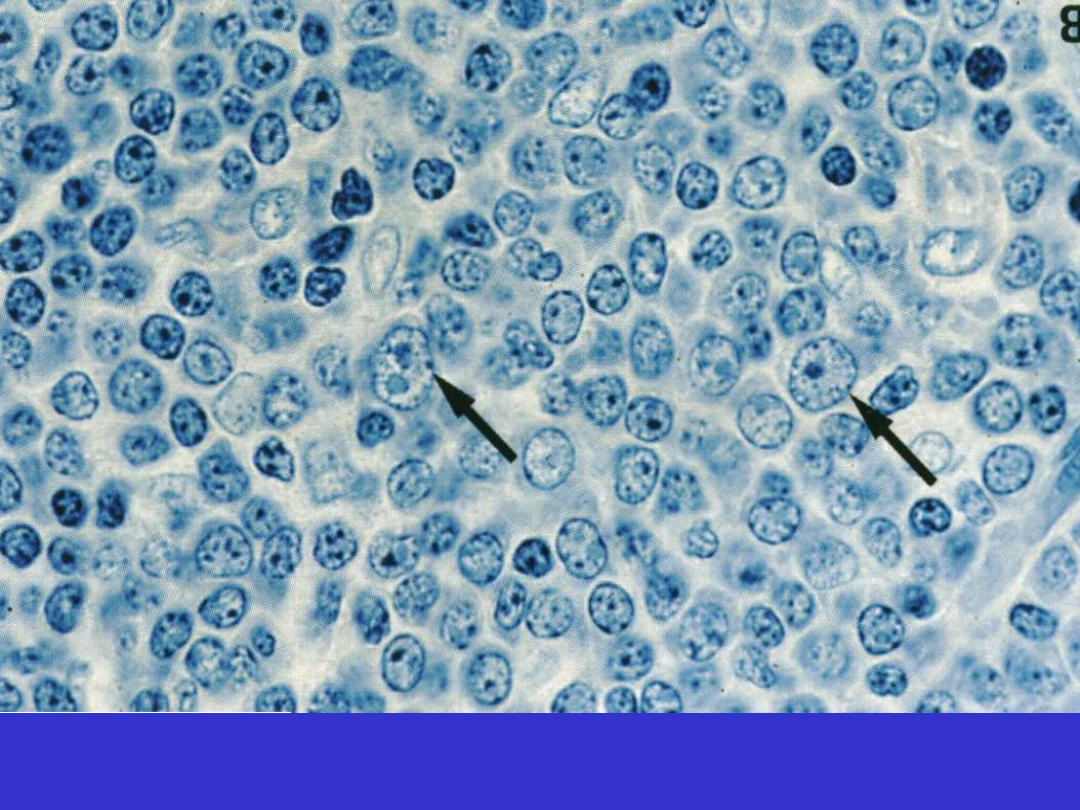
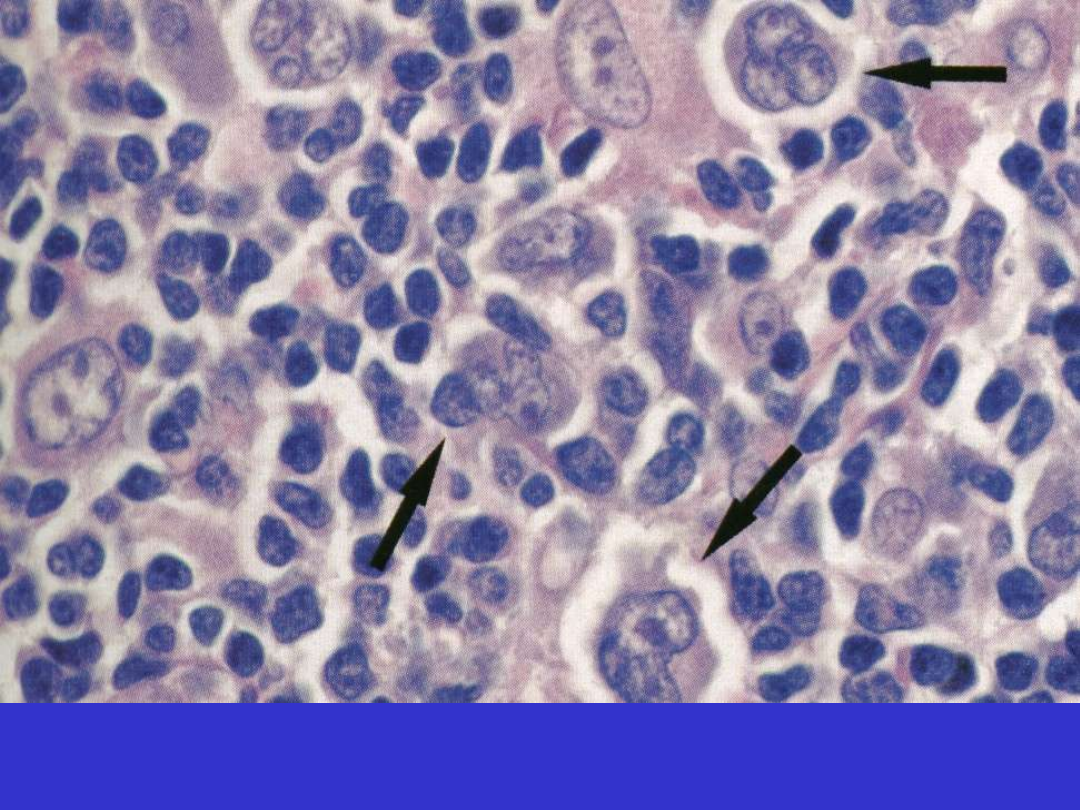
SLL/CLL; Centrum proliferacyjne, prolimfocyty i paraimunoblasty
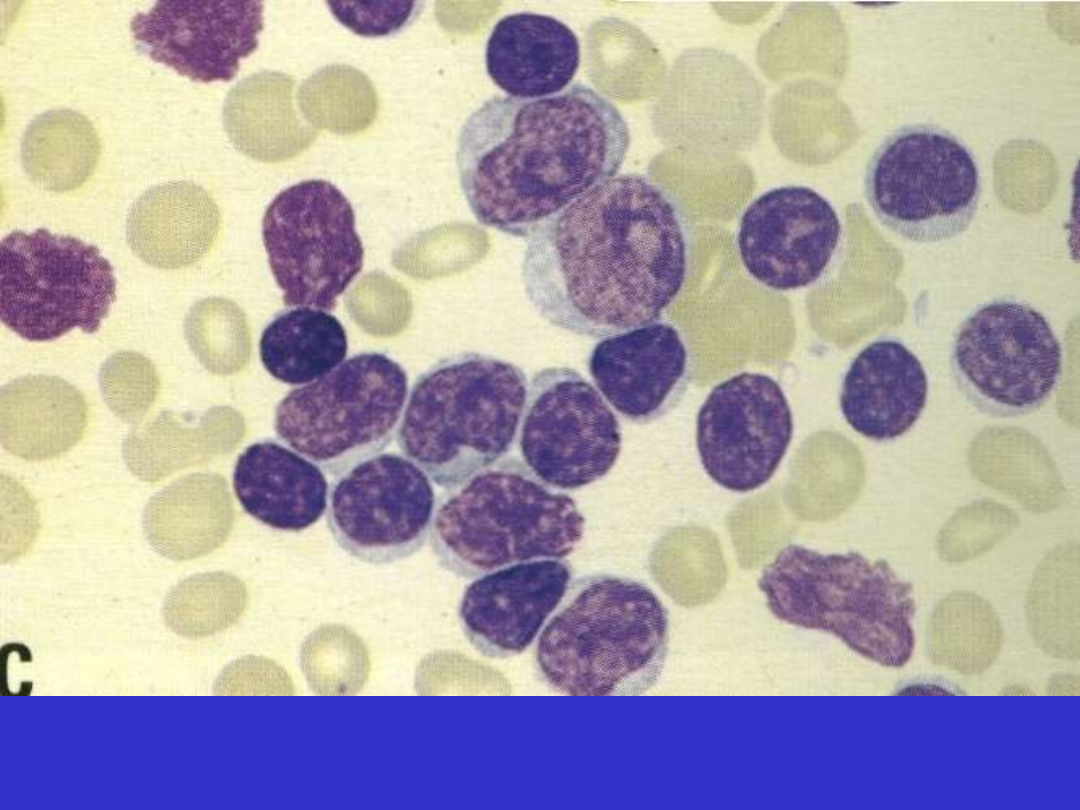
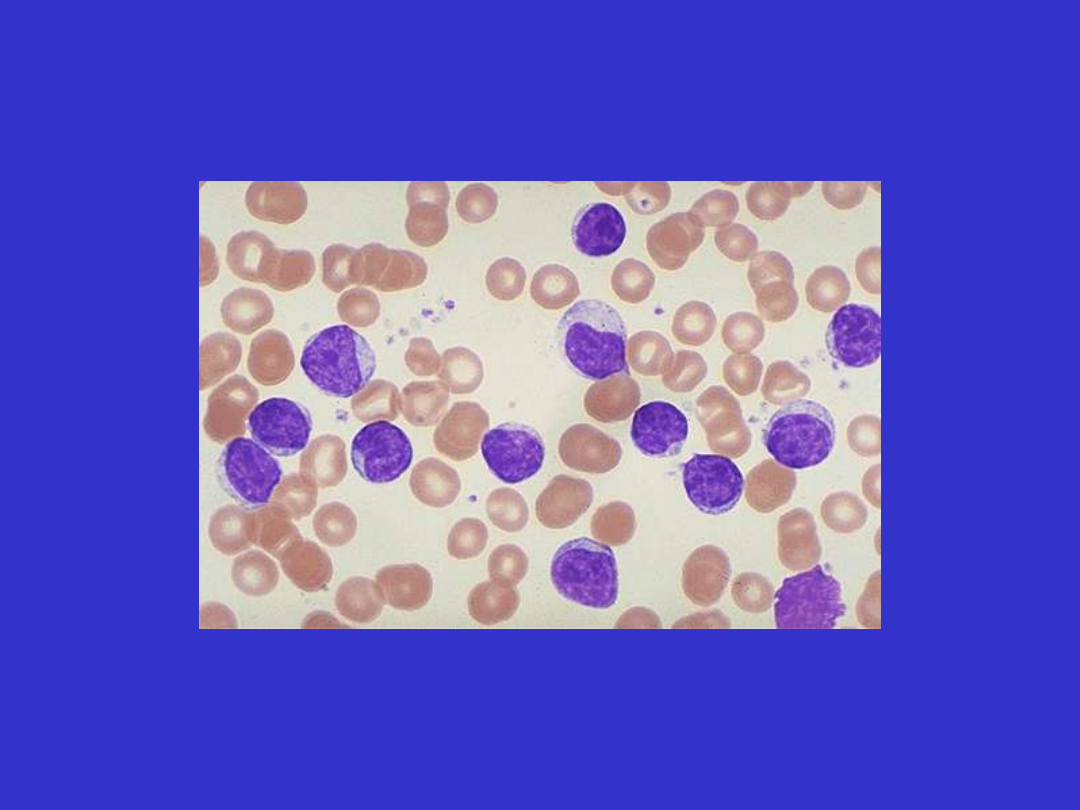
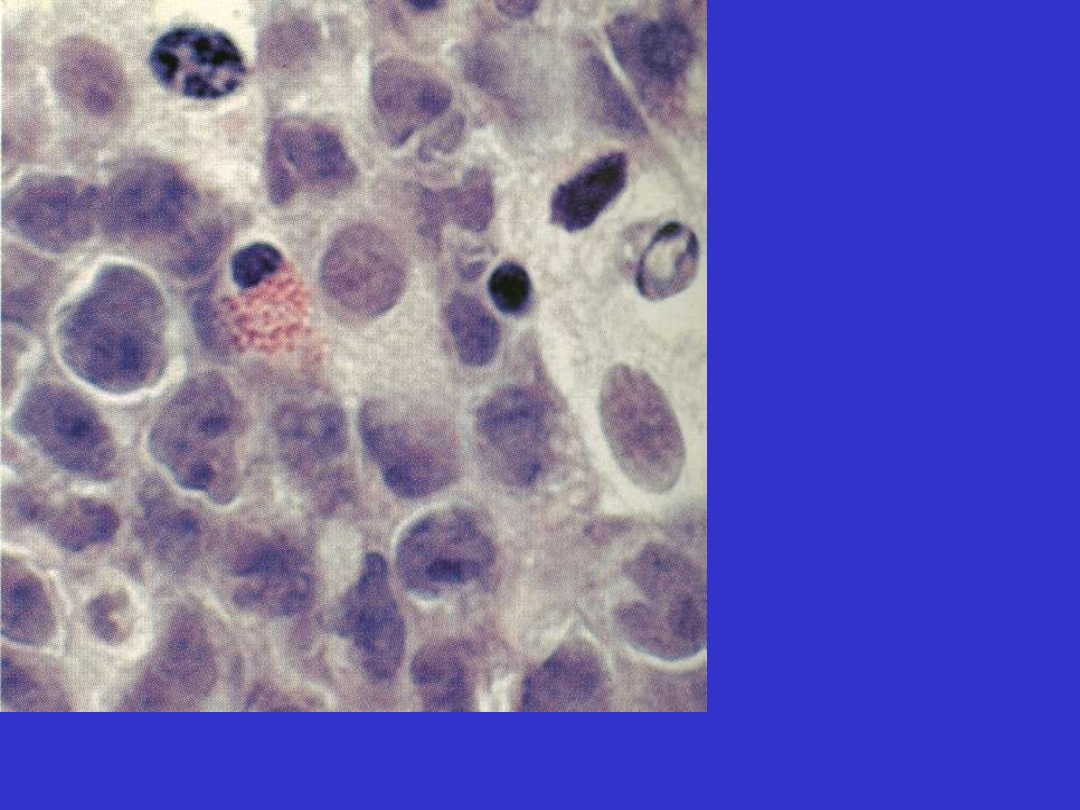
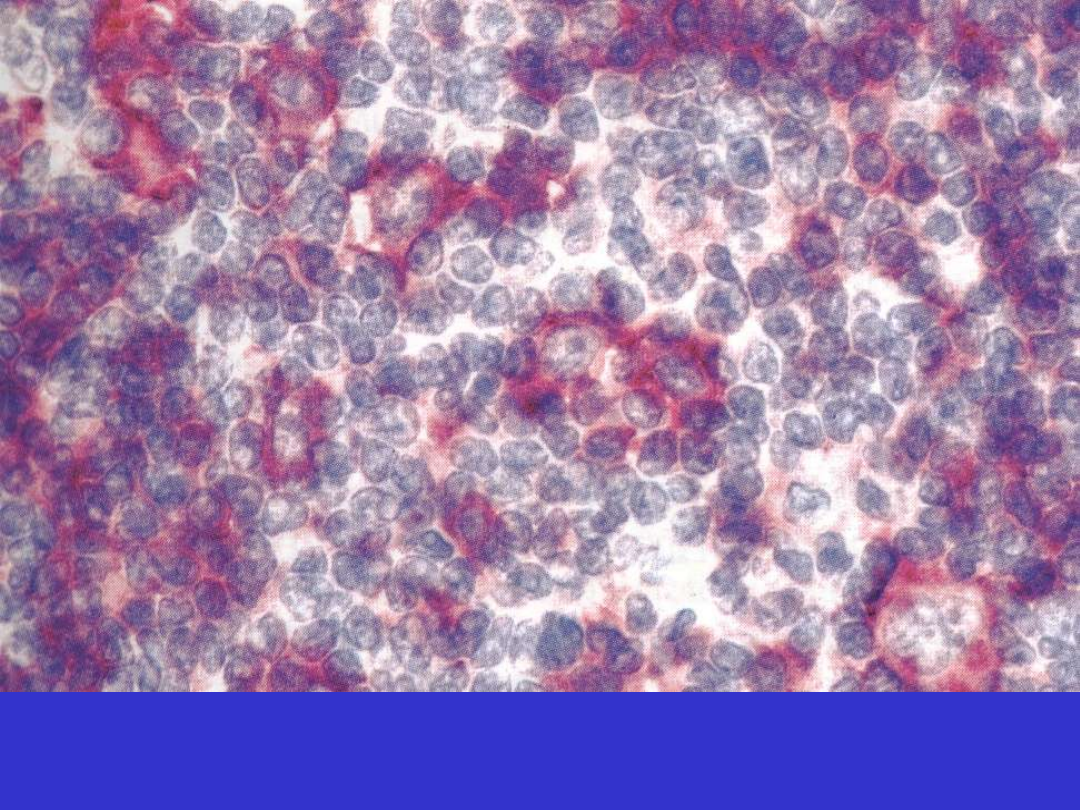
SLL/CLL; rozmaz krwi obwodowej, limfocyty i prolimfocyty
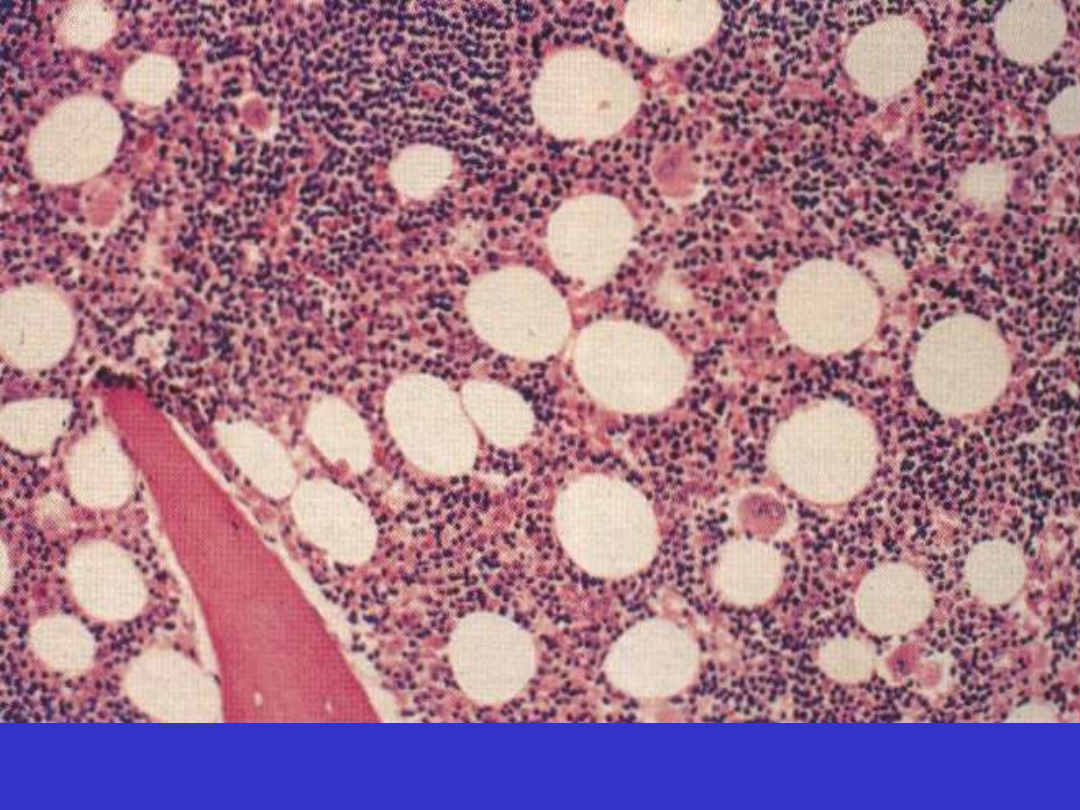
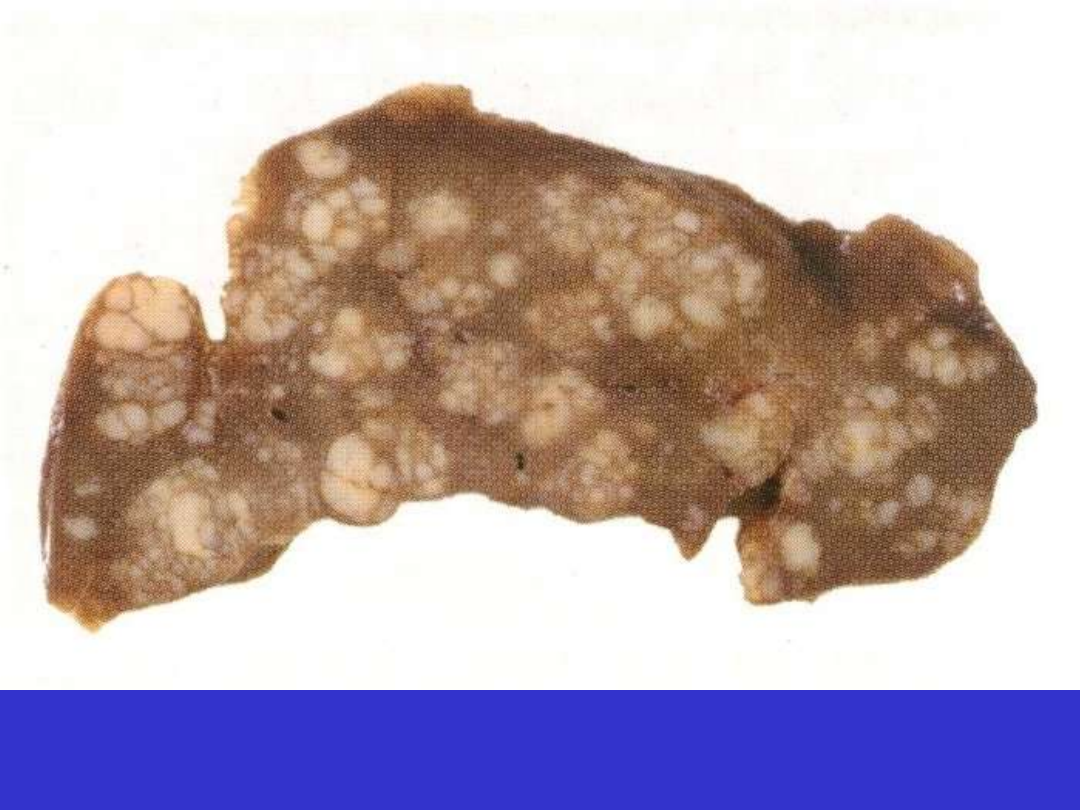
SLL/CLL; trepanobiopsja szpiku

Immunofenotyp:
Markery komórek B; CD19+, CD20+ (słabo), CD23+, CD79a
CD5+, CD10-, cyklina D1-.

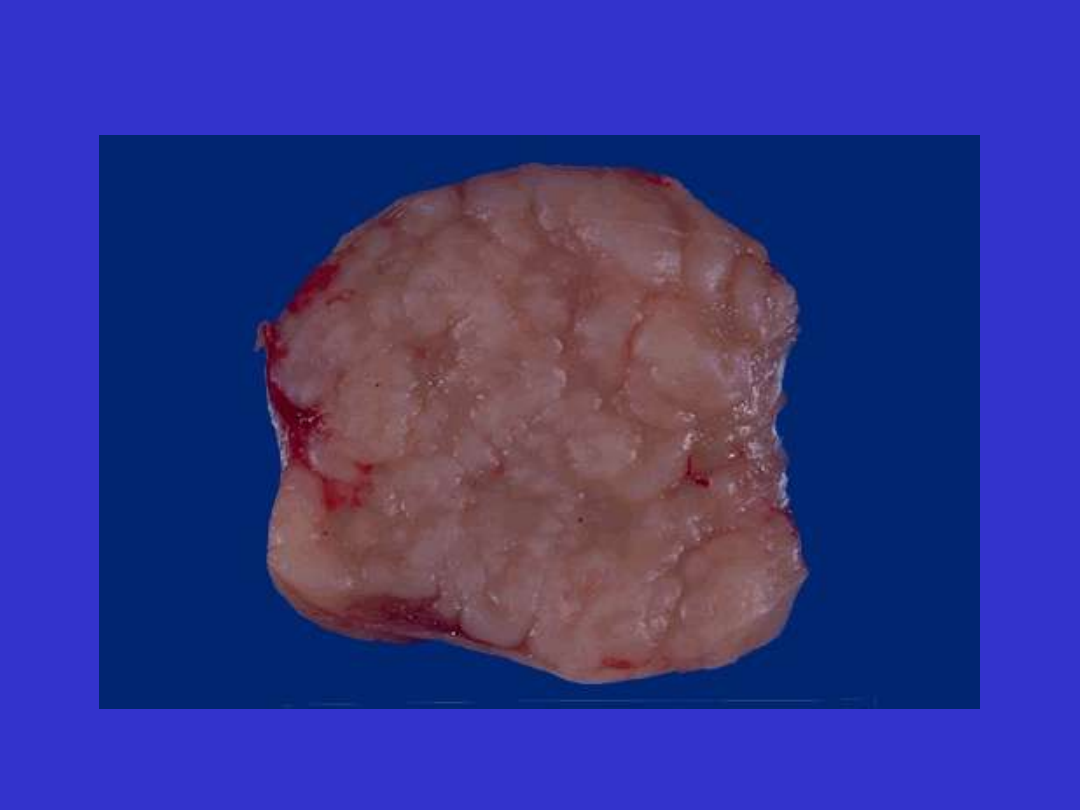
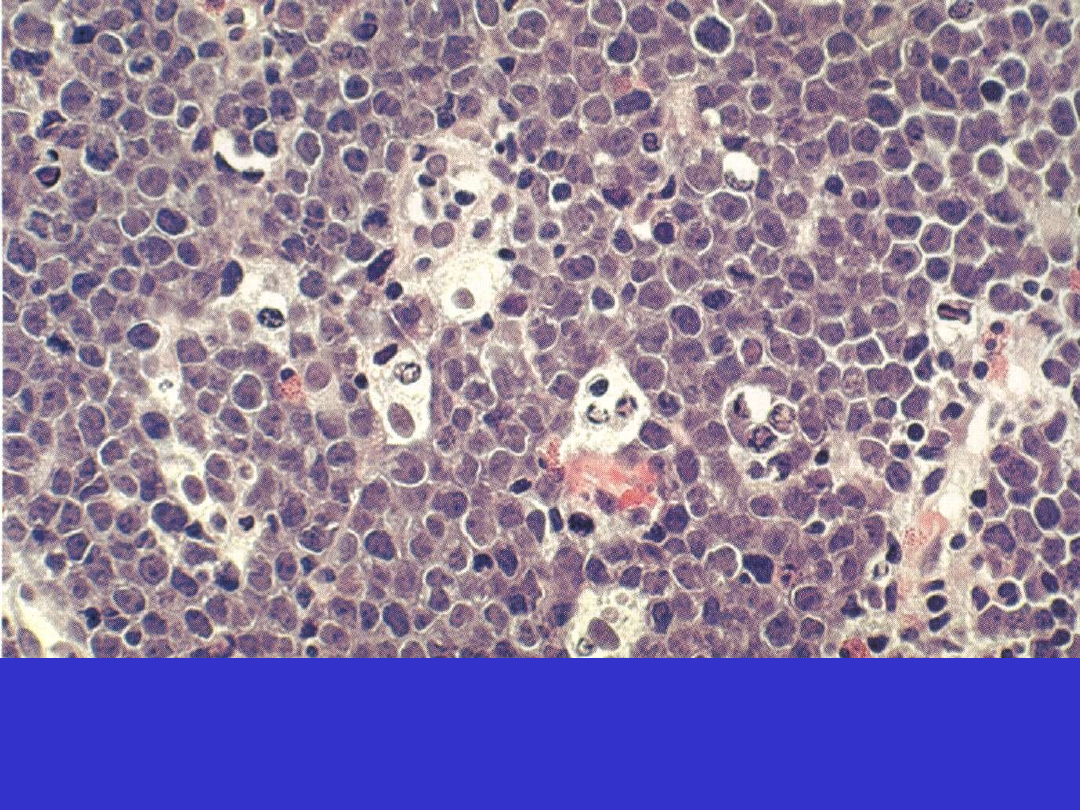
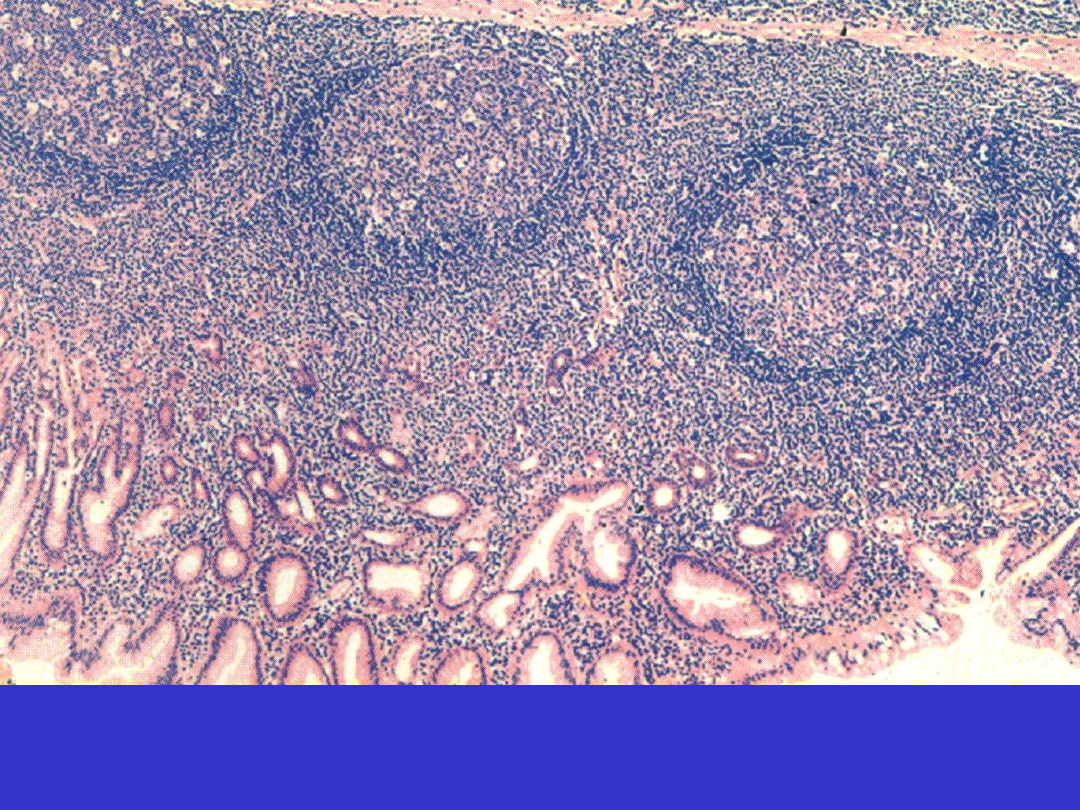
Chłoniak grudkowy (follicular lymphoma) – chłoniak z
komórek tworzących w normie grudki chłonne (CentroCyty,
CentroBlasty),
o przynajmniej częściowo zachowanym
grudkowym sposobie wzrostu (35% w USA, 22% poza USA).
Klinicznie
- Średni wiek ok. 59 r.ż.; M/K = 1/1,7; rzadko przed 20 r.ż.
(choroba ograniczona do głowy i szyi – migdałki).
- Rozsiane zajęcie węzłów chłonnych obwodowych i centralnych,
śledziony, w ok. 40% zajęcie szpiku.
-Tylko ok. 1/3 chorych w I, II stadium; mimo to przebieg z reguły
bezobjawowy (z wyjątkiem uogólnionej limfadenopatii).
- Możliwe pierwotne zajęcie narządów pozawęzłowych (pierścień
Waldeyera) innych (p. pok., tkanki miękkie, skóra) skutkiem
rozsiewu.
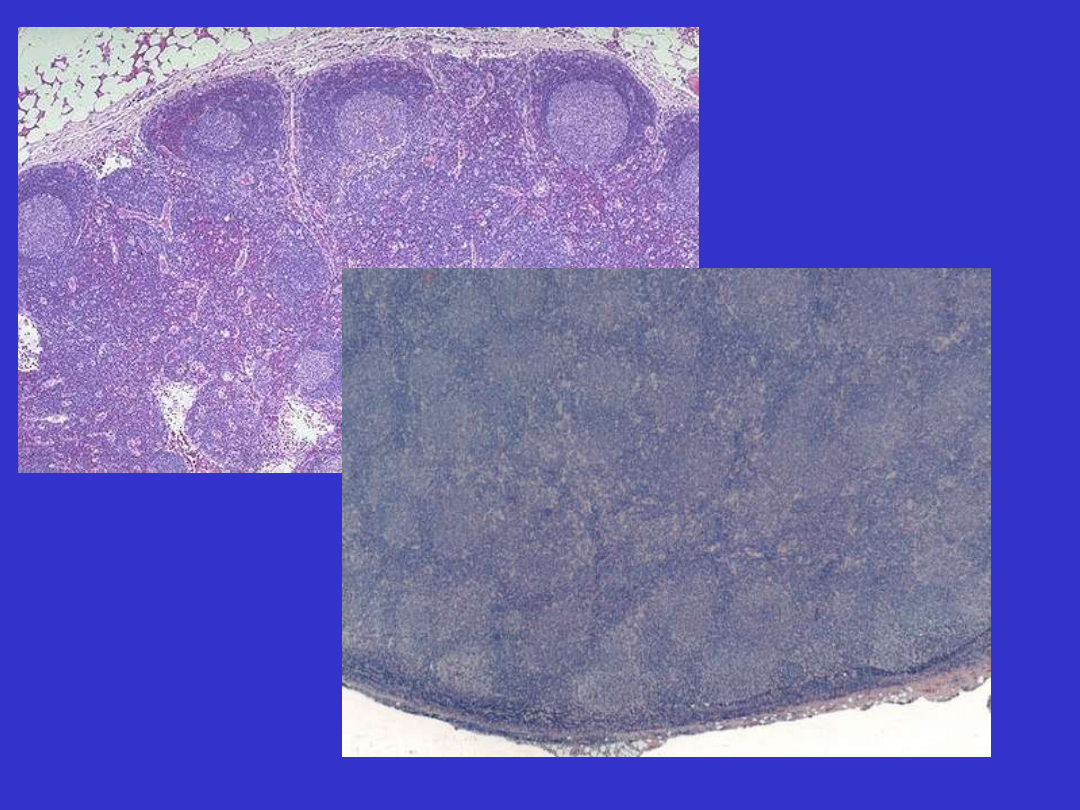

MORFOLOGIA:
- W większości przypadków dominuje obraz grudkowy, nowo-
tworowe grudki słabo odgraniczone, różnej wielkości i kształtu, bez
płaszcza, polaryzacji i makrofagów („obraz gwiaździstego nieba”);
- dwa typy komórek (CC - komórki małe do średnich, z
nieregularnymi jądrami, cytoplazma dość jasna; CB);
- grading na podstawie liczby CB (w polu 40x),

FL Grade 1
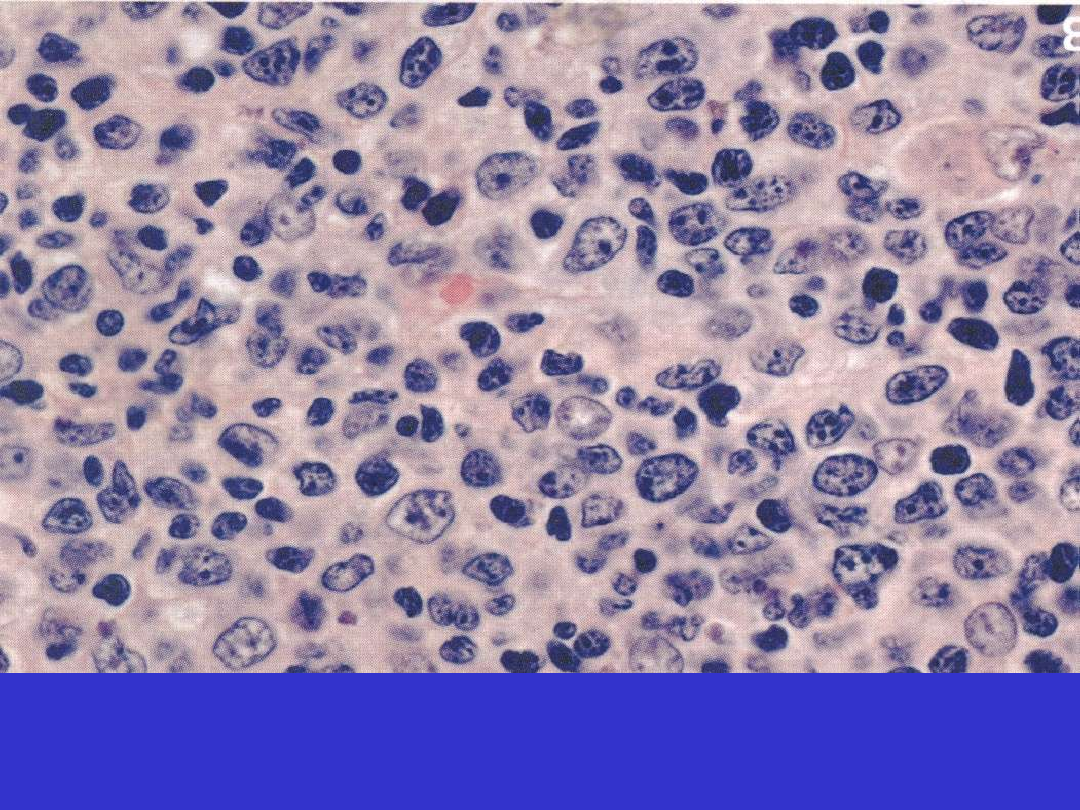
FL Grade 2
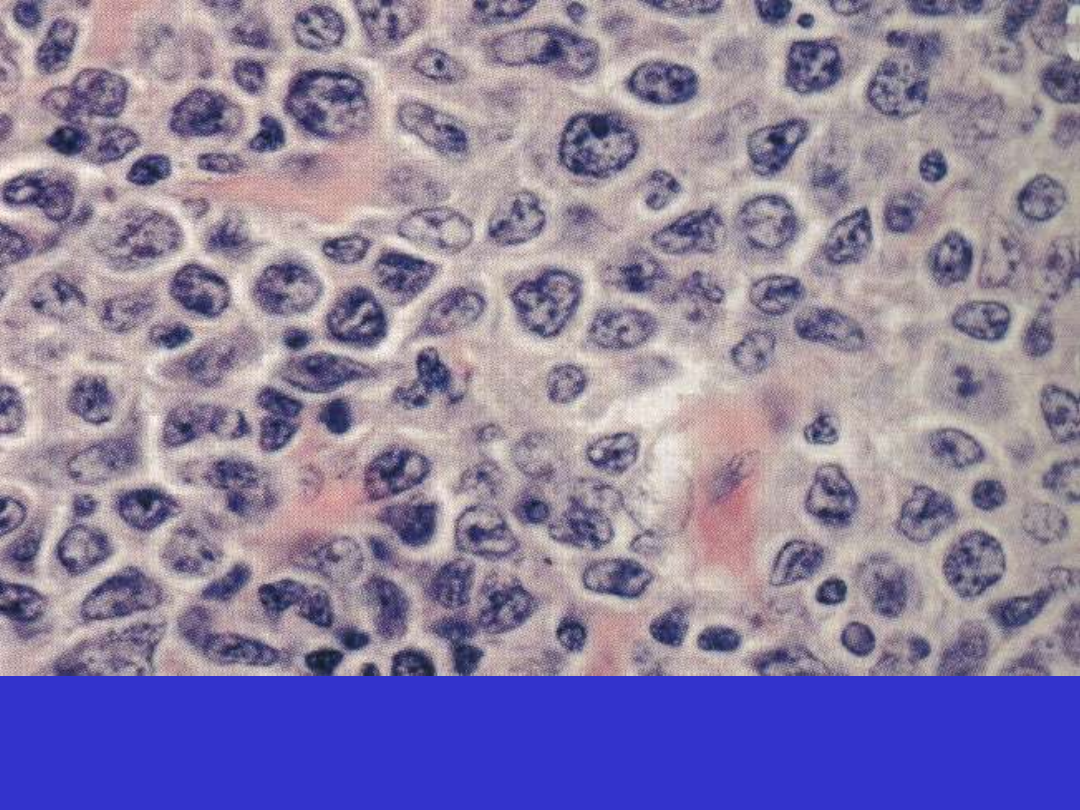
FL Grade 3A
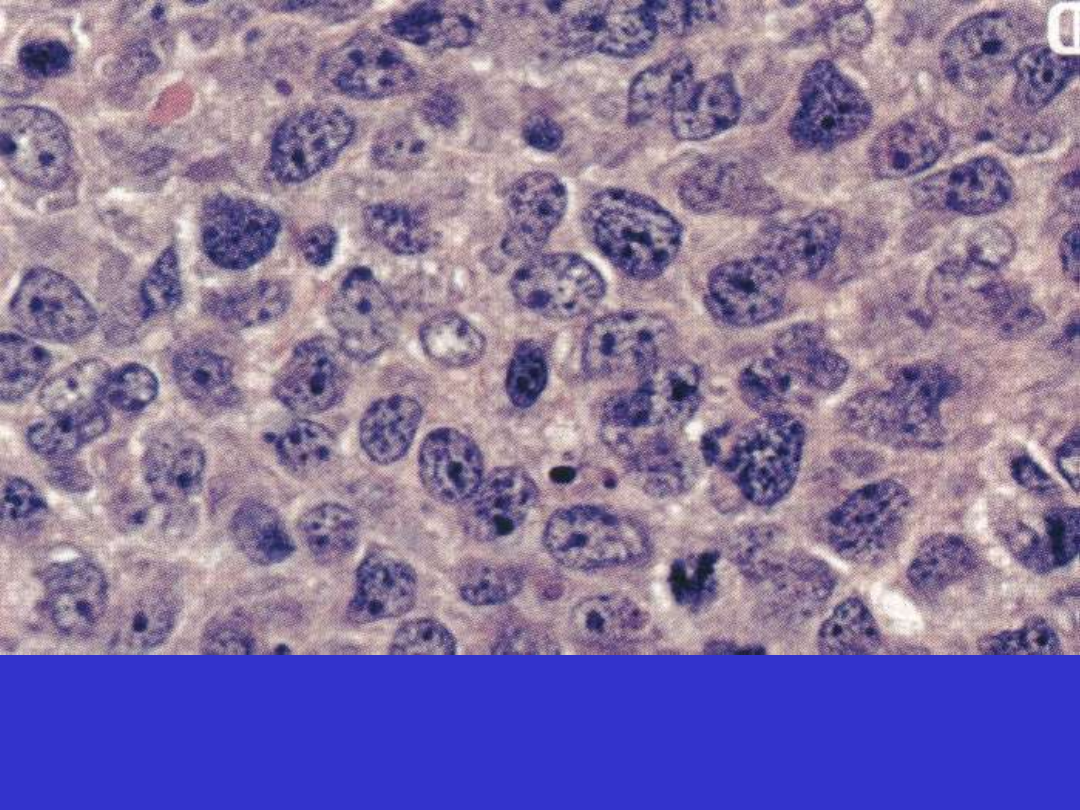
FL Grade 3B
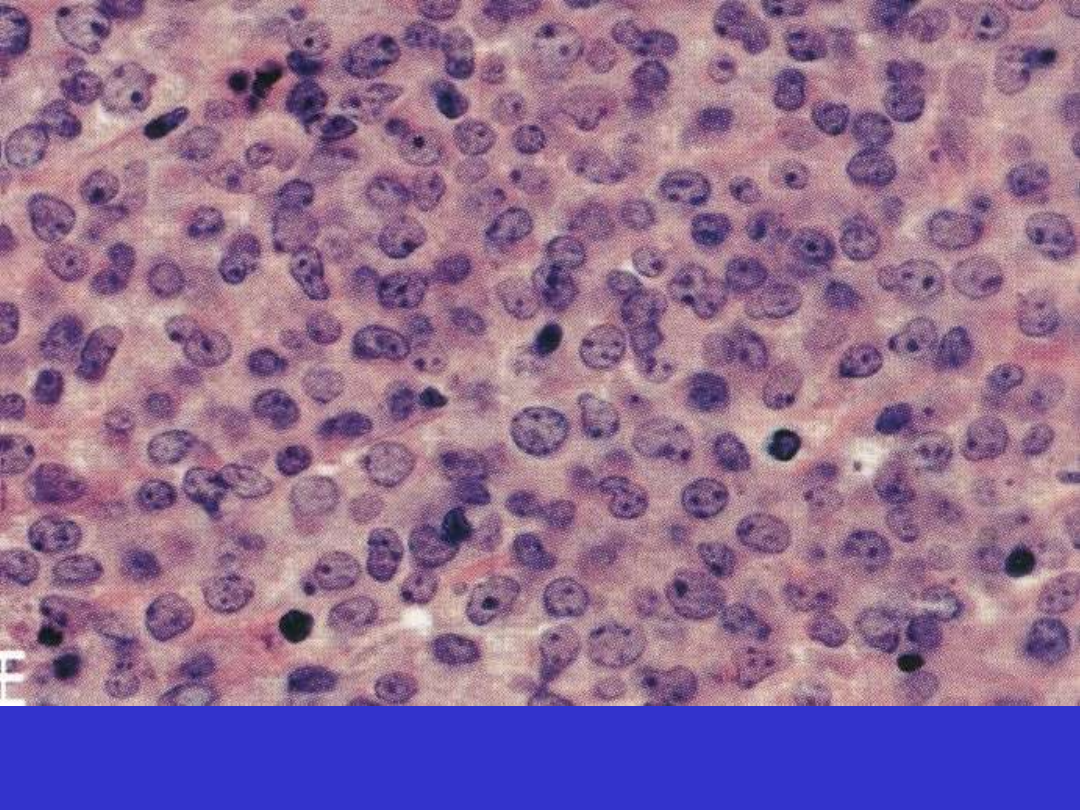
FL Grade 3B

Immunofenotyp:
IgM+, CD20+, CD10+, (CD19+, CD22+), Bcl2+ (czynnik
antyapoptotyczny, różnicuje grudki nowotworowe od reaktywnych,
ale nie od innych choniaków o niskim stopniu złośliwości),
CD79a+; CD5-, CD10+, w odróżnieniu od węzłów reaktywnych
restrykcja łańcuchów lekkich Ig,
ROKOWANIE:
Obraz histologiczny koreluje z rokowaniem, G1 i G2 – przebieg
powolny (7-9 lat), ale nie do wyleczenia; G3 – przebieg bardziej
agresywny, ale możliwe całkowite wyleczenie przy pomocy
chemioterapii (jak w chłoniaku wielkokomórkowym),
Rokowanie u dzieci – dobre.
Transformacja
do
chłoniaka
wielkokomórkowego
(25-35%
chorych) – rokowanie złe; brak odpowiedzi na leczenie.


Chłoniak z komórek płaszcza (mantle cell lymphoma
wg WHO, MCL)
– chłoniak z monomorficznych małych do
średnich komórek B, z nieregularnymi jądrami (CC); brak CB,
immunoblastów (3%-10% NHL);
• występuje u osób w średnim i starszym wieku (średnio ok. 60 r.ż.;
M/K = 2/1).
Klinicznie:
Najczęściej dotyczy węzłów chłonnych, ale także śledzionę, szpik
kostny; mogą być zajęte narządy pozalimfatyczne (przew. pok. –
30% - multiple lymphomatous polyposis, oraz pierścień Walde-
yera);

MCL, lymphomatous polyposis, jelito grube

MCL, lymphomatous polyposis, jelito grube

KLINICZNIE:
Większość chorych w stadium III lub IV, z limfadenopatią,
hepatosplenomegalią (często masywną) oraz zajęciem szpiku (pow.
50%), zajęcie krwi obwodowej w 25%.
MORFOLOGIA:
zatarcie architektoniki, obraz z zajęciem „płaszcza” wokół grudek,
obraz rozlany lub z tworzeniem słabo widocznych grudek z
komórek nowotworowych; grudki słabo odgraniczone, różnej
wielkości i kształtu;
- komórki
małe
do
średnich,
z
nieregularnymi
jądrami,
przypominają CC;
-liczne szkliwiejące naczynia w tle,
-wariant
blastoidny
(komórki większe, polimorfizm, wzrost
wielkości jąder,
- transformacja do chłoniaka wielkokomórkowego nie występuje;
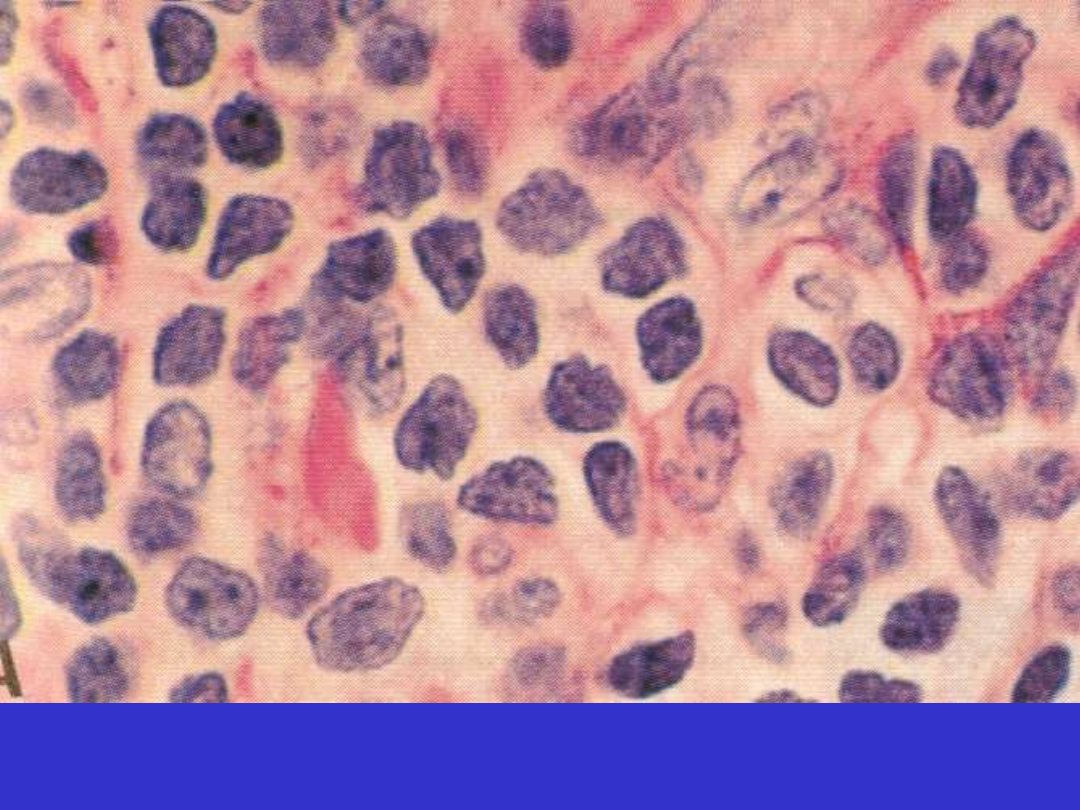
MCL, rozrost strefy płszcza dookoła prawidłowych grudek

MCL, rozrost rozlany i guzkowy
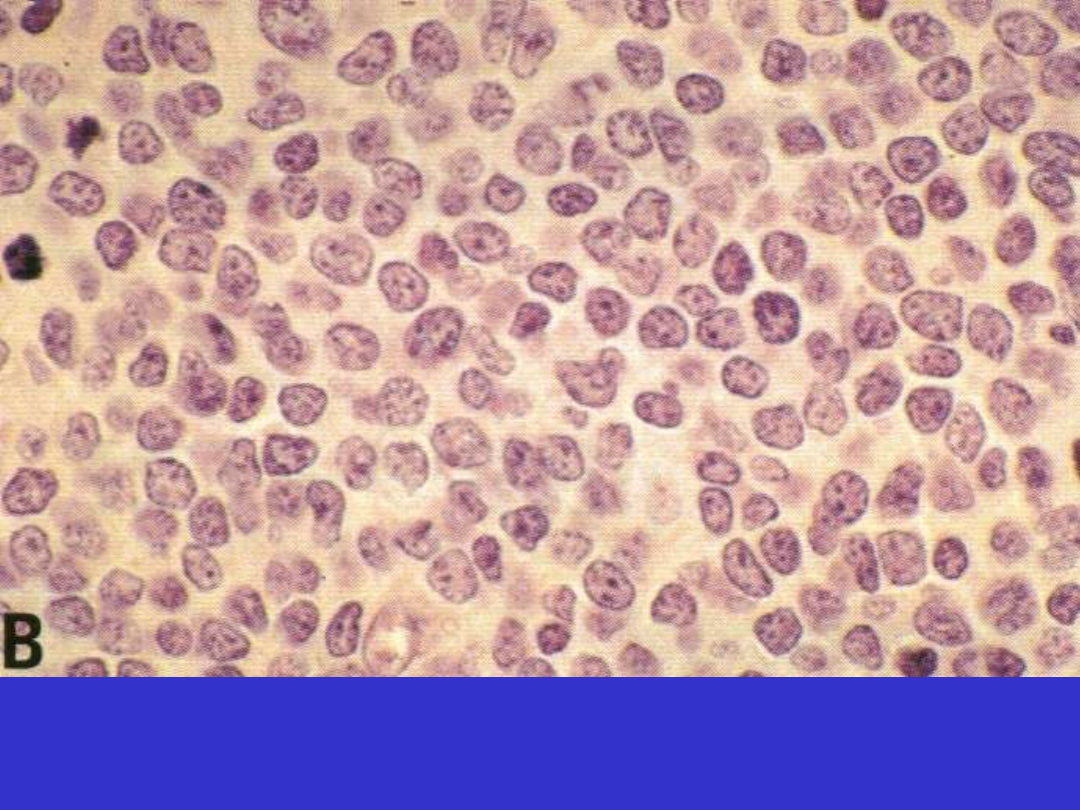
MCL, komórki przypominające centrocyty, szkliwiejące naczynia, paS
MCL, typ blastoidny, komórki przypominają limfoblasty
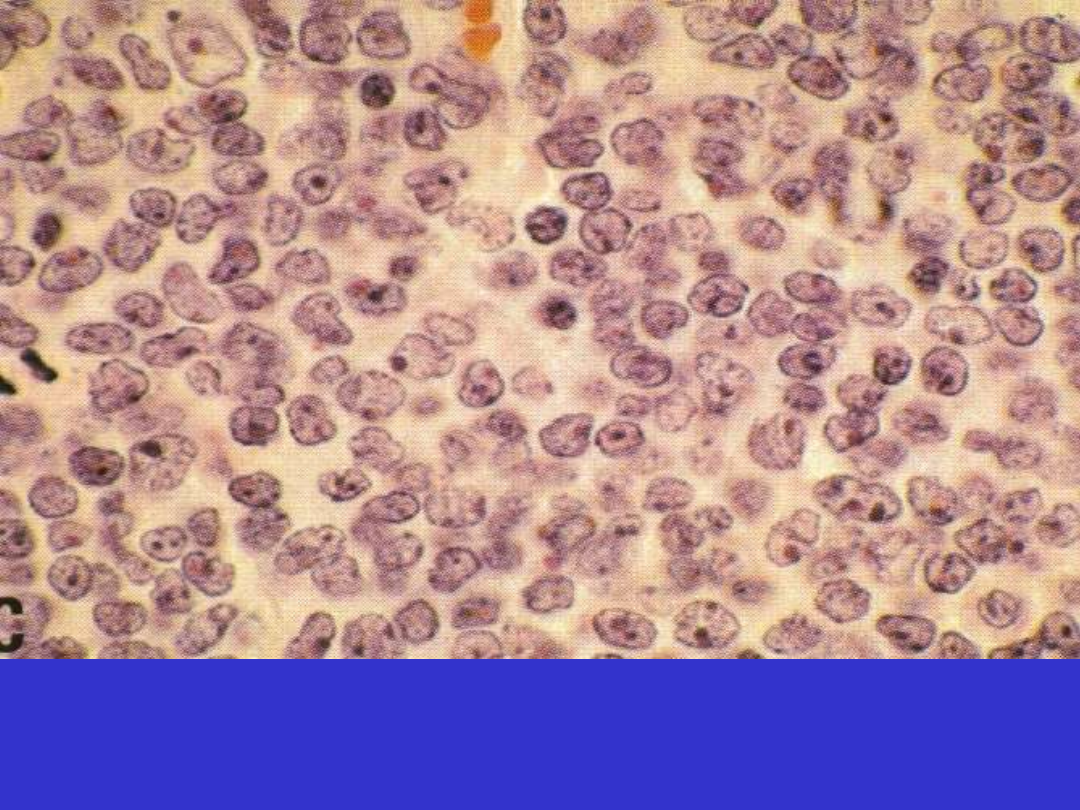
MCL, typ blastoidny; komórki polimorficzne, wydatne jąderka

Immunofenotyp:
Markery komórek B; CD19+, CD20+ (słabo), CD23-, CD79a
CD5+, CD10-, cyklina D1+ (DD – SLL/CLL).
Kariotyp; t(11;14); fuzja genu cykliny D1 (11) z regulatorami
produkcji łańcuchów ciężkich Ig (chromosom 14) nadekspresja
cykliny D1
Prognoza; średnia przeżycia 3-5 lat, wiekszość nieuleczalna
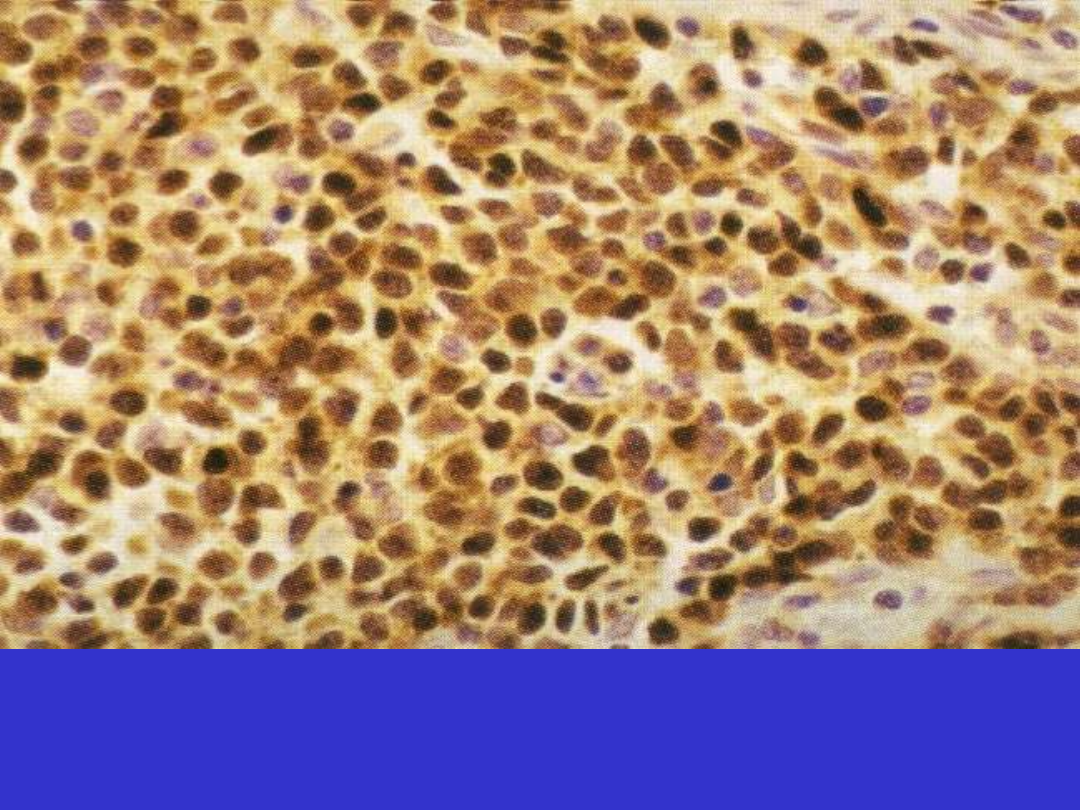
MCL, cyklina D1+ w jądrach komórek nowotworowych

Chłoniak rozlany z dużych komórel B (diffuse large B-cell
lymphoma, DLBCL)
– rozlana proliferacja dużych
nowotworowych limfoidnych komórek B o jądrach mających
wielkość równą lub większą od jądra prawidłowego makrofaga lub
ponad dwukrotnie większych od jąder prawidłowych limfocytów.
- 30%-40% NHL.
- występuje u osób w średnim i starszym wieku (średnio ok. 60 r.ż.;
M/K = 2/1), czasem u dzieci.
Umiejscowienie:
Najczęściej dotyczy węzłów chłonnych, ale w 40% pozawęzłowo;
najczęściej p.pok. (żołądek, ok. krętniczo-kątnicza), ale także skóra,
o.u.n., kości, jądra, tkanki miękkie, ślinianki, żeński układ rozrodczy,
płuca, nerki, wątroba, pierścień Waldeyera, śledziona. Pierwotne zajęcie
szpiku kostnego i/lub krwi obwodowej rzadko spotykane.

Klinicznie: typowy przebieg; szybko powiększający się guz, często
proces rozsiany w momencie diagnozy
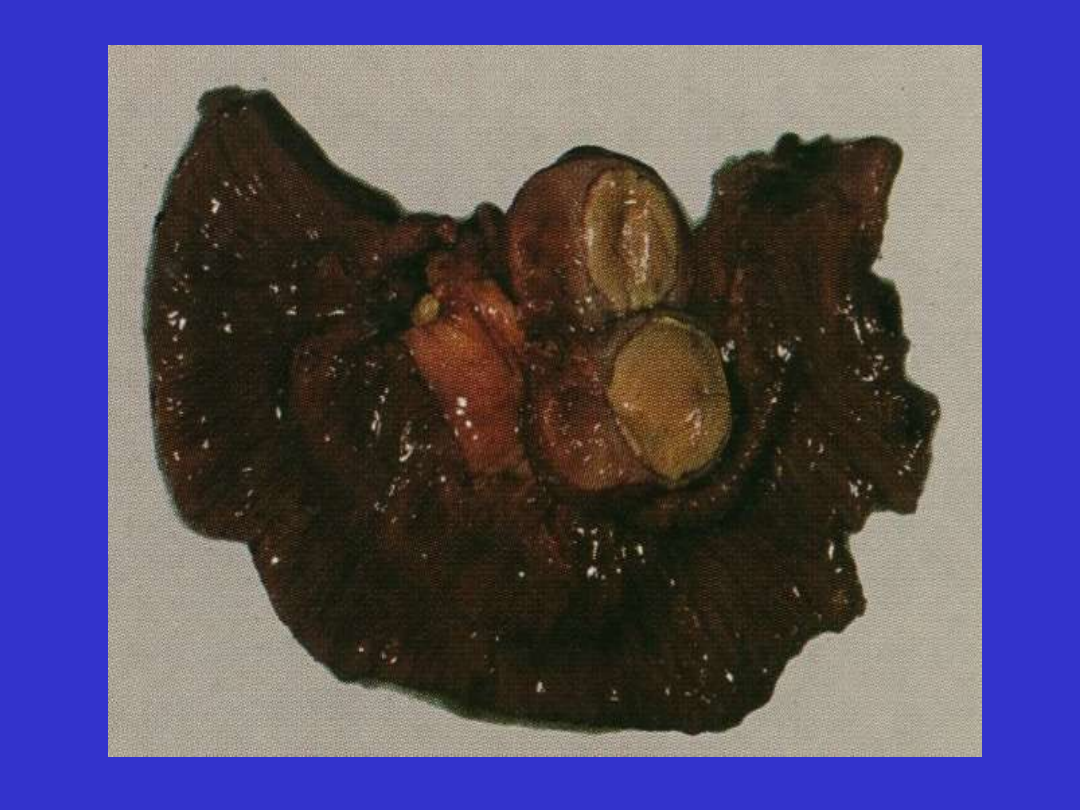
Morfologia; rybie mięso, krwotoki, martwica. Mikroskopowo warianty
centrobalstyczny (najczęściej), immunoblastyczny, bogaty w komórki
T/histiocyty, anaplastyczny.
Immunofenotyp; markery kk. B + (CD19, 20, 22, 79a). Anaplastyczne-
CD30+. Frakcja proliferacyjna (Ki67, MIB) - > 40%, czasem > 90%
Kariotyp; w 30% t(11;18) z rearanżacją BCL2 – jak w chłoniakach
grudkowych
Prognoza; agresywne, możliwe całkowite wyleczenie przy użyciu
wieloskładnikowej chemioterapii
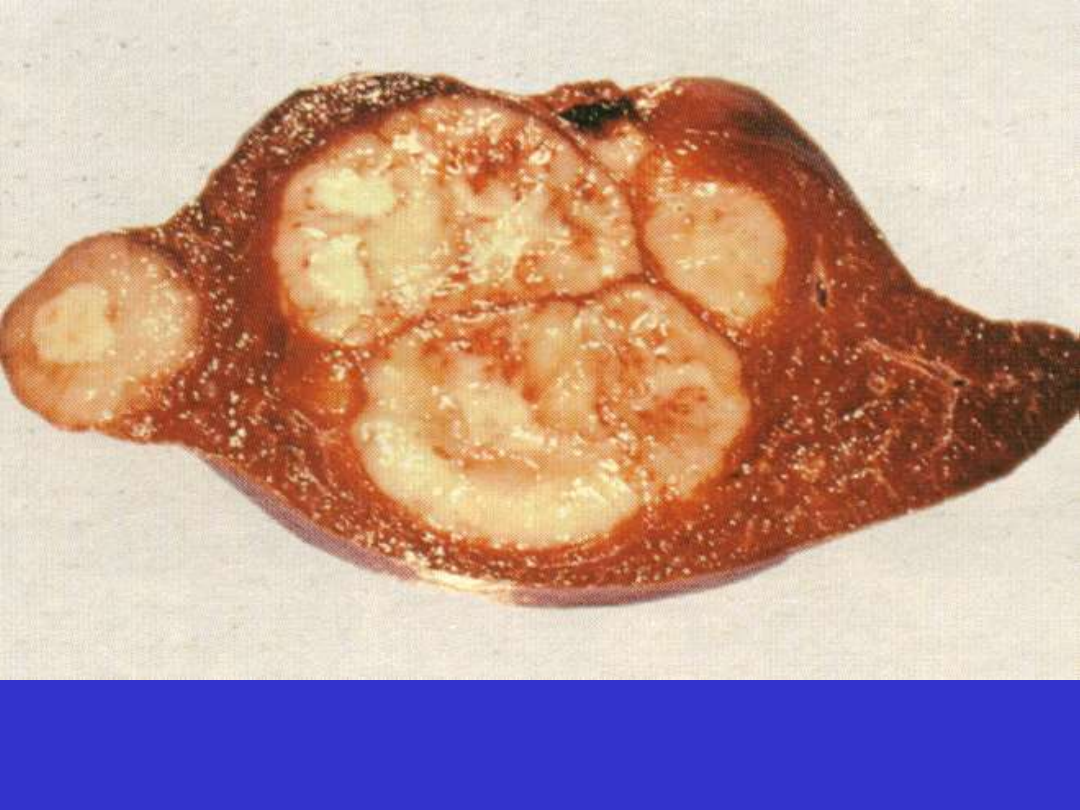
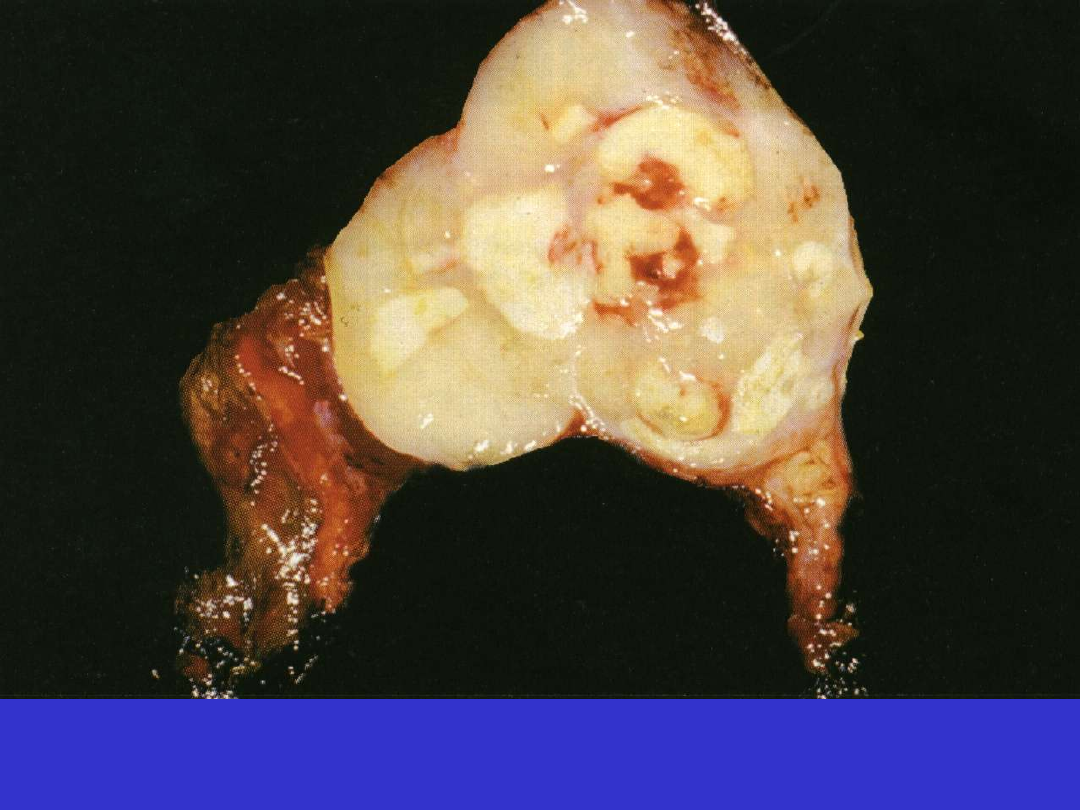
DLBCL, śledziona; rybie mięso, krwotoki, martwica
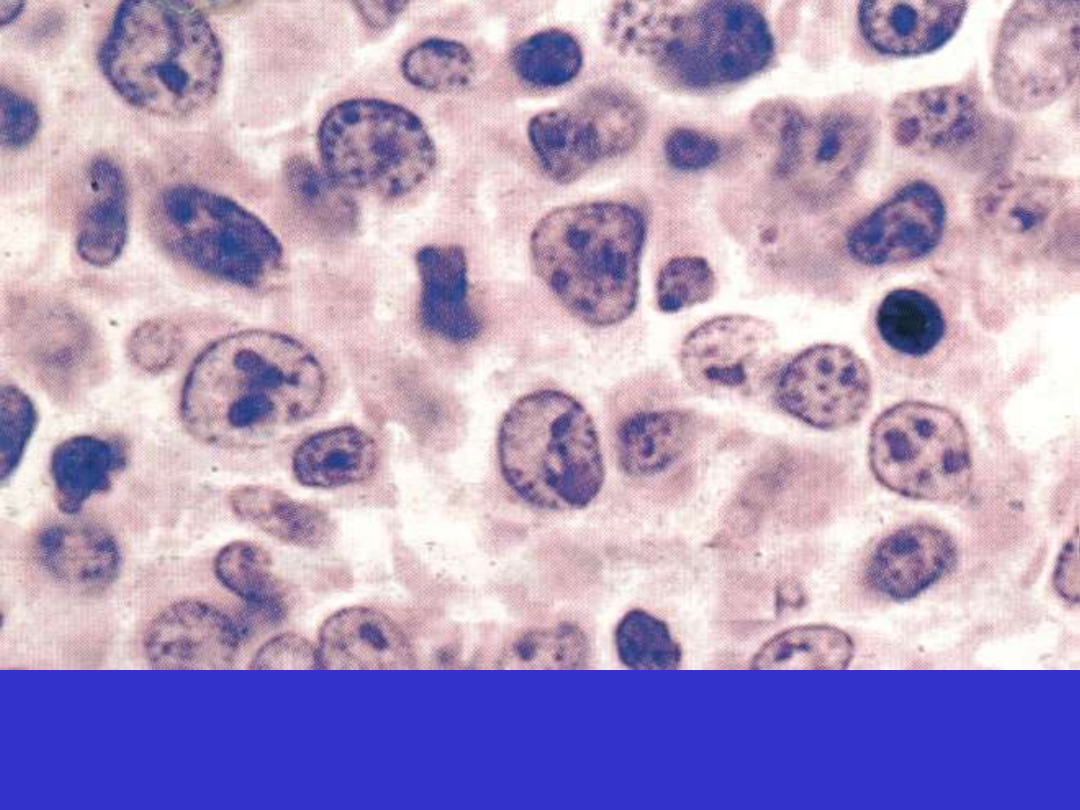
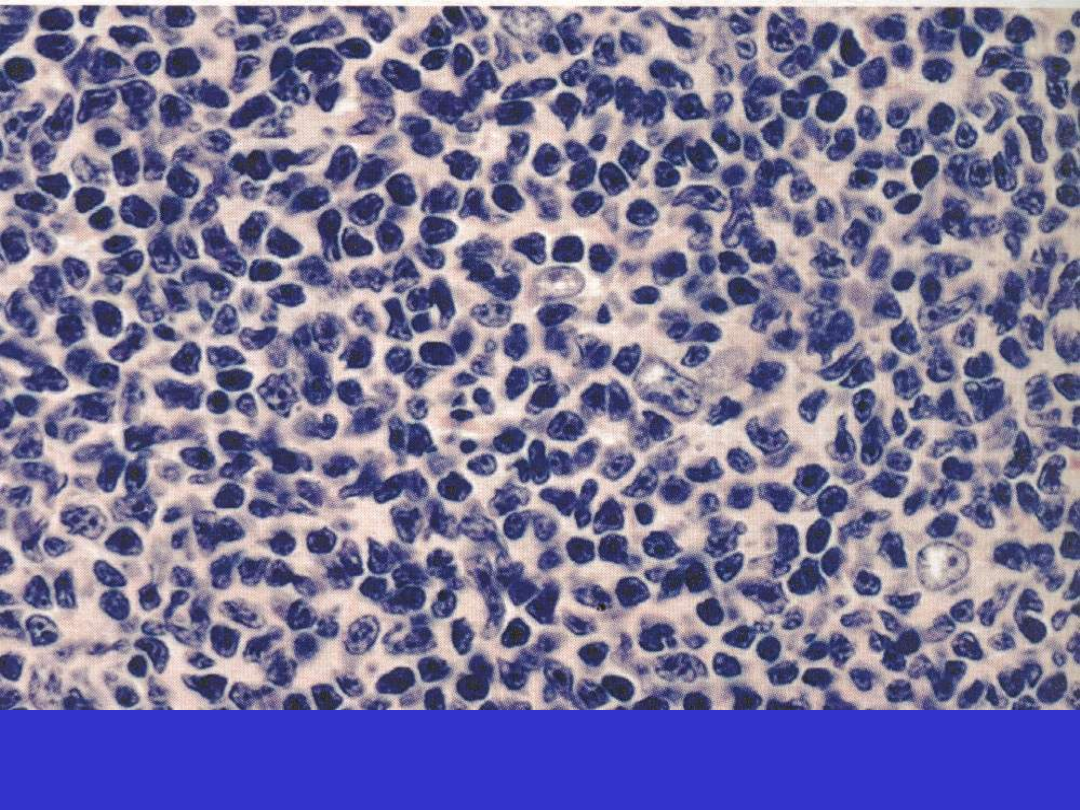
DLBCL; wariant centroblastyczny
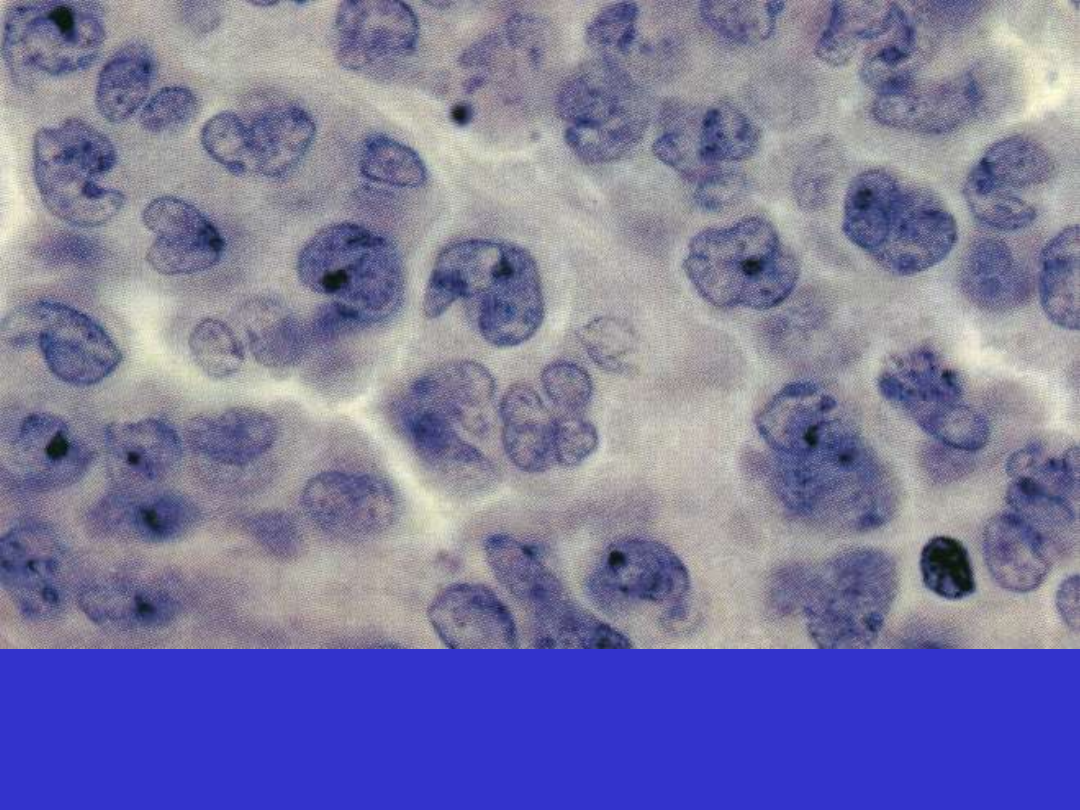
DLBCL; wariant centroblastyczny, jądra płatowate i polimorfizne
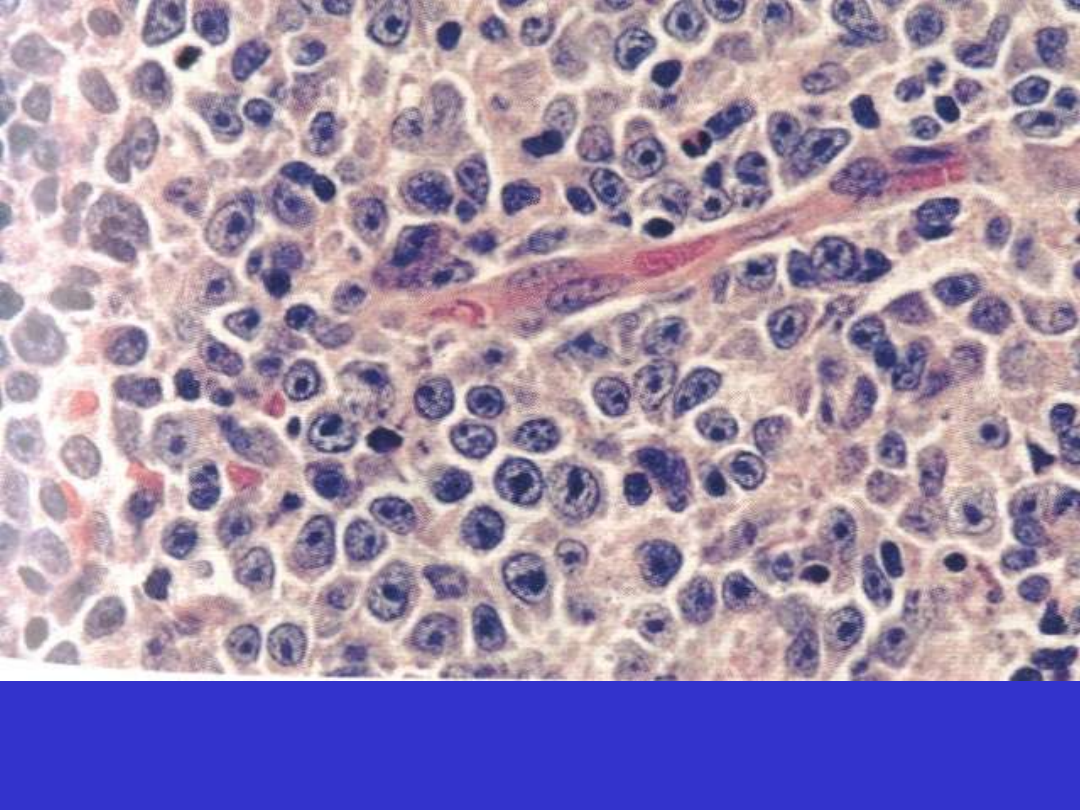
DLBCL; wariant immunoblastyczny
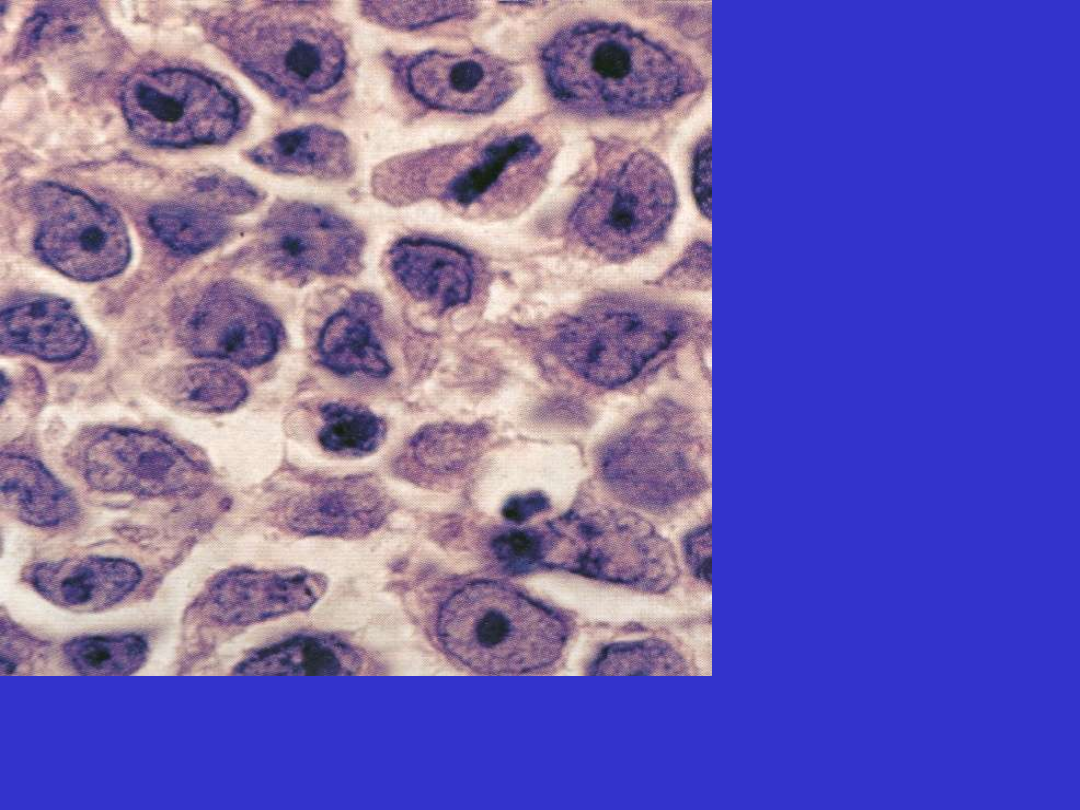
DLBCL; wariant immunoblastyczny
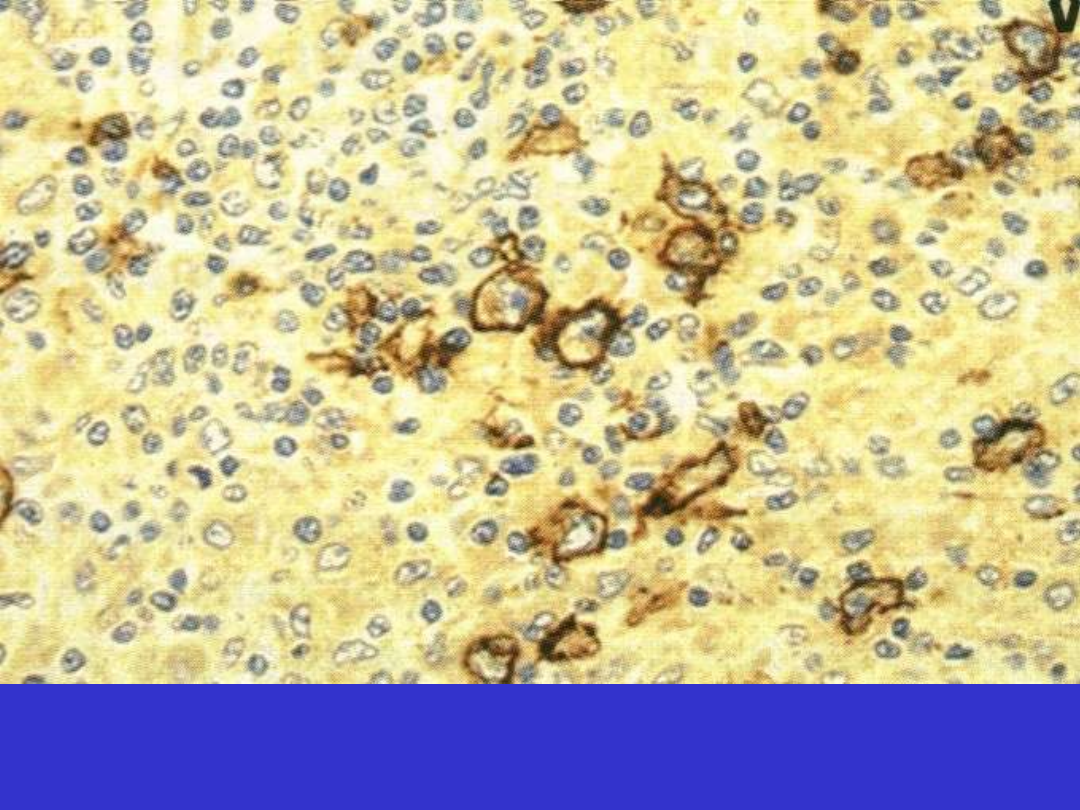
DLBCL; wariant bogaty w limfocyty T/histiocyty, nowotworowe kk. B, CD20+
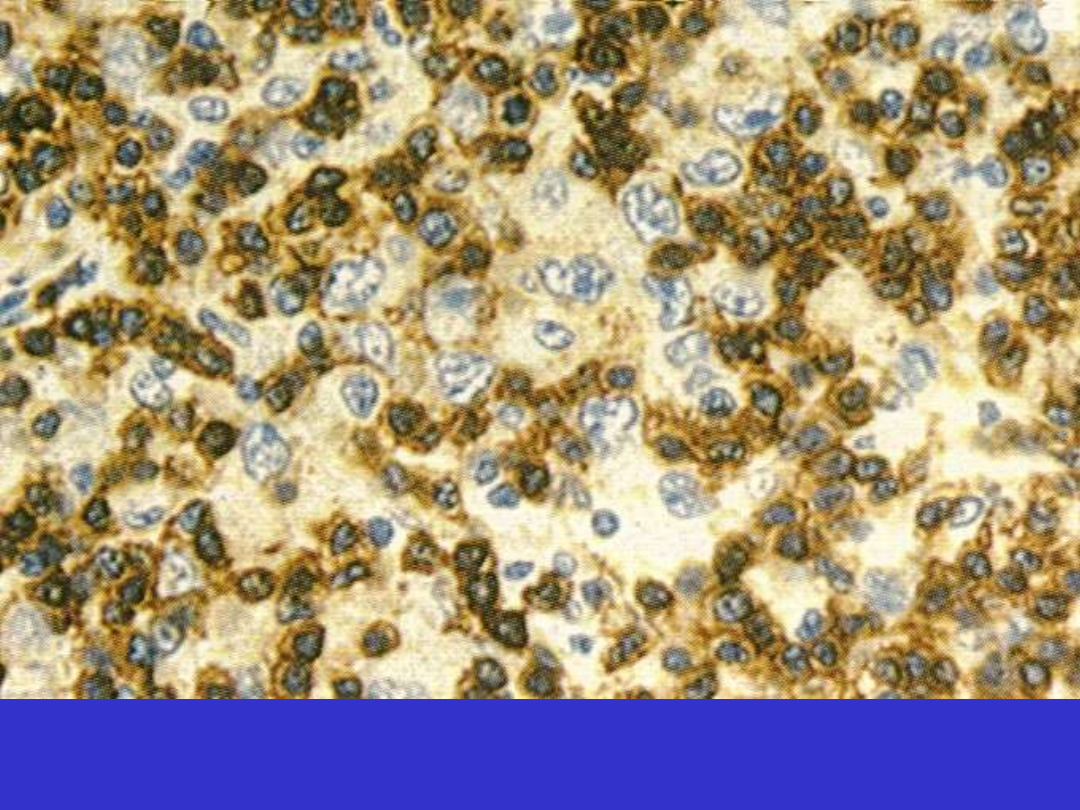
DLBCL; wariant bogaty w limfocyty T/histiocyty, nienowotworowe kk. T CD3+
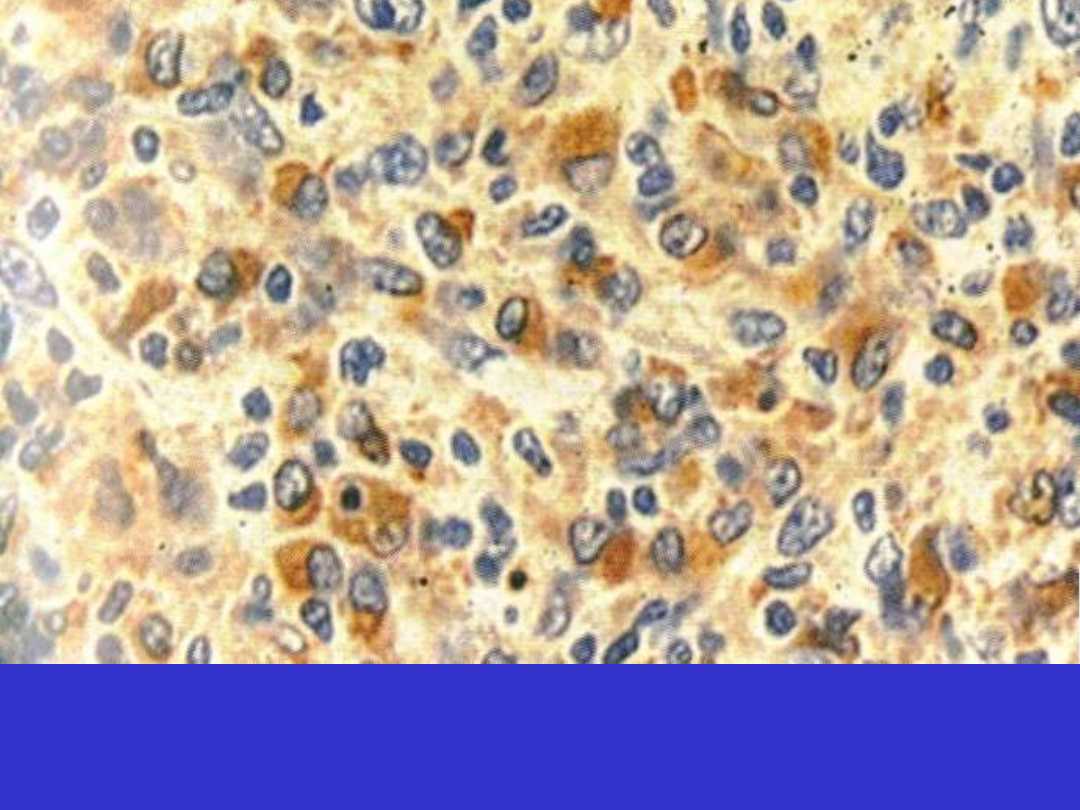
DLBCL; wariant bogaty w limfocyty T/histiocyty, nienowotworowe histiocyty CD68+
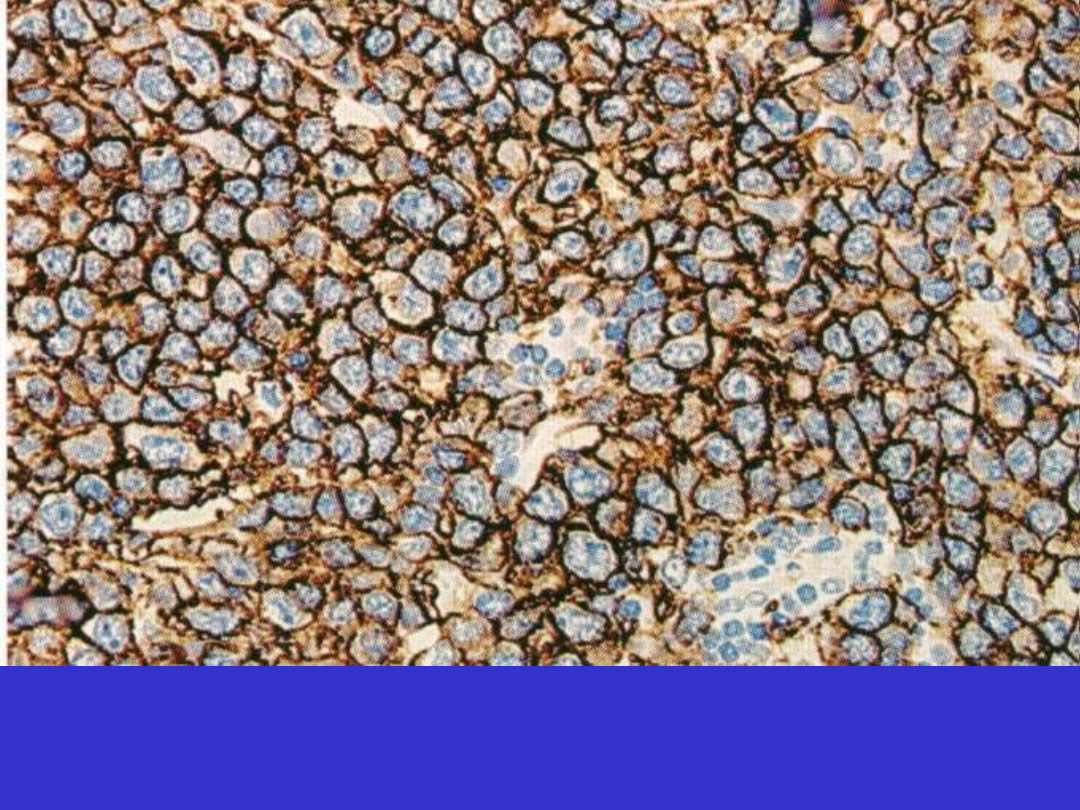
DLBCL; wariant anaplastyczny, nowotworowe kk. B, CD20+

PODTYPY DLBCL
Pierwotny chłoniak śródpiersia (grasicy) z dużych kk. B
Klinicznie: 20-34 lata, głównie kobiety,
typowy przebieg; guz śródpiersia przedniego, zespół żyły głównej
górnej, rozsiew do okolic pozawęzłowych (nerki, nadnercza, wątroba,
skóra, mózg).
Chłoniak na tle nabytych niedoborów immunologiznych
(AIDS, długotrwała immunosupresja), w wyniku infekcji EBV.
Pierwotny wysiękowy chłoniak błon surowiczych – w wyniku
zakażenia wirusem opryszczki typu 8 (HHV8).
Wewnątrznaczyniowy chłoniak z dużych komórek B – komórki
nowotworowe obecne wyłącznie w świetle naczyń.
Mediastinal (thymic) large B-cell lymphoma; rybie mięso, martwica
Mediastinal (thymic) large B-cell lymphoma; duże komórki CD20+
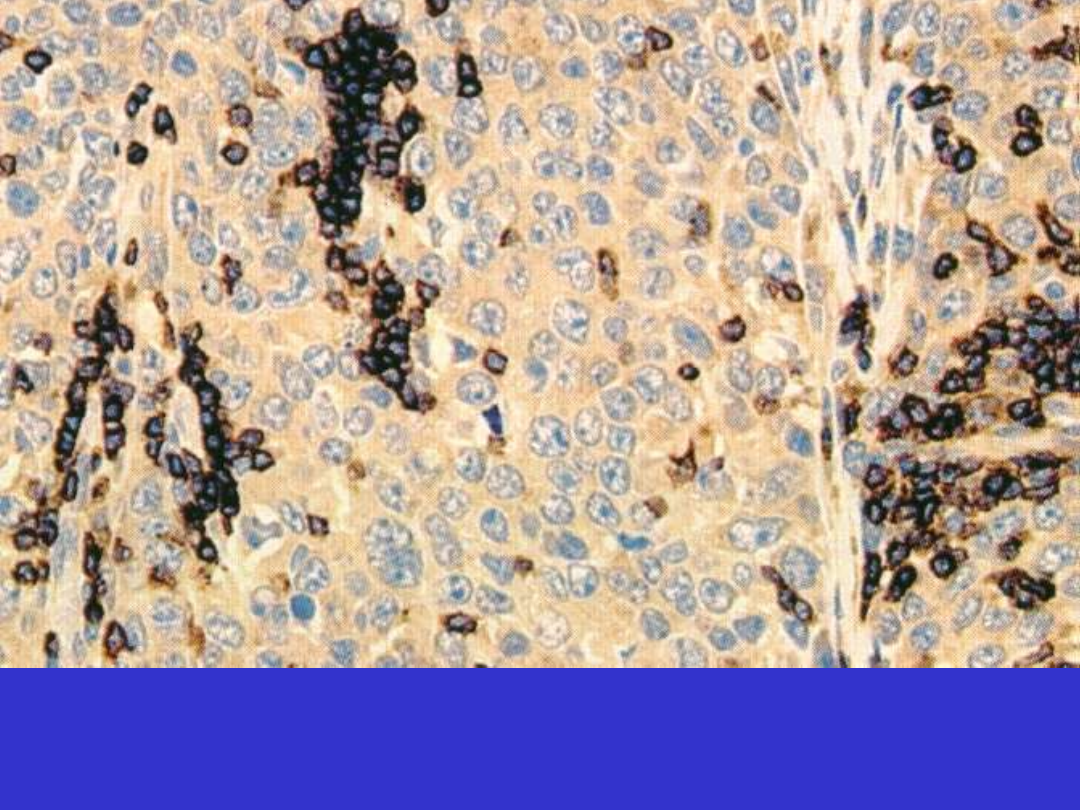
Mediastinal (thymic) large B-cell lymphoma; komórki CD3+ dookoła naczyń

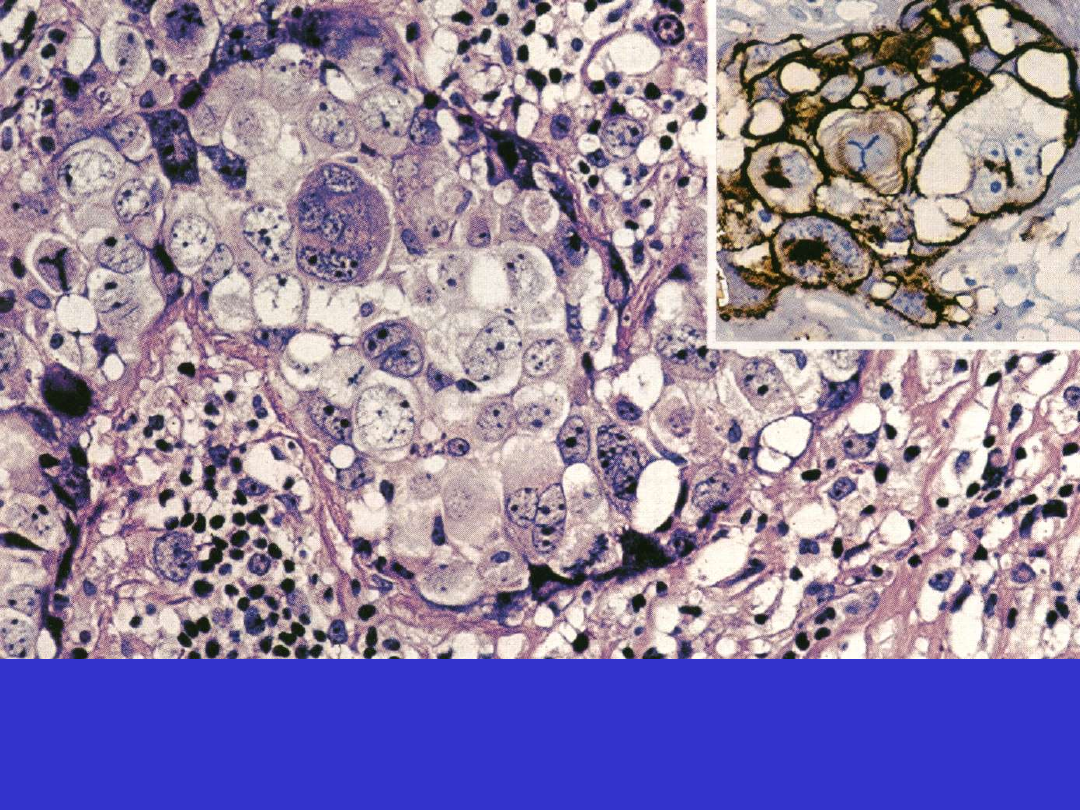
Chłoniak Burkitta (Burkitt lymphoma, BL)
– bardzo
agresywny chłoniak, często rozwijający się pozawęzłowo lub w
formie ostrej białaczki, zbudowany z monomorficznych kk. B
o średniej wielkości, z bazofilną cytoplazmą i licznymi
figurami podziału. Stała cechą genetyczną jest translokacja w
obrębie MYC (chromosom 8). EBV jest identyfikowany w
części przypadków.
3 warianty kliniczne;
1. Endemiczny BL;
w Afryce równikowej, Papui-Nowej Gwinei, 4-7 r.ż., M:K=2:1,
najczęstszy nowotwór złośliwy dzieci. Występuje na tych samych terenach co malaria.
EBV w większości komórek u WSZYSTKICH chorych.
2. Sporadyczny BL;
u dzieci i młodych dorosłych, 1-2% wszystkich chłoniaków w
Europie Zach. i USA, 30-50% chłoniaków u dzieci. M:K=2-3:1. Niski status materialny
i wczesna infekcja EBV podwyższają ryzyko zachorowania. EBV w mniej niż 30%
przypadków.
3. Związany z niedoborami immunologicznymi;
infekcja HIV i AIDS. EBV
stwierdzany u 30-40% chorych.
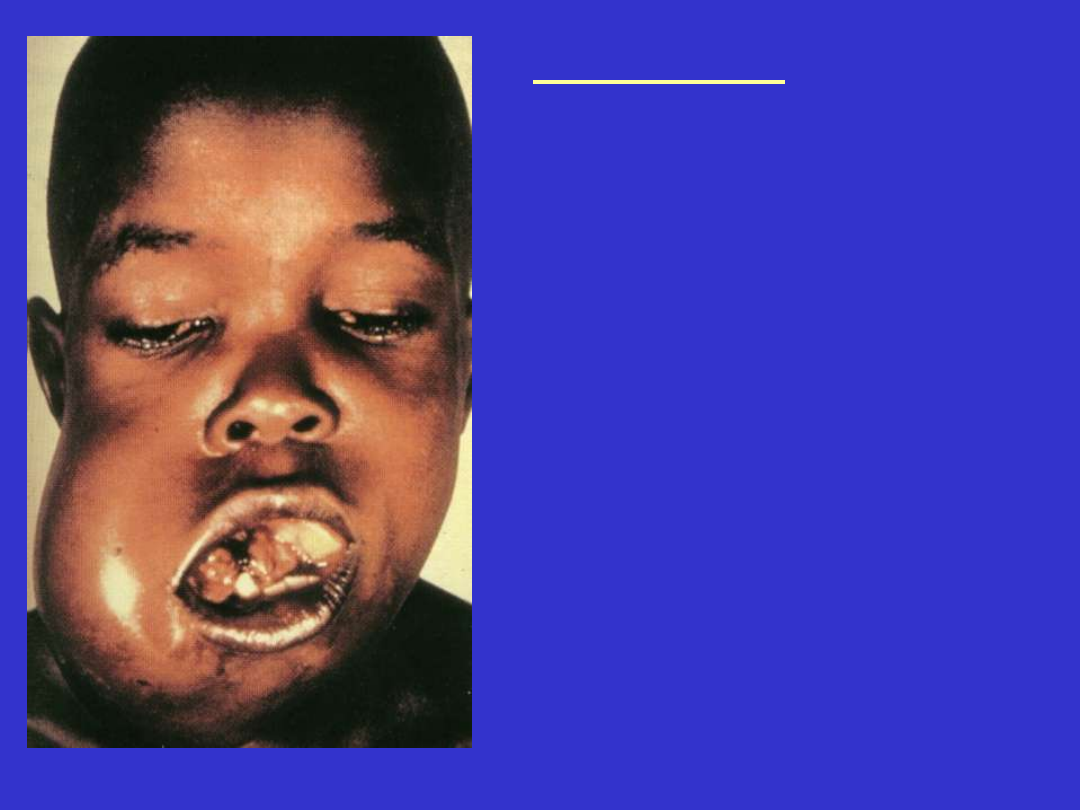
Umiejscowienie:
Ad 1. Żuchwa, inne kości twarzy
(oczodół) – 50% przypadków.
Ok. krętniczo-kątnicza, sieć,
jajnik, pierś.
Ad 2. Jama brzuszna; okolica
krętniczo-kątnicza, sieć, jajnik,
pierś, przestrzeń zaotrzewnowa,
węzły chłonne (dorośli), pierścień
Waldeyera. Rzadko jako
białaczka
Ad 3. Węzły chłonne, szpik.
Ad 1, 2, 3. O.U.N.
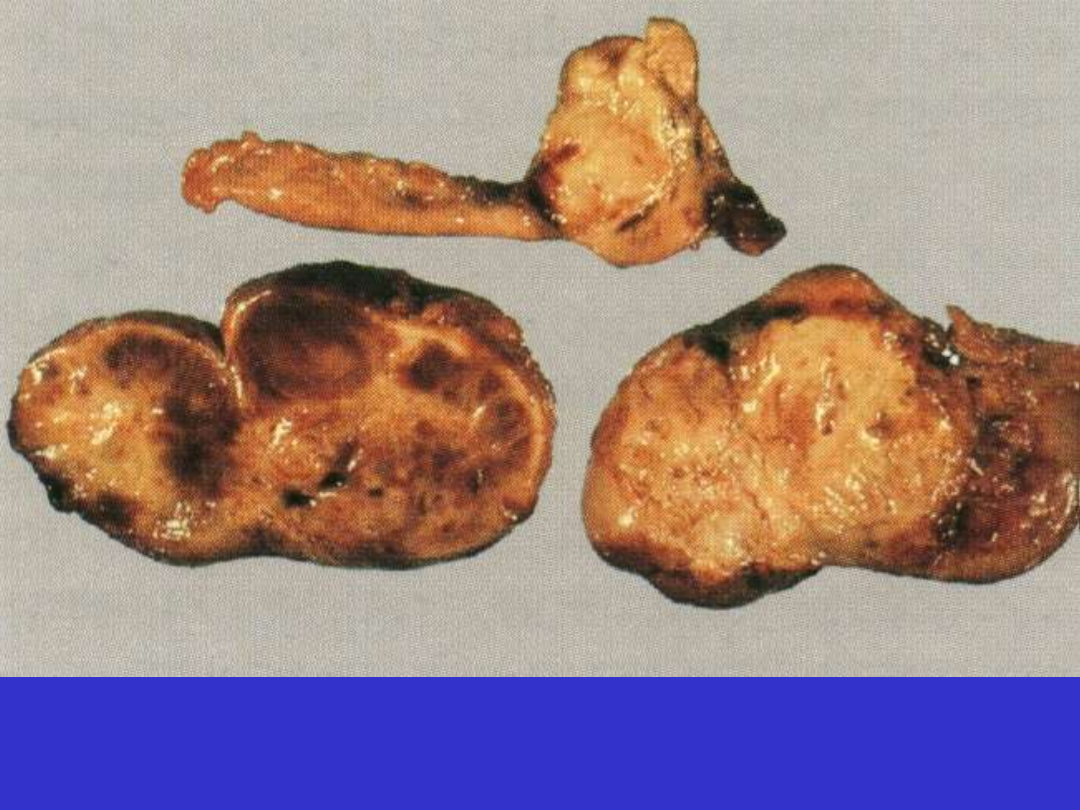
BL; wariant sporadyczny, zajęcie jajników
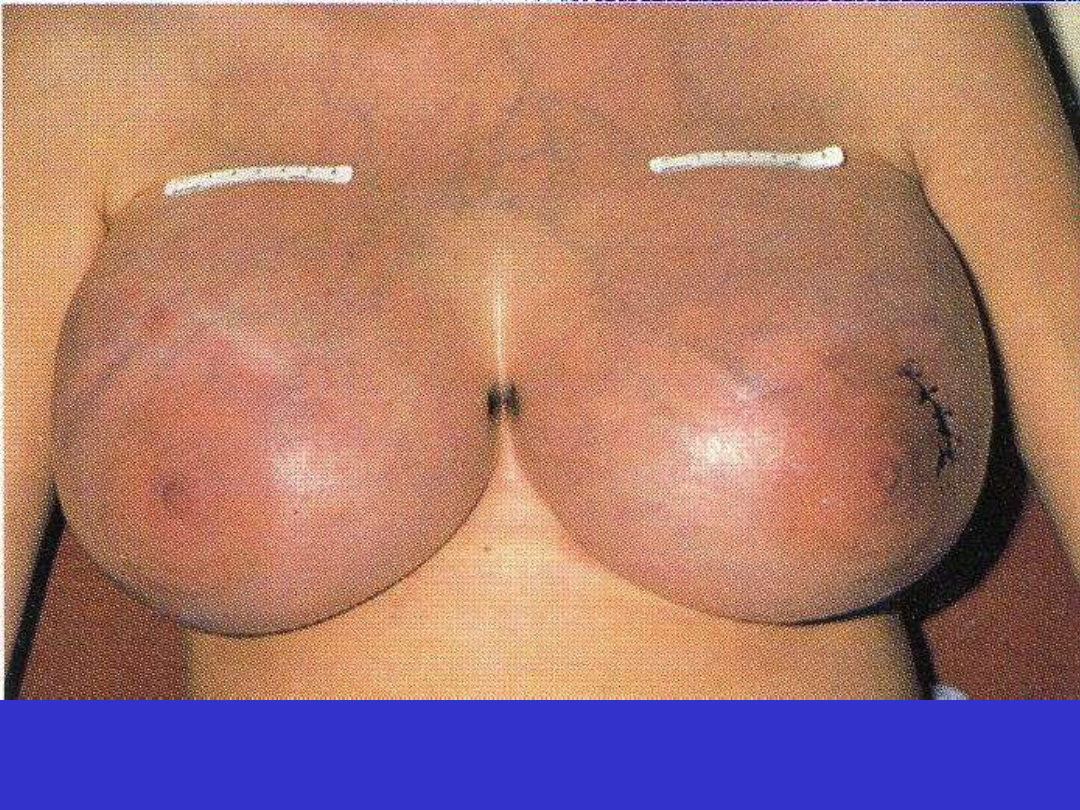
BL; wariant sporadyczny, zajęcie piersi. Komórki chłoniaka mają receptory PRL

Klinicznie: zwykle III lub IV stopień zawansowania (70% chorych)
w momencie zgłoszenia.
Morfologia; rybie mięso, krwotoki, martwica. Mikroskopowo
klasyczny; monotonne, średniej wielkości kk. z mnogimi
bazofilnymi jąderkami, gwiaździste niebo.Warianty; z
różnicowaniem plazmocytoidnym, atypowy/Burkitt-like.
Immunofenotyp; markery kk. B + (CD19, 20, 22). BCL2-, CD5-,
TdT-. Frakcja proliferacyjna (Ki67, MIB) - 100%.
Kariotyp; translokacja MYC z 8q24 na 14 (gen łańcuchów ciężkich
IgH) lub inne (2, 22 – geny łańcuchów lekkich)
Prognoza; agresywna chemioterapia umożliwia całkowite
wyleczenie w formie endemicznej i sporadycznej (80-90% chorych).
Nawrót choroby zwykle w ciągu roku, jeśli nie wystąpi w ciągu 2 lat
– najprawdopodobniej całkowite wyleczenie.
BL; klasyczny
BL; wariant atypowy
BL; wariant atypowy

Pozawęzłowe
chłoniaki
strefy
brzeżnej
(typu
MALT)
(extranodal
marginal
zone
B-cell
lymphoma
of
mucosa-
associated lymphoid tissue – MALT lymphoma) – chłoniak
pozwęzłowy, zbudowany z heterogennych, małych kk. B,
mogących przypominać centrocyty, kk. monocytoidne, małe
limfocyty, + rozproszone immunoblasty i centroblasty. Czasem
różnicowanie w kierunku plazmocytów.
Chłoniak zajmuje strefę
brzeżną reaktywnych grudek oraz strefę międzygrudkową.
- 7-8% wszystkich chłoniaków z kk. B, ale 50% pierwotnych
chłoniaków żołądka: przew. pok. (żołądek – 85%, jelito cienkie,
grube), płuco, głowa i szyja, narząd wzroku, skóra, tarczyca, sutek;
- średni wiek ok. 61 r.ż., M:K = 1/1,2
- zmiany prekursorowe: przewlekłe zapalenia (gastritis chronica +
H. pylori, indukcja remisji po eradykacji H. pylori); choroby
autoimmunizacyjne (z. Sjoegrena. – 44x, wole Hashimoto – 70x
ryzyko chłoniaka tarczycy).

Klinicznie – zwykle w I, II stadium, w ok. 20% zajęcie szpiku,
wieloogniskowe zajęcie węzłów rzadkie.
MORFOLOGIA:
- Naciekanie początkowo dookoła reaktywnych grudek, na zew-
nątrz płaszcza, następnie zajęcie grudek, komórki małe do średnich
(CC), z nieregularnymi jądrami, cytoplazma dość jasna (obraz
monocytoidny);
- W 1/3 przypadków różnicowanie plazmatycznokomórkowe;
- W tkankach z obecnością gruczołów LEL (laesio lymphoepi-
thelialis) naciekanie nabłonka gruczołów!
Chłoniak MALT, żołądek; kk. nowotworowe otaczją reaktywne grudki i naciekają
śluzówkę
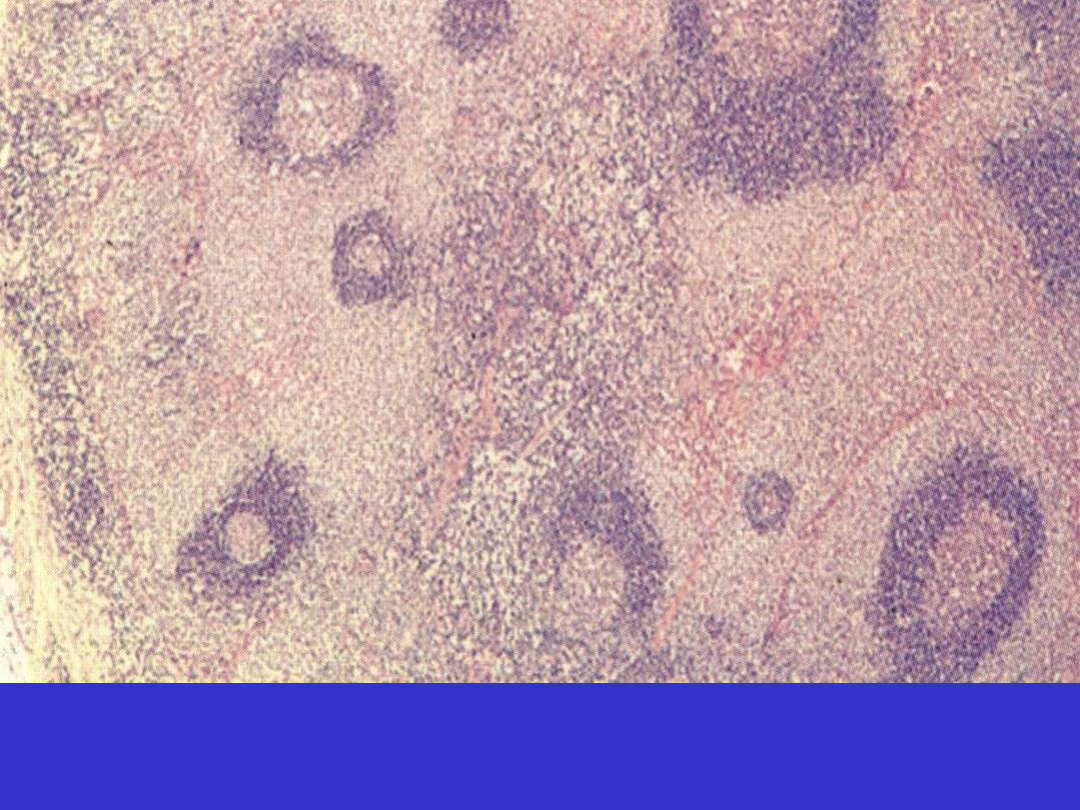
Chłoniak MALT, zajęcie węzła chłonnego; kk. nowotworowe naciekają strefę brzeżną i
obszar międzygrudkowy
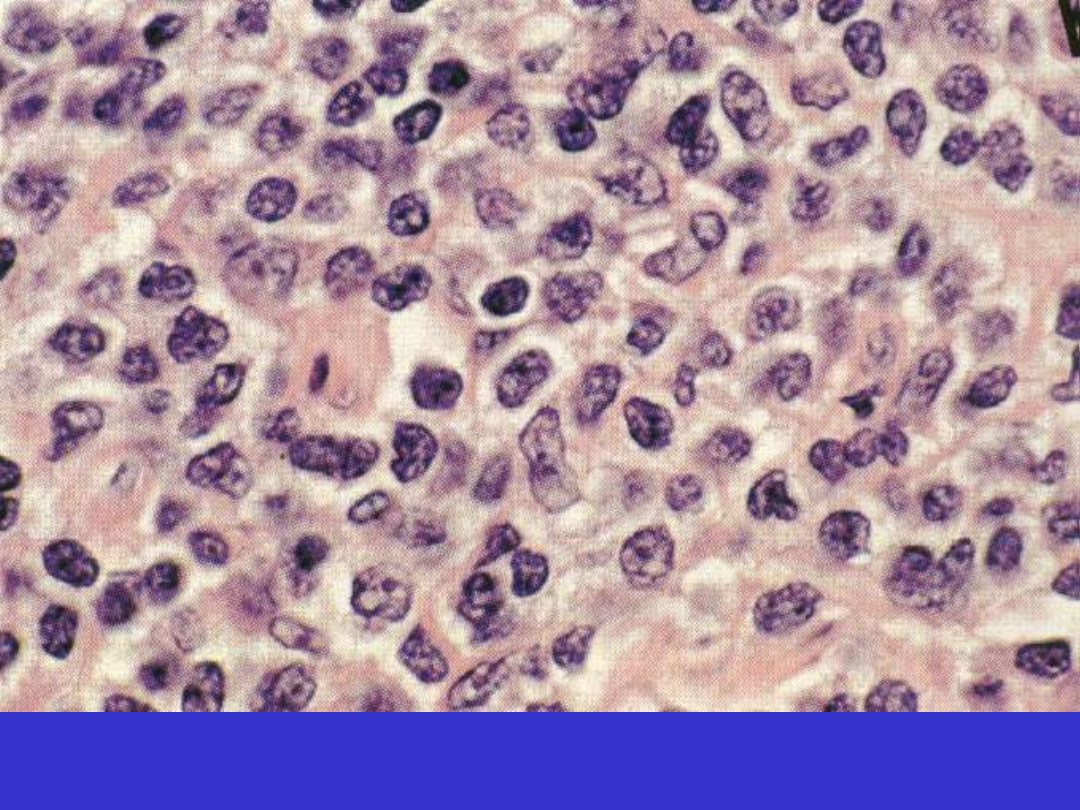
Chłoniak MALT, komórki przypominające centrocyty
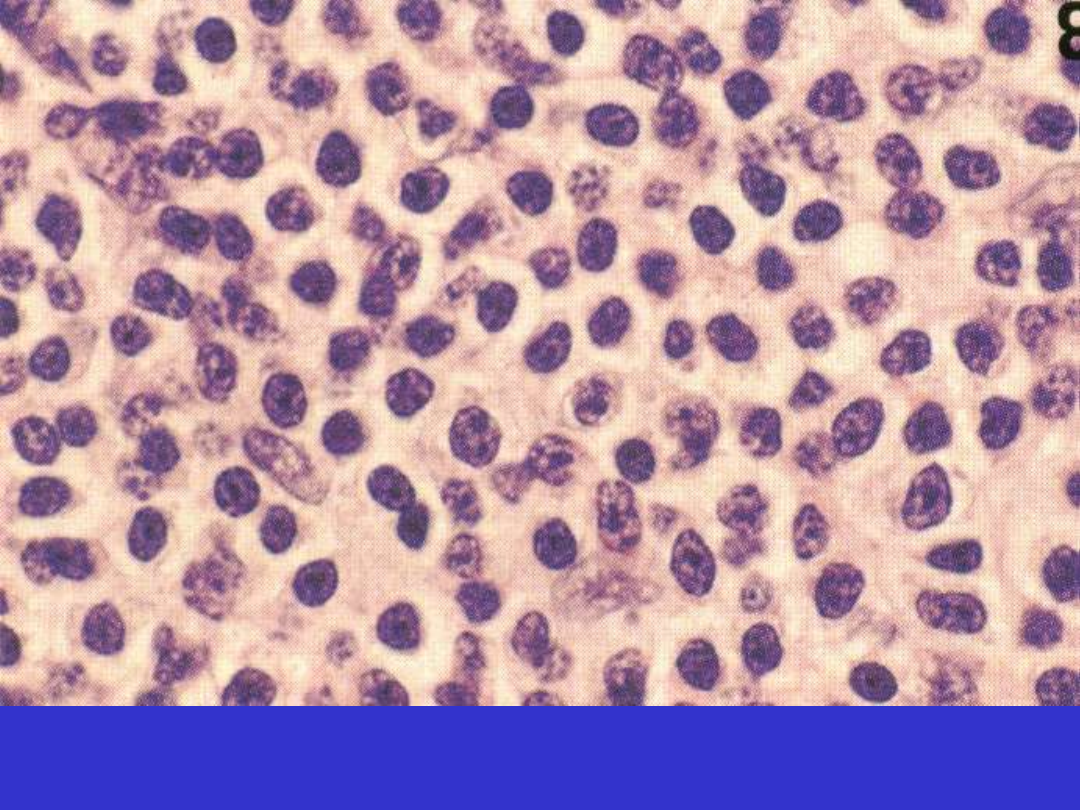
Chłoniak MALT, komórki monocytoidne
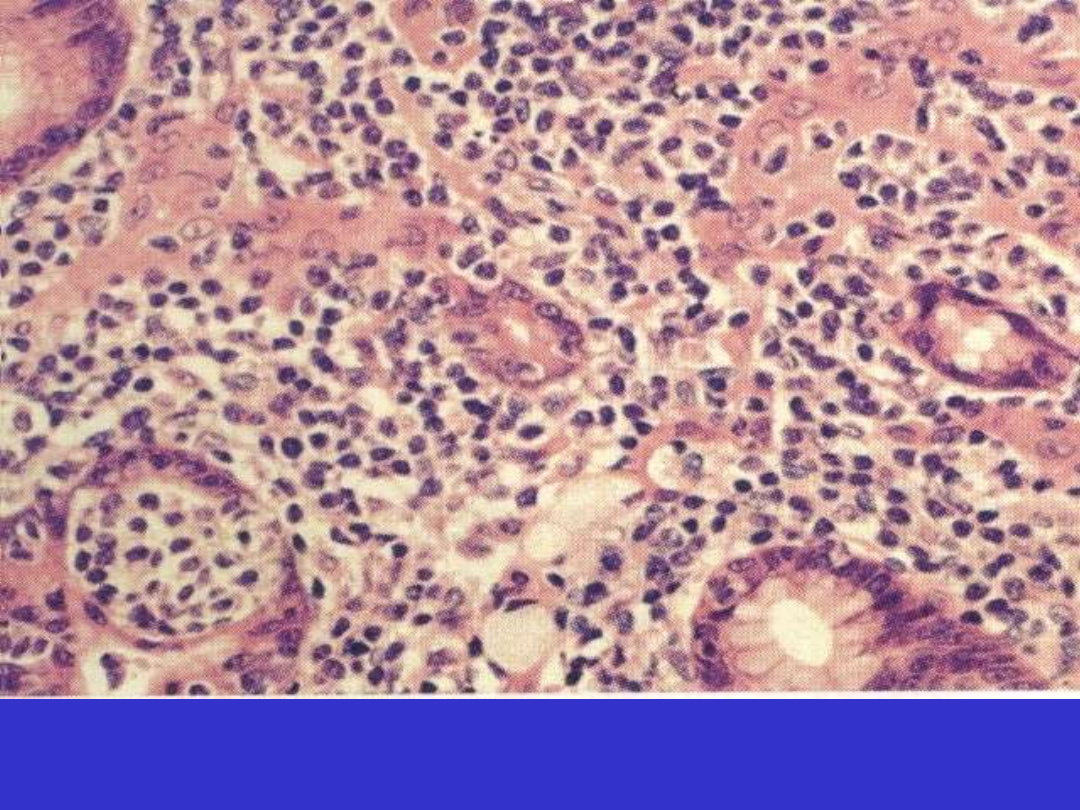
Chłoniak MALT, żołądek, laesio lymphoepithelialis
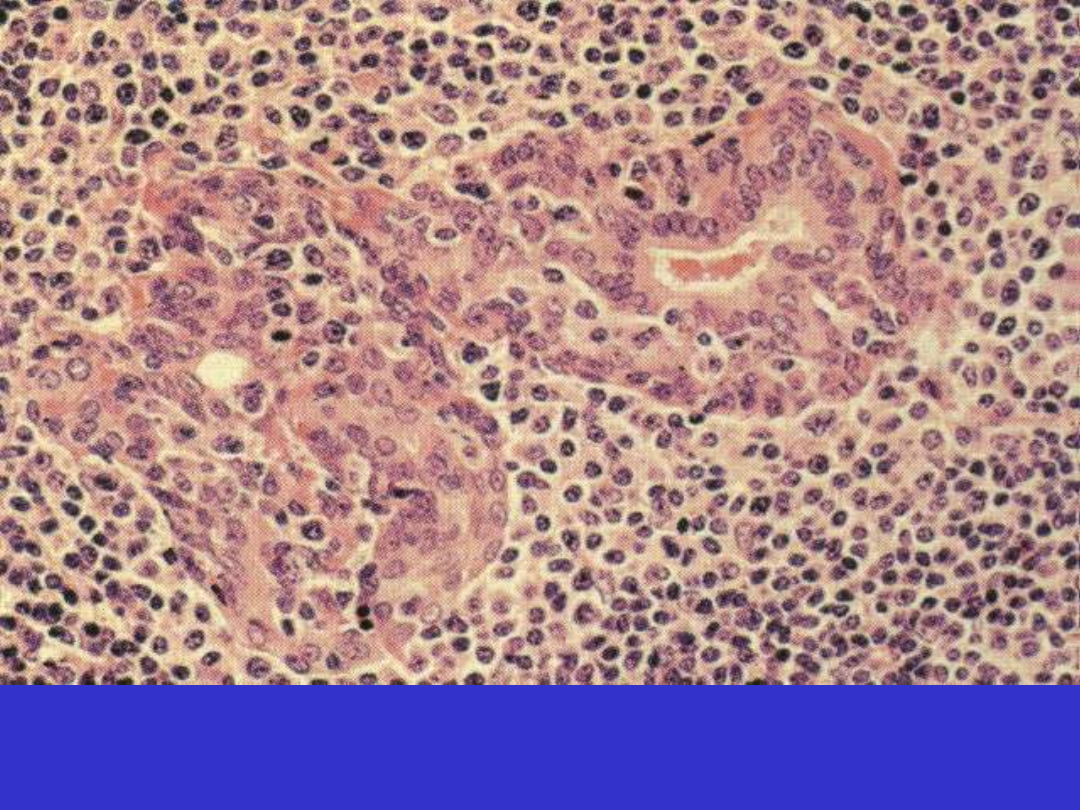
Chłoniak MALT, ślinianka, laesio lymphoepithelialis
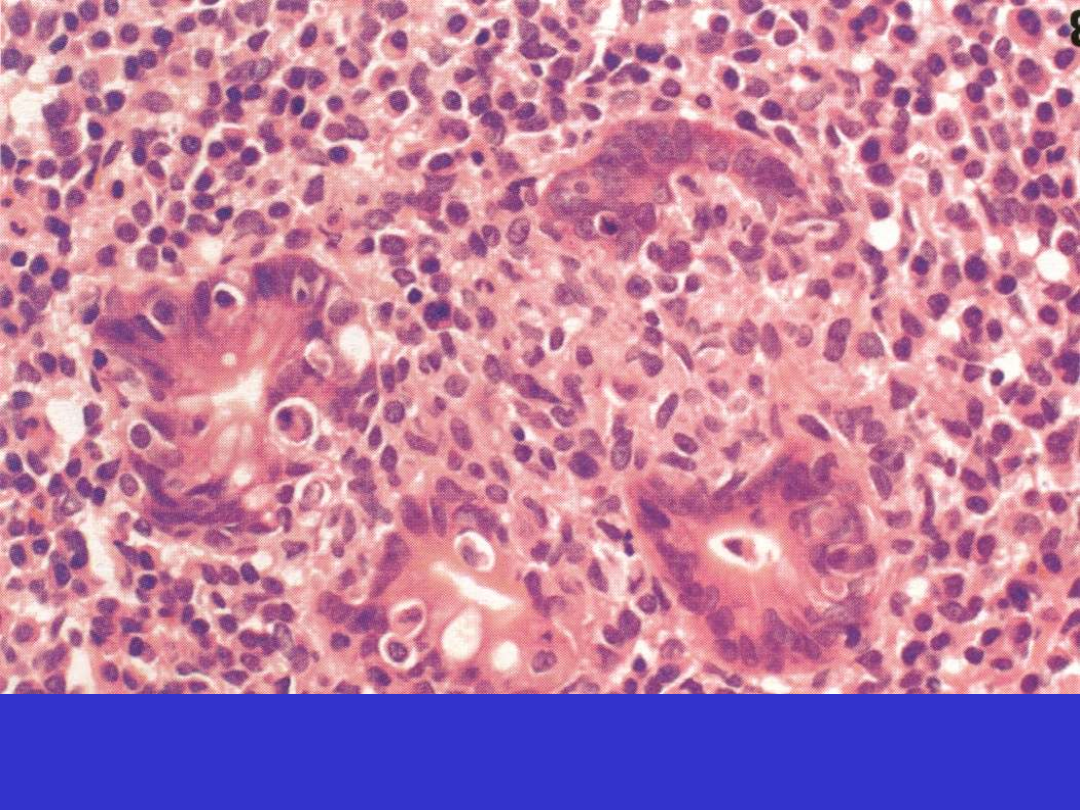
Chłoniak MALT, jelito cienkie, laesio lymphoepithelialis

Transformacja do chłoniaka wielokomórkowego – mogą być
obecne pojedyncze czy nawet dość liczne transformowane komórki
przypominające immunoblasty, centroblasty, ale nie mogą tworzyć
litych wysp (wtedy rozpoznanie chłoniaka wielokomórkowego na
podłożu MALT).
Immunofenotyp:
IgM+, CD20+, CD79a+; CD5-, CD10-, CD23-; w odróżnieniu od
węzłów reaktywnych restrykcja łańcuchów lekkich Ig,
CD5- odróżnienie od SLL/CLL i chłoniaka z komórek płaszcza
(także cyklina D1), a CD10- od chłoniaka grudkowego.

Rokowanie:
Przebieg powolny, z powolnym rozsiewem;
- wrażliwy na leczenie miejscowe (eradykacja H.pylori) czy rtg-
terapię, po których zwykle występuje dość długi okres remisji.
Transformacja do chłoniaka wielkokomórkowego – rokowanie złe;
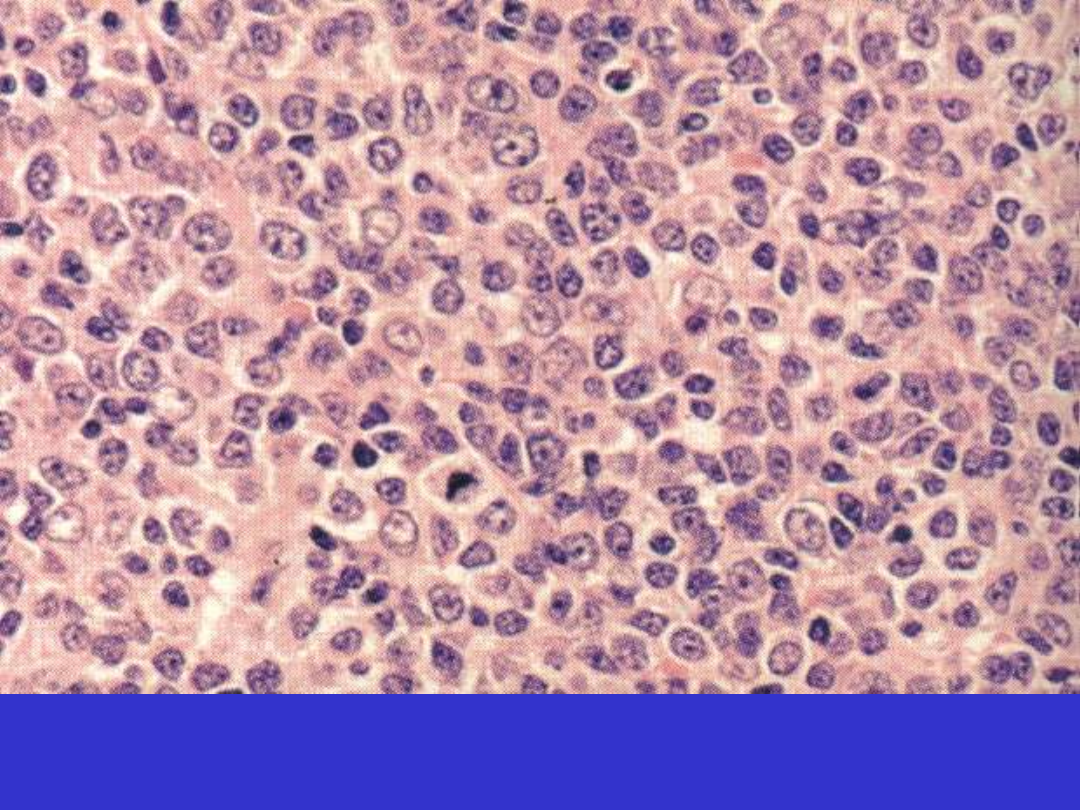
Chłoniak MALT ze zwiększoną liczbą dużych komórek
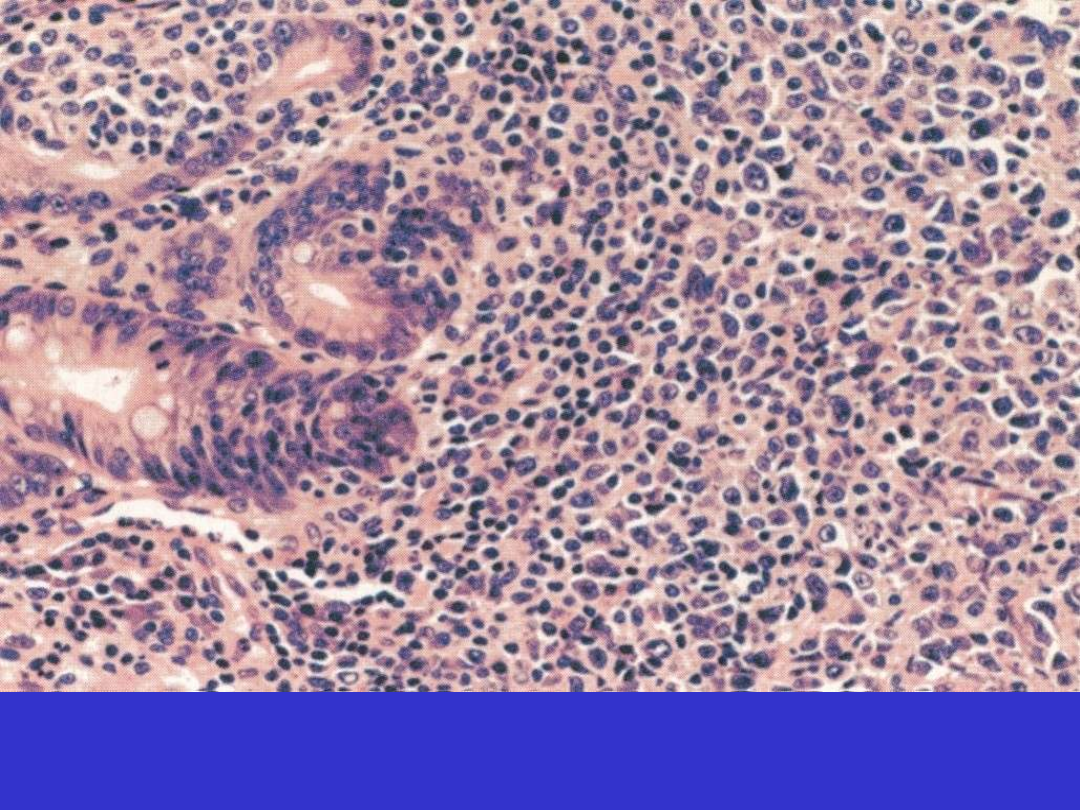
DLBCL (po prawej) z resztkami chłoniaka MALT naciekającego śluzówkę

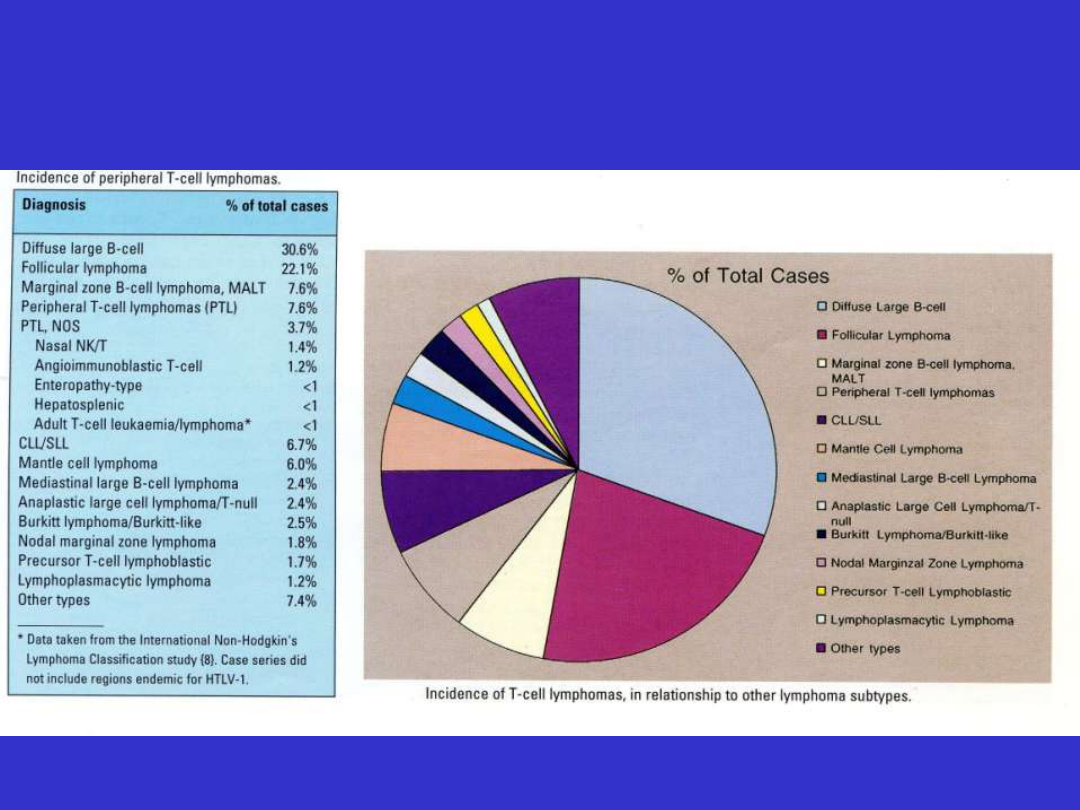
12% wszystkich NHL

Ziarniniak grzybiasty (mycosis fungoides, MF)
– chłoniak
z dojrzałych limfocytów T, objawiający się skórnymi zmianami
naciekowymi i guzowatymi. W skórze i naskórku obecne są
nacieki z małych i średnich komórek T z jądrami o
pofałdowanych konturach (nuclei cerebriformes)
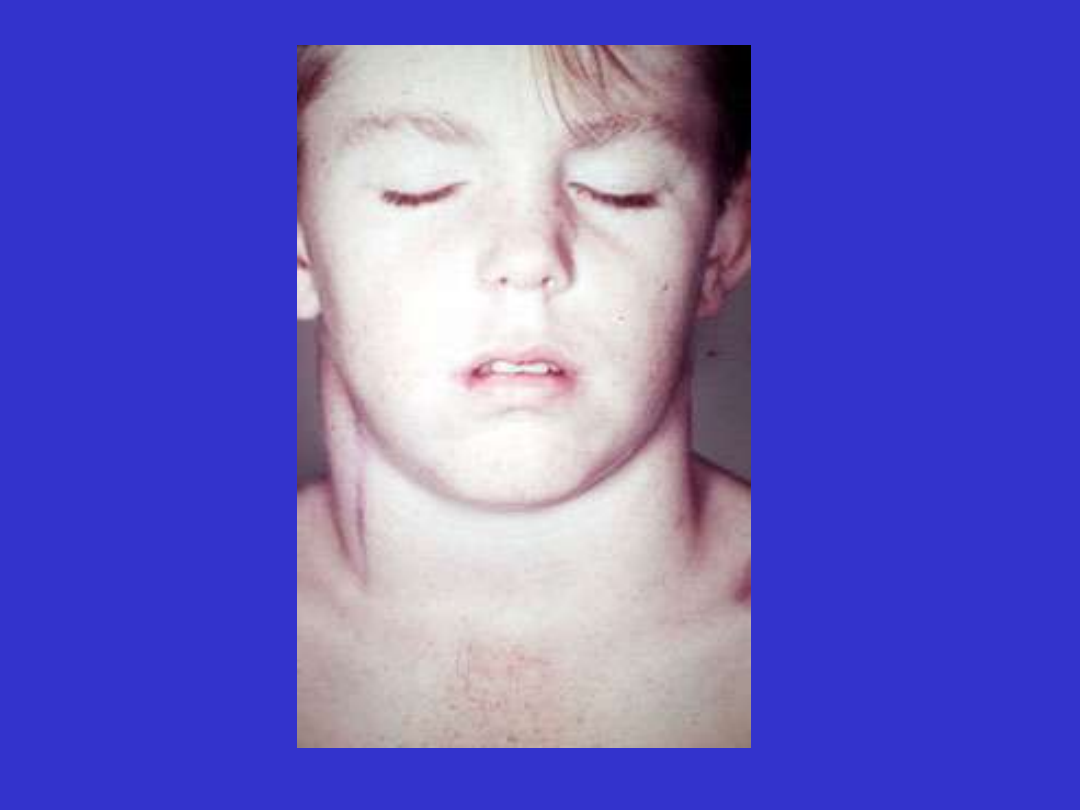
Zespół Sezary’ego (Sezary syndrome, SS)
– chłoniak z
dojrzałych limfocytów T, charakteryzujący się uogólnioną
erythrodermią, lymfadenopatią i obecnością komórek nowotwo-
rowych we krwi obwodowej. Komórki z jądrami jak w MF.
- MF nie więcej niż 0,5% NHL. Najczęstszy pierwotny chłoniak T
skóry. SS jeszcze rzadziej.
- występują u osób w średnim i starszym wieku, M/K = 2/1.
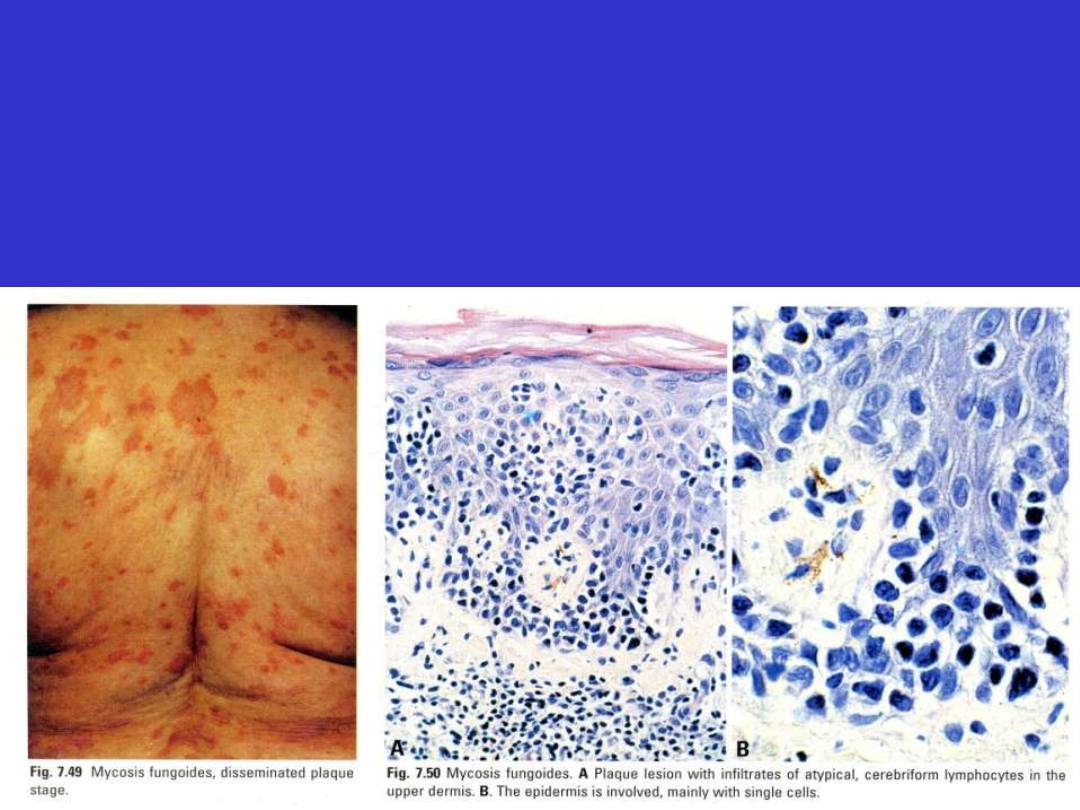
Klinicznie:
MF – przez wiele lat zmiany ograniczone do skóry (na tułowiu!),
początkowo nieswoiste, łuszczące się wykwity, później płaskie nacieki, w końcu guzy.
Rzadko dochodzi do uogólnionej erytrodermii. W zaawansowanej chorobie mogą być
zajęte węzły chłonne, wątroba, śledziona, płuca i krew. Zajęcie szpiku kostnego bardzo
rzadko spotykane.
Mikroskopowo; nacieki z limfocytów nowotworowych w skórze i naskórku
(exocytosis)
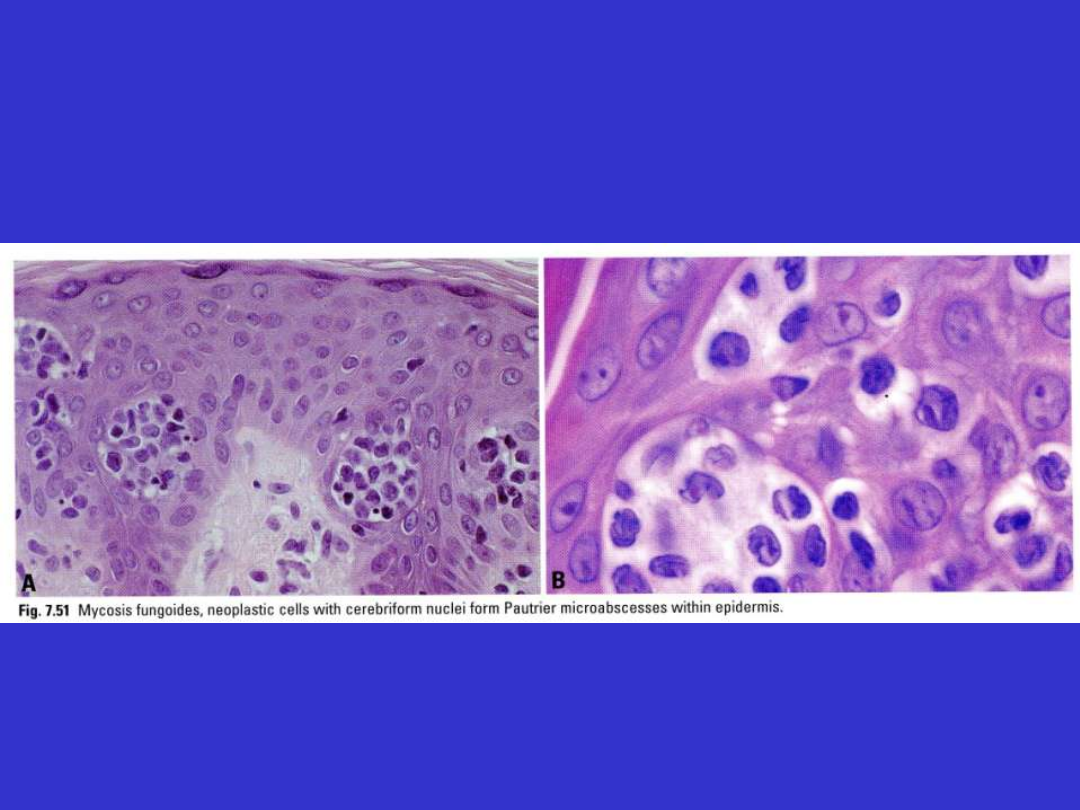
Mikroskopowo; nacieki z limfocytów nowotworowych w naskórku
z formowaniem mikroropni Pautrier’a
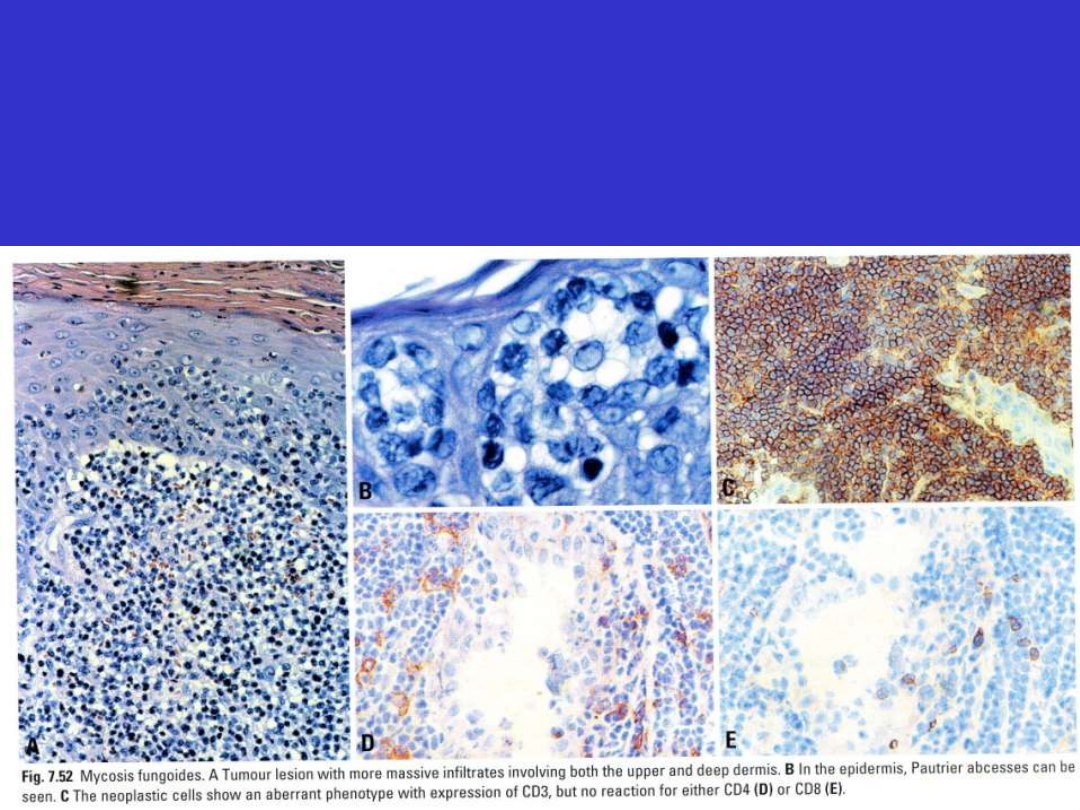
Mikroskopowo; w fazie guzowatej naciek bardziej intensywny.
Immunofenotyp; typowo CD3+, CD4+, CD8-.
Prognoza; w stadium zmian ograniczonych do skóry bardzo dobra, w
bardziej zaawansowanych - zła

SS – uogólnione zajęcie skóry (erytrodermia), węzłów chłonnych i krwi.
Ponadto świąd skóry, łysienie, nadmierne rogowacenie na dłoniach i
podeszwach, onychodystrofia.
Mikroskopowo; komórki o pofałdowanych jądrach, CD3+, CD4+.
Rokowanie; 5-letnie przeżycie 10-20%

30% wszystkich chłoniaków

NLPHL
NLPHL CD20
NLPHL, popcorn cells
NLPHL, CD20, reakcja błonowa z komórkami typu popcorn
Klasyczny HL, śledziona
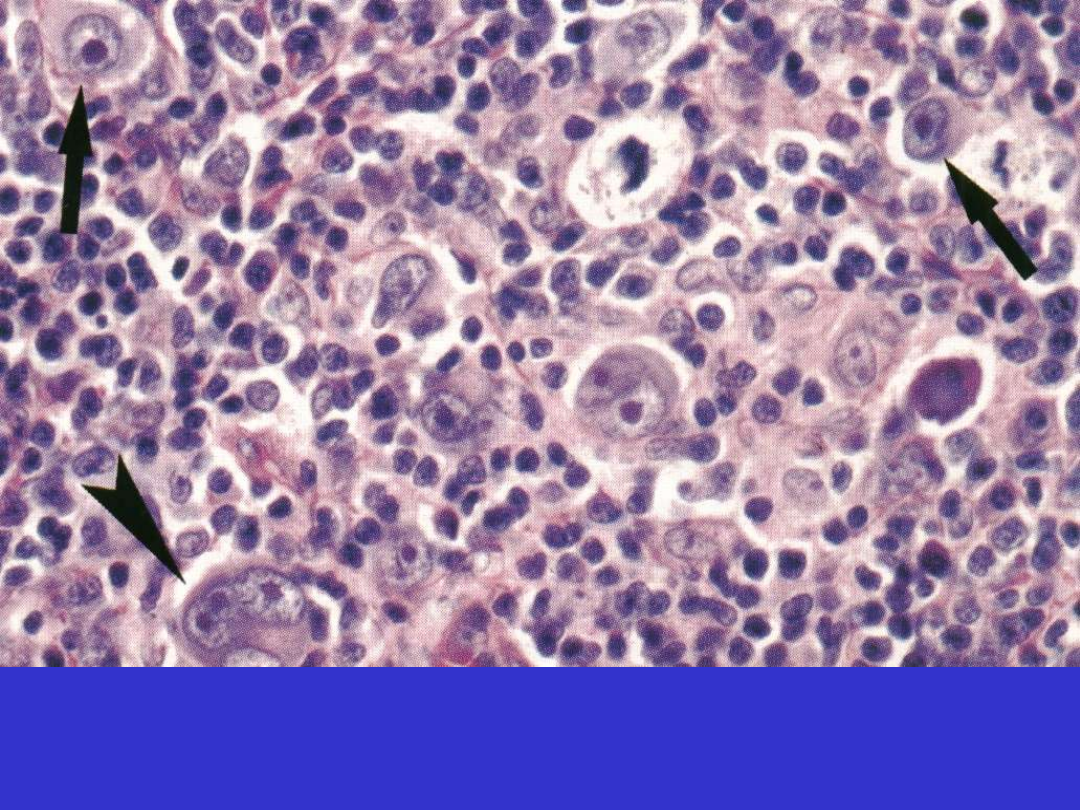
Klasyczny HL, komórki Hodgkina i Reed-Sternberga
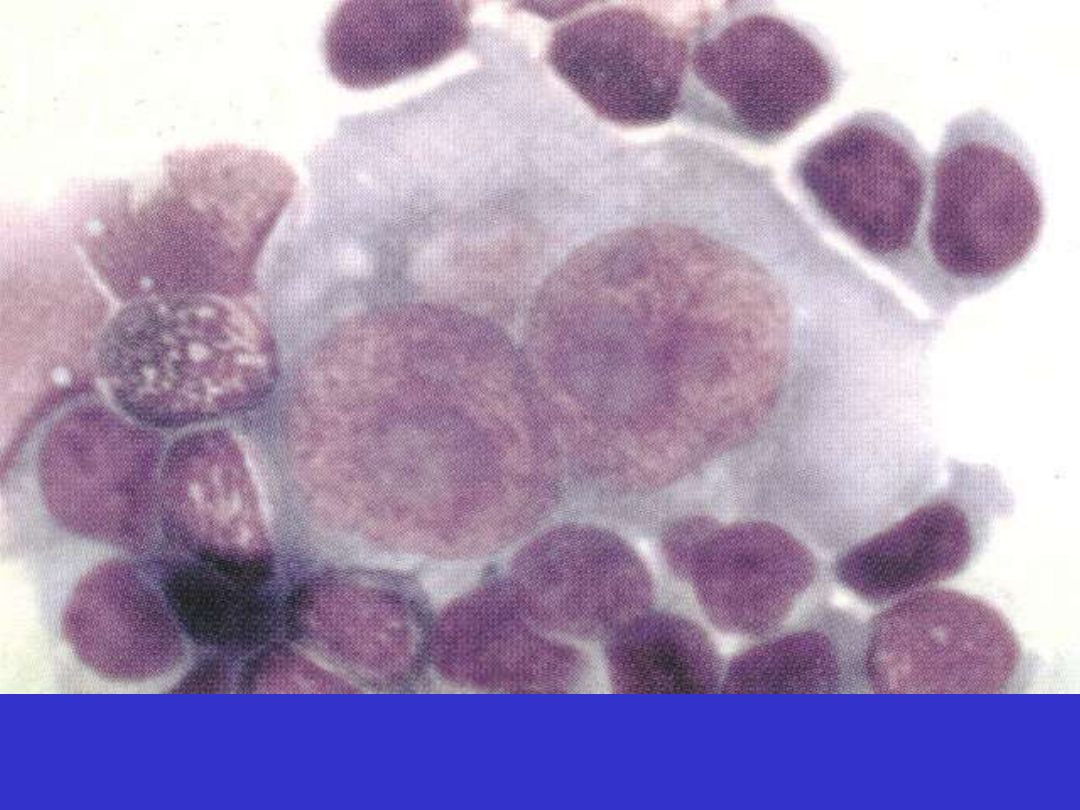
Klasyczny HL, komórka RS
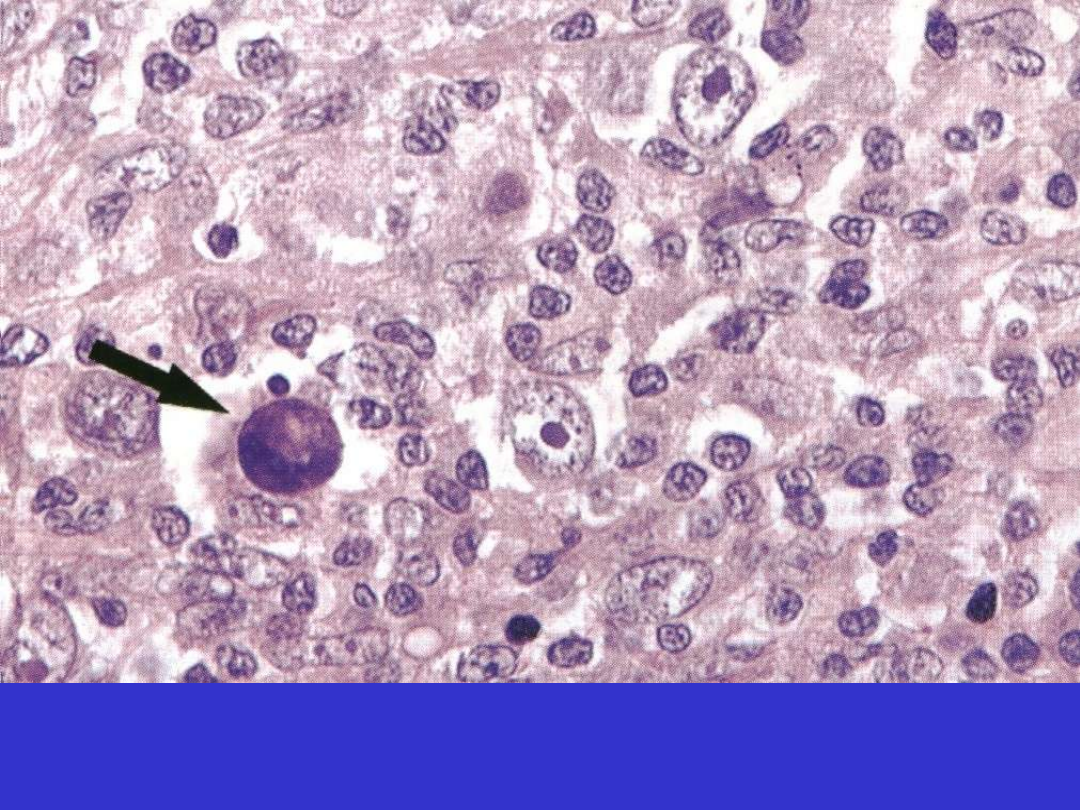
Klasyczny HL, komórki Hodgkina (jedna zmumifikowana)
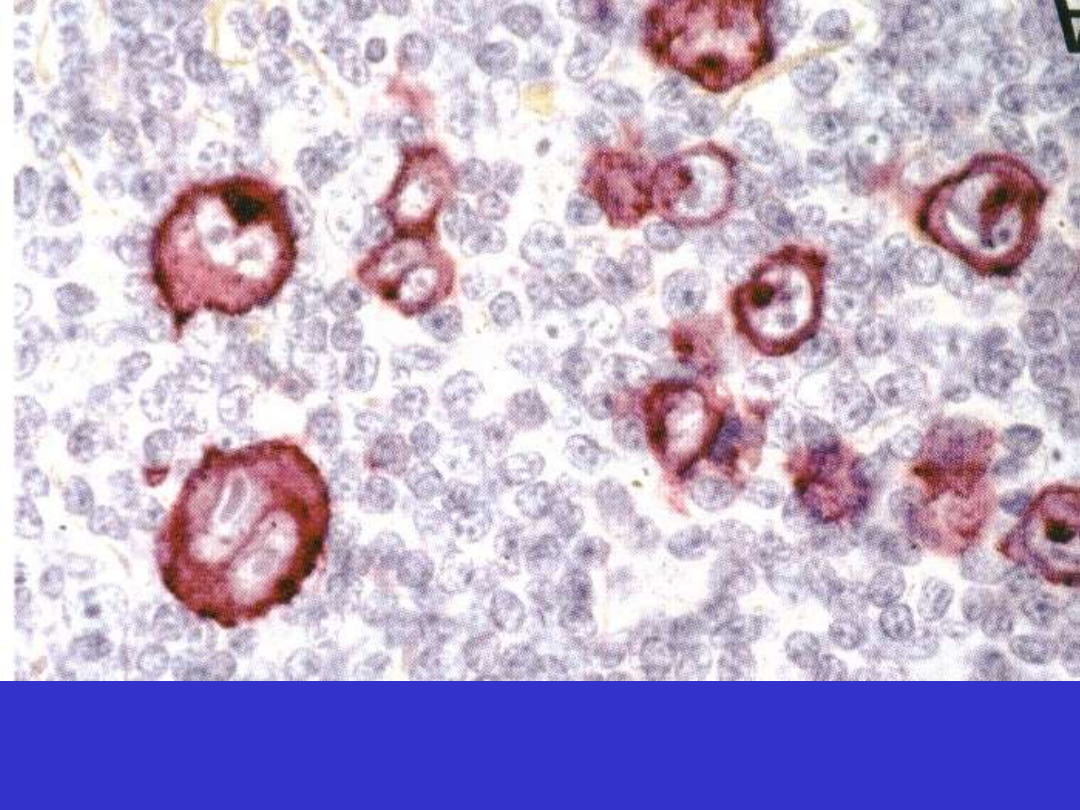
Klasyczny HL, CD30; komórki Hodgkina i Reed-Sternberga
,
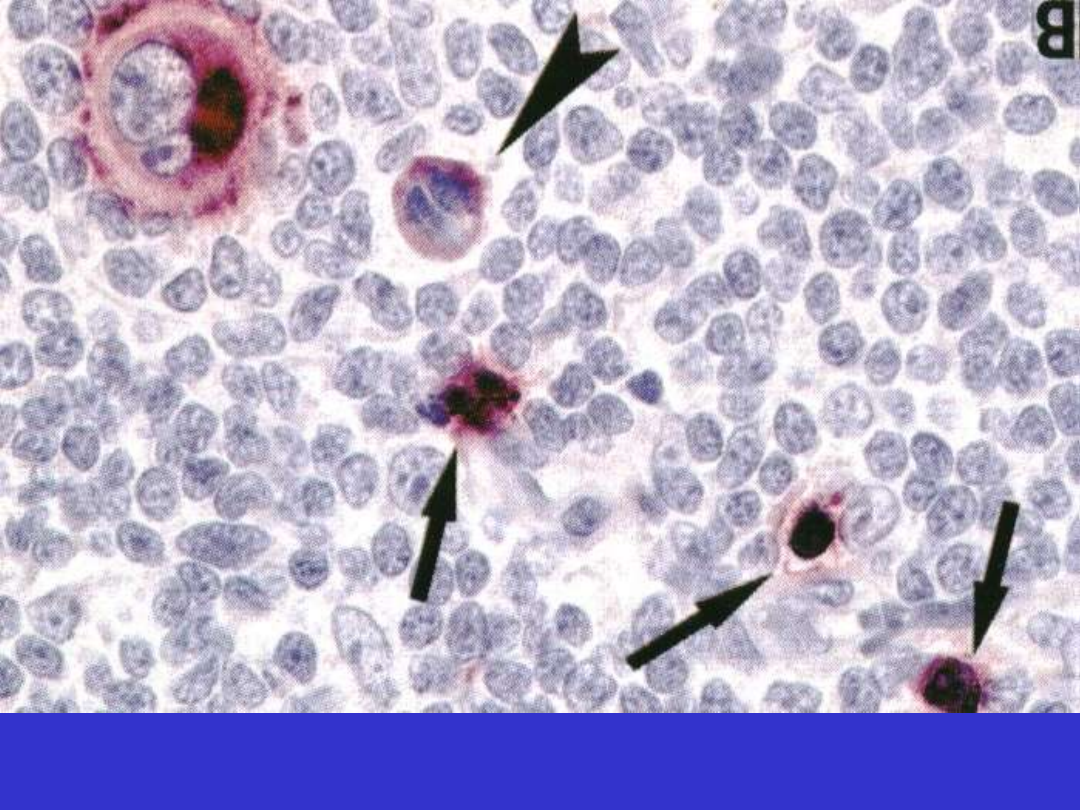
Klasyczny HL, CD15; komórki Reed-Sternberga, neutrofile
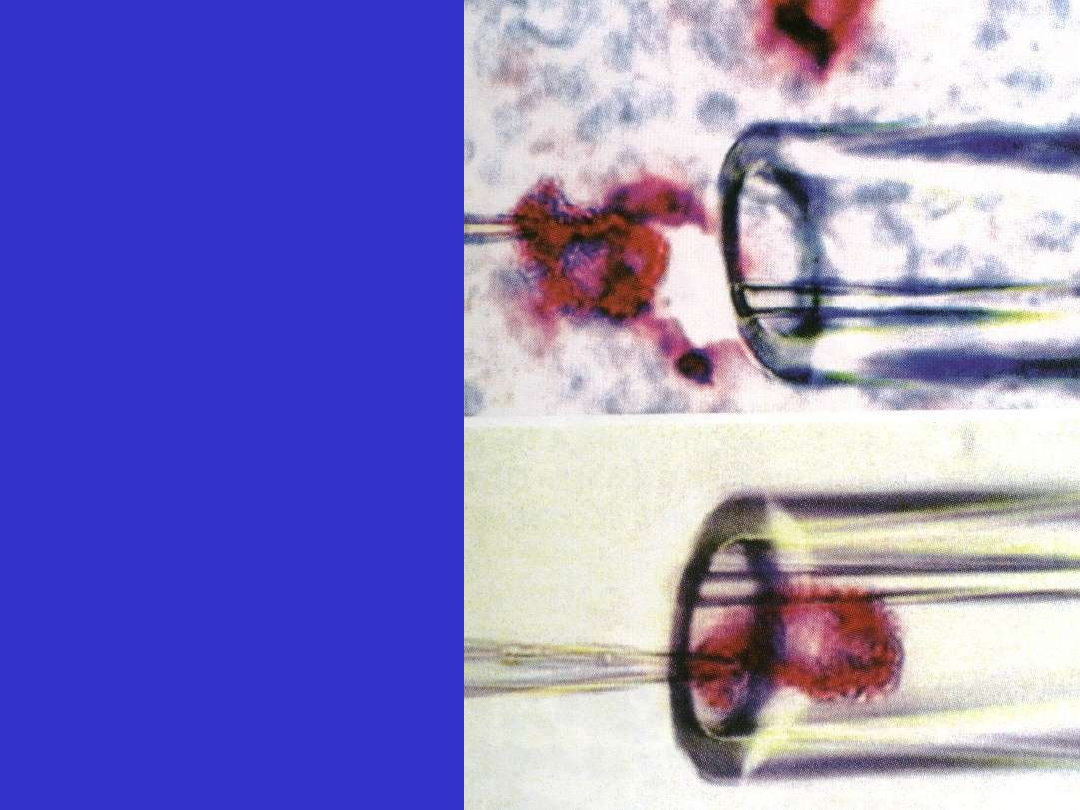
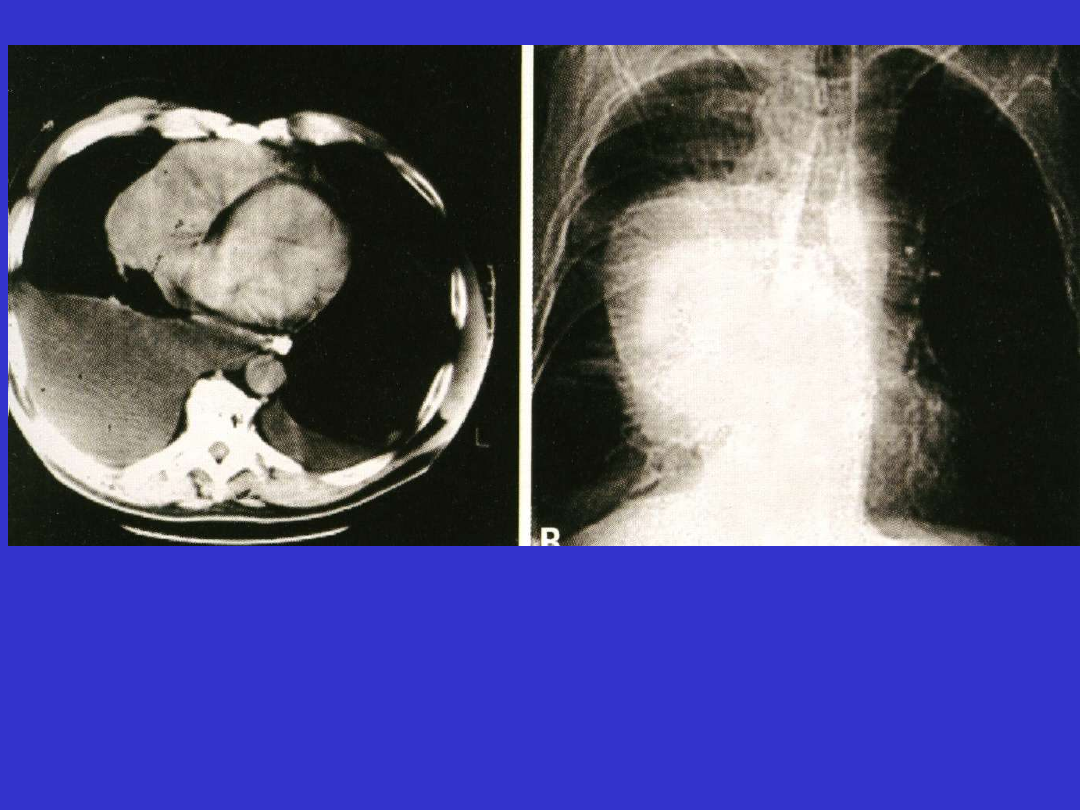
Klasyczny HL, stwardnienie guzkowe (NS); KT i Rtg, śródpiersie
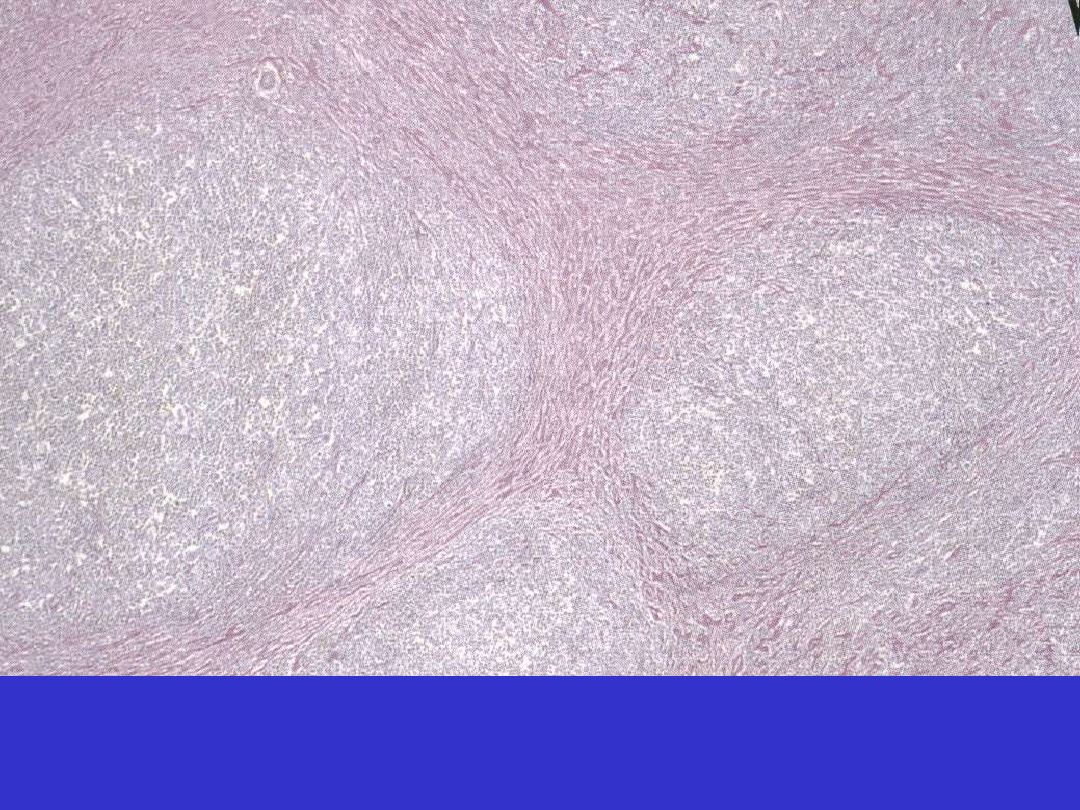
Klasyczny HL, stwardnienie guzkowe (NS)
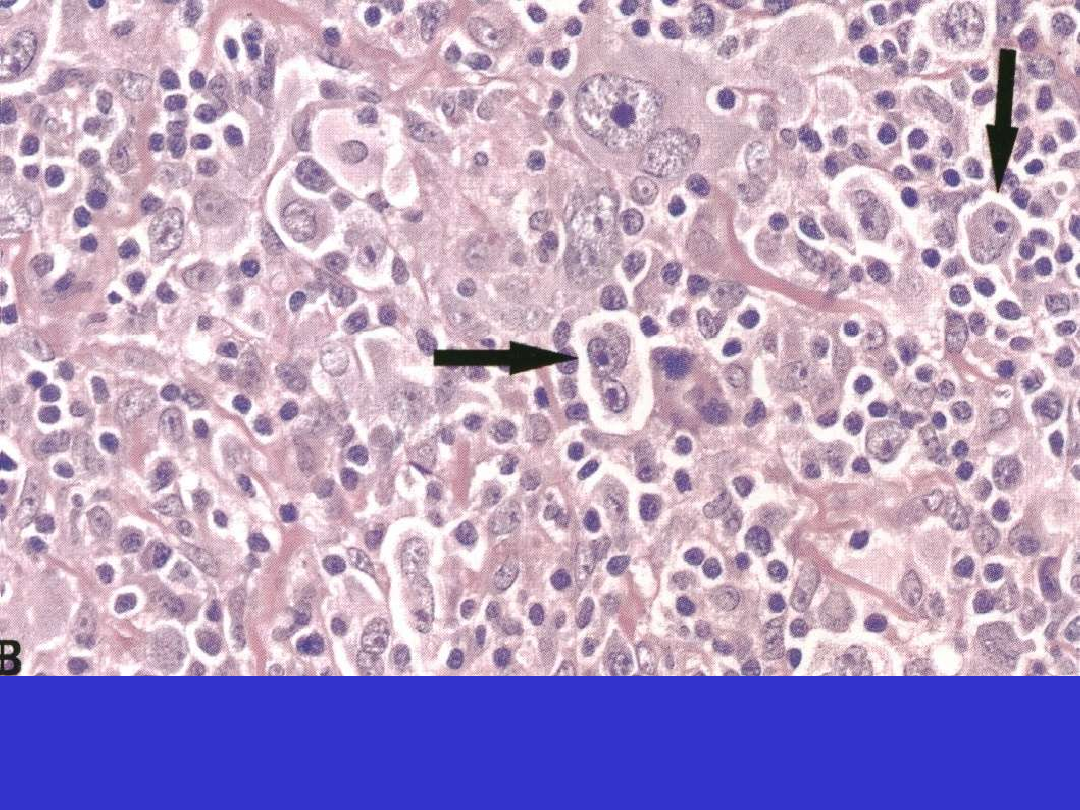
Klasyczny HL, stwardnienie guzkowe (NS); komórki lakunarne
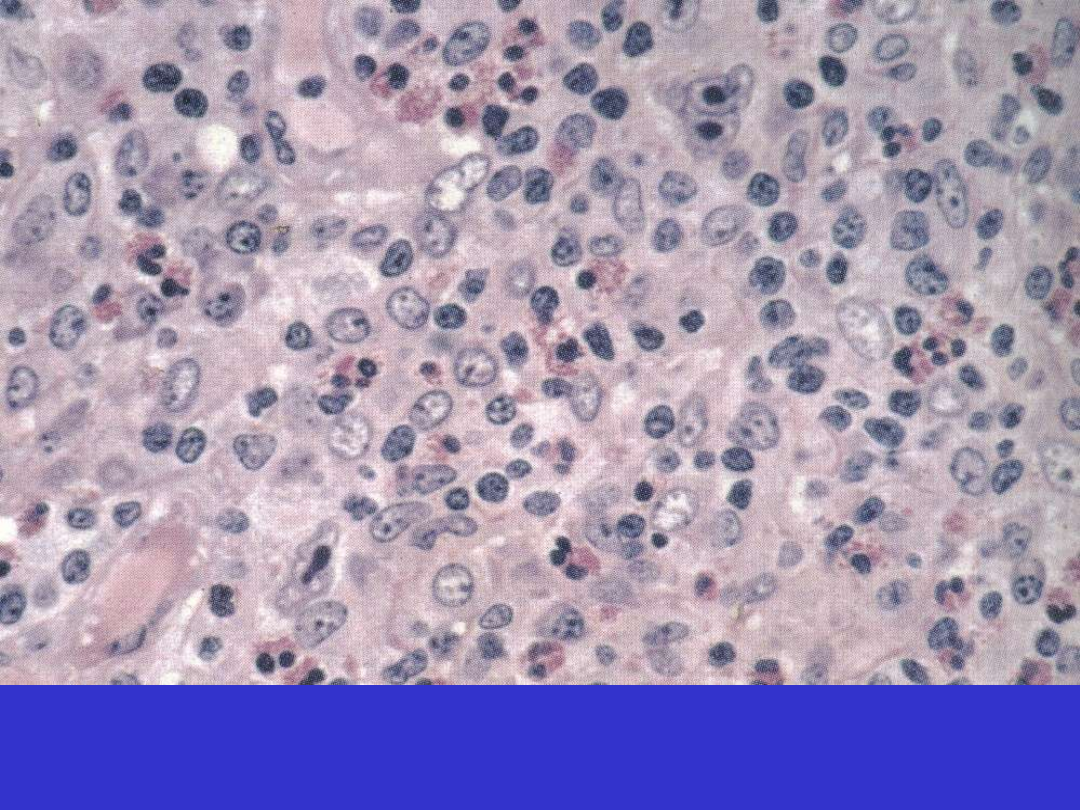
Klasyczny HL, typ mieszanokomórkowy (MC)
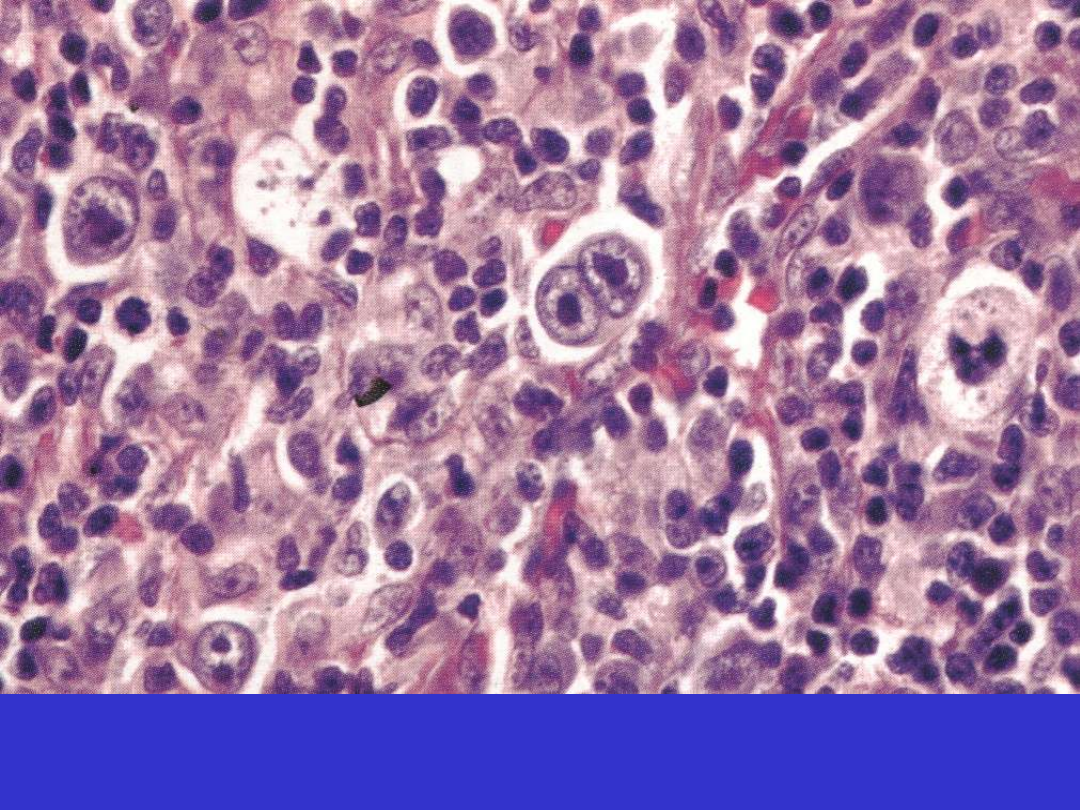
Klasyczny HL, typ mieszanokomórkowy (MC), typowa komórka RS
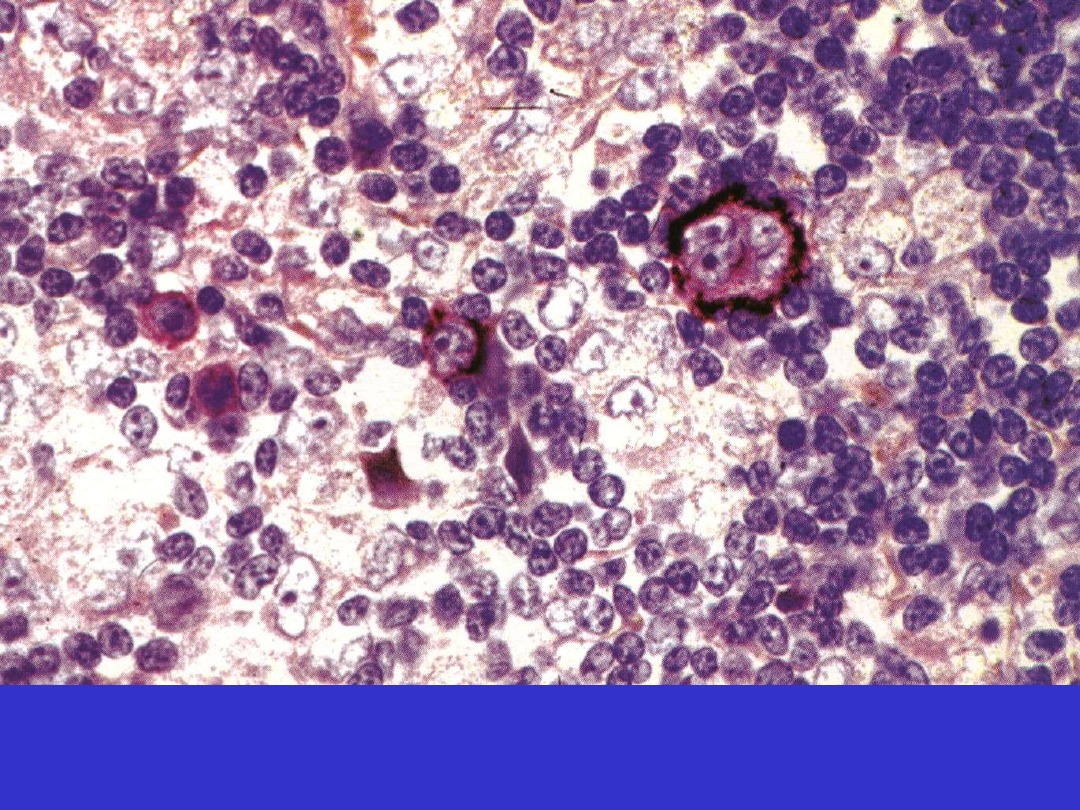
Klasyczny HL, CD30, typ mieszanokomórkowy (MC)

Klasyczny HL, typ bogaty w limfocyty (lymphocyte-rich, LR)
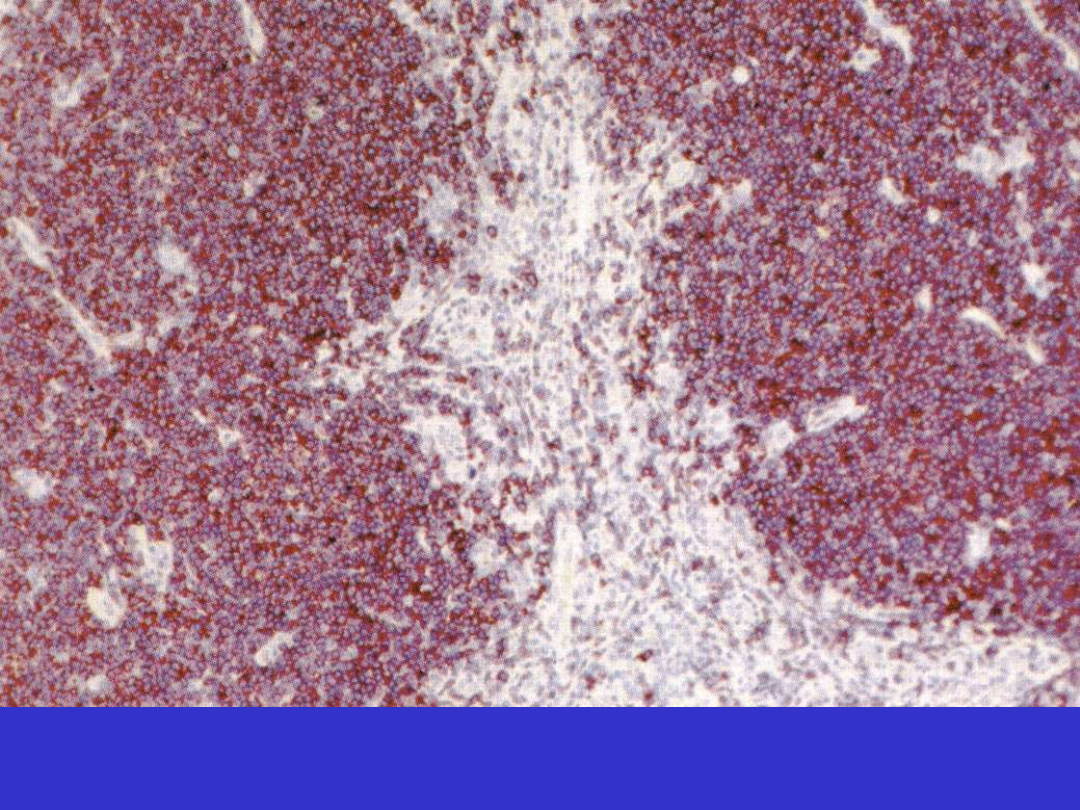
Klasyczny HL, typ bogaty w limfocyty (lymphocyte-rich, LR), CD20
Klasyczny HL, typ bogaty w limfocyty (lymphocyte-rich, LR), CD20
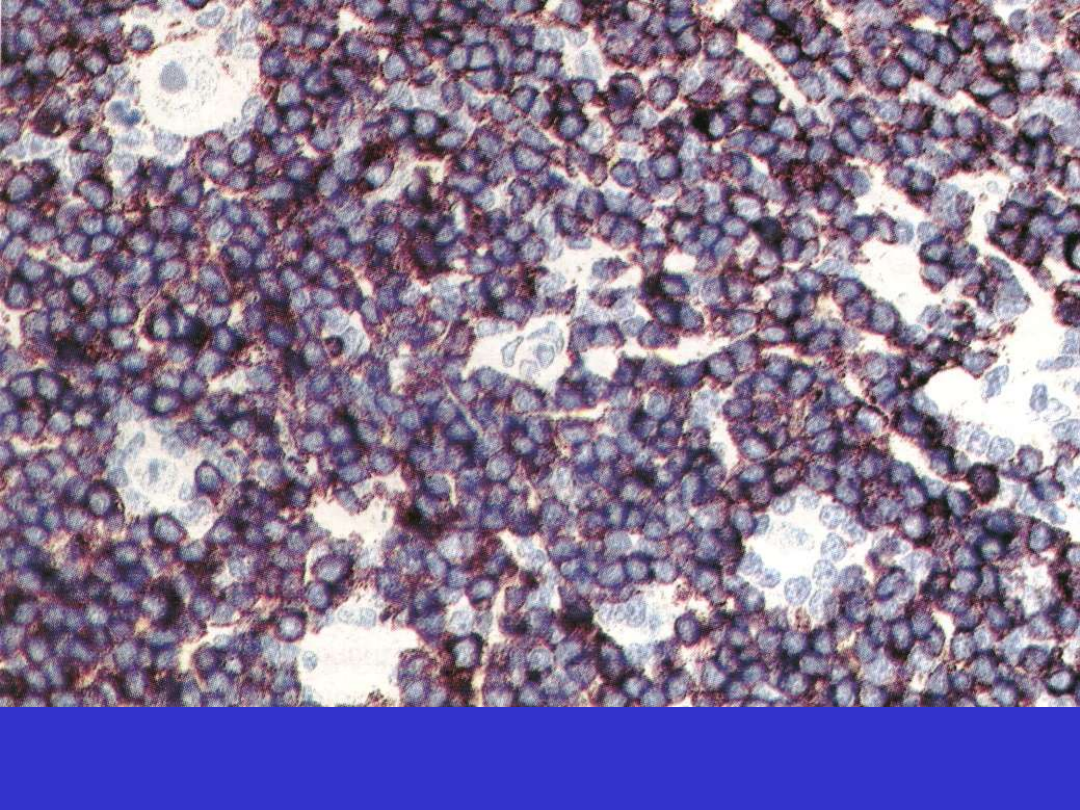
Klasyczny HL, typ z zanikiem limfocytów (lymphocyte-depleted, LD).
Liczne kk. Hodgkina, limfocyty pojedyncze
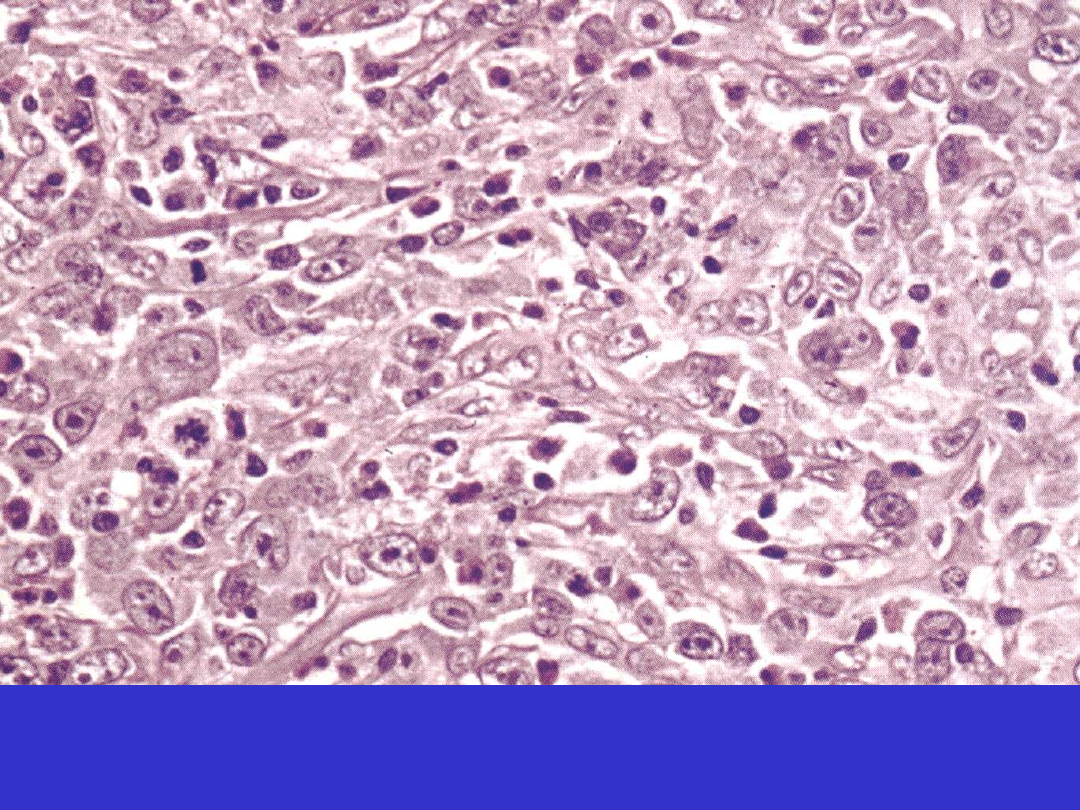
Klasyczny HL, typ z zanikiem limfocytów (lymphocyte-depleted, LD).
Włokienkowa macierz z licznymi fibroblastami.
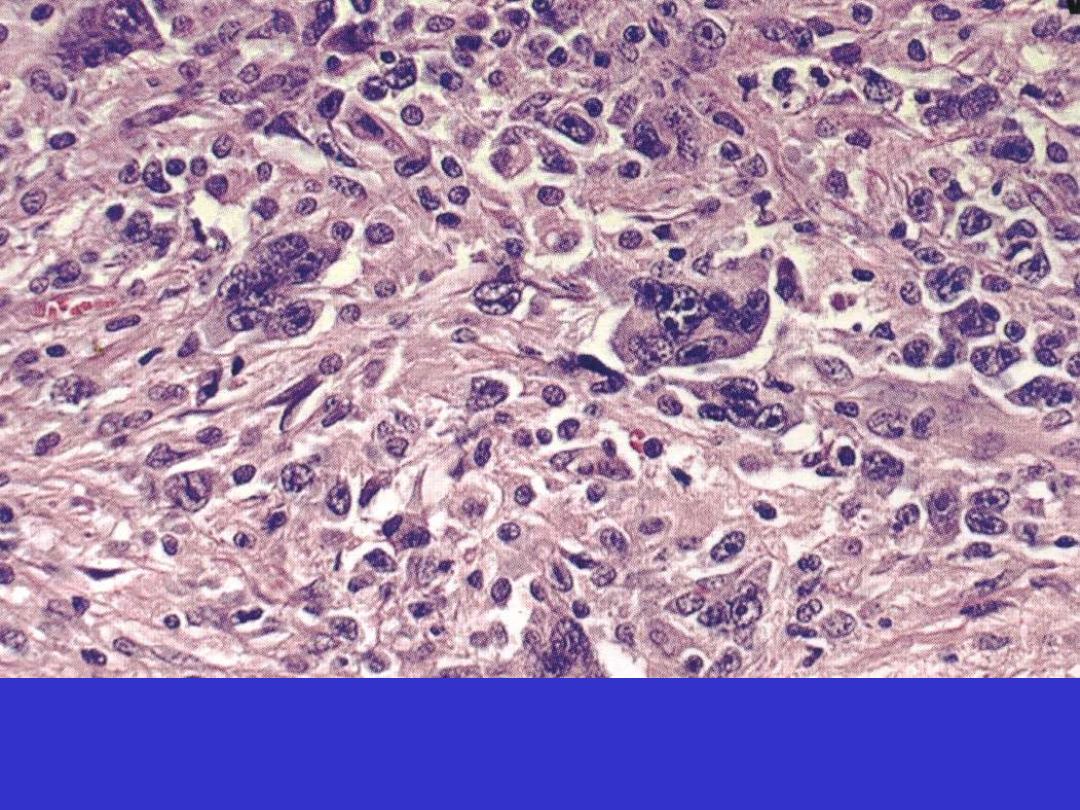
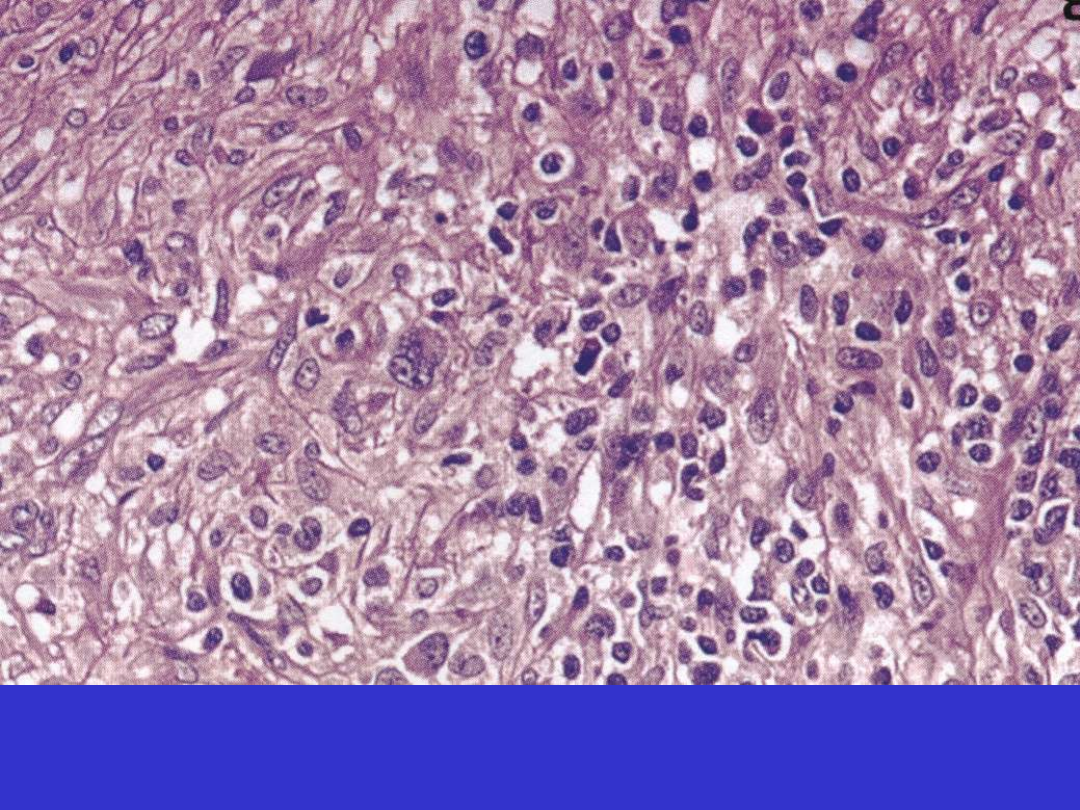
Klasyczny HL, typ z zanikiem limfocytów (lymphocyte-depleted, LD).
Liczne dziwaczne kk. HRS.
Wyszukiwarka
Podobne podstrony:
chloniaki komplet
CHŁONIAKI ZŁOŚLIWE
Chłoniaki nieziarnicze wykład 2007
chloniaki zlosliwe 2
KOMPLEKSY POLAKOW wykl 29 03 2012
pytania nowe komplet
zwiazki kompleksowe 2
8 kompleksy
Chłoniaki złośliwe tzw niehodgkinowskie
W19 kompleksonometria, wska«niki i krzywe miareczkowania kompleks i
Bliskowschodni kompleks bezpieczeństwa Przyczyny destabilizacji w regionie
Kompleksowa ocena geriatryczna
chloniak ziarniczy
Kopia Chłoniak
Chloniaki Vrok
Komplementarnosc
Kompleksowa rozgrzewka z pilkam Nieznany
więcej podobnych podstron