3. HALOGENOALKANY, REAKCJE SUBSTYTUCJI
Związki organiczne zawierające halogen należą do trzech grup różniących się właściwościami chemicznymi. Pierwsza z nich, najbardziej reaktywne, to halogenoalkany, czyli halogenki alkilowe, w których atom halogenu powiązany jest atomem węgla o hybrydyzacji sp3. Pozostałe dwie to halogenki winylowe i halogenki arylowe, w których atom halogenu przyłączony jest to atomu Csp2. W tym rozdziale omówione zostaną głównie halogenki alkilowe.
3.1 Występowanie i wykorzystanie
Halogenoalkany są powszechnie spotykane, ponieważ odgrywają znaczącą rolę jako rozpuszczalniki (CH2Cl2, CH3Cl, CCl3CH3 i inne) oraz jako surowce przemysłu chemicznego i odczynniki laboratoryjne. Stosowany dawniej w pralniach chemicznych toksyczny i kancerogenny rozpuszczalnik - 1,1,2-trichloroeten (znany pod skrótową nazwą TRI - CCl2=CHCl) został zastąpiony przez mniej szkodliwy 1,1,1-trichloroetan (CCl3CH3). Bromek metylu jest używany do odkażania ziemi w sadownictwie i uprawie warzyw, a także do niszczenia termitów. Obecnie jednak, zgodnie z tendencją do ochrony atmosfery ziemskiej dąży się do ograniczania stosowania tego typu rozpuszczalników, ponieważ są niebezpieczne i trudne do utylizacji. Wypuszczone do atmosfery wolno ulegają rozkładowi, a w wyniku spalania powstają z nich groźne dla życia halogenowodory.
Pośród pestycydów jest wiele związków organicznych zawierających atom lub atomy chloru. Do najbardziej znanych należą: DDT (dichlorodifenylotrichloroetan) i lindan (γ-heksachlorocykloheksan). W wielu krajach ze względu na szkodliwe działanie i na inne organizmy zakazano ich stosowania.
Halogenoalkany służą jako skuteczne środki gaśnicze wykorzystywane, np. w gaśnicach (CCl4). Stosuje się też do tego celu mieszane pochodne bromochlorowe, np. bromochlorometan. Ze względu na dużą gęstość jest on skuteczniejszym środkiem gaśniczym niż tetrachlorometan. Związki halogenoorganiczne stanowią cenne półprodukty do otrzymywania polimerów (polichlorku winylu), leków, barwników, środków higieny, ochrony roślin i wielu innych. Wykorzystywane są do alkilowania (wprowadzania reszt alkilowych) w przemyśle i w laboratoriach.
Złą sławą okryte są freony - związki fluoroorganiczne (perfluorochlorometany i perfluorochloroetany). Jeszcze do niedawna stosowane na dużą skalę jako media w chłodziarkach, czy sprężarkach zanieczyściły atmosferę ziemską do tego stopnia, że spowodowały duże ubytki ozonu - czynnika chroniącego ziemię przed szkodliwym promieniowaniem UV. Perfluorowane polimery, np. teflon znalazły zastosowanie w życiu codziennym jako hydrofobowy materiał, bardzo odporny na działanie czynników chemicznych i podwyższonej temperatury. Powleka się nim patelnie i inne naczynia kuchenne, z niego wyrabia się elementy aparatury chemicznej, a także części zastępujące tkanki biologiczne. Wyroby z teflonu są bardzo gładkie, służą wobec tego też do wytwarzania łożysk oraz jako dodatek do smarów, polepszający ich smarowność.
Znaczne ilości lotnych halogenowodorów, np. bromek metylu i etylu są wytwarzane przez organizmy żyjące. Szczególnie aktywne w emitowaniu ich do atmosfery są mikroorganizmy morskie.
3.2 Nomenklatura
Halogenoalkany nazywane są dwoma sposobami, Zgodnie z zasadami IUPAC ich nazwy tworzy się dodając do nazwy węglowodoru nazwę halogenu wraz z lokantami.
1-chloropentan 2-chloropentan 3-chloropentan
Drugi sposób przypomina nazywanie soli. Polega on na dodaniu do słowa halogenek (chlorek, itp.) nazwę reszty alkilowej, do której przyłączony jest halogen, np. chlorek etylu (CH3CH2Cl), bromek izopropylu (CH3CHBrCH3), chlorek metylenu (CH2Cl2), itd.
3.3 Otrzymywanie
3.3.1 Halogenowanie wolnorodnikowe
W podwyższonej temperaturze lub w obecności światła alkany reagują z chlorem lub bromem tworząc mieszaninę halogenowanych produktów różniących się położeniem atomu halogenu w cząsteczce lub/i liczbą jego atomów. Brom jest znacznie mniej reaktywny niż chlor i reakcja z nim wymaga drastyczniejszych warunków. Jod jest nieaktywny, a fluor reaguje wybuchowo z alkanami nawet w obniżonej temperaturze i w ciemności; szybkość tej reakcji można spowolnić poprzez rozcieńczenie reagentów gazem obojętnym, np. azotem. Reakcje alkanów z halogenami są wysoko egzotermiczne; wydziela się też halogenowodór, jako produkt uboczny.
3.3.2 Chlorowanie utleniające
Chlorowanie utleniające umożliwia zużytkowanie chlorowodoru powstającego jako produkt uboczny w reakcjach chlorowania za pomocą chloru lub w innych procesach. Prowadzi się je za pomocą chlorowodoru w atmosferze utleniającej. Z metanu w tych warunkach powstaje chlorometan i wyższe produkty chlorowania.
3.3.3 Addycja halogenowodoru do podwójnego (potrójnego) wiązania
Addycja halogenowodorów do alkenów jest ważnym sposobem otrzymywania halogenków alkilowych; przyłączenie jednego mola halogenowodoru do alkinów prowadzi do halogenków winylowych, a dwóch moli daje dihalogenoalkany.
3.3.4 Addycja halogenu do podwójnego (potrójnego) wiązania
W wyniku addycja chloru, bromu lub jodu do alkenów powstają wicinalne dihalogenowodory; wicinalne - oznacza na sąsiadujących atomach C. Halogenowanie alkenów biegnie łatwo, nie wymaga wspomagania katalizatorami.
3.3.5 Substytucja grupy hydroksylowej przez halogen
Alkohole pod wpływem halogenków fosforu, a nawet w wyniku reakcji z kwasem bromowodorowego ulegają przekształceniu w halogenki alkilowe.
3.3.6 Substytucja halogenu przez inny halogen
Jest to często stosowana metoda otrzymywania fluoropochodnych z chloro lub bromozwiązków.
Można również w ten sposób przekształcać chlorki lub bromki alkilowe w jodki alkilowe za pomocą jodku potasu w acetonie. Siłą napędową tej reakcji jest wytrącanie się nierozpuszczalnego w acetonie chlorku potasu.
3.4 Mechanizm reakcji halogenowania alkanów
W reakcji metanu z chlorem w obecności światła lub w podwyższonej temperaturze tworzą się produkty, w których zamiast atomu (atomów) wodoru znajduje się atom (atomy) chloru. Powstają w ten sposób chloroalkany, przy czym wydziela się chlorowodór:
W reakcji tej atom chloru nie zostaje dodany do cząsteczki metanu, lecz wchodzi w miejsce atomu wodoru - wymienia atom wodoru. O takiej przemianie mówimy, że jest to reakcja wymiany lub częściej - substytucji.
Sądząc po liczbie produktów reakcja jest skomplikowana. Pojawiają się przy tym pytania:
- dlaczego powstaje mieszanina produktów,
- z ilu etapów składa się reakcja;
- jaka jest kolejność przemian prowadzących od substratów do produktów i
- jakie znaczenie dla tej reakcji ma światło lub ciepło?
Pytania te dotyczą mechanizmu reakcji. Odpowiedź na nie otrzymuje się na podstawie kojarzenia wniosków wynikających z obserwacji eksperymentów. Poznanie mechanizmów reakcji ułatwia zrozumienie chemii organicznej, ponieważ opisują one kolejno, krok po kroku zmiany, jakie następują w obrębie wiązaniach reagentów - ich rozrywanie i tworzenie nowych. W rzeczywistości zmiany te dotyczą przemieszczania się elektronów tworzących wiązania i wolne par elektronowe.
Badając reakcję chlorowania metanu można zauważyć, że:
- metan i chlor nie reagują z sobą w ciemności;
- do reakcji w ciemności dochodzi, ale po ogrzaniu reagentów powyżej 250oC;
- reakcja biegnie również w temperaturze pokojowej pod wpływem światła UV;
- obecność nawet małej ilości tlenu zmniejsza na chwilę szybkość reakcji,
jednak po pewnym czasie wraca ona do poprzedniej szybkości;
- czas spowolnienia szybkości reakcji (okres inhibicji) zależy od ilości dodanego tlenu.
Obserwacje te ułatwiają zaproponowanie mechanizmu reakcji, a przyjęty mechanizm nie może być w sprzeczności z żadnym faktów dotyczących reakcji.
Zarówno inicjowanie reakcji przez światło lub podwyższoną temperaturę oraz inhibitujące działanie tlenu sugerują, że jest to reakcja rodnikowa. Zaczyna się ona od homolitycznego rozpadu wiązania w jednym z reagentów:
W wyniku homolizy dochodzi do rozpadu wiązania, w wyniku którego powstają fragmenty (atomy lub grupy atomów) zawierające niesparowany elektron. Takie fragmenty nazywane są rodnikami. Jeżeli niesparowany elektron przypisany jest do atomu węgla to mamy do czynienia z karborodnikiem.
Znana jest również heteroliza, czyli taki rozpad wiązania, w wyniku którego para elektronów tworząca wiązanie pozostaje przy jednym z fragmentów. Ten fragment nosi nazwę anionu (jest obdarzony ładunkiem ujemnym), a drugi fragment z ładunkiem dodatnim nazywany jest kationem.
Fragmenty heterolizy, w których ładunki znajdują się przy atomach węgla nazywane są odpowiednio karboanionem i karbokationem.
Każde wiązanie ma określoną specyficzną siłę i do jego zerwania potrzebna jest energia. Im większa energia wiązania, tym większej potrzeba energii do jego rozerwania. Minimalna energia potrzebna do rozerwania wiązania nazywa się energią dysocjacji. Może ona być dostarczona w postaci ciepła (odpowiednio wysokiej temperatury reakcji), promieniowania (np. UV) lub w inny sposób.
Entalpia (Ho) - energia dysocjacji (homolizy) wybranych wiązań A−B _→ A. + B. Tabela 3.1
.
Wiązanie pomiędzy |
Ho [kcal/mol] |
Wiązanie pomiędzy |
Ho [kcal/mol] |
Wiązanie pomiędzy |
Ho [kcal/mol] |
jednakowymi atomami A-A |
atomem węgla i wodoru |
atomami węgla |
|||
H−H D−D |
104 106 |
NC−H |
130 |
|
102 |
Cl−Cl |
58 |
HC≡C−H |
125 |
H3C−CH3 |
88 |
Br−Br |
46 |
|
112 |
CH3H2C−CH3 |
85 |
F−F |
38 |
H2C=CH−H |
111 |
(CH3)2HC−CH3 |
84 |
I−I |
36 |
H3C−H |
104 |
H3CH2CO−CH3 |
81 |
atomami wodoru i halogenem |
CH3CH2−H |
98 |
H2C=CHH2C−CH3 |
74 |
|
H−F |
136 |
(CH3)2HC−H |
95 |
|
72 |
H−Cl |
103 |
(CH3)3C−H |
91 |
H2C=CHH2C−Cl |
69 |
H−Br |
88 |
H2C=CHH2C−H |
87 |
|
77 |
H−I |
71 |
|
86 |
atomami wodoru i heteroatomami |
|
|
|
|
85 |
HO−H |
119 |
atomami węgla i halogenem |
H2N−H |
103 |
|||
CH3−F |
109 |
H3C−Br |
70 |
H3CO−H |
102 |
H2C=CH−Cl |
88 |
|
70 |
EtO−H |
103 |
H3C−Cl |
84 |
(CH3)2HC− Br |
68 |
H3C−OH |
91 |
|
|
|
|
H3CS−H |
88 |
|
82 |
CH3CH2−Br |
68 |
heteroatomami |
|
(CH3)2HC−Cl |
80 |
(CH3)3C−Br |
65 |
|
112 |
(CH3)3C−Cl |
79 |
CH3CH2−I |
53 |
H3C−NH2 |
80 |
|
|
(CH3) 3C−I |
50 |
HO−OH |
51 |
3.4.1 Mechanizm reakcji rodnikowych
Reakcje rodnikowe zaczynają się od utworzenia rodnika. Rodniki powstają najczęściej pod wpływem
podwyższonej temperatury, światła lub substancji rodnikotwórczych, np. nadtlenków.
W obecności tych czynników najsłabsze wiązanie w jednym z reagentów ulega homolizie i tworzą się rodniki. Ten etap reakcji nazywa się inicjacją (rozpoczęciem). W reakcji chloru z metanem najsłabszym wiązaniem jest wiązanie Cl−Cl (58 kcal/mol), podczas gdy C− H w metanie wynosi 104 kcal/mol.
Cząsteczka chloru absorbując kwant światła o odpowiedniej energii ulega dysocjacji. Powstające rodniki chloru reagują z drugim z substratów generując rodniki metylowe oraz chlorowodór. Rodniki metylowe łatwo reagują z chlorem tworząc chlorek metylu i kolejny rodnik chlorkowy.
Rodnik chlorkowy może wejść w reakcję z następną cząsteczką metanu i tak krok po kroku w łańcuchu przemian tworzy się wiele cząsteczek chlorku metylu. Ten etap reakcji nazywa się propagacją (rozwijaniem reakcji), a cała reakcja określana jest łańcuchową, ponieważ z jednego rodnika wytworzonego na początku w etapie inicjacji powstaje łańcuch cząsteczek CH3Cl.
Takich pojedynczych łańcuchów w każdej reakcji jest wiele, a każdy z nich rozwija się aż do wyczerpania substratów lub zostaje przerwany na skutek zderzenia dwóch rodników, albo też przez wytracenie energii, np. poprzez zderzenie rodnika ze ścianką naczynia. Etap zakończenia łańcucha nazywany jest terminacją. Inne łańcuchy mogą być rozwijane dalej, są również inicjowane nowe łańcuchy. W reakcji łańcuchowej etap terminacji poprzez zderzenia dwóch rodników nie odgrywa większej roli, ponieważ ze względu na krótki okres życia (istnienia) rodników ich stężenie jest małe, a przez to prawdopodobieństwo ich zderzenia jest niskie.
Mechanizm reakcji rodnikowej tłumaczy, dlaczego w takich reakcjach, jak chlorowanie alkanów wystarcza jeden foton do wytworzenia wielu (setek, a nawet tysięcy) cząsteczek produktu, w powyższym przykładzie - chlorku metylu.
Reakcja nie zatrzymuje się na etapie monochlorowania. W miarę wzrostu stężenia CH3Cl również ten związek staje się konkurencyjnym substratem w wychwytywaniu rodników chlorkowych i następuje wymiana atomu wodoru na chlor. W ten sposób tworzy się chlorek metylenu (CH2Cl2), który z kolei po dalszym chlorowaniu ulega przekształceniu w chloroform (CHCl3), a następnie powstaje tetrachlorek węgla. Tak więc produktem chlorowania metanu jest mieszanina chlorometanów, o różnym stopniu wysycenia chlorem, zależnym przede wszystkim od nadmiaru jednego z reagentów:
metan chlorek metylu chlorek metylenu chloroform tetrachlorek węgla
Zadanie: napisz etapy powstawania chlorku metylenu w reakcji chlorowania chlorku metylu
W reakcji, w której użyto 1 mol chloru na 1 mol metanu powstaje mieszanina składająca się z 37% CH3Cl, 41% CH2Cl2, 19% CHCl3 i 3% CCl4. Niektóre z tych produktów można otrzymać z większą wydajnością zmieniając proporcje reagentów. Nadmiar użytego w reakcji metanu zapewnia dużą wydajność chlorku metylu, a nadmiar chloru prowadzi do tetrachlorometanu. Maksymalną wydajność chlorku metylu (23%) osiąga się wówczas, kiedy nadmiar metanu w stosunku molowym dochodzi do 5. Natomiast najwięcej dichlorometanu otrzymuję się, kiedy stosunek chloru do metanu wynosi 1,6 : 1. Gdy stosunek molowy chloru do metanu sięga czterokrotnego nadmiaru, jedynym produktem chlorowania jest tetrachlorometan. Chloroform otrzymuje się w reakcji haloformowania acetonu.
Reakcja halogenowania alkanów jest silnie egzotermiczna - na każdym etapie wydziela się po około 100 kJ/mol. Bez odprowadzania ciepła może dojść do przegrzania, prowadzącego do gwałtownego przebiegu reakcji z wydzieleniem węgla.
CH4 + 2 Cl2 _→ C + 4 HCl
3.4.2 Regiospecyficzność reakcji halogenowania alkanów
Podczas chlorowania etanu tworzy się mieszanina zawierająca głównie monochloro-, 1,1-dichloro- i 1,1,1-trichloroetan.
Cl2
CH3CH3 _→ CH3CH2Cl + CH3CHCl2 + CH3CCl3 + ....
300oC
etan chloroetan 1,1-dichloroetan 1,1,1-trichloroetan
Taki przebieg reakcji spowodowany jest osłabieniem wiązania C−H po przyłączeniu atomu chloru.
W propanie atomy wodoru są nierównocenne z powodu różnicy energii wiązania C−H. Dla atomów wodorów 1o wynosi ona 98 kcal/mol, podczas gdy dla 2o 95 kcal/mol. Ta niewielka zdawałoby się różnica znacząco zwiększa szybkość wymiany atomu H 2o na atom Cl. Podczas chlorowania propanu 1-chloropropan powstaje z mniejszą wydajnością, niż 2-chloropropan, pomimo tego, że stężenie atomów wodoru 1o jest 3 razy większe (6 atomów H 1o i 2 atomy 2o). Biorąc pod uwagę tę proporcję (3 : 1) oraz różnice wydajności obu produktów (40% i 60% odpowiednio) można obliczyć, że podstawienie 2o atomu wodoru przez atom chloru biegnie 4,5 razy szybciej niż 1o atomu H; innymi słowy w tej reakcji 2o atomy wodoru są 4,5 razy reaktywniejsze od 1o atomów H.
propan 1-chloropropan (40%) 2-chloropropan (60%)
Zadanie: oblicz różnicę w reaktywności 1o i 2o atomów wodoru w propanie.
Chlorowanie propanu jest reakcją bardzo nieselektywną, ponieważ nawet przy stosunku molowym chloru do propanu 1 : 1, oprócz monochloropropanów powstaje znaczna ilość dichloropropanów.
Podczas chlorowania butanu wydajność 2-chlorobutanu jest znacznie większa niż 1-chlorobutanu. Ten wzrost wydajności izomeru 2 w porównaniu do chlorowania propanu wynika z większego udziału drugorzędowych atomów wodoru.
n-butan 1-chlorobutan (28%) 2-chlorobutan (72%)
Różnica reaktywności atomów H 1o i 3o jest jeszcze większa (Ho odpowiednio 98 i 91 kcal/mol) i dlatego w mieszaninie produktów monochlorowania tert-butylu jest aż 36% 2-chloro-2-metylopropanu, pomimo 9-krotnej liczbowej przewagi 1o atomów wodoru w substracie.
Różnica w podatności na oderwanie atomu wodoru z atomów węgla o różnej rzędowości wynika z trwałości powstających rodników. Najtrwalsze są rodniki 3o i dlatego tworzą się najłatwiej w wyniku oderwania atomu wodoru przy 3ooatomie węgla. Innymi słowy, utworzenie 2o rodnika wymaga więcej energii niż oderwanie atomu H z 3o atomu C. Jeszcze więcej energii wymaga utworzenie rodnika 1o, ale z kolei tworzy się on łatwiej niż rodnik metylowy. Porównaj odpowiednie wartości Ho w Tabeli 3.1.
Brom jest znacznie mniej reaktywniejszy od chloru, dlatego też bromowanie alkanów biegnie z mniejsza szybkością, a przez to z większą selektywnością niż chlorowanie:
propan 1-bromopropan (3%) 2-bromopropan (97%)
n-butan 1-bromobutan (2%) 2-bromobutan (98%)
Z wykresów zmiany energii w reakcjach chlorowania i bromowania metanu wynika, że reakcja bromowania wymaga znacznie większej energii aktywacji, a więc będzie wolniejsza niż chlorowanie w tej samej temperaturze. Z tego powodu staje się ona selektywniejsza, gdyż rodniki bromu będą statystycznie częściej wybierać najbardziej reaktywne miejsca, czyli wg wzrastającej kolejności aktywności atomy wodoru przy atomach węgla 1o, 2o i 3o.
Zmiany energii w procesie halogenowania metanu Wykres 3.1.
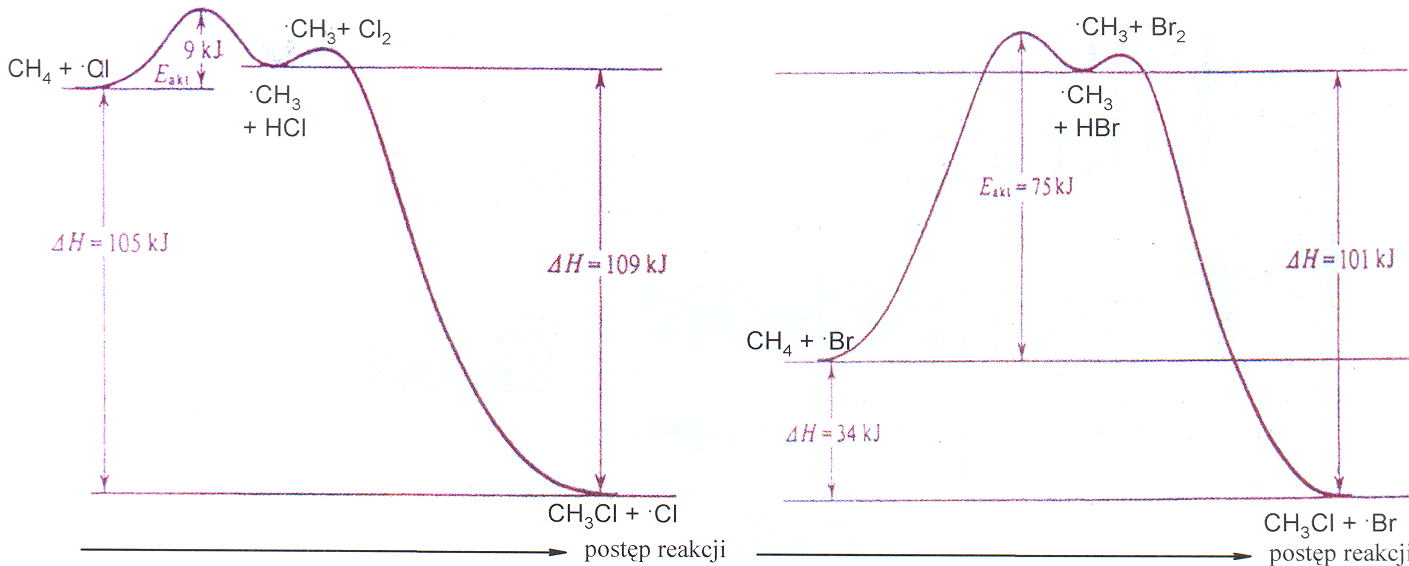
chlorowanie .CH3 + Cl2 bromowanie .CH3 + Br2
Rodnik chloru potrzebuje niewielkiej energii aktywacji do wyrwania atomu wodoru z cząsteczki węglowodoru, żeby utworzyć rodnik alkilowy; w przypadku metanu jest to jedynie 4 kcal/mol (17 kJ/mol). Natomiast energia aktywacji metanu do reakcji z rodnikiem bromu i utworzenie takiego samego rodnika metylowego jest kilkakrotnie większa, wynosi bowiem 18 kcal/mol (75 kJ/mol). W reakcjach z węglowodorami, w których znajdują się atomy wodoru różnej rzędowości rodnik chloru obdarzony wysoką energią może oderwać prawie równie łatwo atom wodoru 1o czy 2o, natomiast dla mniej aktywnego rodnika .Br znacznie łatwiejsze jest oderwanie atom wodoru o wyższej rzędowości. W reakcji propanu z chlorem różnica energii aktywacji w oderwaniu 1o i 2o atomu wodoru wynosi jedynie 4 kcal/mol (17 kJ/mol), podczas gdy w reakcji z bromem ta różnica dochodzi do 9 kcal/mol (38 kJ/mol).
Z rzędowością rodników związana jest także ich trwałość: wraz ze wzrostem rzędowości rodników rośnie ich trwałość. Prawie wszystkie rodniki obdarzone są wysoką energią, a przez to są bardzo reaktywne. Jednak reszty alkilowe związane z atomem węgla posiadającym niesparowany elektron stabilizują go poprzez rozłożenie tej energii w przestrzeni. Im więcej reszt alkilowych związanych z rodnikowym atomem węgla (wyższa jego rzędowość) tym rodnik jest stabilniejszy.
Względna trwałość rodników węglowych:
Szybkość wymiany wodoru na halogen w reakcjach substytucji rodnikowej zależy od trwałości tworzącego się przejściowo rodnika alkilowego. Im trwalszy rodnik, tym szybciej (łatwiej) się tworzy, po czym może ulegać dalszym przemianom. Z wykresu 2. widać, że rodnik izopropylowy (2o) będzie się tworzył trudniej (wolniej) niż 3o tert-butylowy, ponieważ do jego utworzenia potrzebna jest większa energia aktywacji.
Energia przemian bromowania propanu i tert-butanu Wykres 3.2.
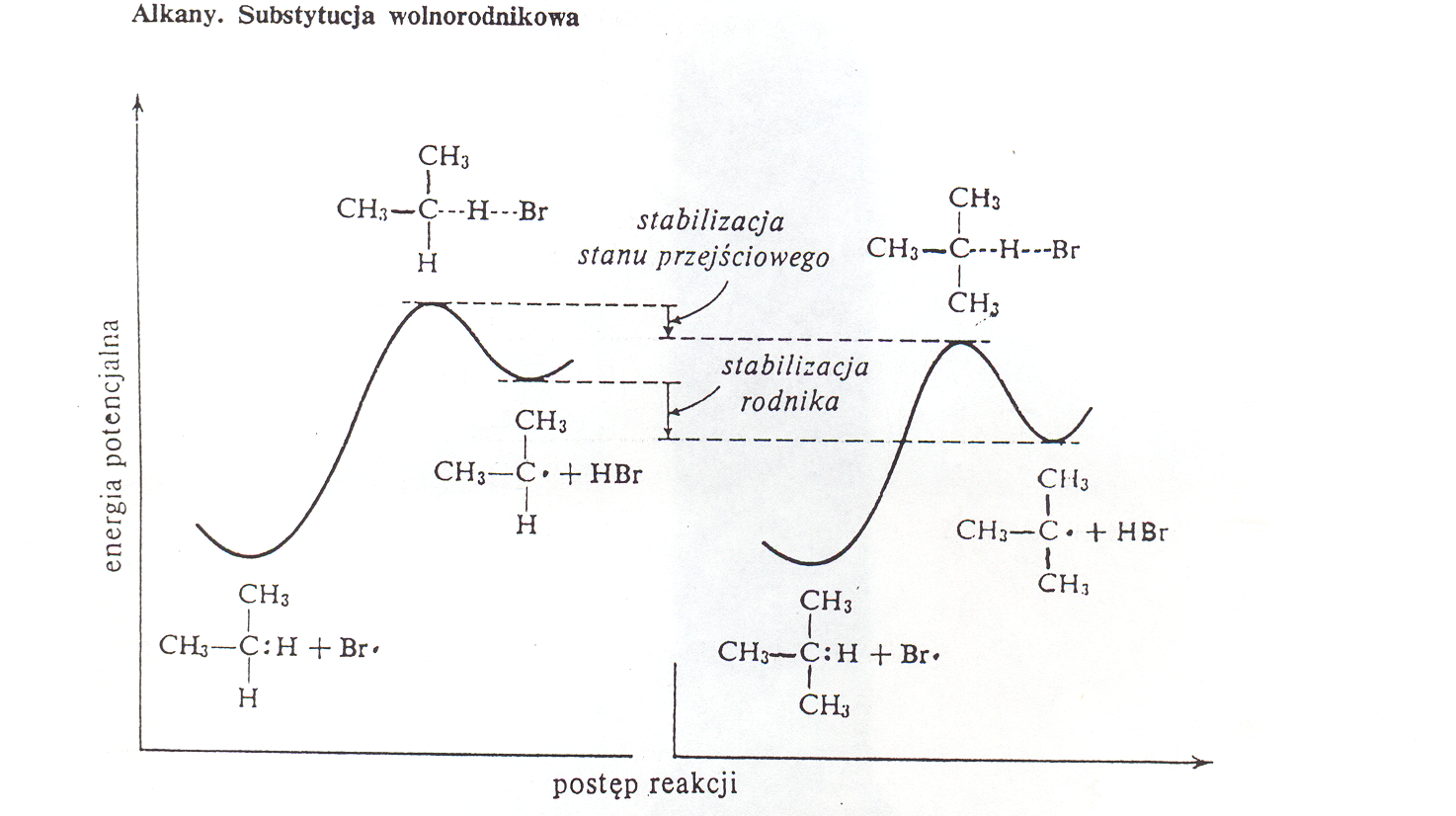
3.4.3 Halogenowanie cykloalkanów
Cyklopropan w reakcji z chlorem także tworzy rodnik cyklopropylowy, który łatwo przekształca się w znacznie trwalszy (stabilizowany mezomerycznie) rodnik allilowy, a ten w dalszej reakcji z chlorem zostaje przechodzi w chlorek 1-propylu. Równocześnie, chociaż z mniejszą wydajnością rodnik cyklopropylowy tworzy z chlorem chlorek cyklopropylu:
Cyklopropan jest pod wpływem bromu w obecności AlBr3 przekształcany w 1,3-dibromopropan, a katalityczne wodorowanie zarówno cyklopropanu, jak i cyklobutanu prowadzi do rozerwania pierścieni i utworzenia odpowiednich alkanów:
Wyższe cykloalkany reagują z halogenami podobnie jak alkany - ulegają halogenowaniu w reakcji substytucji rodnikowej:
cyklopentan chlorocyklopentan cykloheksan bromocykloheksan
(nadmiar) (nadmiar)
Mogą się tworzyć polihalogenoalkany. Nadmiar węglowodorów zmniejsza ich wydajność.
Alkilocykloalkany tworzą z halogenami mieszaninę izomerycznych monopochodnych:
Halogenoalkany - halogenki alkilowe
Halogenoalkany są powszechnie spotykane, ponieważ odgrywają znaczącą rolę jako rozpuszczalniki (CH2Cl2, CH3Cl i inne) oraz surowce przemysłu chemicznego. Należą do skutecznych środków gaśniczych wykorzystywanych w gaśnicach (CCl4). Stanowią cenne półprodukty do otrzymywania polimerów (polichlorku winylu), leków, barwników, środków higieny, ochrony roślin i wielu innych. W życiu codziennym perfluorowane polimery, np. teflon znalazły zastosowanie jako hydrofobowy materiał, bardzo odporny na działanie czynników chemicznych i podwyższonej temperatury. Powleka się nim patelnie i inne naczynia kuchenne, z niego wyrabia się elementy aparatury chemicznej, a także części zastępujące tkanki biologiczne. Wyroby z teflonu są bardzo gładkie, służą wobec tego do wytwarzania łożysk oraz jako dodatek do smarów, polepszający ich smarowność. Złą sławą okryte są inne fluoroorganiczne związki - freony (perfluorochlorometany i perfluorochloroetany). Jeszcze do niedawna stosowane na dużą skalę jako media w chłodziarkach, czy sprężarkach zanieczyściły atmosferę ziemską do tego stopnia, że spowodowały duże ubytki chroniącą ziemię warstwę ozonu. Znaczne ilości lotnych halogenowodorów, np. bromek etylu wytwarzane są przez organizmy żyjące, szczególnie mikroorganizmy morskie i wydzielane do atmosfery.
3.5 I Z O M E R I A
Izomerami nazywane są związki chemiczne posiadające taki sam wzór sumaryczny, różniące się jednak konstytucją (budową, tj. rozmieszczeniem atomów i wiązań w cząsteczce) lub ułożeniem atomów w przestrzeni.
Rozróżniane są izomery konstytucyjne i stereoizomery.
3.5.1 Izomery konstytucyjne
Różnica w konstytucji cząsteczki (jej budowie) wynikająca z innej kolejności atomów ją tworzącą czy innych (inaczej rozmieszczonych wiązań) jest przyczyną istnienia izomerów konstytucyjnych. Przykładem może być prosty (normalny) i rozgałęziony łańcuch węglowy lub usytuowanie wiązań wielokrotnych.
Istnieją trzy izomery pentanu, wszystkie mają ten sam wzór sumaryczny C5H12:
Węglowodór o wzorze sumarycznym C5H10 może być cyklicznym węglowodorem nasyconym: cyklopentanem, metylocyklobutanem lub dimetylocyklopropanem (we wzorach kreska oznacza grupę metylową).
Może być również jednym z wielu izomerów alkenów (węglowodorów zawierających 1 podwójne wiązanie C=C):
Równie dobrze znane są izomery konstytucyjne chlorowcopochodnych, alkoholi i innych związków.
Zadanie: Narysuj i nazwij izomery związku o wzorze sumarycznym C6H13Br.
3.5.2 Stereoizomery
Tego typu izomery mają nie tylko taki sam wzór sumaryczny, ale również identyczną konstytucję, czyli rozmieszczenie atomów i wiązań w cząsteczce, różnią się natomiast przestrzennym (sterycznym) ułożeniem atomów. Różnice w przestrzennym ułożeniu atomów występują w cząsteczkach zawierających cztery różne podstawniki przy tym samym atomie (np. atomie węgla) lub w innych przypadkach, kiedy cząsteczka pozbawiona jest niektórych elementów symetrii.
1-bromo-1-chloroetan ma cztery różne podstawniki przy C1 (brom, chlor, metyl i wodór) dlatego może występować w postaci dwóch streoizomerów, które w typowych warunkach mają identyczne właściwości fizyczne (np. tw., tt. czy gęstość), takie same właściwości chemiczne (reaktywność), identyczną wartość bezwzględną skręcalności właściwej [], ale o przeciwnych znakach, różnią się również reaktywnością w stosunku do innych stereoizomerów.
Stereoizomeria jest ważną częścią chemii organicznej ponieważ wiele związków biologicznie czynnych jest stereoizomerami. Właściwości biologiczne stereoizomerów mogą być różne, ponieważ w organizmie oddziałują z innymi stereoizomerami. Stereoizomerami są cukry, niektóre alkohole, aminy, większość aminokwasów oraz tworzone z nich peptydy czy białka.
Ciała czynne żywego organizmu (enzymy, hormony, receptory) są zbudowane z białek, a więc jako stereoizomery inaczej będą reagować ze stereoizomerycznymi substratami. Z tego powodu odczuwamy inny smak dwóch różnych stereoizomerów tego samego aminokwasu (reakcja z białkowymi kubkami smakowymi), inaczej też będą oddziaływać na organizm stereoizomeryczne substancje lecznicze.
Drastycznym przykładem takich różnic jest tragedia spowodowana thalidomidem, lekiem uspakajającym i przeciwbólowym wprowadzonym do użytku w II połowie XX w. Kobiety ciężarne przyjmujące ten lek rodziły dzieci bez kończyn lub z kończynami bardzo zdeformowanymi. Lek okazał się czynnikiem teratogennym. Badania wykazały, że był on mieszaniną dwóch stereoizomerów, przy jeden z nich wykazywał oczekiwane właściwości lecznicze a drugi, niespodziewany w otrzymywanym produkcie był teratogenem.
(S)-(-)-thalidomid (R)-(+)-thalidomid
teratogen lek
Do syntezy użyto właściwego substratu, a podczas badań klinicznych i przedklinicznych nie stwierdzono żadnych niepożądanych działań preparatu. Skąd się więc wziął drugi izomer w preparacie leczniczym? Otóż drugi stereoizomer pojawił się w znaczących ilościach dopiero podczas produkcji leku na dużą skalę. Większa skala wymagała przedłużenia czasu reakcji, co spowodowało, że drugi izomer tworzący się w wyniku tzw. racemizacji pojawił się w większym stężeniu. W tym czasie nikt nie znał zależności stopnia racemizacji od czasu reakcji i nie spodziewano się takich komplikacji w wyniku zwiększenia skali syntezy.
Innym przykładem dramatycznej różnicy we właściwościach biologicznych stereoizomerów, tym razem cis i trans, są kwasy buteno-1,4-diowe. Kwas cis-butenodiowy, zwany kwasem maleinowym, jest substancją toksyczną, drażniącą tkanki, natomiast kwas trans-butenodiowy, zwany kwasem fumarowym, występuje jako ważny półprodukt w procesach biochemicznych, zachodzących w organizmach zwierzęcych i roślinnych. Z kolei tłuszcze zawierające nienasycone kwasy tłuszczowe cis stanowią cenne produktu spożywcze, podczas, gdy spożywanie izomerów trans tych kwasów jest szkodliwe dla zdrowia.
Właściwa konfiguracja (ułożenie atomów w przestrzeni) substratów ma istotne znaczenie w reakcjach biochemicznych, ponieważ hormony, enzymy i inne ciała czynne są stereoizomerami, i tylko odpowiednia konfiguracja substratów zapewnia powodzenie przemian. Substraty o innej konfiguracji nie będą brały udziału w reakcji, lub co gorsze mogą wywołać niepożądane, szkodliwe działanie. Prostą ilustracją wymaganej konfiguracji (kształtu) cząsteczek jest zabawka dla małych dzieci, której elementy o różnych kształtach (kule, sześciany, ostrosłupy itp.) należy włożyć do pojemnika przez specjalne otwory (Rys. 3.1). Tylko te przedmioty uda się włożyć do pojemnika, które pasują kształtem do otworu. Podobnie, tylko ten związek będzie substratem enzymu, którego kształty pasują do rozmieszczonych odpowiednio miejsc reaktywnych enzymu.
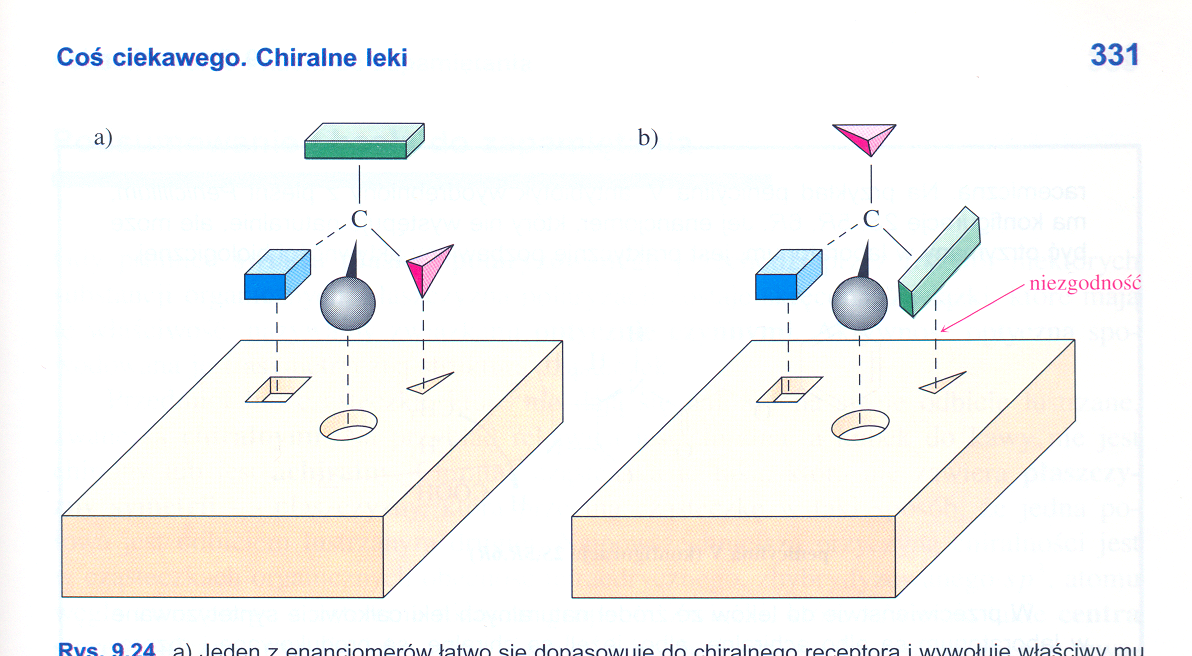
Rys. 3.1 Do reakcji pomiędzy receptorem, a substancją aktywną dochodzi w przypadku zgodności sterycznej
3.5.3 Sposoby rozróżniania stereoizomerów
Związki organiczne, w których atom węgla (lub inny czterokoordynacyjny atom) związany jest z dwoma (lub więcej) takimi samymi podstawnikami (atomami lub grupami atomów) będą identyczne po każdorazowej zamianie podstawników w przestrzeni.
Odbicia lustrzane takich związków są identyczne z oryginałami (przedmiotami przed lustrem):
etan chloroetan
W przypadku, kiedy atom węgla (lub inny) jest związany z czteroma różnymi podstawnikami, każda pojedyncza zamiana dwóch podstawników w przestrzeni prowadzi do utworzenia stereoizomeru, który jest odbiciem lustrzanym pierwszego stereoizomeru. Taki atom nazywamy jest atomem (centrum) chiralnym lub stereogenicznym.
lustro
↓
(R)-1-bromo-1-chloroetan (S)-1-bromo-1-chloroetan
Powyższe dwa stereoizomery (odbicia lustrzane) nie da się nałożyć na siebie tak, żeby jedna cząsteczka miała pod sobą te same podstawniki drugiej cząsteczki. Stereoizomery, które są do siebie jak odbicia lustrzane nazywane są enancjomerami. (R)-1-bromo-1-chloroetan i (S)-1-bromo-1-chloroetan są enancjomerami. Enancjomery należą do stereoizomerów.
W życiu spotykamy się nie tylko ze stereoizomerycznymi substancjami, ale również z przedmiotami o podobnych właściwościach. Dwa buty czy dwie rękawiczki tej samej pary też nie można nałożyć na siebie (dlatego nie da się włożyć prawej nogi do lewego buta). Podobnie jest ze stopami i dłońmi, są one w parze odbiciami lustrzanymi. Właśnie od greckiej nazwy dłoni - cheir pochodzi określenie tego typu przedmiotów i związków chemicznych - chiralne. Warto zdać sobie sprawę z tego, że przedmiotami chiralnymi są te, których odbicie lustrzane różnią się od obiektów oryginalnych (nie da się ich nałożyć na siebie). Takimi popularnym przedmiotami chiralnymi są, np. śruby, a nawet samochody.
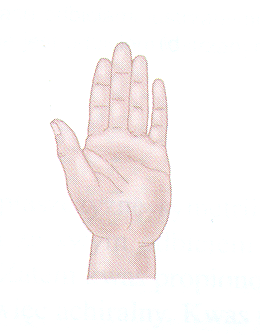
Rys. 3.2. Ręce są „chiralne”; nie można nałożyć na siebie lewej i prawej ręki (czy też ich odbić lustrzanych)
Atom węgla związany z czteroma różnymi podstawnikami jest nie tylko atomem chiralnym, ale również centrum chiralnym, jako że dzięki niemu cząsteczka nabiera właściwości związanych z chiralnością. Litery (R) i (S) pisane w nawiasach, kursywą to jeden ze sposobów różnicowania enancjomerów na piśmie. Określają one konfigurację absolutną enancjomerów. Ustala się ją w oparciu o reguły pierwszeństwa wg Cahna, Ingolda i Preloga.
3.5.4. Reguły pierwszeństwa wg Cahna, Ingolda i Preloga
1. W celu przypisania centrum chiralnemu odpowiedniej konfiguracji, np. literek R lub S, należy podstawniki uszeregować wg ważności (starszeństwa), tj wg wartości liczby atomowej atomu związanego z chiralnym (stereogenicznym) atomem węgla od 1 (dla atomu o najwyższej liczbie atomowej) poprzez 2 i 3 do 4 (dla atomu o najniższej liczbie atomowej). Jeżeli atomy są izotopami tego samego pierwiastka o kolejności decyduje liczba masowa. Zwykle chiralny atom oznaczany jest gwiazdką - *.
2. Jeżeli atomy związane z centrum chiralnym są takie same o ważności podstawników decydują atomy z drugiego szeregu, ewentualnie trzeciego lub dalszych; tak żeby można było wyznaczyć starszeństwo - dla różnych podstawników musi ono zaistnieć.
Przykład:
Powyższym pochodnym pentanu łatwo przypisać starszeństwo atomom wodoru i chloru, odpowiednio 1 i 4. Natomiast pozycje pozostałych podstawników (etylu i izopropylu) zależą od tego, ile jest i jakie są atomy w drugim rzędzie. W 3-chloro-2-metylopentanie izopropyl ma 2 atomy C i dlatego otrzymuje drugą pozycję (2) przed etylem (3), który w drugim szeregu ma tylko jeden atom węgla. Natomiast w przypadku drugiego związku kolejność na drugiej i trzeciej pozycji ulega odwróceniu ponieważ w grupie etylowej w drugim rzędzie atomów w odniesieniu do centrum chiralnego znajduje się chlor. Jeden atom o większej liczbie atomowej jest ważniejszy od dwóch, a nawet trzech atomów o mniejszych liczbach atomowych.
3. W związkach zawierających wiązania wielokrotne oba atomy powiązane w ten sposób liczy się odpowiednio podwójnie lub potrójnie. Reguła ta wynika z formalnego rozerwania wiązania wielokrotnego i dodaniu do nowoutworzonych wiązań tych samych atomów, które tworzą wiązanie wielokrotne.
Przykłady:
Po ustaleniu pierwszeństwa podstawników ustawia się cząsteczkę w ten sposób, żeby najmłodszy podstawnik (oznaczony liczbą 4) znalazł się najdalej od obserwatora. Bardzo często tym podstawnikiem jest atom wodoru. Następnie zakreślamy krąg od podstawnika najstarszego 1 do trzeciego 3 poprzez podstawnik 2; podstawnik 4 jest ignorowany. Jeżeli ruch po zakreślanym kręgu jest zgodny z kierunkiem wskazówek zegara to ten enancjomer jest określany jako (R) - rectus (łac. prawy), a jeżeli przeciwnie do ruchu wskazówek zegara to mamy do czynienia z enancjomerem (S) - sinister (łac. lewy).
W literaturze można spotkać różne sposoby (reguły) określania konfiguracji enancjomerów, ale wszystkie prowadzą do tych samych ustaleń. Znana jest reguła kciuka (J. McMurry Chemia organiczna), reguła kierownicy samochodu i inne.
Konfiguracja określona za pomocą symboli (R) i (S) nazywana jest konfiguracja absolutną, ponieważ odpowiada ona rzeczywistej konfiguracji związków organicznych. Znana jest również konfiguracja względna, gdyż została ona wyznaczona względem jakiegoś związku przyjętego za wzorzec.
Przykład: określić konfigurację absolutną obu enancjomerów 2-bromobutanu.
Narysujmy wzory enancjomerów 2-bromobutanu, tak żeby widać było przestrzenne ułożenie podstawników, ponumerujmy podstawniki wg starszeństwa i umieśćmy najmłodsze najdalej od siebie:
Powyższy sposób przedstawienia przestrzennej budowy związków organicznych nazywa się projekcją Newmana. Wzór jest tak zapisywany, jakby obserwator patrzył na cząsteczkę wzdłuż wiązania atomu C z najmłodszym podstawnikiem, tzn. oznaczonym cyfrą 4.
3.5.5 Projekcja Fischera - konfiguracja względna
Istnieją jeszcze inne sposoby przestrzennego zapisu cząsteczek chiralnych, pośród nich projekcja Fischera. Cząsteczkę ustawia się tak, żeby najdłuższy łańcuch węglowy znalazł się w jednej płaszczyźnie. Następnie robi się jego pionowy (od góry kartki papieru w dół) rzut na papier (tablicę czy ekran). W każdym centrum chiralnym wiązania pionowe oznaczają wiązania znajdujące się pod płaszczyzną kartki, a wiązania boczne nad płaszczyzną. Nawet jeżeli tego rozróżnienia nie widać na przedstawionym wzorze (projekcji) to należy o tym pamiętać - wiązania poziome są nad płaszczyzną, a pionowe pod płaszczyzną.
2-bromobutan w projekcji Fischera
Pojedyncza (lub nieparzysta) zamiana podstawników w projekcji Fischera, np. atomu Br z atomem H powoduje zmianę konfiguracji z (R) na (S) lub z (S) na (R).
Parzysta zamiana podstawników w projekcji Fischera, np. atomu Br z atomem H i następnie Br z CH3 zachowuje pierwotną konfigurację.
W celu przypisania konfiguracji absolutnej należy przejść z projekcji Fischera do Newmana. W projekcji Newmana najmłodszy podstawnik (w tym przypadku H) powinien znajdować się za atomem C, czyli pod płaszczyzną kartki. Jeżeli go zamienimy, np. z grupą metylową nastąpi zmiana konfiguracji. W celu zachowania konfiguracji należy dokonać jeszcze jednej zamiany podstawników, np. bromu z grupą etylową:
pojedyncza zamiana podstawników
powoduje zmianę konfiguracji
parzysta zamiana podstawników zachowuje wyjściową konfigurację
(S)-2-bromobutan
Ten sposób określania konfiguracji absolutnej na podstawie projekcji Fischera jest dość czasochłonny, wymaga bowiem dwukrotnej zamiany podstawników, jeżeli najmłodszy podstawnik znajduje się nad płaszczyzną kartki. Prościej jest oznaczyć konfiguracje bez zamian podstawników i tylko zamienić otrzymany wynik na przeciwny, tzn. (R) na (S) lub (S) na (R):
określoną niezgodnie z zasadami konfigurację (S),
należy odwrócić - otrzymuje się wówczas prawidłową konfigurację (R)
(R)-2-bromobutan
Projekcję Fischera wprowadzono wcześniej niż Newmana i była przeznaczona głównie dla związków zawierających kilka grup funkcyjnych; dlatego obowiązywała w niej jeszcze jedna reguła: rzut cząsteczki na płaszczyznę należy tak wykonać, że najbardziej utleniony atom węgla znajdował się u góry. W cząsteczce kwasu mlekowego w projekcie Fischera grupę karboksylowę należy umieścić u góry. Dla oznaczenia konfiguracji absolutnej nie ma znaczenia, gdzie znajduje się grupa karboksylowa, byleby wszystkie podstawniki miały określone usytuowanie przestrzenne. Wymóg zapisania najbardziej utlenionej grupy u góry wzoru potrzebny jest do przypisania konfiguracji względnej D lub L.
kwas: D-mlekowy ≡ kwas (R)-mlekowy L-mlekowy ≡ (S)-mlekowy
Konfiguracja względna określana literami D lub L oznacza położenie grupy funkcyjnej (w kwasie mlekowym -OH) po prawej (D) lub po lewej (L) stronie chiralnego atomu węgla, we wzorze prawidłowo zapisanym wg reguł projekcji Fischera.
Konfiguracja nazywa się względną, ponieważ odnosi się do konfiguracji względem aldehydu glicerynowego - CH2CH(OH)CHO - przyjętego za związek modelowy.
3.5.6 Konfiguracja cząsteczek zawierających dwa centra chiralne
Określanie konfiguracji cząsteczek zawierających dwa lub więcej chiralnych atomów węgla polega na przypisaniu konfiguracji kolejno poszczególnym centrom chiralnym.
Zadanie: Oznaczyć konfigurację względną 2,3-dibromopentanu - CH3CHBrCHBrCH2CH3.
Pierwszą czynnością jest zapisanie wzoru przestrzennie, np. za pomocą projekcji Fischera. Zgodnie z regułami otrzymujemy wzory czterech stereoizomerów:
W 2,3-dibromopentanie są dwa centra chiralne: atomy C2 i C3. We wszystkich wzorach najmłodsze podstawniki - atomy H są zapisane poziomo, czyli znajdują się nad płaszczyzną kartki, dlatego stosując uproszczoną procedurę należy po oznaczeniu konfiguracji kolejno dla C2 i C3 przyjąć konfigurację odwrotną jako rzeczywistą.
(2R,3R)-2,3-dibromopentan (2S,3S)-2,3-dibromopentan
(2R,3S)-2,3-dibromopentan (2S,3R)-2,3-dibromopentan
Stereoizomery a i b czyli (2R,3R)-2,3-dibromopentan oraz (2S,3S)-2,3-dibromopentan są enancjomerami (mają się do siebie jak odbicia lustrzane).
Stereoizomery nie będącymi do siebie jak odbicia lustrzane nazywane są diastereoizomerami; powyżej są to parami „a i c”, „a i d”, „b i c” oraz „b i d”.
Zdarza się, że cząsteczki posiadające po dwa lub więcej elementów chiralnych tracą chiralność z uwagi na symetrię. W 2,3-dibromobutanie płaszczyzna symetrii przechodzącą pomiędzy C2 i C3 powoduje, że cząsteczka jest achiralna, a w związku z tym staje się optycznie nieczynna (nie skręca płaszczyzny światła spolaryzowanego). Jej odbicie lustrzane da się na siebie nałożyć. Stereoizomery zawierające centra chiralne oraz płaszczyznę symetrii nazywane mezo, inaczej są to achiralne związki zawierające centra chiralne.
(2R,3S)-2,3-dibromobutan (2S,3R)-2,3-dibromobutan
W rzeczywistości jest to jeden i ten sam związek mezo-dibromobutan.
Płaszczyzna symetrii dzieli stereoizomer na pół jest, jak zwierciadło, w którym odbijają się obie połowy związku.
3.5.7 Enancjomery
Reguły dotyczące chiralnych związków.
Chiralność cząsteczki jest warunkiem koniecznym i wystarczającym do zaistnienia enancjomerów;
Obecność chiralnego atomu węgla nie jest warunkiem koniecznym ani wystarczającym do wystąpienia enancjomerów;
Liczba enencjomerów zależy od liczby centrów asymetrii w cząsteczce. Maksymalna liczba stereoizomerów LS = 2n, gdzie n oznacza liczbę chiralnych atomów.
Enancjomery nie różnią się właściwości fizycznymi i chemicznymi w środowisku achiralnym, mają np. identyczną gęstość, rozpuszczalność, temperatury topnienia czy wrzenia, wykazują też taką sama reaktywność. Różnią się natomiast w środowisku chiralnym, np. z różną szybkością reagują z odczynnikami chiralnymi, w tym z enzymami, mogą również różnić się rozpuszczalnością w chiralnych rozpuszczalnikach. Inaczej oddziałują z chiralnym podłożem, dlatego można je rozdzielić za pomocą chromatografii na złożach chiralnych. Udaje się je też rozdzielić stosując chiralną fazę ruchomą lub fazę zawierająca chiralne dodatki. Enancjomery inaczej reagują ze światłem spolaryzowanym - skręcają jego płaszczyznę polaryzacji o ten sam kąt, ale w przeciwnych kierunkach. Związki, które skręcają płaszczyznę światła spolaryzowanego nazywane są optycznie czynnymi. Na zjawisku optycznej czynności oparte polarymetria, czyli technika mierzenia kąta skręcania płaszczyzny światła spolaryzowanego. Można tym sposobem zidentyfikować dany stereoizomer lub oznaczyć jego stężenie.
Równowagowa mieszanina enancjomerów nazywana jest mieszaniną racemiczną. Może się ona różnić właściwościami fizycznymi od enencjomerów, a na pewno jest optycznie nieczynna. W pewnych warunkach enancjomery mogą racemizować częściowo lub całkowicie w następstwie zmiany konfiguracji po wpływem różnych czynników.
3.5.8 Stereochemia związków cyklicznych
Stereoizomerię związków cyklicznych warto rozpatrywać na przykładzie cyklopentanu, którego pierścień uważa się za płaski. Wystąpi ona wówczas, kiedy w cyklopentanie znajdują się co najmniej dwa podstawniki. 1,2-Dimetylocypentan występuje w formie dwóch enancjomerów (R,R) i (S,S), które są zarazem izomerami trans, oraz w formie mezo (cis):
Atomy węgla C1 i C2 są chiralne. Dla streoizomeru a konfigurację można oznaczyć w następujący sposób:
(R,R)-1,2-dimetylocyklopentan
Izomer b ma konfigurację (S,S), a c jest związkiem mezo (R,S) i (S,R), ponieważ ma płaszczyznę symetrii.
Podobne zależności występują dla 1,3-dipodstawionych pochodnych cyklopentanu. Identycznie postępuje się przy określaniu konfiguracji stereoizomerów pochodnych cyklopropanu i cyklobutanu. Cząsteczka cykloheksanu nie jest co prawda płaska, ale dla oznaczania kofiguracji występujących w niej poszczególnych chiralnych atomów węgla można rysować ją w postaci płaskiej.
3.6 Właściwości fizykochemiczne halogenoalkanów
Fluoro-, chloro- i bromometan są gazami, natomiast jodometan, dichlorometan są niskowrzącymi cieczami (42 i 40oC, odpowiednio). Chloroform jest niskowrzącą cieczą (61oC), bromoform wrze w temp. około 150 oC, a jodoform jest ciałem stałym w temp. pokojowej; jego t.t. wynosi ~ 120oC. Temperatury wrzenia wyższych halogenolkanów rosną wraz ze wzrostem ich masy molowej.
Halogenoalkany należą do substancji hydrofobowych, trudno rozpuszczalnych w wodzie, a bardzo dobrze w rozpuszczalnikach organicznych. Ich gęstość zależy od udziału masy halogenu w masie molowej związku. Fluorometan, chlorometan, fluoroetan, chloroetan, difluoroetan i chloropropany są lżejsze od wody, zaś takie pochodne jak bromoetan i polihalogenowodory mają gęstość większą niż woda (dichlorometan - 1,34, chloroform - 1,50, tetrachlorometan - 1,60, zaś jodoform - 4,01 g/mL); wraz ze wzrostem udziału części organicznej maleje gęstość halogenowodorów. Temperatury wrzenia halogenowodorów są zwykle wyższe od temperatury wrzenia alkanów o podobnej masie molowej, ponieważ te pierwsze są bardziej polarne, a przez to siły wzajemnego przyciągania się ich cząsteczek są większe.
Chlororo-, bromo- i jodoalkany są toksyczne i wywierają silne, inne oddziaływania fizjologiczne. Chloroform przez długi czas był stosowany do narkozy, jednak po stwierdzeniu przypadkowych śmiertelnych zejść usypianych pacjentów został wycofany z klinik. Mniej toksyczny jest szeroko używany inny wziewny anestetyk - halotan (2-bromo-2-chloro-1,1,1-trifluoroetan). Tetrachlorek węgla uszkadza wątrobę, nerki i centralny układ nerwowy. Również toksyczne są popularne rozpuszczalniki takie, jak dichlorometan i 1,1,1-trichloroetan; znacznie bardziej szkodliwy jest 1,2-dibromoetan. Duże zastosowanie medyczne ma gazowy chlorek etylu. Po lekkim sprężeniu jest przechowywany w ampułkach w postaci cieczy. W wyniku spryskania taką cieczą niewielkiej powierzchni ciała ulega rozprężeniu i szybko parując ochładza tę część ciała, wywołując znieczulenie w tym miejscu. Wdychany w niewielkich ilościach nie powoduje groźnych następstw.
3.7 Właściwości chemiczne - reaktywność halogenoalkanów
Reaktywność halogenoalkanów wynika z rozkładu ładunków elektrycznych w ich cząsteczkach, czyli efektu polaryzacji i polaryzowalności wiązań.
Cząsteczka, w której są tak rozłożone ładunki jest podatna na atak różnych odczynników. Kwasy Lewisa A (AlCl3, FeBr3) będą atakować miejsce obdarzone ładunkiem ujemnym, czyli halogen (X), nukleofil Nu: ma powinowactwo do dodatnio naładowanego atomu węgla, zaś zasada B: będzie odrywać proton z C2.
Metal (M) odda elektron dodatnio naładowanemu atomowi C, tworząc z nim związki metaloorganiczne -C-M lub -C-M-X, o budowie zależnej od wartościowości metalu. Z metalami grupy I (Li, Na, K) halogen zostanie odłączony, a z metalami grupy II (Mg, Zn, Cd) metal zajmie położenie pomiędzy atomami C i X.
3.7.1 Substytucja nukleofilowa SN
Reakcje substytucji należą obok reakcji eliminacji i addycji do trzech głównych typów reakcji, jakim ulegają związki organiczne. Reakcje substytucji mogą być reakcjami substytucji nukleofilowej lub substytucja elektrofilowej. Substytucja nukleofilowa polega na reakcji nukleofilu (Nu) ze związkiem organicznym zawierającym łatwo odchodzącą elektroujemną grupę (np. halogen) w wyniku, której następuje wymiana grupy odchodzącej na nukleofil. Nukleofilami nazywane są donory elektronów - atomy lub grupy atomów obdarzone ładunkiem ujemnym lub dysponujące wolną parą elektronów; wykazują one powinowactwo do dodatnio naładowanego atomu C. W tabeli 3.2 podane są najczęściej spotykane nukleofile i ich produkty reakcji z bromkiem metylu.
Reakcje bromku metylu z wybranymi nukleofilami Tabela 3.2.
CH3-Br + Nu:- _→ CH3-Nu + Br-
Nukleofil |
Produkt |
||
Wzór |
Nazwa |
Wzór |
Nazwa |
H:- |
anion wodorkowy |
CH4 |
metan |
CH3S- |
anion metylosulfidowy |
CH3SCH3 |
sulfid dimetylowy |
HS- |
anion hydrosulfidowy |
HSCH3 |
metanotiol |
CN- |
anion cyjankowy |
N≡CCH3 |
acetonitryl |
I- |
anion jodkowy |
ICH3 |
jodek metylu |
HO- |
anion wodorotlenkowy |
HOCH3 |
metanol |
H3CO- |
anion metanolanowy |
CH3OCH3 |
eter dimetylowy |
N=N=N- |
anion azydkowy |
N=N=NCH3 |
azydometan |
Cl- |
anion chlorkowy |
ClCH3 |
chlorek metylu |
CH3COO- |
anion octanowy |
CH3COOCH3 |
octan metylu |
H3N: |
amoniak |
H3N+CH3 Br- |
bromek metyloamoniowy |
(CH3)3N: |
trimetyloamina |
(CH3)4N+Br- |
bromek tetrametyloamoniowy |
3.7.1.1 Kinetyka reakcji substytucji nukleofilowej (SN)
Szybkość reakcji SN zależy zarówno od warunków reakcji, jak i od właściwości nukleofila oraz budowy reagenta organicznego. Poniżej podany jest szereg reaktywności najpopularniejszych odczynników nukleofilowych w reakcji z bromkiem metylu:
CH3-Br + Nu:- _→ CH3-Nu + Br-
względna szybkość 1 700 103 1,6.104 2,5.104 105 1,25.105
Nu: HOH NH3 Cl- HO- CH3O- I - NC- HS-
_________________→
mniej reaktywne bardziej reaktywne
Zwykle odczynniki nukleofilowe, jako grupy obdarzone ładunkiem ujemnym są zasadowe, czyli wykazują powinowactwo do protonu. Nukleofilowość nie zawsze pokrywa się z zasadowością, np. jon HO- jest silną zasadą, ale średniej mocy nukleofilem, natomiast HS- jest bardzo silnym nukleofilem, chociaż znacznie słabszą zasadą.
Na szybkość reakcji SN duży wpływ mają właściwości grupy odchodzącej. Najbardziej reaktywną grupą odchodzącą jest anion tosylanowy (p-toluenosulfonowy), a bardzo trudno podstawić anion fluorkowy.
Względna reaktywność grup odchodzących w reakcjach SN:
zasadowość (pKa zasady
skoniugowanej) 35 16 15,7 4,8 3,2 -7 -9 -9,5 -6,5
względna szybkość ∼0 1 2.102 104 3.104 6.104
grupa odchodząca NH2-, RO-, HO- AcO- F- Cl- Br- I - TosO-
________________________→
mniej reaktywne bardziej reaktywne
Aniony silnych kwasów (słabe zasady) są dobrymi grupami odchodzącym, aniony słabszych kwasów (silne zasady) są gorszymi grupami odchodzącymi.
Kinetyka hydrolizy halogenków alkilowych w środowisku zasadowym zależy od rzędowości halogenku. Halogenki metylu i 1o ulegają hydrolizie i reakcji z innymi nukleofilami (substytucji nukleofilowej) zwykle wg kinetyki drugiego rzędu, podczas gdy halogenki 3o wchodzą w reakcję z tymi samymi odczynnikami zgodnie z kinetyką pierwszego rzędu.
CH3-Br + OH- _→ CH3-OH + Br-
Szybkość powyższej reakcji jest zależna od stężenia obu reagentów - V = k[CH3Br].[ OH- ]. Reakcje substytucji nukleofilowej, których szybkość jest zależna od stężenia obu reagentów (wymaga zderzeń dwóch cząsteczek) nazywana jest substytucją nukleofilową dwucząsteczkową i oznacza się ją symbolem SN2.
W reakcji substytucji nukleofilowej halogenków alkilowych 3o, np. ich hydrolizy szybkość reakcji jest zależna jedynie od stężenia halogenku alkilu i dla poniższej reakcji wyraża się wzorem: V = k[(CH3)3C-Br].
(CH3)3C-Br + OH- _→ (CH3) 3C-OH + Br-
Reakcje substytucji nukleofilowej, których szybkość jest zależna od stężenia jednego z reagentów (nie wymaga zderzeń dwóch cząsteczek) nazywana jest substytucją nukleofilową jednocząsteczkową i oznacza się ją symbolem SN1.
3.7.1.2 Mechanizm SN2
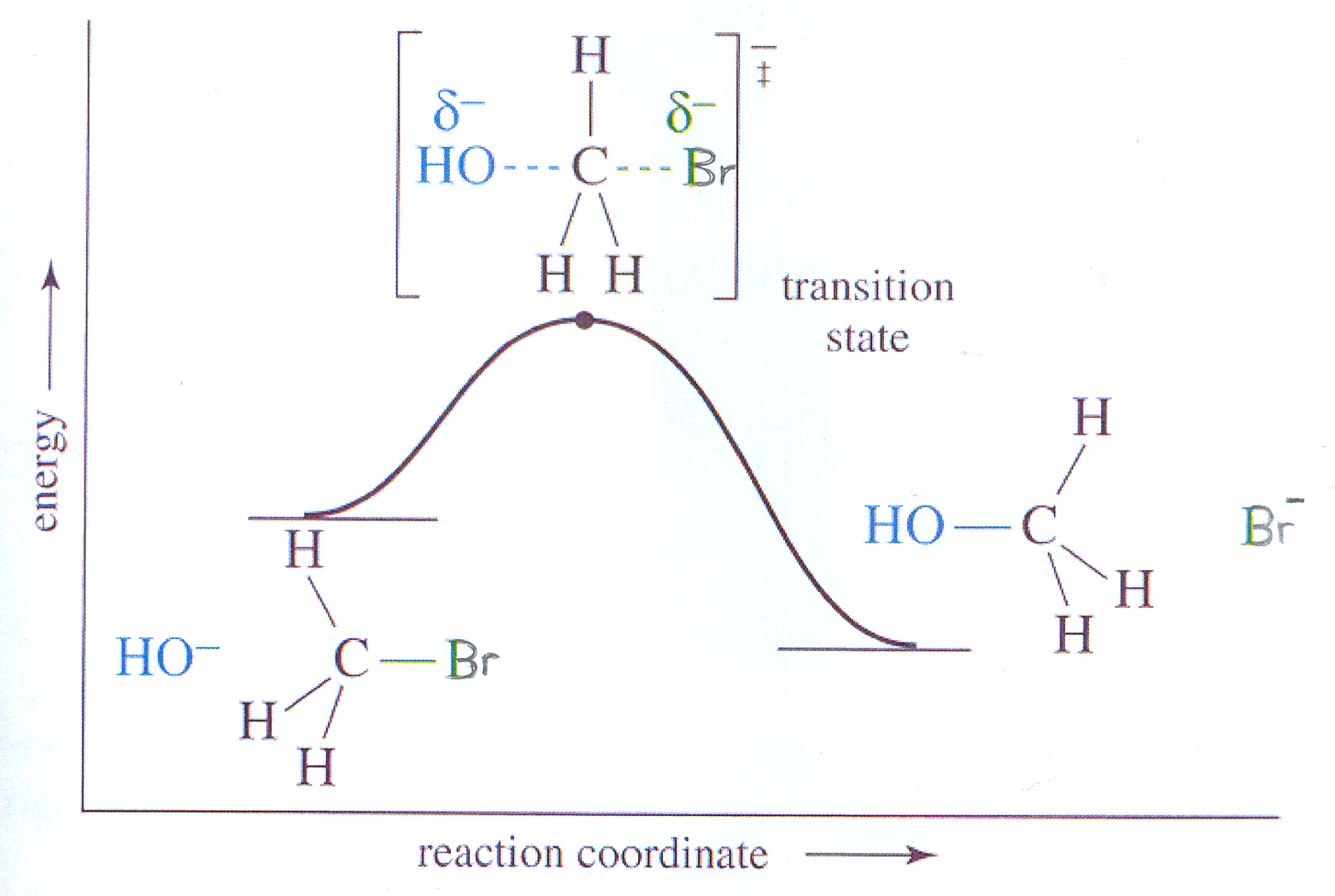
Reakcje substytucji nukleofilowej SN2 zaczynają się od ataku nukleofila na atom węgla obdarzony ładunkiem dodatnim. Zbliżenie się nukleofila do takiego atomu węgla jest możliwe tylko z przeciwnej strony elektroujemnego atomu (grupy) związanego z tym atomem węgla - z tej samej strony nie może ze względów zarówno objętościowych jak i elektrostatycznych.
Ponadto nie każde zbliżanie się obu reagentów kończy się substytucją. Tylko w wyniku zderzenia się nukleofila z cząsteczką halogenku z odpowiednią energią i od właściwej strony umożliwia zapoczątkowanie reakcji poprzez utworzenie więzi pomiędzy nukleofilem a atomem węgla obdarzonym ładunkiem δ+, równoczesnie następuje przy tym rozluźnienie wiązania C-Br; na rysunku oba te wiązanie - tworzone i rozrywane - zobrazowane są przerywaną, niebieską linią. Tworzy się stan przejściowy, w którym atom węgla staje się pięciowiązalny, a podstawniki przy nim (R, R' i R'') zostają zagęszczone w jednej płaszczyźnie.
Stan przejściowy (nie produkt pośredni) jest bardzo nietrwały, wobec czego szybko przekształca się w trwały produkt poprzez odszczepienie anionu bromkowego (jeżeli Br- jest łatwiej odchodząca grupą niż nukleofil) lub wraca do stanu poprzedniego (porównaj względną reaktywność grup odchodzących w reakcjach SN).
Na wykresie zmiany energii w trakcie reakcji widoczne jest maksimum dla stanu pośredniego, a siłą napędową reakcji jest zysk energetyczny - energia wydzielana przy przejściu od substratów do produktów. Reakcja jest jednoetapowa - nie tworzy się produkt pośredni.
Wykres zmiany energii substratu (CH3Br) i produktu w trakcie hydrolizy bromku metylu Wykres. 3.3
(na podstawie L.G. Wade Jr, Organic Chemistry, 6th ed., Pearson Prentice Hall, Upper Saddle River, NJ, 2006
3.7.1.3 Reaktywność halogenków alkilowych
Podatność halogenków alkilowych na substytucję nukleofilową zależy od ich konstytucji, głównie od rzędowości podstawionego atomu C.
Względna szybkość reakcji halogenków alkilowych w reakcji SN2
względna szybkość reakcji SN2 dla R-X ∼0 10-5 0,02 1 30
alkil (R) (CH3)3C- (CH3)3CCH2- (CH3)CH- Et- Me-
rzędowość 3o neopentyl (1o) 2o 1o 0o
__________________→
mniej reaktywne bardziej reaktywne
Do powyższego szeregu nie pasuje halogenek neopentylu, który w reakcjach SN2 jest nieaktywny (prawie zerowa szybkość reakcji), pomimo tego, że halogen związany jest 1o atomem węgla. Otóż duża objętość grupy tert-butylowej związanej z atomem węgla stanowiącym centrum reakcji uniemożliwia atak odczynnika nukleofilowego od strony grupy tert-butylowej, a innej możliwości nie ma.
Rys. 3.3. Możliwość dostępu nukleofila do centrum reakcji zmniejsza się wraz ze wzrostem zatłoczenia wokół atomu węgla związanego z grupą ze odchodzącą, wg J. McMurry, Chemia Organiczna, PWN, Warszawa 2000
3.7.1.4 Inwersja konfiguracji
W trakcie reakcji biegnących mechanizmem SN2 dochodzi do zmiany konfiguracji; zjawisko to zostało nazwane inwersją konfiguracji.
cis-(1R,3S)-1-chloro-3-metylocyklopentan trans-(1S,3S)-1-hydroksy-3-metylocyklopentan
Strzałki na rysunku powyżej i na każdym innym objaśniającym mechanizm reakcji wskazują ruch elektronów: od nukleofila do elektrofila; wobec tego mówimy o ataku nukleofilowym, a nie odwrotnie.
Przyczyną inwersji konfiguracji jest atak odczynnika nukleofilowego (I etap reakcji) z przeciwnej strony cząsteczki w stosunku do grupy odchodzącej, w wyniku czego tworzy się stan przejściowy - nietrwały addukt, zawierający pięciokoordynacyjny atom węgla.
Stan przejściowy reakcji SN2
Następnie, w II etapie zostaje zerwane wiązanie z grupą odchodzącą, a nowy podstawnik zajmuje pozycję po przeciwnej stronie cząsteczki niż grupa, która została podstawiona.
W stanie przejściowym trzy podstawniki na atomie węgla w centrum reakcji zostają stłoczone w jednej płaszczyźnie, zatem nie mogą być objętościowo duże. Najłatwiej stłoczyć atomy H lub podstawniki przy 1o atomie węgla, dlatego tylko pochodne metanu i zawierające 1o grupy alkilowe ulegają łatwo tej reakcji. Reakcja SN2 dla pochodnych 2o zachodzi wolno, a 3o praktycznie nie reagują wg SN2.
Inwersję konfiguracji, towarzyszącą reakcji SN2 można obserwować mierząc skręcalność właściwą substratów i produktów. W trakcie hydrolizy (R)-(-)-2-bromooktanu powstaje 2-oktanol. Reakcja biegnie wg mechanizmu SN2, co wynika z kinetyki: v = k[OktBr].[HO-]. Enancjomery 2-oktanolu mają tę samą tt. i tw., to samo widmo NMR i IR w środowisku achiralnym, różnią się natomiast znakiem skręcalności właściwej []. Wartość [] wynosi -9,9o i 9,9o, odpowiednio dla enancjomerów (R) i (S).
Tworzenie się z (R)-(-)-2-bromooktanu enancjomeru (S)-(+)-2-oktanolu jest dowodem istnienia zjawiska inwersji konfiguracji.
3.7.1.5 Wpływ rozpuszczalnika na reakcję SN2
Środowisko, najczęściej rozpuszczalnik, ma ogromny wpływ na większość reakcji, również na reakcję SN2. Reakcję utrudniają rozpuszczalniki protyczne, czyli takie, które posiadają ruchliwe protony, np. woda, alkohole czy aminy. Solwatują (osłaniają) one cząsteczki nukleofila, utrudniając mu tym samym zbliżenie się miejsca reaktywnego w cząsteczce R-X.
Nukleofil solwatowany przez cząsteczki protycznego rozpuszczalnika jest nie tylko mniej aktywny, ale i większy, przez co trudniej mu podejść do drugiego reagenta. Z tego samego powodu nukleofile o większych rozmiarach (w parach HS- i HO- lub Br- i F-) są aktywniejsze w reakcjach SN2, ponieważ im mniejszy promień jonu tym silniej jest on solwatowany przez cząsteczki protycznego rozpuszczalnika.
Natomiast polarne rozpuszczalniki aprotyczne ułatwiają reakcje SN2 zwiększając jego aktywność, ponieważ solwatują one kation, a nie anion (nukleofil), przez co taki „nagi” anion jest bardziej ruchliwy (aktywny), ma mniejsze rozmiary, czyli łatwiej mu dostać się do miejsca reaktywnego cząsteczki organicznej.
Rys. 3.4. Cząsteczki aprotycznego rozpuszczalnika polarnego
solwatują kation przez co „nagi” nukleofil (towarzyszący anion)
jest bardziej reaktywny
Do rozpuszczalników aprotycznych należą: acetonitryl (CH3CN), dimetyloformamid [(CH3)2NCHO, DMF], sulfotlenek dimetylu [(CH3)2SO, DMSO] czy heksametylofosforotriamid {[(CH3)2N]3PO, HMPA}. Ten ostatni okazał się być kancerogenny i teratogenny, więc należy go stosować z odpowiednią ostrożnością. Wysoka polarność rozpuszczalników jest potrzebna do rozpuszczenia polarnych reagentów.
Z poniższego diagramu widać jak zwiększa się szybkość reakcji SN2 w aprotycznych rozpuszczalnikach polarnych na przykładzie reakcji.
rozpuszczalnik
n-Bu−Br + N3- ____→ n-BuN3 + Br-
rozpuszczalnik: CH3OH HOH DMSO DMF CH3CN HMPA
względna reaktywność 1 7 1300 2800 5000 2.105
Wartości stałej dielektrycznej popularnych rozpuszczalników Tabela 3.3.
Rozpuszczalnik |
Wzór |
Stała dielektryczna |
woda |
HOH |
80 |
kwas mrówkowy |
HCOOH |
59 |
dimetylosulfotlenek (DMSO) |
CH3SOCH3 |
49 |
dimetyloformamid (DMF) |
HCON(CH3)2 |
37 |
acetonitryl |
CH3CN |
36 |
metanol |
CH3OH |
33 |
heksametylofosforotriamid (HMPT) |
[(CH3)2N]3PO |
30 |
etanol |
CH3CH2OH |
24 |
aceton |
CH3COCH3 |
21 |
kwas octowy |
CH3COOH |
6 |
3.7.1.5 Podsumowanie zależności w reakcjach SN2
Reakcję SN2 ułatwia:
brak zawady przestrzennej reagenta organicznego;
duża reaktywność odczynnika nukleofilowego;
podatność grupy opuszczającej na odłączenie się od atomu C;
wysoka polarność rozpuszczalników aprotycznych
3.7.2 R e a k c j e SN1
Szybkość reakcji typu SN1 nie zależy od stężenia odczynnika nukleofilowego, reakcja biegnie zatem zgodnie z kinetyką pierwszego rzędu:
V = k.[RX]
Przykładem takiej reakcji jest hydroliza bromku t-butylu w wodzie:
(CH3)3C-Br + HOH _→ (CH3)3C-OH + HBr
Różnice w kinetyce reakcji hydrolizy bromku n-butylu i t-butylu wynikają z odmiennego mechanizmu obu reakcji. Jak wiadomo halogenki t-butylowe z uwagi na dużą objętość 3o grupy alkilowej są odporne na atak nukleofilowy (reakcję typu SN2), natomiast znacznie łatwiej niż halogenki 1o ulegają dysocjacji z utworzeniem karbokationu. W momencie pojawienia się karbokationu, ulega on w reakcji z cząsteczką wody szybko przekształceniu w alkohol.
- Br- HOH
(CH3)3C-Br _→ (CH3)3C+ _→ (CH3)3C-OH + H+
wolno szybko
Karbokationy 3o są trwalsze niż 2o, a te z kolei przewyższają trwałością kationy 1o. Z tego powodu karbokationy 3o tworzą się najłatwiej. Halogenki 1o nie ulegają reakcjom SN1, ponieważ trudno dysocjują na karbokation i jon halogenkowy, a pochodne 2o reagują mechanizmem mieszanym, częściowo SN1 i częściowo SN2.
Warto zdawać sobie sprawę z tego, że dysocjacja wiązania C−X, nawet dla halogenków 3o jest procesem wolnym, ponieważ oderwanie jonu halogenkowego wymaga dużej energii aktywacji (Rys. 3.6), zatem w reakcjach SN1 stanowi najwolniejszy etap reakcji - on decyduje o szybkości całej reakcji! Ten fakt nie oznacza, że reakcje SN1 są wolne, mogą być szybsze niż SN2.
W reakcjach SN1 szybkość powstawanie kabokationu zależy w danych warunkach tylko od stężenia halogenku alkilu, tym samym od jego stężenia zależy szybkość całej reakcji.
Wykres zmiany energii w reakcji SN1. Energia aktywacji I etapu reakcji, czyli tworzenia Wykres 3.4.
karbokationu jest znacznie większa niż energia aktywacji reakcji pomiędzy karbokationem i wodą
HOH
t-BuCl __→ t-BuOH
Szybkość reakcji wieloetapowej jest zależna od najwolniejszego etapu reakcji. Ten najwolniejszy etap reakcji decyduje o szybkości całej reakcji, podobnie jak najwęższa zwężka w wielokomorowej klepsydrze decyduje o szybkości przesypującego się piasku :
Rys. 3.5 Schemat podwójnej klepsydry obrazujący fakt, że o szybkości przesypywania piasku decyduje najwęższa zwężka
Mechanizm reakcji hydrolizy bromku t-butylu jest następujący:
3.7.2.1 Stereochemia reakcji SN1
Produktem pośrednim w procesie hydrolizy 3o halogenku alkilowego jest płaski karbokation - o hybrydyzacji atomu C sp2. Cząsteczka wody może przyłączyć się do niego z jednej lub z drugiej strony. Wobec czego powstaje mieszanina enancjomerów (R) i (S), często w stosunku 1:1, niezależnie od tego, jaką konfigurację miał substrat. Zjawisko towarzyszące reakcji, w której z jednego enancjomeru tworzy się mieszanina enancjomerów (R) i (S) nazywa się racemizacją, a mieszanina 50% enancjomeru (R) i 50% (S) nazywa się racematem.
Racemat jest optycznie nieczynny; zwykle różni się temperaturą topnienia i innymi właściwościami fizycznymi od enancjomerów.
Rys 3.6. Powstawanie przejściowo płaskiego karbokationu w reakcji SN1 prowadzi do racemizacji
Tylko te halogenki alkilowe, które są zdolne utworzyć płaski karbokation mogą reagować wg mechanizmu SN1.
Są znane halogenki, które nie ulegają ani reakcje SN1 ani SN2. Należą do nich 3o pochodne cykliczne, np. 1-bromobicyklo[2,2,2]oktan - jest nieaktywny w reakcjach SN2 jako halogenek 3o, a ponieważ nie może utworzyć płaskiego karbokationu nie reaguje też wg mechanizmu SN1.
Zdarza się jednak, że w reakcjach SN1 racemizacja nie jest całkowita, dochodzi tylko do częściowej inwersji konfiguracji. Jest ona spowodowana tym, że atak odczynnika nukleofilowego następuje zanim dochodzi całkowitego oddysocjowania anionu. Nukleofil w takim przypadku atakuje tworzący się karbokation z przeciwnej strony do odchodzącego anionu, podobnie jak w trakcie reakcji SN2. Opuszczający anion utrudnia nukleofilowi zbliżenie się do karbokationu z tej samej strony. W reakcji hydrolizy (R)-6-chloro-2,6-dimetylooktanu obserwuje się 20% inwersję konfiguracji.
Rys. 3.7. Hydroliza (R)-6-chloro-2,6-dimetylooktanu prowadzi do produktu częściowo zracemizowanego. Etanol dodaje się dla ułatwia rozpuszczenia substratu
3.7.2.2 Wpływ rozpuszczalnika na reakcję SN1
Rozpuszczalnik ma duży wpływ na szybkość reakcji SN1. Może on ułatwiać dysocjację halogenku alkilowego i stabilizować karbokation. Wpływa więc na pierwszy etap reakcji, tj. stan przejściowy. Rozpuszczalniki polarne, przede wszystkim protyczne solwatując jony sprzyjają reakcjom SN1. Do tego typu rozpuszczalników należą alkohole, kwas mrówkowy i woda. Najkorzystniejszym rozpuszczalnikiem dla SN1 jest woda, jednak wiele substratów organicznych nie rozpuszcza się w niej.
Dodatek rozpuszczalników organicznych ułatwia rozpuszczanie reagentów organicznych, ale zwykle obniża szybkość reakcji.
Rys. 3.8. Cząsteczki polarnego rozpuszczalnika stabilizują karbokation poprzez jego solwatację
3.7.2.3 Wpływ grupy odchodzącej
Podobnie jak w reakcjach SN2 reaktywność substratu zależy od właściwości grupy odchodzącej. Podobny jest też szereg reaktywności, najbardziej aktywne są tosylany:
HOH ≈ Cl-< Br- < I- < < TosO-
3.7.2.4 Wpływ konstytucji substratu
Struktury reszty organicznej stabilizujące karbokation ułatwiają reakcje SN1. Wzrost rzędowości karbokationu wpływa na jego trwałość, dlatego trzeciorzędowe pochodne najłatwiej ulegają reakcjom SN1, 2o trudniej, a 1o są w tego typu reakcjach nieaktywne. Duży wpływ na stabilizację karbokationu ma mezomeria, szczególnie obecność podwójnego wiązania w położeniu , tzw. układ allilowy. Ułatwienie reakcji SN1 obserwuje się nie tylko dla halogenków allilowych (1o czy 2o), ale również dla benzylowych.
stabilizacja kationu allilowego
stabilizacja kationu benzylowego
Wiązania C-X w halogenkach allilowych i benzylowych są osłabione w porównaniu do wiązania, np. w halogenku etylu:
energia wiązania [kJ/mol] 338 289 293
CX [kcal/mol] 81 69 70
Obie grupy związków są również bardzo reaktywne w reakcjach biegnących mechanizmem SN2, tak więc szybkość reakcji substytucji nukleofilowej halogenków allilowych czy benzylowych jest sumą szybkości obu reakcji - SN1 i SN2. Jest to przyczyną wysokiej reaktywności tych związków. Reakcja substytucji bromku allilu biegnie około 40 razy szybciej niż bromku n-propylu z tymi samymi nukleofilami.
3.7.2.5 Wpływ nukleofilu
Nukleofil ma niewielki wpływ na szybkość reakcji SN1, ponieważ nie bierze udziału w najwolniejszym etapie reakcji. Po utworzeniu karbokationu jego reakcja z nukleofilem biegnie szybko i nie wpływa na sumaryczną szybkość reakcji. Halogenowodory (HCl, HBr lub HI) reagują z taką samą szybkością z t-butanolem:
t-butanol halogenek t-butylu (X: Cl, Br lub I)
3.8 Porównanie reakcji biegnących mechanizmem SN1 i SN2
Substratami reakcji SN1 są związki organiczne tworzące stabilne karbokationy, np. halogenki 3o, allilowe czy benzylowe;
Reakcję SN1 ułatwiają polarne rozpuszczalniki protyczne (solwatują aniony i kationy);
Odczynniki nukleofilowe nie wpływają na szybkość reakcji SN1;
Solwoliza halogenków alkilowych jest przykładem reakcji biegnącej wg kinetyki SN1. Nawet jeżeli substratem nie jest halogenek 3o to stężenie rozpuszczalnika użytego w dużym nadmiarze jest w trakcie reakcji stałe i jego wpływ na szybkość reakcji zawiera się w stałej szybkości:
v = k[RX]
Szybkość reakcji maleje wraz ze wzrostem rzędowości: CH3-X < RCH2-X << R2CH-X
Substraty reakcji SN2 nie mogą mieć zawad sterycznych;
Reakcję SN2 ułatwiają polarne rozpuszczalniki aprotyczne, a podwyższenie stężenia odczynnika nukleofilowego zwiększa szybkość reakcji;
Efekt grupy odchodzącej jest taki sam w obu reakcjach: R-OTos > R-I > R-Br > R-Cl >> R-F
1
Wyszukiwarka
Podobne podstrony:
3 Halogenoalkany Substytucja nukleofilowa
Zadania z reakcji substytucji nukleofilowej i eliminacji w halogenkach alkilowych
Halogenki alkilowe – substytucja nukleofilowa – grupa opuszczająca – kwasy i zasady
Substytucje Nukleofilowe w Pochodnych Karbonylowych
Substytucja nukleofilowa SN (2)
substytucja nukleofilowa, ochrona środowiska UJ, IV semestr, chemia ograniczna, sprawozdania
Substytucja Nukleofilowa SN (1)
Substytucje Nukleofilowe w Pochodnych Karbonylowych
09 Halogenki alkilowe substytucja eliminacja 26 05 2011 zadania
chemia, Alkany, Alkany - substytucja [podstawianie] z halogenami [fluorowce]
09 Halogenki alkilowe substytucja eliminacja materialy dodatkowe
aldehydy i ketony addycja nukleofilowa
cykliczne nukleotydy
halogenki alkilowe
Koniec z kacem stworzono substytut alkoholu
Nukleotydy
Ściemniacz lampy halogenowej
Hydroksykwasy i halogenokwasy
SEM-15WF2011 - Metabolizm nukleotydow pur i pyr, Studia, I semestr II rok, Biochemia, Różne
więcej podobnych podstron